

Abstract
The Fc receptor γ-chain (FcRγ), which was first identified as a constituent of the high affinity IgE receptor, associates with various cell surface receptors to mediate intracellular signals. We identified three transcriptional enhancer elements in the 5′ region of the human FcRγ gene; one of the cis-elements was recognized by the transcription factor Sp-1 and another was recognized by GABP or Elf-1. The sequence of the other element was similar to a binding motif of the C/EBP family. Overexpression experiments showed that these transcription factors cooperatively activated the FcRγ promoter. Furthermore, inactivation of the GABP-binding site by nucleotide substitutions as well as repression of GABPα expression by RNA interference reduced Sp1-mediated transactivation of the FcRγ promoter, demonstrating that Sp1 and GABP synergistically activated the FcRγ promoter. This synergistic activation was suggested to require physical interaction between the two transcription factors, because the Ets domain of GABPα was demonstrated to directly bind Sp1. On the other hand, GABP and Elf-1, whose recognition sequences overlapped, were shown to bind the FcRγ gene with similar affinity in the context of chromatin, although Elf-1 exerted weaker enhancer activity for FcRγ gene expression than did GABP. Both were thought to compete for binding to the element, because additional expression of Elf-1 in combination with Sp1 and GABP reduced FcRγ promoter activity. Such functional and physical interactions among transcription factors involved in the cooperative regulation of FcRγ gene expression as revealed in this study will become promising targets for medical applications against various immune diseases involving FcRγ.
The Fc receptor γ-chain (FcRγ),2 which was first identified as a constituent of the high affinity IgE receptor (FcεRI) (1), also associates with other immunoglobulin Fc receptors, including the IgG receptors (FcγRI and FcγRIII) (2–5) and the IgA receptor (FcαR) (6–8). FcRγ possesses an immunoreceptor tyrosine-based activation motif and mediates intracellular signals upon stimulation of a wide variety of cell surface receptors. These receptors play essential roles in immune reactions of the host. However, unusual or excess activation of effector cells through FcRγ often leads to various immune diseases. For instance, cross-linking of FcεRI on mast cells or basophils by antigen (allergen)-IgE complexes triggers an allergic reaction by activating intracellular signal cascades to induce not only the release of various chemical mediators, including histamine and leukotriene in the early phase reaction, but also cytokine gene expression leading to the late-phase reaction. On the other hand, excessive responses to self-antigens through FcγRI or FcγRIII occur in autoimmune diseases. IgE-mediated passive cutaneous anaphylaxis, experimental hemolytic anemia induced by anti-erythrocyte Ab, and the Arthus reaction induced by immune complexes were all inhibited in FcRγ-deficient mice lacking expression of functional FcεRI, FcγRI, and FcγRIII, indicating an essential role of FcRγ for all types of hypersensitivity reactions (9). Therefore, a novel manipulation strategy based on the regulation of FcRγ function is expected to yield medical applications.
In addition to Fc receptors, FcRγ is also reported to associate with the collagen receptor glycoprotein (GP) VI on platelets (10). Binding of collagen to GP VI activates platelets in an FcRγ-dependent manner and induces aggregation of platelets. Moreover, FcRγ was recently reported to associate with the osteoclast-associated receptor on dendritic cells and monocytes, indicating the possible involvement of FcRγ in osteoclast function (11–13).
Because FcRγ associates with various receptors, including Fc receptors, GP VI and osteoclast-associated receptor as described above, functional regulation of FcRγ will control various immune diseases, including allergy, thrombosis, and lupus nephritis involving FcεRI, GP VI, and FcαR, respectively. Here we analyzed the regulatory mechanisms of human FcRγ gene expression, as local and specific regulation of FcRγ gene expression is thought to be one reasonable means of controlling FcRγ function.
The genomic structure of the human FcRγ gene has already been determined (14, 15). However, little analysis has been carried out on the regulatory mechanisms of FcRγ gene transcription. The human FcRγ gene consists of five exons, and its 5′ region contains a GC box and a reversed CAAT box but not a canonical TATA box. As seen in many genes possessing a TATA-less promoter, transcription of the FcRγ gene starts at multiple positions, including a major site 25 bp upstream from the translation start site and several minor sites within 100 bp of the translation start site (15). Brini et al. (16) analyzed the 5′ region of the human FcRγ gene over 2.5 kb and revealed that this region was involved in hematopoietic cell-specific transcriptional activation. We here analyzed a 5′ region of about 450 bp of human FcRγ gene and identified cis-elements within 100 bp upstream of the translation start site. Because synergistic activation by transcription factors binding to these elements was observed, we further analyzed and discussed the synergistic activation of the FcRγ gene promoter by multiple transcription factors.
EXPERIMENTAL PROCEDURES
Cell Culture—THP1 (a human monocyte line), U937 (a human promonocyte line), KU812 (a human basophilic leukemia cell line), and Jurkat cells (a human T cell line) were cultured in RPMI 1640 medium (Sigma) at 37 °C in a humidified incubator with 5% CO2. HeLa (a human epithelial cell line) was cultured in Dulbecco's modified Eagle's medium (Sigma). Both media contained 10% (v/v) fetal bovine serum (JRH Bioscience, Lenexa, KS), 100 units/ml of penicillin (Banyu Pharmaceutical, Tokyo, Japan), and 100 μg/ml of streptomycin (Meiji Seika, Tokyo, Japan).
Plasmid Construction—A DNA fragment corresponding to the nt -450/+861 region (nucleotide numbers start at the translation start site) of the human FcRγ gene was obtained by PCR using the human genomic DNA library (Clontech) as a template. Synthetic oligonucleotides of 5′-GGCGTGGTGGTGCATGCCTGTAATGCCAGCTACTC-3′ and 5′-ACCAAGGGTGCTTTGTATATTAAACCAGCTAGAGA-3′ were used as primers. The amplified product was inserted into a pCR2.1 vector (Invitrogen) for cloning and verifying its nucleotide sequence. The resulting plasmid was named pCR2.1-γ. The DNA fragments corresponding to nt -450/-1 and nt -370/-1 of the FcRγ gene were prepared by PCR employing pCR2.1-γ as a template and inserted into pGL3-Basic vector (Promega, Madison, WI) at the KpnI/XhoI site to obtain pGLγ-(-450/-1) and pGLγ-(-370/-1). The primers used for the PCR are as follows: forward primer for -450/-1, 5′-ggggtaccccGGCGTGGTGGTGCATGCCTG-3′; forward primer for -370/-1, 5′-ggggtaccTGAGCCGAGATGGCGCCATTGCACT-3′ (with the nucleotides represented by lowercase letters added to introduce a KpnI site (underlined)). The reverse primer used was 5′-ccgctcgagCTTGGGCTGGAGATCGGCCGTTCTG-3′ (with the nucleotides represented by lowercase letters added to introduce a XhoI site (underlined)).
