

Abstract
Phenolic glycolipids (PGL) play a major role in the virulence of mycobacteria, notably in strains of the Mycobacterium tuberculosis complex and in Mycobacterium leprae. The structure of the carbohydrate domain of these compounds is highly variable, and the genetic bases for these variations remain unknown. We demonstrated that the monoglycosylated PGL formed by Mycobacterium bovis differs from the triglycosylated PGL synthesized by M. tuberculosis (PGL-tb) because of the following two genetic defects: a frameshift mutation within the gene Rv2958c, encoding a glycosyltransferase involved in the transfer of the second rhamnosyl residue of the PGL-tb, and a deletion of a region that encompasses two genes, which encode a GDP-d-mannose 4,6-dehydratase and a GDP-4-keto-6-deoxy-d-mannose-3,5-epimerase/reductase, required for the formation of activated l-fucose. Expression of these three genes in M. bovis BCG allowed synthesis of PGL-tb in this recombinant strain. Additionally, we showed that all M. bovis, Mycobacterium microti, Mycobacterium pinnipedii, and some Mycobacterium africanum strains harbor the same frameshift mutation in their Rv2958c orthologs. Consistently, the structure of PGLs purified from M. africanum (harboring the Rv2958c mutation) and M. pinnipedii strains revealed that these compounds are monoglycosylated PGL. These findings explain the specificity of PGL-tb production by some strains of the M. tuberculosis complex and have important implications for our understanding of the evolution of this complex.
Phenolic glycolipids (PGL)2 are produced by certain mycobacterial species, most of which are pathogenic for humans (1). These substances are located in the outermost layers of the mycobacterial envelope where they play a key role in the pathogenicity of mycobacteria. For instance, PGL-1, the major PGL synthesized by Mycobacterium leprae, is important for the unique tropism of this bacterium to peripheral nerves through binding to laminin 2 (2). Additionally, PGL-1 synthesis in the leprosy bacillus affects the resistance to intracellular killing by macrophages (3) and modifies binding to complement receptors (4), which may be important for the pathogenesis of leprosy. PGL-1 can also modulate the immune response (5, 6). In Mycobacterium tuberculosis, recent findings reveal that the production of PGL is associated with hypervirulence in mice (7) and in a rabbit model of meningitis (8). This glycolipid seems to inhibit the release of pro-inflammatory mediators (7).
PGL consist of a lipid core formed by a long-chain β-diol, occurring naturally as diester of polymethyl-branched fatty acids (1) (Figs. 1 and 3). The lipid core is ω-terminated by an aromatic nucleus that is glycosylated. The sugar moiety of PGL consists of one to four sugar residues, depending on the species, and most are O-methylated deoxysugars (9, 10). Only a few M. tuberculosis strains synthesize PGL-tb (11). In such strains, the carbohydrate domain of the major form of PGL-tb is 2,3,4-tri-O-methyl-l-fucopyranosyl-(α1->3)-l-rhamnopyranosyl-(α1->3)-2-O-methyl-l-rhamnopyranosyl-(α1->) (12) (Fig. 1). This structure seems specific to the M. tuberculosis and Mycobacterium canettii lineages; other members of the M. tuberculosis complex, which gathers mycobacterial strains very closely related to M. tuberculosis, do not produce the same saccharidic domain (1). For instance, M. bovis and Mycobacterium microti produce a truncated form of PGL-tb, called mycoside B, in which the carbohydrate domain is restricted to 2-O-methylrhamnose (1, 13). In other members of M. tuberculosis complex, such as Mycobacterium africanum and Mycobacterium pinnipedii, production of PGL has not been reported to date.
FIGURE 1.

Identification of the genes required for the production of PGL-tb in M. bovis BCG. A, TLC analysis of lipid extracts from the bacterial cell of recombinant M. bovis BCG. Crude lipid extract was from M. bovis BCG (lane 1), M. bovis BCG::pPET52 (Rv2958c) (lane 2), M. bovis BCG::pPET52:pWM85 (Rv1511, Rv1512, and Rv2958c) (lane 5), M. tuberculosis H37Rv:pPET1 (pks15/1) (lane 6), M. canettii, a natural mycobacterial producer of PGL-tb (lane 7), mycoside B purified from M. bovis BCG (lane 3), PGL-tb purified from M. bovis BCG::pPET52:pWM85 (Rv1511, Rv1512, and Rv2958c) (lane 4). Lipid extracts were dissolved in CHCl3 and run in CHCl3/CH3OH (95:5, v/v). Glycolipids were visualized by spraying the plates with 0.2% anthrone (w/v) in concentrated H2SO4 followed by heating. PGL-2S corresponds to mono-O-methyl-diglycosyl-phenolphthiocerol dimycocerosate as described previously (15). The arrowheads indicate the positions of the purified PGLs. B, structure of the putative PGLs produced by the recombinant M. bovis BCG strains. R1 corresponds to the phthiocerol dimycocerosate core of PGL. Genes transferred in M. bovis BCG are indicated beside the arrows. Other genes such as Rv2957 (encoding the third glycosyltransferase) and those encoding the methyltransferases are required for the synthesis of the complete saccharide moiety of PGL-tb, but our findings demonstrated that they are all already functional in M. bovis BCG.
FIGURE 3.

1H NMR spectrum of the purified PGL from M. bovis BCG::pPET52:pWM85. The spectrum was recorded in CDCl3 at 600, 13 MHz. The structure of the analyzed compound is shown above the spectrum, and the protons corresponding to the main signals are indicated.
The biosynthesis of PGL-tb involves more than 25 enzymatic steps. Genes required for PGL biosynthesis and translocation to the mycobacterial cell surface are clustered on a 73-kb fragment of the M. tuberculosis chromosome (14). The genetic basis for the inability of most M. tuberculosis isolates to synthesize PGL-tb was found to be a mutation within the pks15/1 gene that encodes an enzyme involved in the formation of a precursor of PGL-tb (11). However, it is still unknown why other members of the M. tuberculosis complex, with the exception of M. canettii, produce only mycoside B.
