

Abstract
The destiny and activity of sterol regulatory element-binding proteins (SREBPs) in the nucleus are regulated by modification with ubiquitin, small ubiquitin-like modifier (SUMO), or phosphorus. ERK-dependent phosphorylation causes an increase in their transcriptional activity, whereas SUMO modification halts it. We hypothesized a causal linkage between phosphorylation and sumoylation because their sites are very closely located in SREBP-1 and -2 molecules. When Ser455, a phosphorylation site in SREBP-2, was substituted with Ala, this SREBP-2 mutant was more efficiently modified by SUMO-1. On the other hand, substitution of Asp inhibited SUMO conjugation, mimicking phosphoserine. When cells were cultured with insulin-like growth factor-1, sumoylation of SREBP-2 was decreased with an increase in its phosphorylation, but SREBP-2(S455A) was continuously sumoylated. An ERK cascade inhibitor, U0126, inversely augmented SUMO modification of SREBP-2. Insulin-like growth factor-1 treatment stimulated the expression of SREBP target genes such as the low density lipoprotein (LDL) receptor, squalene synthase, and hydroxymethylglutaryl-CoA synthase genes. These results indicate that growth factor-induced phosphorylation of SREBP-2 inhibits sumoylation, thereby facilitating SREBP transcriptional activity. Glutathione S-transferase pulldown assays revealed that wild-type SREBP-2, but not a mutant lacking Lys464, interacts with HDAC3 preferentially among the histone deacetylase family members. HDAC3 small interfering RNA induced gene expression of the LDL receptor and thereby augmented fluorescently labeled LDL uptake in HepG2 cells. In summary, growth factors inhibit sumoylation of SREBPs through their phosphorylation, thus avoiding the recruitment of an HDAC3 corepressor complex and stimulating the lipid uptake and synthesis required for cell growth.
Sterol regulatory element-binding proteins (SREBPs)2 regulate a wide variety of genes involved in cholesterol and fatty acid synthesis and low density lipoprotein (LDL) uptake (1). SREBPs are synthesized as membrane proteins located on the endoplasmic reticulum (ER) and thereafter are processed to liberate the N-terminal halves that function as transcription factors in the nucleus. The proteolytic processing of SREBPs is highly controlled by the interaction between two ER membrane proteins, the SREBP cleavage-activating protein (SCAP) and insulin-inducing gene (INSIG). Once the content of ER membrane cholesterol increases, SCAP, an SREBP-associated protein that binds cholesterol, induces conformational change and becomes attached to INSIG, thereby remaining on the ER membrane. Because the proteolytic processing occurs on the Golgi membrane where two processing enzymes reside, SREBPs together with SCAP on the ER membrane are never processed, and therefore, cholesterol is a critical determinant of SREBP activation. In contrast, under cholesterol-depleted conditions, an SREBP·SCAP complex is transferred to the Golgi apparatus, and thereafter, the proteolytic activation of SREBPs occurs.
The transcriptional activity of active SREBPs that are translocated into the nucleus is affected by various modifications, including phosphorylation, ubiquitylation, and sumoylation. In cells arrested at G2/M, the transcriptional and DNA-binding activities of the nuclear form of SREBP-1, but not SREBP-2, are increased after being hyperphosphorylated (2). On the other hand, the growth hormone-induced MAPKs ERK1 and ERK2 phosphorylate SREBPs and up-regulate their transcriptional activity (3–5). Moreover, phosphorylation of SREBP-1 is enhanced in response to DNA binding and thereby promotes recruitment of the ubiquitin ligase Fbw7, with degradation carried out by the ubiquitin-proteasome system (6, 7). Independently of ubiquitylation in SREBP-1 and -2, SREBPs are modified by another ubiquitin-like protein, SUMO (8, 9). This modification does not affect a rapid turnover of the transcription factors but rather impairs their transcriptional activity.