A BglII-digested fragment from pGLγ-(-450/-1) was inserted at the BglII site of pGL3-Basic vector to yield pGLγ-(-177/-1). Similarly, a PvuII/BglII-digested fragment from pGLγ-(-450/-1) was inserted at the SmaI/BglII site of pGL3-Basic vector to obtain pGLγ-(-39/-1). The DNA fragments corresponding to nt -133/-1, nt -102/-1, and nt -73/-1 region of FcRγ gene were prepared by PCR using pGLγ-(-177/-1) as a template and introduced into pGL3-Basic vector. The employed PCR primers are as follows: forward primer for -133/-1, 5′-gcggtaccTCTTGTGCAGGAAGGGGAAGGG-3′; forward primer for -102/-1, 5′-gcggtaccTGGGGGAAGGCGTGGCAGGA-3′; forward primer for -73/-1, 5′-gcggtaccGACTCTGTGGTCAGGGAACTGCT-3′ (with the nucleotides represented by lowercase letters added to introduce a KpnI site (underlined)). The reverse primer used was 5′-gcagatctCTTGGGCTGGAGATCGGCCGTTC-3′ (with the nucleotides represented by lowercase letters added to introduce a BglII site (underlined)).
The amplified products were digested with KpnI and BglII to insert at the KpnI/BglII site of the pGL3-Basic vector. A series of mutant plasmids of pGLγ-(-177/-1) carrying a few nucleotide substitutions (mutA, mutB, mutC, and mutD) was constructed using QuickChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). The primers used to introduce nucleotide substitutions by expressing the substituted nucleotides as underlined are as follows: mutA, 5′-GGGCCAAAGCATGTCTGAAGGCGTGGCAGGA-3′; mutB 5′-GGGAAGGCGTGGCACTAAGAGGGGGACTCTG-3′; mutC, 5′-GTGGCAGGAAGAGCGCTACTCTGTGGTCAGG-3′; mutD, 5′-GAGGGGGACTCTGTAGTCAGGGAACTGCTC-3′.
An Sp1 expression plasmid carrying a human Sp1 cDNA on pCL-neo vector (Promega) was kindly provided by Dr. Y. Ikeda (Kochi Medical School, Kochi, Japan) (17). Expression plasmids of GABPα, GABPβ, and deletion mutants of GABPα were prepared as described previously (18). The empty vector pCR3.1-Empt was generated by self-ligation of EcoRI-digested pCR3.1 (Invitrogen). The Elf-1 expression plasmid pcDNA3.1-Elf-1 was constructed as follows. A full-length of human Elf-1 cDNA was prepared by reverse transcription-PCR employing total RNA from Jurkat cells as a template and synthetic oligonucleotides of 5′-ATGGCTGCTGTTGTCCAACAGAAC-3′ and 5′-CTAAAAAGAGTTGGGTTCCAGCAG-3′ as primers and cloned into pcDNA3.1 (Invitrogen). For pGST-GABPα, glutathione S-transferase (GST) was fused to the N terminus of human GABPα and its deletion mutants were generated as described previously (18). To construct siRNA expression plasmid of GABPα, a double-stranded DNA generated by annealing synthetic oligonucleotides of 5′-GATCCGTGTAAGCCAGGCCATAGACACTGTGAAGCCACAGATGGGTGTCTATGGCCTGGCTTACACTTTTTTA-3′ and 5′-AGCTTAAAAAAGTGTAAGCCAGGCCATAGACACCCATCTGTGGCTTCACAGTGTCTATGGCCTGGCTTACACG-3′ was inserted into pBAsi-hU6 Pur (TAKARA BIO, Shiga, Japan) digested with BamHI/HindIII.
Transfection of Cells for Luciferase Assay—Transfection was performed as described previously (19). Briefly, cells were transfected with 5 μg of test construct by electroporation at 300 V, 950 microfarads using a Gene Pulser II (Bio-Rad). For overexpression experiments, 3 μg of expression plasmids of the transcription factors of Sp1, GABPα, GABPβ, and Elf-1 or each corresponding empty vector as control were co-introduced into the cells with 3 μg of the reporter construct. For RNA interference experiments, 3 μg of the expression plasmid of GABPα siRNA was co-introduced into the cells. Twenty five picograms of the plasmid phRL-CMV (Promega) carrying Renilla luciferase gene under the control of the human cytomegalovirus promoter were introduced to normalize transfection and cell lysis efficiency in every experiment. After 20–24 h of culture, cells were harvested and washed with phosphate-buffered saline (PBS) (pH 7.4). Cell lysis and determination of the luciferase activity were carried out using the dual-luciferase assay kit (Promega) according to the manufacturer's instructions. Luminescence was measured with Luminometer (Berthold, Postfach, Germany).
Nuclear Extract Preparation—Nuclear extract was prepared as described previously (20). Briefly, cells were washed with ice-cold PBS and resuspended in ice-cold buffer A (10 mm HEPES (pH 7.9), 10 mm KCl, 0.1 mm EDTA, 1 mm dithiothreitol (DTT)) containing protease inhibitors. Cells were then incubated on ice for 10 min and for an additional 15 min with 0.5% Nonidet P-40. After centrifuging at 6,000 × g for 1 min, the pellet was resuspended in extract buffer (20 mm HEPES (pH 7.9), 400 mm KCl, 4.5 mm MgCl2, 0.2 mm EDTA, 1 mm DTT) containing protease inhibitors and incubated on ice for 1 h. The lysate was centrifuged at 10,000 × g for 10 min. After the addition of 15% glycerol, the supernatant was stored at -80 °C until use.