In this study, we determine the molecular factors explaining why Mycobacterium bovis BCG does not synthesize PGL-tb. We also report the structures of PGL synthesized by additional strains of the M. tuberculosis complex and correlate the structure of these glycolipids with a mutation in a PGL biosynthetic gene.
EXPERIMENTAL PROCEDURES
Bacterial Strains and Culture Conditions—M. africanum strains (RIVM 2005-0479 and RIVM 2005-0419) were obtained from the collection of the National Institute for Public Health and the Environment (Bilthoven, Netherlands). M. pinnipedii and M. microti strains were provided by Dr. A. Cataldi (Instituto Nacional de Tecnologia Agropecuaria, Castelar, Argentina) and Prof. P. Wheeler (London, UK). All the strains used in this study have been extensively characterized using reference typing methods (IS6110 RLFP and spoligotyping), and most of them have been described previously (see Table 1). M. bovis BCG 1173P2 (the Pasteur strain) and the other strains from the M. tuberculosis complex were grown on Middlebrook 7H9 medium (Difco) supplemented with ADC (0.2% dextrose, 0.5% bovine serum albumin fraction V, 0.0003% beef catalase) and 0.05% Tween 80 where indicated or on solid Middlebrook 7H11 medium (Difco) supplemented with OADC (0.005% oleic acid, 0.2% dextrose, 0.5% bovine serum albumin fraction V, 0.005% NaCl, 0.0003% beef catalase). Kanamycin and hygromycin were added when required to final concentration of 40 and 50 μg/ml, respectively. Recombinant M. bovis BCG strains were grown as surface biofilm at 37 °C on Sauton's medium for biochemical analyses.
TABLE 1.
M. tuberculosis complex strains analyzed in this study
| Species | Isolate no. | Host | Country |
|---|---|---|---|
| M. canettii | 10059a | Human | France |
| 971549b | Human | Swiss | |
| 94217b | Human | ||
| M. tuberculosis | H37Rvc | Human | United States |
| H37Rac | Human | United States | |
| CDC1551c | Human | United States | |
| 210c | Human | United States | |
| F11c | Human | South Africa | |
| Tb36a | Human | India | |
| 103a | Human | China | |
| 5116d | Human | ||
| 10270d | Human | ||
| 3046d | Human | ||
| 10775d | Human | ||
| 8128d | Human | ||
| M. bovis (including M. bovis BCG) | AF2122/97c | Cattle | UK |
| 60a | Oryx | Saudi Arabia | |
| 61a | Oryx | Netherlands | |
| AN5 | |||
| ATCC19210 | Cattle | United States | |
| BCG 1173P2 | Vaccine | ||
| M. microti | OV254a | Vole | UK |
| 56a | Vole | UK | |
| 15496e | Vole | UK | |
| M. pinnipedii | 1886f | Seal | Argentina |
| 0217f | Seal | Argentina | |
| M. africanumg | 2005-0479f | Human | |
| 2005-0419f | Human | ||
| IS960097a | Human | Guinea | |
| IS960102a | Human | Guinea | |
| IS960090a | Human | Sierra Leone | |
| IS960092a | Human | Sierra Leone | |
| IS960093a | Human | Sierra Leone |
Strains were previously described in Brosch et al. (20)
Strains were previously described in Pfyffer et al. (29)
Sequenced strains are available through the NCBI web site (www.ncbi.nlm.nih.gov/)
Strains were previously described in Deshayes et al. (30)
Strains were previously described in Kremer et al. (28)
Strains are from this study
M. africanum isolates IS960090 and IS960092 belong to the subgroup containing the RD7, RD8, RD10, and RD11 region, whereas the others isolates are deleted for these regions (20)
Construction of the M. bovis BCG Recombinant Strains—The cloning of Rv2958c was performed previously (15). Briefly, the Rv2958c gene was amplified by PCR and cloned under the control of promoter phsp60 into the mycobacterial integrative vector pMV361H to give the plasmid pPET52 (15).
For the cloning of Rv1511 and Rv1512, a 2-kb fragment encompassing these two genes was amplified by PCR using primers 1511A (5′-TTATACATATGAAGCGAGCGCTCATCAC-3′) and 1512A (5′-AATATACTAGTCATTGCCGAACCGTTCCC-3′). The PCR was performed in a final volume of 50 μl containing 2.5 units of Pfu DNA polymerase (Promega, Lyon, France), 10% Me2SO, and 1 μm of each primer. The amplification program consisted of 1 cycle of 5 min at 95 °C followed by 30 cycles of 30 s at 95 °C, 30 s at 57 °C, and 2 min at 72 °C. A final extension of 10 min at 72 °C was then applied. The PCR product was analyzed by electrophoresis in a 0.8% agarose gel. The 2-kb fragment was purified using the QIAquick purification kit (Qiagen, Courtaboeuf, France). It was digested with NdeI and SpeI and inserted between the NdeI and SpeI sites of pMIP12d vector to give pWM85. Plasmid pMIP12d was derived from plasmid pMIP12 (16). First, the km cassette in a fragment flanked by DraI and MscI restriction sites was removed from pMIP12 by DraI and MscI digestion and replaced by the km cassette from plasmid pET26b (Novagen, Darmstadt, Germany). This resulted in removal of one HindIII restriction site. The resulting plasmid was then digested by HindIII, filled in, and then religated to eliminate the remaining HindIII restriction site. A polylinker allowing the use of the NdeI restriction site for in-frame insertion of gene was then inserted into the modified pMIP12 vector to give pMIP12d plasmid. In pWM85, genes Rv1511 and Rv1512 were placed under the control of the mycobacterial promoter pblaF*. The two plasmids were used for electrotransformation of M. bovis BCG. Transformants were selected on plates containing either kanamycin, hygromycin, or both.