Here, we report a novel link between the phosphorylation and sumoylation of SREBPs in response to growth hormone stimuli. Based on the fact that the ERK-induced phosphorylation sites in SREBP-1 and -2 are located very close to sumoylation sites (4, 5, 8), we hypothesized that there might be a causal linkage between these two modifications because of their opposite effects on transcriptional activity. We found that mutation of the phosphorylation site stimulated sumoylation and that insulin-like growth factor-1 (IGF-1) treatment reduced sumoylation along with an increase in the phosphorylation of SREBPs. Sumoylation of SREBPs triggered recruitment of a corepressor complex containing HDAC3, thereby impairing their transcriptional activity. HDAC3 knockdown elevated gene expression of the LDL receptor (LDLR) and the uptake of LDL in HepG2 cells. Thus, these results demonstrate that growth hormones stimulate the lipid synthesis and uptake required for cell growth through an increase in SREBP activity because of a reduction in sumoylation.
EXPERIMENTAL PROCEDURES
Materials—Cholesterol, 25-hydroxycholesterol, lipoprotein-deficient serum, N-ethylmaleimide (NEM), protease inhibitor mixture, and trichostatin A (TSA) were purchased from Sigma. IGF-1 was from R&D Systems. Calpain inhibitor I (N-acetyl-Leu-Leu-norleucinal) was from Nacalai Tesque, and U0126 was from Calbiochem.
Cultured Cells—COS-1, HepG2, and HEK293 cells were maintained in medium A (Dulbecco's modified Eagle's medium containing 100 units/ml penicillin and 100 μg/ml streptomycin) supplemented with 10% fetal bovine serum at 37 °C under 5% CO2 atmosphere.
Antibodies—Anti-FLAG (M2), anti-glutathione S-transferase (GST), and anti-HDAC3 antibodies were purchased from Sigma. Anti-SREBP-1 (2A4) antibody was from Santa Cruz Biotechnology. Anti-hemagglutinin (HA) antibody was from Covance. Anti-phospho-p44/42 ERK (Thr202/Tyr204) and anti-p44/42 ERK (Thr202/Tyr204) antibodies were from Cell Signaling Technology. Anti-SREBP-2 polyclonal antibody has been described previously (22).
Plasmids—An expression plasmid for HA-HDAC3 (pcDNA-HDAC3-HA) was kindly provided by Dr. Minoru Yoshida (RIKEN). An expression plasmid for HA-SUMO-1 (pCAG-HA-SUMO-1) (23) was kindly provided by Dr. Takayuki Ohshima (Tokushima Bunri University). The expression plasmids for SREBP-1a and -2 (pME-GST-SREBP-1a and -2, pCMV-3×FLAG-SREBP-1a and -2, and pGAL4-SREBP-1a and -2) were described previously (8). Expression plasmids for various mutant versions of SREBP-1a and -2 were generated using a site-directed mutagenesis kit (Stratagene).
In Vivo GST Pulldown and Immunoprecipitation Experiments—COS-1 and HepG2 cells (100-mm dishes) were transfected with the indicated plasmids (6 μg each) using Lipofectamine 2000 (Invitrogen). 36 h later, the cells were harvested and lysed with radioimmune precipitation assay buffer (50 mm Tris-HCl (pH 7.4), 1 mm EDTA, 150 mm NaCl, 1% Nonidet P-40, and 0.25% sodium deoxycholate) supplemented with 1 mm phenylmethylsulfonyl fluoride, 10 mm NEM, 50 μm N-acetyl-Leu-Leu-norleucinal, and 0.1% protease inhibitor mixture on ice for 30 min. The lysates were centrifuged at 15,000 rpm for 10 min at 4 °C, and the supernatant was incubated with 50 μl of a 50% slurry of glutathione-Sepharose 4B (GE Healthcare) or immunoprecipitated with the indicated antibody with 50 μl of a 50% slurry of protein G-Sepharose CL-4B (GE Healthcare). The beads were washed three times with 500 μl of radioimmune precipitation assay buffer, and the specifically bound proteins were pelleted, resuspended with sample buffer, and subjected to immunoblot analysis.