Electrophoretic Mobility Shift Assay (EMSA)—Double-stranded DNAs were prepared as probe A and probe B by annealing FITC-labeled synthetic oligonucleotides. Nucleotide sequences of the probes are as follows: probe A, 5′-ATGGGGGAAGGCGTG-3′; probe B, 5′-GCGTGGCAGGAAGAGG-3′. Nonlabeled, double-stranded oligonucleotides carrying the same sequence as the probe were prepared to use as competitors. Similarly, mutant competitors with a few nucleotide substitutions were generated. Their nucleotide sequences were as follows with the substituted nucleotides underlined: mutant competitor A, 5′-ATGGTCTAAGGCGTG-3′; mutant competitor B, 5′-GCGTGGCAACTAGAGG-3′. Thirty micrograms of nuclear extract and 5 pmol of DNA probe were incubated at room temperature with 5–125 pmol of competitors in 10 mm HEPES buffer (pH 7.9) containing 400 ng of poly(dI-dC), 1 mm MgCl2, 30 mm KCl, 1 mm DTT, and 5% glycerol for 20 min. For the supershift or inhibition experiments by Abs, 2.0 μg of Abs were added to the reaction mixtures and incubated for an additional 20 min. Rabbit polyclonal Abs raised against chicken GABPα and GABPβ, which cross-react with human GABP, were kindly provided by Dr. S. Toku (University of Ryukyus, Okinawa, Japan). The other Abs were all purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The mixtures were separated by electrophoresis on 4% polyacrylamide gels at 120 V for 2.0–2.5 h in 0.25× TBE buffer (22.5 mm Tris, 22.5 mm boric acid, 0.5 mm DTT). FITC fluorescence was detected by Fluor Imager 595 (Amersham Biosciences).
Chromatin Immunoprecipitation (ChIP) Assay—ChIP assays were performed using the ChIP assay kit (Upstate Biotechnology, Inc., Lake Placid, NY). Cells were exposed to 1% formaldehyde for 10 min to obtain cross-linked chromatins. After quenching the reaction with 0.125 m glycine, cells were washed with ice-cold PBS, solubilized with SDS lysis buffer containing protease inhibitors, and sonicated to shear the genomic DNA to an average length of 500–1000 bp. After centrifuging, the supernatants were diluted 10-fold in ChIP dilution buffer and immunoprecipitated with anti-GABPα rabbit polyclonal Ab (provided by Dr. S. Toku), anti-Elf-1 rabbit polyclonal Ab (Santa Cruz Biotechnology), or rabbit IgG as control (Santa Cruz Biotechnology) and salmon sperm DNA/protein A-agarose (Upstate Biotechnology, Inc.). The immunoprecipitated chromatins were washed, eluted from beads, and incubated at 65 °C to reverse the cross-links. After treatment with proteinase K, DNA was recovered by QIAquick PCR DNA purification kit (Qiagen, Hilden, Germany) and subjected to PCR analyses using synthetic oligonucleotide primers of 5′-AGCTGCACAGTGCTGTCAGAACGGCCGATC-3′ and 5′-CCCTCACCAAACCCTCTTACCTGCTTGTTC-3′ specific for nt -41/-12 and nt +40/+69 regions of the FcRγ gene, respectively. Primers of 5′-ACCAGGAGACTTACGAGACTCTGAAGCATG-3′ and 5′-AGAATATAAATATCCGTAAACAGCATCTGA-3′ corresponding to nt +3576/+3605 and nt +3796/+3825 regions of the FcRγ gene, respectively, were used as control. A thermal cycle of 94 °C for 40 s, 64 °C for 40 s, and 72 °C for 1 min was repeated 32 times to amplify the nt -41 + 69 region and that of 94 °C for 40 s, 57 °C for 40 s, and 72 °C for 1 min was repeated 32 times to amplify the nt +3576/+3825 region.
Western Blotting—Cells were transfected with the expression plasmids of Sp1, GABPα, GABPβ, and Elf-1 or each corresponding empty vector as control and cultured for 16 h. After adding G418 to select the transfected cells, cells were cultured for an additional 28–32 h. Nuclear extracts were prepared for immunoblotting with anti-Sp1 (Santa Cruz Biotechnology), anti-GABPα (provided by Dr. S. Toku), anti-GABPβ (provided by Dr. S. Toku), anti-Elf-1 (Santa Cruz Biotechnology), and anti-β-actin Abs (Abcam, Cambridge, UK). Alternatively, cells were transfected with the siRNA expression plasmid for GABPα or the empty vector as control and cultured for 24 h. After selecting the transfected cells with puromycin for an additional 24 h, cell lysates were prepared and subjected to immunoblotting with anti-GABPα Ab or anti-β-actin Ab.
Pulldown Assay—Pulldown assay was performed as described previously (18). In brief, GABPα, its deletion mutants, and GABPβ fused to GST were respectively expressed in E. coli. The recombinant proteins in the sonicated E. coli lysates were immobilized on glutathione-Sepharose 4B beads (Amersham Biosciences). Five microliters of beads bound to 10 μg of GST-fused protein were incubated with [35S]methionine-labeled Sp1 prepared by in vitro transcription/translation employing TnT QuickCoupled Transcription/Translation Systems (Promega) in the presence of 100 μg/ml ethidium bromide. After washing, the proteins bound to the beads were eluted by boiling in SDS sample buffer and subjected to SDS-PAGE.
RESULTS
An Approximate 100-bp Region Upstream of the Translation Start Codon Is Commonly Required for Activation of the Human FcRγ Gene Promoter in Various Types of FcRγ-expressing Cells—The 5′ regions of the human FcRγ gene were inserted upstream of a luciferase gene and analyzed for their transcriptional regulatory activity by reporter gene assays employing various FcRγ expressing human cell lines as follows: THP1 (a monocyte line), U937 (a promonocyte line), Jurkat (a T cell line), and KU812 (a basophilic leukemia cell line). (Note, nucleotide numbers are counted from the translation start site as +1.) The regions nt -450/-1, nt -370/-1, and nt -177/-1 activated the FcRγ gene promoter to almost the same extent but nt -39/-1 hardly activated it in all cell lines used for the assay (Fig. 1). Standard deviations were large in some cell lines, because the levels of luciferase activity from FcRγ constructs compared with those from the promoter-less construct varied widely among experiments probably depending on cell conditions, although the relative patterns among the FcRγ constructs were very reproducible in each cell line. These results indicated that the region nt -177/-40 contained core enhancer elements required for the activation of the FcRγ gene promoter. We further specified the cis-acting elements in the region nt -177/-40 by a reporter gene assay employing a series of deletion constructs. About 30–40 bp of the nt -177/-40 region of the FcRγ gene was sequentially deleted from the 5′ end. Deletion of nt -102/-74 resulted in almost undetectable transcriptional activation in all cell lines, indicating that the region of nt -102/-74 contained common enhancer elements essential for activation of the FcRγ promoter (Fig. 2). Moreover, the regions nt -133/-103 and nt -177/-134 were shown to contain additional enhancer elements crucial for THP1, Jurkat, and U937 cells and for U937 and KU812 cells, respectively.