PCR Amplification of the Rv1511/Rv1512 and Rv2958c Regions—Mycobacterial genomic DNA was extracted from each strain of the M. tuberculosis complex as described previously (17). For the analysis of the Rv1511/Rv1512 locus, PCR amplification was performed using primers 1511A and 1512A as described above. We determined the size of the amplified fragments on a 0.8% agarose gel. The Rv2958c ortholog was amplified from genomic DNA respectively using primers Rv2958A (5′-TATATCATATGGAGGAAACAAGCGTCG-3′) and Rv2958B (5′-TATATACTAGTTAGCAGACGAGCCGCAGC-3′). The PCR was performed in a final volume of 50 μl containing 2.5 units of Pfu DNA polymerase (Promega, Lyon, France), 10% Me2SO, and 1 μm of each primer. The amplification program consisted of one cycle of 5 min at 95 °C followed by 30 cycles of 30 s at 95 °C, 30 s at 57 °C, 2 min at 72 °C. A final extension of 10 min at 72 °C was then applied. The PCR products were analyzed by electrophoresis in a 0.8% agarose gel. The 1.3-kb fragments were purified using the QIAquick purification kit (Qiagen, Courtaboeuf, France) and inserted into pGEM-T vectors (Promega). Inserts were then sequenced using SP6 and T7 primers (Millegen, Labège, France). For sequencing the Rv2958c fragment containing the frameshift mutation, 400 bp was amplified using primers Rv2958D (5′-AGCTGGTGCCGACATACAACC-3′) and Rv2958E (5′-GCCTCCATGTTCAAGTGCTG-3′). The PCR was performed as described above, and the PCR products were sequenced directly using primer Rv2958S (5′-GCCAACCATCGATATCTCGG-3′) (Millegen).
Extraction and Purification of Glycolipids—Glycolipids were extracted and analyzed as described previously (11). Briefly, mycobacterial cells obtained from culture on Sauton's medium were left in 60 ml of CHCl3/CH3OH (1:2 v/v) for 24 h to kill bacteria. Lipids were then extracted once with CHCl3/CH3OH 1:1 (v/v) for 24 h and twice with CHCl3/CH3OH 2:1 (v/v) for 24 h each, washed twice with water (50 ml), and dried.
Production of glycolipids by the various strains was examined by thin layer chromatography (TLC). Briefly, extracts were dissolved in CHCl3 to give a final lipid concentration of 20 mg/ml. Equivalent volumes of each extract were deposited on Silica Gel G60 plates (0.3 mm, 20 × 20 cm, Merck) and run in CHCl3/CH3OH (95:5 v/v). Glycolipids were visualized by spraying the plates with 0.2% anthrone (w/v) in concentrated H2SO4, followed by heating. Glycolipids were purified as described previously (12). Crude lipid extracts from cells were subjected to chromatography on a Florisil (60–100 mesh) column and eluted with a series of concentrations of CH3OH (0, 1, 2, 3, 4, 5, 10, and 50%) in CHCl3. Each fraction was analyzed by TLC on Silica Gel G60 using CHCl3/CH3OH (95:5, v/v) as the solvent system. Glycolipids were visualized as described above. When necessary, glycolipids were additionally purified by preparative chromatography on Silica Gel G60 plates (0.3 mm, 20 × 20 cm, Merck) using CHCl3/CH3OH (95:5, v/v) as the developing solvent.
Structural Analysis—Purified molecules were analyzed by matrix-assisted laser desorption-ionization time-of-flight (MALDI-TOF) mass spectrometry as described previously (18). The spectra were acquired in reflectron mode with an Applied Biosystems 4700 analyzer mass spectrometer (Applied Biosystems, Framingham, MA) equipped with an Nd:YAG laser (wavelength 355 nm; pulse <500 ps; repetition rate 200 Hz). A total of 2500 shots were accumulated in positive ion mode, and mass spectrometry data were acquired using the instrument default calibration. NMR spectroscopy experiments were carried out at 295 K on a Bruker AVANCE spectrometer operating at 600,13-MHz with a 5-mm triple resonance TCI 1H 13C 15N pulsed field z-gradient cryoprobe. Samples were dissolved in 99.9% CDCl3. Chemical shifts were expressed in parts/million by using chloroform signal as an internal reference (7.23 ppm).
RESULTS
Identification of M. tuberculosis Genes Required for PGL-tb Synthesis in M. bovis BCG—The structure of the saccharidic moiety of PGL-tb was established as 2,3,4-tri-O-methylfucose-(α1->3)-rhamnose-(α1->3)-2-O-methylrhamnose (12). We have previously shown that M. bovis BCG does not synthesize PGL-tb because of several genetic defects. Indeed, M. bovis contains a frameshift mutation within its ortholog of Rv2958c, which encodes the glycosyltransferase involved in the transfer of the second rhamnosyl residue of PGL-tb in M. tuberculosis (15). However, transfer of a functional Rv2958c gene in M. bovis BCG led to the production of di-glycosylated PGL but not of PGL-tb (15). Thus additional genetic defects exist that are also responsible for the inability of M. bovis to synthesize PGL-tb.
We thus hypothesized that a possible genetic defect may prevent the formation of fucose, the last residue of the PGL-tb carbohydrate domain. l-Fucose is a 6-deoxyhexose synthesized in bacteria from GDP-d-mannose in a three-enzymatic step process (for review, see Ref. 19). GDP-d-mannose is first converted into GDP-4-keto-6-deoxy-d-mannose by a GDP-d-mannose 4,6-dehydratase. This intermediate undergoes epimerization at positions C-3 and C-5 to form GDP-4-keto-6-deoxy-l-galactose from which the keto group at the C-4 position is then reduced, giving GDP-l-fucose. These last two steps are catalyzed by a bifunctional enzyme, a GDP-4-keto-6-deoxy-d-mannose-3,5-epimerase/reductase. To test the hypothesis, we compared the sequenced genomes of M. tuberculosis and M. bovis. These are known to differ in several regions deleted in the M. bovis-related strains (20). One of these deleted regions (RD4) contains two genes Rv1511 (gmdA) and Rv1512 (epiA) encoding proteins exhibiting very high similarities to GDP-d-mannose 4,6-dehydratase and GDP-4-keto-6-deoxy-d-mannose-3,5-epimerase/reductase respectively (21). This observation suggested that the RD4 might be a second genetic defect preventing the formation of PGL-tb in some M. bovis-related strains, including the various M. bovis BCG isolates.