[32P]Orthophosphate Labeling Experiments—COS-1 cells transfected with various expression plasmids were precultured with a phosphate-free medium for 6 h and then radiolabeled with [32P]orthophosphate (300 μCi/ml; GE Healthcare) for several hours. The cells were harvested and lysed with phosphate buffer containing phosphatase inhibitors. The lysates were subjected to immunoprecipitation with anti-FLAG antibodies. The signals on the membrane were quantified using a Fujifilm FLA-3000 image analyzing system.
Luciferase Assays—Reporter assays were performed as described previously (24, 25).
Chromatin Immunoprecipitation Assays—Chromatin immunoprecipitation assays were performed as described previously (26). PCR was performed with the following primers: LDLR forward, 5′-CTCTTCACCGGAGACCCAAA-3′; and LDLR reverse, 5′-GGCCCACGTCATTTACAGCA-3′. The primers generate a 226-bp fragment containing a sterol regulatory element (SRE) of the human LDLR promoter. Fragments amplified by PCR were analyzed on 1% agarose gel.
Small Interfering RNA Experiments—The small interfering RNA (siRNA; 150 pmol/6-well plate, 30 pmol/24-well plate) for human HDAC3 (nucleotides 1170–1190 (GCUGAACCAUGCACCUAGUGU) in NM_003883) (14) and the control (GCGCGCUUUGUAGGAUUCG, the sequence of Scramble II duplex by Dharmacon) were transfected using Lipofectamine RNAiMAX (Invitrogen) into HepG2 cells according to the manufacturer's instructions. 72 h later, the cells were harvested and subjected to immunoblot analysis, luciferase assays, and LDL uptake assays.
LDL Uptake Assays—HepG2 cells transfected with siRNA against human HDAC3 68 h prior to assays were incubated with 10 μg/ml DiI-labeled LDL (Molecular Probes) for 4 h and washed with phosphate-buffered saline. Intracellular fluorescence staining was visualized using an Olympus IX70 microscope. The fluorescence intensity was measured by Fluoroskan Ascent (Thermo Electron Corp.).
RESULTS
Ser455 of SREBP-2 Is Critical for SUMO-1 Conjugation at Lys464—We have reported previously that the human nuclear form of SREBP-1 has two sumoylation sites and that SREBP-2 has one site (8). Fig. 1A shows that these sumoylation sites are located close to the phosphorylation sites of ERK1/2 that are activated by growth factors such as platelet-derived growth factor and insulin, as reported previously (4, 5). To examine whether there is any causal linkage between the phosphorylation and sumoylation occurring at these closely situated amino acid residues, we focused on analyzing SREBP-2 in the subsequent experiments simply because it is less cumbersome and complicated to analyze a single sumoylation site localized in SREBP-2 and because the SUMO-conjugated band of SREBP-2 on immunoblots is more sharply distinct than the SUMO-conjugated bands of SREBP-1. When various mutant versions of SREBP-2 lacking one or two phosphorylation sites were expressed in COS-1 cells in the presence of [32P]orthophosphate, the less phosphorylated SREBP-2 was immunoprecipitated by an Ala substitution for Ser432 and/or Ser455 (Fig. 1B), suggesting that they are major phosphorylation sites. The calculated molecular mass of these phosphorylated forms of SREBP-2 is close to that of FLAG-tagged SREBP (∼68 kDa), but distinct from that of the sumoylated form (∼79 kDa) (Fig. 2A), indicating that the phosphorylated forms of SREBP-2 are not sumoylated. Next, we examined sumoylation at Lys464 of these mutant versions of SREBP-2 in cells that expressed HA-tagged SUMO-1. GST pulldown experiments revealed that Ala substitution for Ser455 augmented the SUMO-1 conjugation of SREBP-2 by almost 5-fold (Fig. 1C). Mutation of another phosphorylation site (Ser432) had no effect, but Asp substitution for Ser455 reduced SUMO-1 conjugation by 10% of wild-type SREBP-2. These results suggest that Ser455 is critical for sumoylation at Lys464 and that phosphorylation at Ser455 and an Asp substitution that mimics phosphoserine might negatively regulate SUMO-1 conjugation.