FIGURE 1.

The 5′ region nt -177/-1 is required for transcriptional activation of the human FcRγ gene. Four hundred and fifty bp of the human FcRγ gene 5′ region and its deletion mutants were evaluated for their transcriptional regulatory activities by reporter gene assays employing γ-chain expressing human cell lines (THP1, U937, Jurkat, and KU812). Luciferase (LUC) activities relative to the control without promoter are shown. Results are expressed as means ± S.D. of three or four independent experiments. Nucleotide numbers are counted from the translation start site as +1.
FIGURE 2.

The region nt -74/-102 of the FcRγ gene contains common transcriptional enhancer elements. Transcriptional enhancer elements in the region nt -177/-1 of γ-chain gene were further mapped by reporter gene assays using a series of deletion constructs. Luciferase (LUC) activities are expressed as the ratios to that of pGLγ-(-177/-1). Results are represented as means ± S.D. of three independent experiments.
Three Enhancer Elements Are Identified in the Promoter Proximal Region of FcRγ Gene—To identify cis-acting elements, we introduced nucleotide substitutions in the sequences similar to transcription factor-binding motifs in the nt -102/-1 region (nt -101/-95 (motif A), nt -87/-80 (motif B), nt -77/-71 (motif C), and nt -67/-57 (motif D)), which was found by the assistance of the TFSEARCH program which is owned by the TRANSFAC databases (21). As shown in Fig. 3, substitution of nucleotides in motifs A (mutA), B (mutB), and D (mutD) reduced the luciferase activity but that in motif C (mutC) did not affect it. These results indicated that motif A and motif B functioned as transcriptional enhancer elements in the region of nt -102/-74, which was shown to commonly activate the FcRγ promoter in various cell lines. In addition, motif D was also revealed to activate the FcRγ promoter probably by cooperating with the elements in the region of nt -102/-74.
FIGURE 3.

The regions nt -101/-95 and nt -87/-80 act as enhancer elements. Mutant reporter plasmids were constructed by introducing nucleotide substitutions into motif A of nt -101/-95 (mutA), motif B of nt -87/-80 (mutB), motif C of nt -77/-71 (mutC), and motif D of nt -67/-57 (mutD) of the FcRγ gene. Cells were transfected with the constructs for a transient expression assay. Luciferase (LUC) activities are expressed as the ratios to those of the wild type construct pGLγ-(-177/-1). Results are represented as means ± S.D. of three independent experiments.
Two of the Enhancer Elements Are Recognized by Transcription Factors of Sp1 and GABP/Elf-1, Respectively—We next identified by EMSA the transcription factors binding to these motifs using nuclear extracts from FcRγ expressing cells and FITC-labeled double-stranded oligonucleotide probes containing motif A or motif B (Fig. 4). Among several shifted bands that appeared upon the addition of the nuclear extracts to a probe containing motif A, the band indicated by an arrow disappeared when nonlabeled, double-stranded oligonucleotide competitor with the same sequence of the probe was added (Fig. 4a). In contrast, this band was not affected by a mutant competitor with the same nucleotide substitutions in motif A as the mutant reporter plasmid (mutA) used in Fig. 3, indicating that this band represented a sequence-specific binding of a nuclear protein to motif A. Furthermore, because the band disappeared only upon the addition of anti-Sp1 antibody and was not affected by other antibodies against USF1, USF2, and Ikaros, the binding protein was identified as Sp1 (Fig. 4b). Several shifted bands also appeared on the addition of nuclear extracts when a probe containing motif B was employed for the assay. The double band indicated by the arrows was revealed to depend on the sequence-specific binding between motif B and nuclear proteins by a similar competition assay using nonlabeled, double-stranded DNA (Fig. 4c). Addition of antibodies specific for some transcription factors, including PU.1, Fli1, Elf-1, GABPα, and GABPβ, revealed that the binding factor forming the lower band was an α/β heteromer of GABP, as the band disappeared following the addition of either anti-GABPα or anti-GABPβ Ab (Fig. 4d). On the other hand, the binding factor forming the upper band was identified as Elf-1 (Fig. 4d). Similar results were obtained when employing nuclear extracts from THP1, U937, and KU812 cells. We also tried to identify by EMSA the transcription factor that bound to motif D, but we were unsuccessful in our attempts to do this (data not shown).
FIGURE 4.