To address this question, we cloned the three M. tuberculosis genes, Rv2958c, Rv1511, and Rv1512, and inserted them into two plasmids. The plasmids were then either independently or simultaneously transferred into M. bovis BCG. The PGL produced by the various recombinant BCG strains were then analyzed on TLC plates (Fig. 1).
The expression of a functional Rv2958c gene in M. bovis BCG led to the synthesis of two glycoconjugates, a minor one corresponding to 2-O-methyl-rhamnosyl-phenolphthiocerol dimycocerosate (mycoside B) and a major one exhibiting a lower mobility and corresponding to a mono-O-methyl-diglycosyl-phenolphthiocerol dimycocerosate or PGL-2S (Fig. 1, lane 2) in agreement with our previous observations (15). When genes Rv1511 and Rv1512 were concomitantly expressed with Rv2958c, the recombinant M. bovis BCG strain now synthesized a new glycoconjugate exhibiting the same mobility on TLC plates as that exhibited by PGL-tb (Fig. 1, lane 5). In contrast, the recombinant BCG strain carrying only plasmid pWM85 (containing Rv1511 and Rv1512) synthesized just mycoside B (data not shown). Therefore, the production of the new compound was dependent on the presence of the glycosyltransferase encoded by gene Rv2958c.
Structural Analysis of the New Glycolipid Produced in the Recombinant Strain M. bovis BCG::pPET52:pWM85—The new glycoconjugate produced by the recombinant strain expressing the three genes Rv2958c, Rv1511, and Rv1512 (Fig. 1, lane 4) was purified by chromatography on a Florisil column and structurally characterized.
The MALDI-TOF mass spectrum of this compound showed a series of pseudomolecular ion (M + Na)+ peaks at 1780, 1794, 1808, 1822, 1836, 1850, 1864, 1878, 1892, and 1906 m/z (Fig. 2A). These mass values were 334 mass units higher than those observed for mycoside B, produced by the parental strain M. bovis BCG (Fig. 2B). This difference corresponded to the mass of the terminal disaccharide composed of tri-O-methyl-deoxyhexosyl linked to an unmethylated deoxyhexosyl as found in PGL-tb, i.e. tri-O-methyl-fucosyl-(1->3)-rhamnosyl. Consistently, the mass values were also 188 mass units higher, i.e. the mass of the terminal tri-O-methyl-deoxyhexosyl moiety, than those observed for the diglycosylated PGL synthesized by M. bovis BCG expressing the glycosyltransferase-encoding gene Rv2958c from the plasmid pPET52 (15). Therefore, these findings were consistent with the new compound produced by M. bovis BCG::pPET52:pWM85 having the same saccharide moiety as PGL-tb. This was confirmed by the one-dimensional 1H NMR spectrum of the purified compound, which exhibited the same signal resonances (Fig. 3) as those described for PGL-tb (11, 12): (i) two doublets at δ 6.97 (signal g) and δ 7.08 (h) assigned to phenolic proton resonances; (ii) three anomeric protons seen at δ 5.50 (signal i, 1H) and δ 5.15 (signal i′, 2H); (iii) the multiplet centered at 4.83 ppm (signal a) attributable to the resonance of the methine protons of the esterified β-diol of PGLs and related phthiocerol DIM; (iv) resonances of several terminal methyl protons seen at 0.8–1.0 ppm (signals e and e′), consistent with the presence of multimethyl-branched fatty acyl residues; (v) resonances at δ 1.15 ppm (signal f) and δ 2.55 (signal d) corresponding to those of the methyl groups located on the α position of multimethyl-branched fatty acyl residues and of the methine protons in these α-carbons, respectively; (vi) a broad signal resonance at 1.29 ppm (signal k) attributable to polymethylenic (CH2) units; (vii) the five singlets characteristic of PGL-tb: four signals at 3.48, 3.51, 3.57, and 3.60 ppm (signals j) attributable to the proton resonances of methoxyl groups linked to the sugar moiety and one signal at 3.32 ppm (signal b) corresponding to the resonance of the methoxyl group of phenolphthiocerol; and (viii) the resonance at 2.85 ppm (signal c) corresponding to that of the methine proton of the carbon bearing the methoxyl group on phenolphthiocerol.
FIGURE 2.
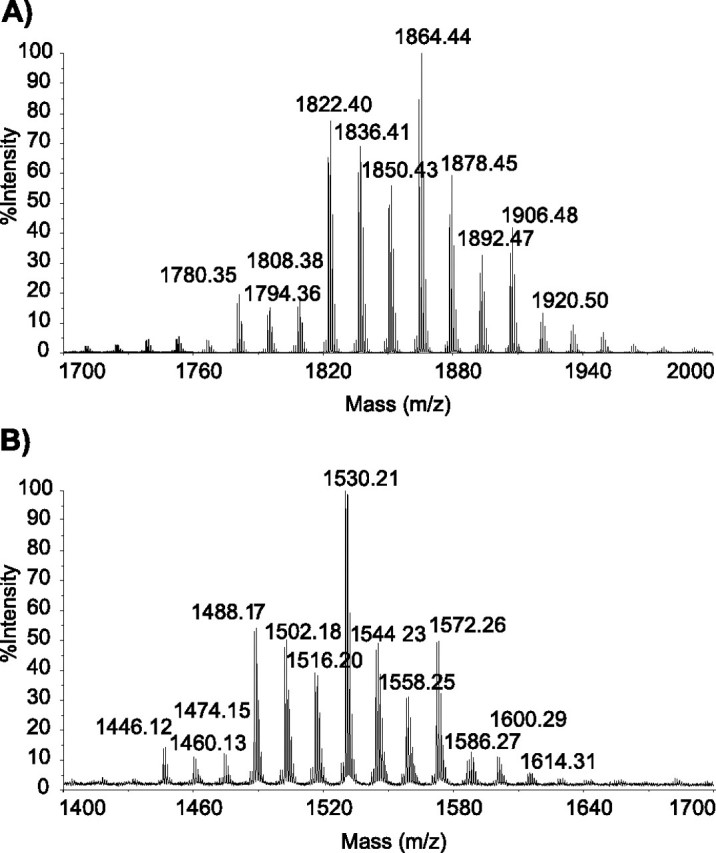
MALDI-TOF mass spectra for the PGL produced by M. bovis BCG::pPET52:pWM85 (A) and the parental strain M. bovis BCG (B).