FIGURE 1.
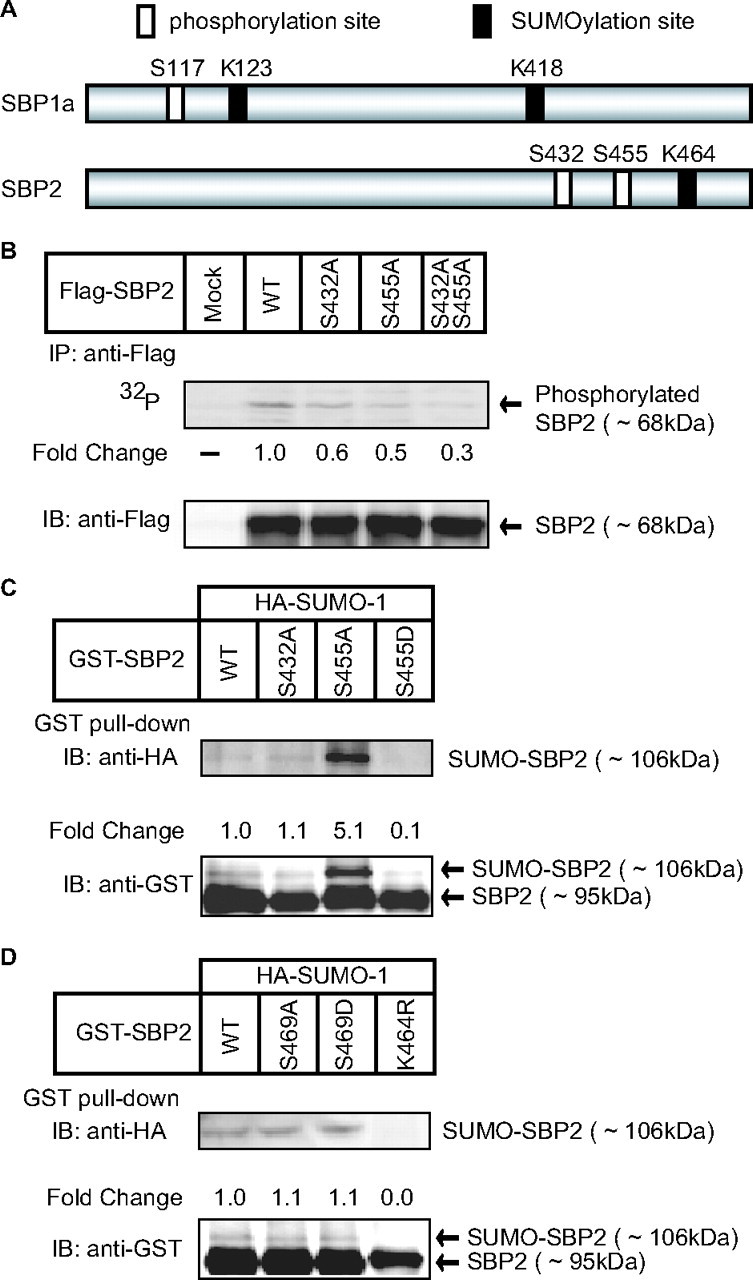
Importance of Ser455 for sumoylation at Lys464 in SREBP-2. A, a schematic diagram of SREBP-1 and -2 is shown. White rectangles indicate the major phosphorylation sites by ERK, and black rectangles indicate the sumoylation sites (4, 5, 8). B, COS-1 cells were transfected with one of the expression plasmids for FLAG-SREBP-2. After a 12-h preincubation with a phosphate-free medium, the cells were radiolabeled with [32P]orthophosphate for 4 h. The cell lysates were subjected to immunoprecipitation (IP) with anti-FLAG antibodies, and the signals were quantified with a Fujifilm FLA-3000 image analyzing system. Aliquots of pellets of immunoprecipitates were subjected to immunoblotting (IB) with anti-FLAG antibodies. The signals were quantified with a Fujifilm LAS-3000 LuminoImager. -Fold change was calculated by the ratio of the intensity between the immunoprecipitation and immunoblot signals. The intensity of wild-type (WT) SREBP-2 was set as 1. C and D, COS-1 cells were transfected with expression plasmids for HA-SUMO-1 and GST-SREBP-2 (wild-type or mutant) as indicated. 36 h later, the cells were harvested, lysed, and subjected to GST pulldown with glutathione-Sepharose resins as described under “Experimental Procedures.” Aliquots of GST pulldown were subjected to SDS-PAGE and immunoblotting with anti-HA or anti-GST antibodies. -Fold change was calculated by the ratio of the intensity between the SUMO-conjugated (∼106 kDa) and unconjugated (∼95 kDa) SREBP-2 signals. The ratio of wild-type SREBP-2 was set as 1. The same results were obtained in more than three separate experiments.