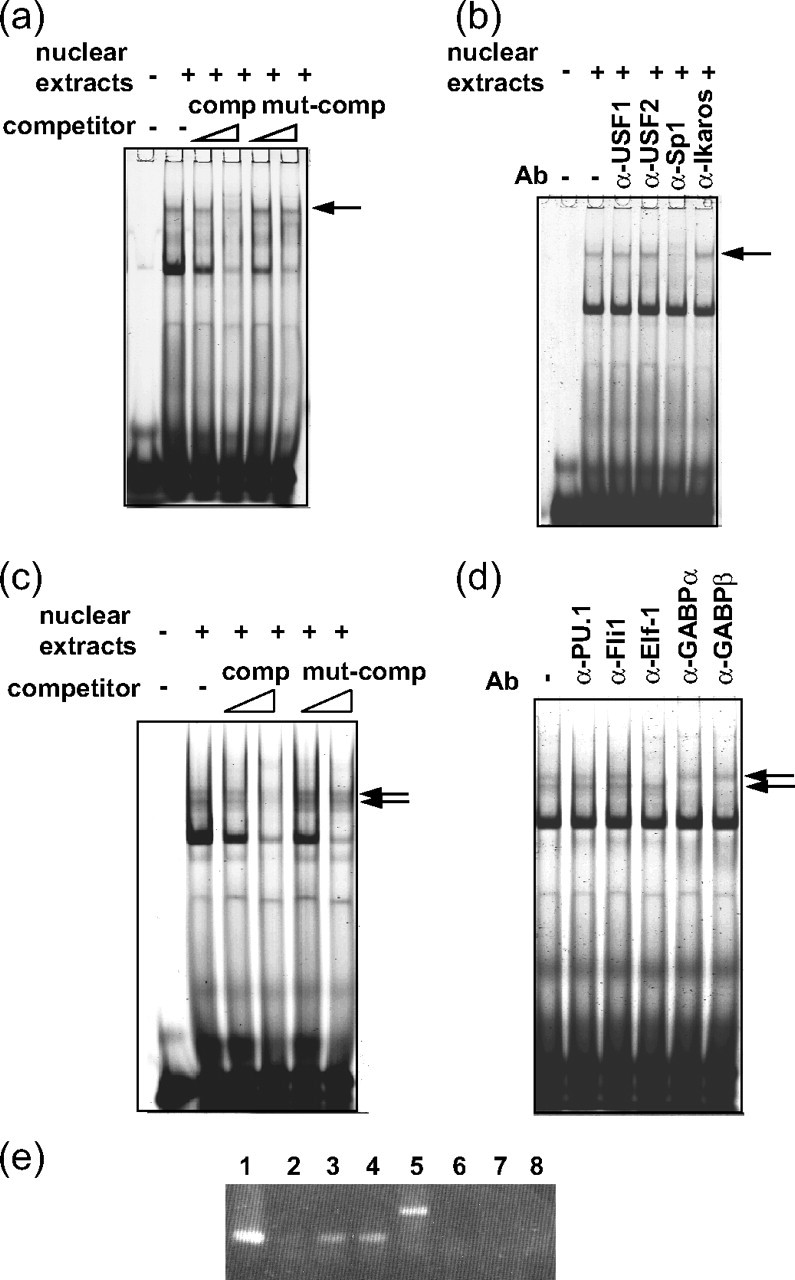
Motif A (nt -101/-95) is recognized by the transcription factor Sp1, and motif B (nt -87/-80) is recognized by GABP or Elf-1. a–d, EMSA was performed employing the nuclear extracts prepared from Jurkat cells and FITC-labeled, double-stranded oligonucleotide probes that corresponded to the sequences of nt -102/-88, including motif A (a and b), and nt -93/-76, including motif B (c and d). Unlabeled double-stranded oligonucleotide competitors with the same sequence of the probe (comp) and with three nucleotide substitutions in motif A or motif B (mut-comp) were used for the assays (a and c). Antibodies specific for various transcription factors were added in b and d. Similar results were obtained using the nuclear extracts prepared from THP1, U937, and KU812 cells. e, ChIP assays of the endogenous FcRγ gene in Jurkat cells were carried out employing anti-Elf-1 and anti-GABPα Abs. Rabbit IgG was used as control. The region nt -41/+69 of FcRγ gene proximal to the element of nt -87/-80 was amplified by PCR from the immunoprecipitated chromatins (lanes 1–4). As control, nt +3576/+3605 region of the FcRγ gene, which was distal to the nt -87/-80 element, was amplified (lanes 5–8). Lanes 1 and 5, input (diluted fractions of nonimmunoprecipitated chromatins were employed as templates for PCR to quantify the amount of DNA present in each sample before immunoprecipitation.); lanes 2 and 6, rabbit IgG; lanes 3 and 7, anti-GABPα Ab; lanes 4 and 8, anti-Elf-1 Ab. Similar results were obtained when using THP1 cells.
Because the sequences required for GABP and Elf-1 binding almost completely overlapped in motif B, it was predicted that these transcription factors could not bind to motif B together. To compare the affinity of GABP and Elf-1 for the nt -87/-80 element in the context of chromatin in vivo, ChIP assays of the endogenous FcRγ gene were performed employing anti-GABP and anti-Elf-1 Abs (Fig. 4e). The nt -41/+69 region of the FcRγ gene, proximal to the element of nt -87/-80, was amplified from the chromatins immunoprecipitated with anti-GABP and anti-Elf-1 Abs (Fig. 4e, lanes 3 and 4) to almost the same extent but scarcely amplified from those immunoprecipitated with control rabbit IgG (lane 2). In contrast, the region nt +3576/+3605, distal to the element of nt -87/-80, as control was not amplified from the chromatins immunoprecipitated with either Ab (Fig. 4e, lanes 7 and 8). These results demonstrated that GABP and Elf-1 bound to motif B with similar affinity in vivo. Collectively, these results demonstrated that Sp1 specifically recognized motif A and that Elf-1 or GABP bound specifically to motif B.
Synergistic Activation of the γ-Chain Gene Promoter by Multiple Transcription Factors—To examine whether the identified transcription factors actually activated the FcRγ promoter, expression plasmids of Sp1, GABPα, GABPβ, and Elf-1 were introduced into KU812 cells with the reporter plasmid pGLγ-(-177/-1) carrying the nt -177/-1 region of the FcRγ gene upstream of a luciferase gene (Fig. 5a). Overexpression of each transcription factor increased the luciferase activity, indicating that these transcription factors respectively activated the FcRγ promoter. Coexpression of Sp1 and GABP caused synergistic activation of the FcRγ promoter, whereas that of Sp1 and Elf-1 only additively activated the promoter, consistent with the result that Elf-1 itself exerted smaller enhancing effect than GABP. Overexpression of the transcription factors by the introduction of expression plasmids was detected by Western blotting as shown in Fig. 5d.
FIGURE 5.

Sp1, GABP, and Elf-1 cooperatively activate the FcRγ promoter. a, expression plasmids of GABPα, GABPβ, Elf-1, and Sp1 (filled bars) or corresponding empty vectors as control (hatched bars) were introduced into KU812 cells with the reporter plasmid pGLγ-(-177/-1) carrying the FcRγ nt -177/-1 region upstream of a luciferase gene for a transient expression assay. b, KU812 cells were co-transfected with an expression plasmid of Sp1 and the reporter plasmid pGLγ-(-177/-1) with or without nucleotide substitutions at the GABP-binding site. c, siRNA expression plasmid of GABPα was co-introduced with the Sp1 expression plasmid and pGLγ-177/-1 in KU182 cells. a–c, relative luciferase (Luc) activities to that of pGLγ-(-177/-1) alone are shown. Results are represented as means ± S.D. of three independent experiments. d, expression of GABPα, GABPβ, Elf-1, and Sp1 in the cells transfected with indicated combinations of expression plasmids and empty vectors was detected by Western blotting. e, expression of GABPα in the cells transfected with GABPα siRNA was analyzed by Western blotting. Lane 1, no treatment; lane 2 and 3,GABPα siRNA; lane 4, control siRNA.