These findings clearly establish that the new glycoconjugate produced by the recombinant strain M. bovis BCG:: pPET52:pWM85 is structurally identical to PGL-tb. They thus demonstrate that deletion RD4, in particular the absence of the Rv1511 and Rv1512 genes, and the frameshift mutation within the M. bovis BCG Rv2958c ortholog underlie the inability of this strain to synthesize and transfer the terminal disaccharide onto mycoside B to form PGL-tb.
Occurrence of the Rv2958c Frameshift Mutation and Rv1511/Rv1512 Deletion in M. tuberculosis Complex Strains—We extended our study to the analysis of 33 strains of the M. tuberculosis complex (Table 1). These strains are representative of the various subspecies defining the M. tuberculosis complex and were isolated from various regions of the world (Table 1). We screened these isolates for both the presence of the frameshift mutation previously identified in M. bovis by sequence analysis of the Rv2958c orthologs (15) and the occurrence of the Rv1511/Rv1512 deletion. Accordingly, a fragment of the chromosome was amplified from each of these strains by PCR with primers Rv2958D and Rv2958E and sequenced. Comparison of the resulting sequences with those of M. tuberculosis revealed that all the M. pinnipedii, M. microti, and M. bovis isolates analyzed, as well as some M. africanum strains, had the same mutation as M. bovis AF2122/97 and M. bovis BCG 1173P2 (Fig. 4A). This mutation corresponds to a single nucleotide insertion at position 867 and leads to a nonfunctional allele as demonstrated for M. bovis BCG. In contrast, all the sequenced M. tuberculosis strains (available at the NCBI web site) and the M. tuberculosis and M. canettii isolates analyzed in this study did not harbor this frameshift mutation. Two M. africanum isolates, IS960090 and IS960092, also harbored a sequence identical to that of M. canettii. These results demonstrated that M. pinnipedii, M. microti, M. bovis, and some M. africanum strains harbor a nonfunctional allele of Rv2958c.
FIGURE 4.

Occurrence of a frameshift mutation in the Rv2958c orthologs from M. tuberculosis complex strains. A, alignment of the partial sequence of the Rv2958c orthologs from M. tuberculosis complex strains. The corresponding amino acid sequences are indicated above and below the nucleotide sequences. For M. tuberculosis, M. canettii, and M. africanum (RD9), the amino acid sequences are identical. B, phylogenetic tree of M. tuberculosis complex strains (adapted from Ref. 20) and occurrence of the RD4, RD7, RD8, RD9, and RD10 deletions and Rv2958c frameshift mutation (Rv2958c*).
In contrast, the Rv1511/Rv1512 deletion was not detected in any of the M. microti, M. africanum, and M. pinnipedii strains analyzed (data not shown). This deletion was just detected in some M. bovis strains, including the vaccine strains M. bovis BCG (20). Accordingly, M. microti, M. africanum, and M. pinnipedii strains are putatively equipped with the machinery responsible for the production of fucose but deficient (except some M. africanum isolates) for the glycosyltransferase Rv2958c involved in the transfer of the second rhamnosyl residue.
Occurrence of PGL in Strains of the M. tuberculosis Complex—Production of PGL has been described for M. canettii, some M. tuberculosis isolates, M. bovis, and M. microti (1, 7, 13). In the last two species, the carbohydrate domain corresponds to 2-O-methylrhamnose (1, 13). These results are consistent with the occurrence of the Rv2958c frameshift mutation in these strains. To correlate the occurrence and structures of PGL potentially synthesized by other strains of the M. tuberculosis complex with their genotype, we analyzed the lipid contents of four strains: two M. africanum isolates and two M. pinnipedii isolates.
The lipids produced by the four selected strains were analyzed by TLC (Fig. 5A). All but one of the strains (M. pinnipedii strain 1886; Fig. 5A, lane 2) synthesized glycolipids. The two M. africanum (Fig. 5A, lanes 4 and 5) produced compounds with a mobility similar to that of mycoside B, whereas the M. pinnipedii strain 0217 contained a more polar glycolipid (Fig. 5A, lane 1). These results suggested that the M. africanum and M. pinnipedii strains may produce PGL.
FIGURE 5.

Analysis of the PGL produced by some M. africanum and M. pinnipedii isolates. A, TLC analysis of lipid extracts from M. pinnipedii 0217 (lane 1), M. pinnipedii 1886 (lane 2), M. bovis BCG (lane 3), M. africanum 2005-0479 (lane 4), M. africanum 2005-0419 (lane 5), and of purified compounds from M. pinnipedii 0217 (lane 6), M. bovis BCG (lane 7), M. africanum 2005-0479 (lane 8), and M. africanum 2005-0419 (lane 9). The purified PGL spotted on lanes 6–9 corresponded to compounds indicated by arrows on lanes 1–5. Lipid extracts were dissolved in CHCl3 and were run in CHCl3/CH3OH (95:5, v/v). Glycolipids were visualized by spraying the plates with 0.2% anthrone (w/v) in concentrated H2SO4 followed by heating. Origins of the TLCs are indicated by the dotted line. B, MALDI-TOF mass spectrum for the PGL produced by M. africanum 2005-0479. C, MALDI-TOF mass spectrum for the PGL produced by M. pinnipedii 0217.