FIGURE 2.

Effect of ERK activation by IGF-1 on the sumoylation of SREBP-2. A, COS-1 cells were transfected with expression plasmids for HA-SUMO-1 and FLAG-SREBP-2 (wild-type (WT) or mutant). The cells were treated with 30 nm IGF-1 for the indicated periods after a 6-h preincubation with a serum-free medium. The whole cell extracts (WCE) were subjected to immunoprecipitation (IP) with anti-FLAG antibodies. Aliquots of whole cell extracts and pellets of immunoprecipitates were subjected to SDS-PAGE and immunoblot (IB) analysis. The whole cell extracts were immunoblotted with antiphospho-ERK (p-ERK) or anti-ERK antibodies. The pellets of immunoprecipitates were immunoblotted with anti-HA or anti-FLAG antibodies. -Fold change was calculated by the ratio of the intensity between the SUMO-conjugated (∼79 kDa) and unconjugated (∼68 kDa) SREBP-2 signals. The ratio at 0 time was set as 1. B, COS-1 cells were transfected with an expression plasmid for FLAG-SREBP-2. After a 12-h preincubation in a phosphate-free medium, the cells were radiolabeled with [32P]orthophosphate for 6 h in the presence of 30 nm IGF-1 for the indicated periods. The cell lysates were subjected to immunoprecipitation with anti-FLAG antibodies. Aliquots of pellets of immunoprecipitates were subjected to immunoblotting with anti-FLAG antibodies. -Fold change was calculated by the ratio of the intensity between the immunoprecipitation and immunoblot signals. The ratio at 0 time was set as 1. The same results were obtained in more than three separate experiments.
A previous study demonstrated that SREBP-2 has a highly conserved motif (ΨKXEXXSP; Lys464 and Ser469) for phosphorylation-dependent SUMO modification of multiple targets such as GATA-1 and heat shock factors (10). To explore the possibility that Ser469 is involved in SUMO-1 modification of SREBP-2, mutant versions of GST-SREBP-2 were expressed in COS-1 cells, and their sumoylation was analyzed. Neither Ala nor Asp substitution for Ser469 had any effect on SUMO modification at Lys464 (Fig. 1D). Taken together, it is likely that phosphorylation at Ser455, which is thought to be enhanced by growth factor stimuli (5), but not at Ser469, affects SUMO-1 conjugation at Lys464 of SREBP-2. In the case of SREBP-1a, we found that Ser117, a phosphorylation site targeted by growth factor stimuli (4), is also critical for SUMO-1 conjugation at Lys123, which is a predominant sumoylation site of SREBP-1a (supplemental Fig. S1).