To further confirm the synergistic activation by Sp1 and GABP, transactivation of the FcRγ promoter by overexpressed Sp1 was compared by employing the wild type reporter pGLγ-(-177/-1) and a mutant reporter plasmid that had nucleotide substitutions at the GABP-binding site (Fig. 5b). Sp1-mediated transactivation was reduced from 3.61- to 1.76-fold by the mutation at the GABP-binding site. Furthermore, repression of endogenous GABPα expression by siRNA also reduced Sp1-mediated transactivation (from 4.09- to 2.25-fold) in addition to FcRγ promoter activity itself (Fig. 5c). The effect of siRNA on the expression of GABPα was verified by Western blotting (Fig. 5e). These results indicated that full activation of the FcRγ promoter by the overexpressed Sp1 was achieved by functional interaction with endogenous GABP. Collectively, Sp1 and GABP were demonstrated to synergistically activate the FcRγ promoter.
GABPα Interacts with Sp1 through Its ETS Domain—Because it was suggested that functional interaction between Sp1 and GABP was required for full activation of the FcRγ promoter in Fig. 5, b and c, we next analyzed whether the two transcription factors physically interacted by a pulldown assay using 35S-labeled Sp1 prepared by in vitro transcription/translation and recombinant GABPα or GABPβ fused to GST. The results demonstrated that GABPα but not GABPβ directly interacted with Sp1 (Fig. 6b). Sp1 has been reported to bind GABPα through its zinc finger motifs (22), although regions of GABPα interacting with Sp1 remain to be analyzed. Consequently, a series of deletion mutants of GABPα was constructed and employed to address this (Fig. 6c). The amino acid 318–399 region of GABPα corresponding to the ETS domain known to be essential for DNA binding interacted with Sp1. These results showed that GABPα physically interacted with Sp1 through its ETS domain.
FIGURE 6.
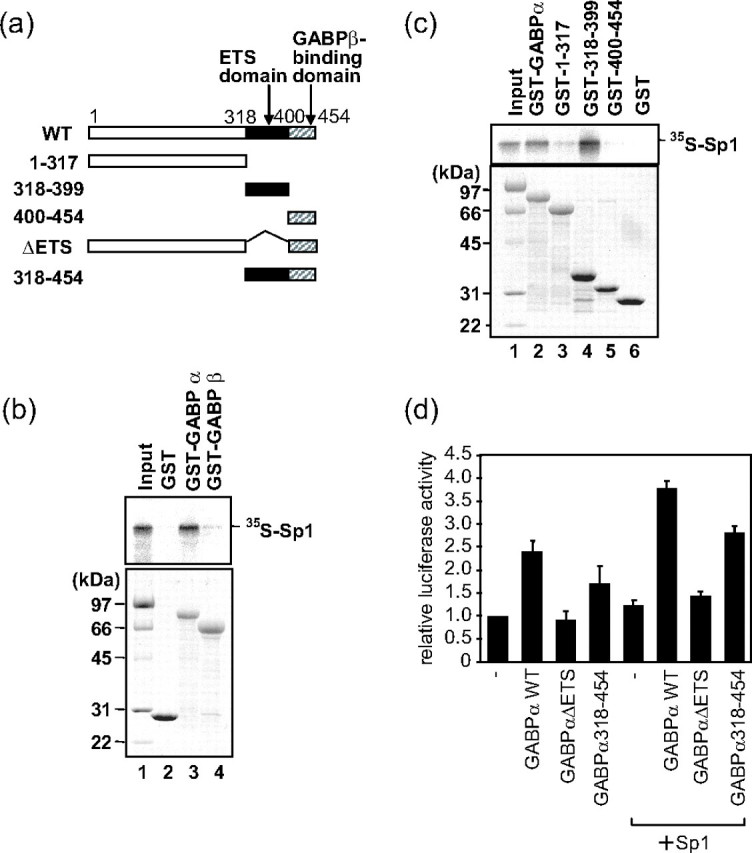
The ETS domain of GABPα physically interacts with Sp1. a, schematic drawing of wild type GABPα (WT) and its deletion mutants employed for the experiments. b, [35S]methionine-labeled Sp1 was prepared by in vitro transcription/translation and mixed with recombinant GABPα or GABPβ fused to GST, which was expressed in E. coli for a pulldown assay using glutathione-Sepharose beads. The proteins bound to the beads were subjected to SDS-PAGE and analyzed by autoradiography (top) or Coomassie Blue staining (bottom). c, [35S]methionine-labeled Sp1 prepared by in vitro transcription/translation and recombinant GABPα or its deletion mutants were mixed for a pulldown assay employing glutathione-Sepharose beads. The proteins bound to the beads were subjected to SDS-PAGE and analyzed by autoradiography (top) or Coomassie Blue staining (bottom). d, 3 μg of expression plasmids of wild type GABPα or its deletion mutants and the reporter plasmid pGLγ-(-177/-1) were co-introduced into HeLa cells with or without 0.3 μg of an expression plasmid for Sp1. Relative luciferase activities to that of pGLγ-(-177/-1) alone are shown. Results are represented as means ± S.D. of more than two independent experiments.
Functions of deletion mutants of GABPα were further analyzed by co-introducing their expression plasmids with the reporter plasmid pGLγ-(-177/-1) and the expression plasmids of GABPβ and Sp1 (Fig. 6d). To clearly evaluate the synergistic activation of Sp1 and GABP, Sp1 was expressed at a low level where the expressed Sp1 by itself hardly activated the FcRγ promoter by introducing a small amount of Sp1 expression plasmid into HeLa cells which expressed no hematopoietic transcription factors. GABPαΔEts, which lacked the Ets domain, could neither activate the FcRγ promoter by itself nor give synergistic activation of FcRγ promoter with Sp1, whereas the wild type GABPα (GABPαWT) activated the promoter by itself and synergistically with Sp1. On the other hand, GABPα-(318–454), which lacked the N-terminal portion and consisted of the Ets domain and GABPβ binding domain, showed reduced enhancing effects on the FcRγ promoter compared with GABPαWT but caused the synergistic activation of the promoter with Sp1. These results were in accordance with the findings that the Ets domain of GABPα was required for both DNA binding and interaction with Sp1.
DISCUSSION
In this study we revealed that expression of the FcRγ gene is regulated by multiple transcription factors, including Sp1, GABP, and Elf-1, which recognize elements in the 5′ region within about 100 bp upstream of the transcription start site. The nucleotide sequence of the region is highly homologous between human and mice, leading us to believe that regulation mechanisms by these transcription factors are therefore possibly common beyond the species.