Structural Analyses of the Glycolipids Produced by M. africanum and M. pinnipedii—PGL production has not been described previously for either M. africanum or M. pinnipedii. Therefore, glycolipids produced by the three strains examined herein were purified by preparative TLC and structurally characterized.
MALDI-TOF mass spectrometry analyses of the glycolipids purified from the two M. africanum strains showed a series of pseudomolecular ion (M + Na)+ peaks at 1488, 1502, 1516, 1530, 1544, 1558, 1572, 1586, 1600, 1614, 1628, 1642, and 1656 m/z (Fig. 5B). These spectra were identical to that observed for the 2-O-methyl rhamnosyl-phenolphthiocerol dimycocerosate, which was produced by the M. tuberculosis H37Rv mutant strain PMM19: pPET1 deficient in the transfer of the second rhamnosyl residue of PGL-tb (15). Interestingly, this series is 42 mass units higher than the one observed for the M. bovis BCG-derived mycoside B, consistent with our previous observation that M. bovis BCG produced mycocerosates shorter than those synthesized by other members of the M. tuberculosis complex (22).
The mass spectrum of the glycolipid produced by M. pinnipedii 0217 showed a series of pseudomolecular ion (M + Na)+ peaks at 1474, 1488, 1502, 1516, 1530, 1544, 1558, 1572, 1586, 1600, and 1614 m/z (Fig. 5C). The major peak was observed at 1558 m/z, 14 mass units lower than the one observed for the two M. africanum strains. This observation, together with the higher polarity of the compound observed on TLC, suggested that a methyl group may be missing either on the sugar residue or on the lipid part of PGL.
To fully determine the structure of the compounds produced by the various strains, the purified glycolipids were analyzed by 1H NMR spectroscopy (Table 2). The proton resonances characteristic of mycoside B (15, 23) were all identified in the spectra of PGL purified from the two M. africanum isolates; thus these PGL are structurally identical to mycoside B. Analysis of the 1H NMR spectrum for the compound purified from M. pinnipedii strain 0217 showed the absence of the resonance signal at 3.49 ppm. This signal has been previously assigned to the methoxyl group linked to the sugar residue of mycoside B (23), confirming our hypothesis based on TLC and MALDI-TOF analyses. Therefore, M. pinnipedii strain 0217 produces unmethylated rhamnosyl-phenolphthiocerol dimycocerosate.
TABLE 2.
1H NMR analysis of the PGLs produced by the various M. tuberculosis complex strains The columns correspond to the various proton resonances (in ppm). Attributions of the various resonance signals of PGL-tb were described previously (11, 12, 15).
| aa | ba | ca | da | e and e′a | fa | ga | ha | ia | i′a | ja | ka | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M. tuberculosis H37Rv:pPET1 | ||||||||||||
| PGL-tb |
4.83 |
3.32 |
2.85 |
2.55 |
0.8–1.0 |
1.15 |
6.97 |
7.08 |
5.50 |
5.17; 5.15 |
3.48; 3.51; 3.57; 3.60 |
1.29 |
| M. bovis BCG | ||||||||||||
| Mycoside B |
4.83 |
3.32 |
2.85 |
2.55 |
0.8–1.0 |
1.14 |
6.9 |
7.1 |
5.5 |
3.51 |
1.29 |
|
| M. africanum | ||||||||||||
| 4.81 |
3.30 |
2.83 |
2.52 |
0.8–1.0 |
1.15 |
6.95 |
7.07 |
5.51 |
3.49 |
1.29 |
||
| M. pinnipedii | ||||||||||||
| 4.81 | 3.30 | 2.83 | 2.51 | 0.8–1.0 | 1.12 | 6.93 | 7.06 | 5.45 | 1.29 | |||
The letters refer to the protons of the various molecules (see Fig. 3) whose resonances are visible on the spectra
DISCUSSION
Several recent studies have demonstrated that PGL is an important virulence factor of pathogenic mycobacteria (2, 7). This compound has several structural variations depending on the species producing it. The impact of these structural modifications has not yet been evaluated but may account for different features of mycobacteria-induced diseases. In this study, we investigated the structural diversity of PGL among strains of the M. tuberculosis complex and its genetic bases. We demonstrated that the RD4 deletion and a frameshift mutation within Rv2958c gene are jointly responsible for the inability of M. bovis BCG to synthesize PGL-tb. Within the RD4 region, only two genes, Rv1511 and Rv1512, are required for PGL-tb synthesis. This was demonstrated by the fact that expression in M. bovis BCG of these two genes in addition to Rv2958c endows the recombinant strain with the capacity to synthesize PGL-tb. This result is consistent with the bioinformatic predictions suggesting that Rv1511 encodes the GDP-d-mannose 4,6-dehydratase and Rv1512 encodes a GDP-4-keto-6-deoxy-d-mannose-3,5-epimerase/reductase, the two enzymes required for the formation of l-fucose from d-mannose. In addition, these results established that M. bovis BCG contains and expresses all the other genes required for the transfer and methylation of the terminal fucosyl residue. One of them, Rv2957, encodes a glycosyltransferase required for the transfer of the terminal sugar residue of PGL-tb. The two genes, Rv1511 and Rv1512, are not located at the PGL + DIM locus, which was shown to harbor many of the known genes required for PGL-tb and DIM synthesis (14); instead, they are found at a locus recently associated with the synthesis of lipooligosaccharides (LOS) in Mycobacterium marinum (24, 25). In the M. tuberculosis complex, very few strains synthesize LOS; interestingly, however, in those that do synthesize LOS, such as M. canettii, the oligosaccharide moiety was found to contain l-fucose (26). In Mycobacterium avium, orthologs of Rv1511 and Rv1512 are involved in the formation of serotype 2 glycopeptidolipids that also contain l-fucose (27). Therefore, these studies suggest that the two enzymes encoded by Rv1511 and Rv1512 orthologs provide activated l-fucose in the biosynthetic pathways of various l-fucose containing glycolipids in mycobacteria such as glycopeptidolipids in M. avium, LOS in M. marinum, and PGL-tb and LOS in M. canettii.