IGF-1-activated ERK Inhibits SUMO-1 Modification of SREBP-2—Next, we examined which growth factor efficiently stimulates the ERK cascade in COS-1 cells and found that IGF-1 effectively activated the ERK pathway (Fig. 2A). Activation of ERK was observed by 6 h after IGF-1 treatment. When cells expressed FLAG-tagged wild-type SREBP-2, SUMO-1 modification of SREBP-2 was diminished after IGF-1 treatment in a time-dependent manner (Fig. 2A). Because wild-type SREBP-2 was highly (5-fold) phosphorylated at 3 and 6 h after IGF-1 treatment (Fig. 2B), it is likely that the preceding phosphorylation at Ser455 in turn inhibited the sumoylation at Lys464 observed at 6 h (Fig. 2A). On the other hand, mutant SREBP-2 with an Ala substitution for Ser455 was continuously sumoylated, even after the activation of ERK (Fig. 2A). The S455A mutant was slightly phosphorylated (∼1.5-fold) in response to IGF-1 treatment (Fig. 2B).
When cells were treated with the ERK cascade inhibitor U0126, SUMO-1 modification of SREBP-2 was augmented time-dependently (Fig. 3A). In this experiment, HepG2 cells, in which the basal level of phosphorylated ERK was higher than in COS-1 cells, were used to inactivate the ERK activities by U0126. There was no alteration in SUMO-1 modification of SREBP-2(S455A). When COS-1 cells were treated with both IGF-1 and U0126, the IGF-1 effect on sumoylation was abolished (Fig. 3B). Furthermore, sumoylation of endogenous SREBP-2 was observed in HepG2 cells cultured under cholesterol-depleted conditions to increase in the amount of the nuclear active form of SREBP-2. Immunoblot analysis of nuclear extracts using anti-SREBP-2 antibodies revealed that the bands corresponding to the mature form of SREBP-2 and sumoylated SREBP-2 were observed only when cells were cultured under the cholesterol-depleted conditions (Fig. 3C, first and second lanes). In the presence of U0126, the upper band increased, as shown in Fig. 3A, but significantly decreased in the absence of NEM, a desumoylation inhibitor (third and fourth lanes), suggesting that the upper band is likely to correspond to sumoylated SREBP-2. These results clearly indicate that phosphorylation at Ser455 stimulated by IGF-1 treatment strongly inhibits SUMO-1 conjugation at Lys464 of SREBP-2.
FIGURE 3.
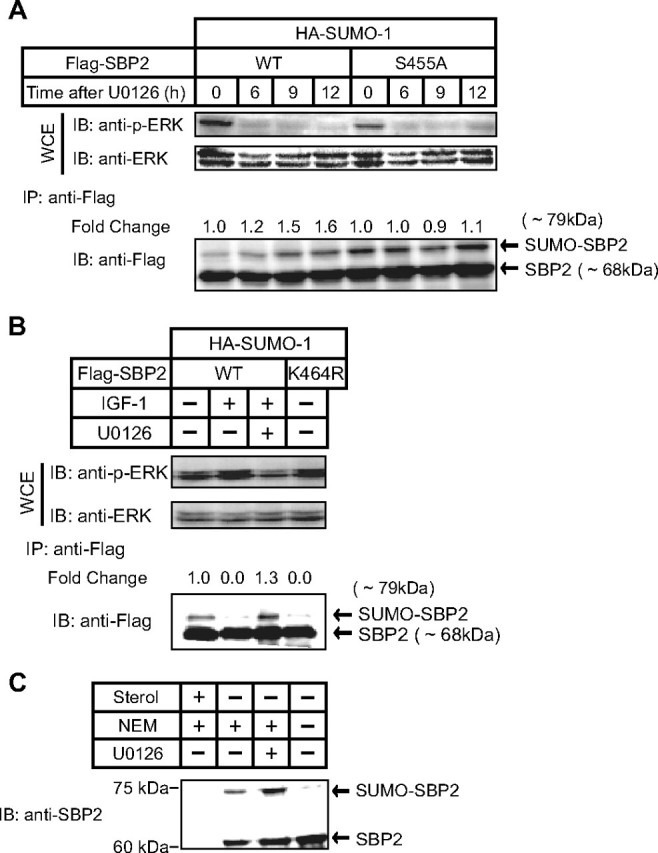
Effect of an ERK inhibitor (U0126) on the sumoylation of SREBP-2. A, HepG2 cells were transfected with expression plasmids for HA-SUMO-1 and FLAG-SREBP-2 (wild-type (WT) or mutant). The cells were treated with 20 μm U0126 for the indicated periods, and the whole cell extracts (WCE) were subjected to immunoprecipitation (IP) with anti-FLAG antibodies. Aliquots of whole cell extracts and pellets of immunoprecipitates were subjected to SDS-PAGE and immunoblot (IB) analysis. The whole cell extracts were immunoblotted with anti-phospho-ERK (p-ERK) or anti-ERK antibodies. The pellets of immunoprecipitates were immunoblotted with anti-FLAG antibodies. -Fold change was calculated by the ratio of the intensity between the SUMO-conjugated (∼79 kDa) and unconjugated (∼68 kDa) SREBP-2 signals. The ratio