Sp1 and GABP physically interacted with each other and synergistically activated the FcRγ promoter. It has been reported that Sp1 cooperatively activates transcription with GABP in several types of genes (23–28) and physically interacts with GABPα through its zinc finger motifs (22). We demonstrated that GABPα physically interacts with Sp1 through its Ets domain, which is known to be essential for DNA binding, indicating that the Ets domain of GABPα is required for binding to both of DNA and Sp1. This is the first study to determine the domain of GABPα that is involved in the interaction with Sp1. The amino acid residues crucial for the binding to DNA and those crucial for the binding to Sp1 can be separated, because GABP interacts with DNA and Sp1 at the same time. Determination of those amino acid residues will enable us to modify the transcriptional activation of the FcRγ gene by inhibiting the interaction of GABPα with protein (Sp1) but not with DNA, for example. Because both Sp1 and GABP are essential transcription factors, absolute functional inhibition of Sp1 or GABP will not be acceptable to the host. Therefore, specific and precise modulation of FcRγ expression depending on the partial or moderate inhibition of the function of these transcription factors is required for medical applications. A mutant form of GABPα with amino acid residue substitutions or chemical compounds that inhibit the protein-protein interaction is expected to yield such a precise modulation of FcRγ expression.
In addition to Sp1, GABP, and Elf-1, a nuclear factor binding to motif D was suggested to be involved in the regulation of FcRγ expression, although we failed to identify it. Although it remains to be analyzed what nuclear factors actually bind to the element in the nucleus of FcRγ-expressing cells, overexpression of C/EBPα increased FcRγ promoter activity (data not shown), suggesting that a nuclear factor similar to C/EBP family bound to motif D and activated the FcRγ promoter. Because substitution of nucleotides in motif D decreased FcRγ promoter activity (Fig. 3) but nt -1/-73 containing motif D hardly activated transcription (Fig. 2), motif D was thought to activate FcRγ promoter by cooperating with motif A recognized by Sp1 and/or motif B recognized by GABP or Elf-1. Actually, cooperative transcriptional enhancing effects by the combination of C/EBP and Sp1 (29–31) and by C/EBP and GABP (18, 32) were previously shown in several reports. The transcriptional enhancing effects of the combination of C/EBPα and GABP on the FcRγ promoter were not only additive but synergistic, with the maximum synergistic effect achieved by the coexpression of Sp1, GABP, and C/EBPα (data not shown). The trimeric transcriptional activating complex consisting of Sp1, GABP, and some C/EBPα-like factor might be formed on the 5′ region of the FcRγ gene, as every physical interaction between Sp1 and GABP (22), GABP and C/EBP (18), and Sp1 and C/EBP (33) has been reported. Modification of the formation of such a regulatory complex will be one promising strategy to specifically and moderately modulate FcRγ expression.
On the other hand, because the recognition sequences of GABP and Elf-1 overlapped, GABP and Elf-1 were thought to compete for binding to the element. The two transcription factors seemed to activate the FcRγ promoter at different levels (Fig. 5a), although they were shown to have similar affinity for the element in vivo by ChIP assays (Fig. 4e). Elf-1, which exerted weaker enhancing activity than GABP, might reduce the FcRγ promoter activity by competitively inhibiting the binding of GABP to the overlapped element, because additional overexpression of Elf-1 in combination with Sp1 and GABP reduced FcRγ promoter activity compared with the case of the Sp1/GABP coexpression (Fig. 5a). Therefore, the ratio of expression levels of GABP and Elf-1, which differs depending on cell types or conditions, might affect the expression level of FcRγ. Consistent with this, Juang et al. (34) recently reported that Elf-1 bound to GGAA elements on the FcRγ promoter and repressed its expression. In their study, Elf-1 bound to multiple elements, including motif A in the region of nt -141/-66 (corresponding to nt -121/-46 in their study), whereas Sp1 bound to motif A in our study. There is a possibility that both Sp1 and Elf-1 can bind to motif B depending on the conditions of cells. GABP activated FcRγ promoter more strongly than Elf-1 in KU812 cells which express FcRγ (Fig. 5a), but GABP and Elf-1 activated the promoter almost equally in HeLa cells which do not express FcRγ (data not shown). The difference of the effects of Elf-1 overexpression on KU812 and HeLa cells is thought to result from the different levels of endogenous expression of Elf-1 in the two cell lines, because the endogenous expression level of Elf-1 is higher in the hematopoietic cell line KU812 than in the nonhematopoietic cell line HeLa. Moreover, conditions of Elf-1 modification, including its phosphorylation level, might be different among the cell lines.
Because FcRγ is expressed in various but limited types of cells, it is predicted that elements responsible for the cell type-specific expression exist. However, activation of the core promoter was essential to FcRγ expression but was mediated by transcription factors such as Sp1 and GABP that are ubiquitously expressed, indicating the possibility that further upstream 5′ regions, introns, and 3′ regions include the elements that determine the cell type-specific expression of FcRγ.
More detailed study will reveal the entire regulatory mechanisms of FcRγ gene expression, and concrete and specific information about the molecules and their interactions that participate in the regulation of FcRγ gene expression will contribute to medical applications for various immune diseases.
Acknowledgments
We are grateful to Dr. Y. Ikeda (Kochi Medical School) and Dr. S. Toku (University of Ryukyus) for kindly providing the Sp1 expression plasmid and antisera against GABP. We thank the members of the Department of Molecular Cell Immunology and Allergology for helpful discussions.
This work was supported in part by a grant-in aid from the Ministry of Education, Culture, Sports, Science, and Technology of Japan. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: FcRγ, Fc receptor γ-chain; FcεRI, high affinity IgE receptor; FcγR, IgG receptor; FcαR, IgA receptor; GP VI, collagen receptor glycoprotein VI; GST, glutathione S-transferase; nt, nucleotide; ChIP, chromatin immunoprecipitation; DTT, dithiothreitol; FITC, fluorescein isothiocyanate; PBS, phosphate-buffered saline; Ab, antibody; siRNA, small interfering RNA; EMSA, electrophoretic mobility shift assay.