In this study, we also demonstrated that the same Rv2958c frameshift mutation was found in M. bovis, M. pinnipedii, and M. microti lineages. Concerning the M. africanum strains, the Rv2958c sequence analysis defined two groups: one exhibiting the Rv2958c frameshift mutation and the second not harboring this mutation. Interestingly this grouping is consistent with the results of Brosch et al. (20) who identified the same two groups of M. africanum isolates based on insertion/deletion analysis as follows: one group (including the IS960090 and IS960092 isolates) lacking only the RD9 region and a second (including the other isolates of this study) lacking the RD7, RD8, and RD10 regions in addition to RD9. Therefore, according to the phylogenetic tree proposed by Brosch et al. (20), the Rv2958c frameshift mutation probably occurred after the RD9 deletion and before the separation of the second M. africanum lineage (Fig. 4B). In contrast to M. bovis BCG and some M. bovis isolates, the M. microti, M. africanum, and M. pinnipedii strains analyzed have retained the genes Rv1511 and Rv1512, required for the formation of fucose. Therefore, we propose that the absence of a functional Rv2958c glycosyltransferase underlies the inability of strains of the M. bovis, M. africanum (RD7-, RD8-, and RD10-), M. microti and M. pinnipedii lineages to synthesize PGL-tb. This conclusion is supported by the structure of the PGLs produced by M. bovis (1), M. microti (13), M. africanum (RD7-, RD8-, and RD10-), and M. pinnipedii, which are all monoglycosylated mycoside B-like substances. In addition, our analyses revealed that other genetic defects affecting the synthesis of PGL are also found in M. tuberculosis complex strains. For instance, one of the two M. pinnipedii isolates analyzed in this study did not synthesize PGL, as most of the M. tuberculosis isolates. The second isolate, M. pinnipedii strain 0217, produces an unmethylated rhamnosyl-phenolphthiocerol dimycocerosate. In this strain, a mutation within gene ortholog of Rv2959c, encoding the enzyme responsible for methylation of the first rhamnosyl residue, may explain the methylation defect (data not shown). Thus other genetic defects occurred within isolates of M. pinnipedii, and probably also in other strains of the M. tuberculosis complex, suggesting that the structure of PGL is more diverse than initially anticipated.
To conclude, our study reveals that among the M. tuberculosis complex strains, only strains of the M. canettii and M. tuberculosis lineages and some M. africanum isolates have retained the ability to synthesize the 2,3,4-tri-O-methyl l-fucopyranosyl-(α1->3)-l-rhamnopyranosyl-(α1->3)-2-O-methyl l-rhamnopyranosyl-(α1->) carbohydrate domain found in PGL-tb and the related molecule, 2,3,4-tri-O-methyl-fucosyl-(α1->3)-rhamnosyl-(α1->3)-2-O-methyl-rhamnosyl-α-p-hydroxybenzoic acid methyl ester (also called p-HBAD II) (11). This finding deserves consideration because this domain was shown to be of key importance for the modulation of the innate immune response by some M. tuberculosis strains (7). In addition to the functional genomics aspect of this work, the construction of a recombinant M. bovis BCG strain producing PGL-tb makes it possible to compare the immune response induced by two isogenic strains that differ only by their capacity to synthesize PGL-tb or mycoside B. This kind of analysis may shed light on the structure/activity relationship of a key lipidic virulence factor of M. tuberculosis.
Acknowledgments
We thank Roland Brosch (Institut Pasteur, Paris, France), Paul Wheeler (London, UK), Kristin Kremer (National Institute for Public Health and the Environment, Bilthoven, The Netherlands), and Dick van Soolingen (National Institute for Public Health and the Environment, Bilthoven, The Netherlands) for providing us with the M. tuberculosis complex strains. We are grateful to Françoise Laval (mass spectrometry) and Anne Lemassu (NMR spectroscopy) for their valuable assistance. The NMR spectrometers were financed by the CNRS, the University Paul Sabatier, the Région Midi-Pyrénées, and the European Structural Funds (FEDER).
This work was supported by the Agence Nationale de la Recherche Grant ANR-06-MIME-032. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: PGL, phenolglycolipid; DIM, phthiocerol dimycocerosates; MALDI-TOF, matrix-assisted laser desorption-ionization time-of-flight; LOS, lipooligosaccharides.
References
- 1.Daffé, M., and Lanéelle, M. A. (1988) J. Gen. Microbiol. 134 2049-2055 [DOI] [PubMed] [Google Scholar]
- 2.Ng, V., Zanazzi, G., Timpl, R., Talts, J. F., Salzer, J. L., Brennan, P. J., and Rambukkana, A. (2000) Cell 103 511-524 [DOI] [PubMed] [Google Scholar]
- 3.Neill, M. A., and Klebanoff, S. J. (1988) J. Exp. Med. 167 30-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schlesinger, L. S., and Horwitz, M. A. (1991) J. Exp. Med. 174 1031-1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Charlab, R., Sarno, E. N., Chatterjee, D., and Pessolani, M. C. V. (2001) Lepr. Rev. 72 63-69 [PubMed] [Google Scholar]
- 6.Mehra, V., Brennan, P. J., Rada, E., Convit, J., and Bloom, B. R. (1984) Nature 308 194-196 [DOI] [PubMed] [Google Scholar]
- 7.Reed, M. B., Domenech, P., Manca, C., Su, H., Barczak, A. K., Kreiswirth, B. N., Kaplan, G., and Barry, C. E., III (2004) Nature 431 84-87 [DOI] [PubMed] [Google Scholar]
- 8.Tsenova, L., Ellison, E., Harbacheuski, R., Moreira, A. L., Kurepina, N., Reed, M. B., Mathema, B., Barry, C. E., III, and Kaplan, G. (2005) J. Infect. Dis. 192 98-106 [DOI] [PubMed] [Google Scholar]
- 9.Daffé, M., and Lemassu, A. (2000) in Glycomicrobiology (Doyle, R. J., ed) pp. 225-273, Plenum Publishing Corp., New York
- 10.Brennan, P. J. (1988) in Microbial Lipids (Ratledge, C., and Wilkinson, S. G., eds) pp. 203-298, Academic Press, London
- 11.Constant, P., Perez, E., Malaga, W., Lanéelle, M.-A., Saurel, O., Daffé, M., and Guilhot, C. (2002) J. Biol. Chem. 277 38148-38158 [DOI] [PubMed] [Google Scholar]
- 12.Daffé, M., Lacave, C., Lanéelle, M.-A., and Lanéelle, G. (1987) Eur. J. Biochem. 167 155-160 [DOI] [PubMed] [Google Scholar]
- 13.Thurman, P. F., Chai, W., Rosankiewicz, J. R., Rogers, H. J., Lawson, A. M., and Draper, P. (1993) Eur. J. Biochem. 212 705-711 [DOI] [PubMed] [Google Scholar]
- 14.Guilhot, C., Chalut, C., and Daffé, M. (2008) in The Mycobacterial Cell Envelope (Daffé, M., and Reyrat, J. M., eds) pp. 273-285, American Society for Microbiology, Washington, DC
- 15.Perez, E., Constant, P., Lemassu, A., Laval, F., Daffe, M., and Guilhot, C. (2004) J. Biol. Chem. 279 42574-42583 [DOI] [PubMed] [Google Scholar]
- 16.Le Dantec, C., Winter, N., Gicquel, B., Vincent, V., and Picardeau, M. (2001) J. Bacteriol. 183 2157-2164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Belisle, J. T., and Sonnenberg, M. G. (1998) in Mycobacteria Protocols (Parish, T., and Stoker, N. G., eds) pp. 31-44, Humana Press Inc., Totowa, NJ
- 18.Laval, F., Lanéelle, M.-A., Deon, C., Montsarrat, B., and Daffé, M. (2001) Anal. Chem. 73 4537-4544 [DOI] [PubMed] [Google Scholar]
- 19.Mäki, M., and Renkonen, R. (2004) Glycobiology 14 R1-R15 [Google Scholar]
- 20.Brosch, R., Gordon, S. V., Marmiesse, M., Buchrieser, C., Eiglmeier, K., Garnier, T., Gutierrez, C., Hewinson, G., Kremer, K., Parsons, L. M., Pym, A. S., Samper, S., van Soolingen, D., and Cole, S. T. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 3684-3689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cole, S. T., Brosch, R., Parkhill, J., Garnier, T., Churcher, C., Harris, D., Gordon, S. V., Eiglmeier, K., Gas, S., Barry, C. E., III, Tekaia, F., Badcock, K., Basham, D., Brown, D., Chillingworth, T., Connor, R., Davies, R., Devlin, K., Feltwell, T., Gentles, S., Hamlin, N., Holroyd, S., Hornsby, T., Jagels, K., Krogh, A., McLean, J., Moule, S., Murphy, L., Oliver, K., Osborne, J., Quail, M. A., Rajandream, M.-A., Rogers, J., Rutter, S., Seeger, K., Skelton, J., Squares, R., Squares, S., Sulston, J. E., Taylor, K., Whitehead, S., and Barrell, B. G. (1998) Nature 393 537-544 [DOI] [PubMed] [Google Scholar]
- 22.Camacho, L. R., Constant, P., Raynaud, C., Lanéelle, M.-A., Triccas, J.-A., Gicquel, B., Daffé, M., and Guilhot, C. (2001) J. Biol. Chem. 276 19845-19854 [DOI] [PubMed] [Google Scholar]
- 23.Daffé, M., Lanéelle, M.-A., Lacave, C., and Lanéelle, G. (1988) Biochim. Biophys. Acta 958 443-449 [DOI] [PubMed] [Google Scholar]
- 24.Burguière, A., Hitchen, P. G., Dover, L. G., Kremer, L., Ridell, M., Alexander, D. C., Liu, J., Morris, H. R., Minnikin, D. E., Dell, A., and Besra, G. S. (2005) J. Biol. Chem. 280 42124-42133 [DOI] [PubMed] [Google Scholar]
- 25.Ren, H., Dover, L. G., Islam, S. T., Alexander, D. C., Chen, J. M., Besra, G. S., and Liu, J. (2007) Mol. Microbiol. 63 1345-1359 [DOI] [PubMed] [Google Scholar]
- 26.Daffé, M., McNeil, M., and Brennan, P. J. (1991) Biochemistry 30 378-388 [DOI] [PubMed] [Google Scholar]
- 27.Miyamoto, Y., Mukai, T., Maeda, Y., Nakata, N., Kai, M., Naka, T., Yano, I., and Makino, M. (2007) J. Bacteriol. 189 5515-5522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kremer, K., van Soolingen, D., Frothingham, R., Haas, W. H., Hermans, P. W. M., Martin, C., Palittapongarnpim, P., Plikaytis, B. B., Riley, L. W., Yakrus, M. A., Musser, J. M., and van Embden, J. D. A. (1999) J. Clin. Microbiol. 37 2607-2618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pfyffer, G. E., Auckenthaler, R., van Embden, J. D. A., and van Soolingen, D. (1998) Emerg. Inf. Dis. 4 631-634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deshayes, C., Perrodou, E., Euphrasie, D., Frappy, E., Poch, O., Bifani, P. J., Lecompte, O., and Reyrat, J. M. (2008) BMC Evol. Biol. 8 78. [DOI] [PMC free article] [PubMed] [Google Scholar]