References
- 1.Blank, U., Ra, C., Miller, L., White, K., Metzger, H., and Kinet, J. P. (1989) Nature 337 187-189 [DOI] [PubMed] [Google Scholar]
- 2.Ra, C., Jouvin, M. H. E., Blank, U., and Kinet, J. P. (1989) Nature 341 752-754 [DOI] [PubMed] [Google Scholar]
- 3.Ernst, L. K., Duchemin, A. M., and Anderson, C. L. (1993) Proc. Natl. Acad. Sci. U. S. A. 90 6023-6027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scholl, P. R., and Geha, R. S. (1993) Proc. Natl. Acad. Sci. U. S. A. 90 8847-8850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Masuda, M., and Roos, D. (1993) J. Immunol. 151 7188-7195 [PubMed] [Google Scholar]
- 6.Pfefferkorn, L. C., and Yeaman, G. R. (1994) J. Immunol. 153 3228-3236 [PubMed] [Google Scholar]
- 7.Saito, K., Suzuki, K., Matsuda, H., Okumura, K., and Ra, C. (1995) J. Allergy Clin. Immunol. 96 1152-1160 [DOI] [PubMed] [Google Scholar]
- 8.Morton, H. C., van den Herik-Oudijk, I. E., Vossebeld, P., Snijders, A., Verhoeven, A. J., Capel, P. J. A., and van de Winkel, J. G. J. (1995) J. Biol. Chem. 270 29781-29787 [DOI] [PubMed] [Google Scholar]
- 9.Takai, T. (1996) Int. Rev. Immunol. 13 369-381 [DOI] [PubMed] [Google Scholar]
- 10.Tsuji, M., Ezumi, Y., Arai, M., and Takayama, H. (1997) J. Biol. Chem. 272 23528-23531 [DOI] [PubMed] [Google Scholar]
- 11.Kim, N., Takami, M., Rho, J., Josien, R., and Choi, Y. (2002) J. Exp. Med. 195 201-209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ishikawa, S., Arase, N., Suenaga, T., Saita, Y., Noda, M., Kuriyama, T., Arase, H., and Saito, T. (2004) Int. Immunol. 16 1019-1025 [DOI] [PubMed] [Google Scholar]
- 13.Mócsai, A., Humphrey, M. B., Van Ziffle, J. A. G., Hu, Y., Burghardt, A., Spusta, S. C., Majumdar, S., Lanier, L. L., Lowell, A. A., and Nakamura, M. C. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 6158-6163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Le Coniat, M., Kinet, J. P., and Berger, R. (1990) Immunogenetics 32 83-86 [DOI] [PubMed] [Google Scholar]
- 15.Kuster, H., Thompson, H., and Kinet, J. P. (1990) J. Biol. Chem. 265 6448-6452 [PubMed] [Google Scholar]
- 16.Brini, A. T., Lee, G. M., and Kinet, J. P. (1993) J. Biol. Chem. 268 1355-1361 [PubMed] [Google Scholar]
- 17.Osaki, F., Ikeda, Y., Suehiro, T., Ota, K., Tsuzura, S., Arii, K., Kumon, Y., and Hashimoto, K. (2004) Atherosclerosis 176 279-287 [DOI] [PubMed] [Google Scholar]
- 18.Shimokawa, T., and Ra, C. (2005) Blood 106 2532-2542 [DOI] [PubMed] [Google Scholar]
- 19.Takahashi, K., Hayashi, N., Kaminogawa, S., and Ra, C. (2006) J. Immunol. 177 4605-4611 [DOI] [PubMed] [Google Scholar]
- 20.Takahashi, K., Nishiyama, C., Hasegawa, M., Akizawa, Y., and Ra, C. (2003) J. Immunol. 171 2478-2484 [DOI] [PubMed] [Google Scholar]
- 21.Heinemeyer, T., Wingender, E., Reuter, I., Hermjakob, H., Kel, A. E., Kel, O. V., Ignatieva, E. V., Ananko, E. A., Podkolodnaya, O. A., Kolpakov, F. A., Podkolodny, N. L., and Kolchanov, N. A. (1998) Nucleic Acids Res. 26 364-370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Galvagni, F., Capo, S., and Oliviero, S. (2001) J. Mol. Biol. 306 985-996 [DOI] [PubMed] [Google Scholar]
- 23.Rosmarin, A. G., Luo, M., Caprio, D. G., Shang, J., and Simkevich, C. P. (1998) J. Biol. Chem. 273 13097-13103 [DOI] [PubMed] [Google Scholar]
- 24.Shirasaki, F., Makhluf, H. A., LeRoy, C., Watson, D. K., and Trojanowska, M. (1999) Oncogene 18 7755-7764 [DOI] [PubMed] [Google Scholar]
- 25.Jiang, P., Kumar, A., Parrillo, J. E., Dempsey, L. A., Platt, J. L., Prinz, R. A., and Xu, X. (2002) J. Biol. Chem. 277 8989-8998 [DOI] [PubMed] [Google Scholar]
- 26.Gyrd-Hansen, M., Krag, T. O., Rosmarin, A. G., and Khurana, T. S. (2002) J. Neurol. Sci. 197 27-35 [DOI] [PubMed] [Google Scholar]
- 27.Rudge, T. L., and Johnson, L. F. (2002) Exp. Cell Res. 274 45-55 [DOI] [PubMed] [Google Scholar]
- 28.Hampel, N., Wang, H., LeCluyse, E. L., McManus, M. E., and Negishi, M. (2004) Mol. Pharmacol. 66 1690-1701 [DOI] [PubMed] [Google Scholar]
- 29.Khanna-Gupta, A., Zibello, T., Simkevich, C., Rosmarin, A. G., and Berliner, N. (2000) Blood 95 3734-3741 [PubMed] [Google Scholar]
- 30.Tzeng, S. J., Chang, W. C., and Huang, J. D. (2005) J. Biol. Sci. 12 741-761 [DOI] [PubMed] [Google Scholar]
- 31.Payton, S. G., Whetstine, J. R., Ge, Y., and Matherly, L. H. (2005) Biochim. Biophys. Acta 1727 45-57 [DOI] [PubMed] [Google Scholar]
- 32.Nuchprayoon, I., Simkevich, C. P., Luo, M., Friedman, A. D., and Rosmarin, A. G. (1997) Blood 89 4546-4554 [PubMed] [Google Scholar]
- 33.Chiang, B. T., Liu, Y. W., Chen, B. K., Wang, J. M., and Chang, W. C. (2006) J. Biomed. Sci. 13 621-635 [DOI] [PubMed] [Google Scholar]
- 34.Juang, Y. T., Sumibcay, L., Tolnay, M., Wang, Y., Kyttaris, V. C., and Tsokos, G. C. (2007) J. Immunol. 179 4884-4889 [DOI] [PubMed] [Google Scholar]