at 0 time was set as 1. B, COS-1 cells were transfected with expression plasmids for HA-SUMO-1 and FLAG-SREBP-2 (wild-type or mutant). The cells were treated with 30 nm IGF-1 and/or 20 μm U0126 for 4 h, and the whole cell extracts were analyzed in the same manner as described for Fig. 2C. -Fold change was calculated by the ratio of the intensity between the SUMO-conjugated (∼79 kDa) and unconjugated (∼68 kDa) SREBP-2 signals. The ratio without IGF-1 and U0126 was set as 1. C, HepG2 cells were cultured with medium A containing 5% lipoprotein-deficient serum supplemented with either 1 μg/ml 25-hydroxycholesterol plus 10 μg/ml cholesterol (cholesterol-loaded conditions; Sterol+) or 50 μm pravastatin (HMG-CoA reductase inhibitor) plus 50 μm sodium mevalonate (cholesterol-depleted conditions; Sterol-) for 12 h to increase the amount of endogenous active SREBPs in the nucleus. The cells (Sterol-) were treated with or without 20 μm U0126 for 6 h and harvested with nuclear extraction buffer with or without 10 mm NEM (8, 9). The nuclear extracts were subjected to SDS-PAGE and immunoblotted with anti-SREBP-2 antibodies. The same results were obtained in more than three separate experiments.
IGF-1 Treatment Induces the Expression of SREBP Target Genes through Stimulation of SREBP Transcriptional Activities—We have reported previously that SUMO-1 modification of SREBP-1 and -2 suppresses their transcriptional activities (8). Based on the finding that IGF-1 reduces SUMO modification of SREBP-2, it seems likely that IGF-1 treatment of COS-1 cells enhances the expression of SREBP target genes. To investigate this possibility, luciferase assays using various reporter genes (the human LDLR, squalene synthase, and hydroxymethylglutaryl (HMG)-CoA synthase promoters) were performed in COS-1 cells cultured under sterol-depleted conditions to increase the amount of nuclear forms of endogenous SREBPs. The promoter activities of three SREBP target genes were highly up-regulated under the sterol-depleted condition (3–8-fold induction by depletion) (data not shown) and were further elevated by IGF-1 treatment for 12 h (Fig. 4A). When the mutant reporter gene of the LDLR promoter lacking an SRE sequence was used, there was not any induction of luciferase activities for 12 h (data not shown), suggesting that the observed increase in the promoter activities in Fig. 4A was caused by SREBP effects. Although a previous report demonstrated that growth factors increased the amount of the nuclear SREBPs by up-regulating SREBP processing through the action of phosphatidylinositol 3-kinase (11), in the current experiment, the amounts of nuclear SREBP-1 and -2, which were fully increased by sterol depletion, were not further elevated by IGF-1 treatment (Fig. 4B, second and third lanes in all panels). Therefore, it is likely that the augmented SREBP activities occur by blocking SUMO conjugation rather than stimulating SREBP processing, which is responsible for the IGF-1-induced luciferase activities observed in Fig. 4A. Although we observed nonspecific bands (marked by asterisks) just above the mature SREBP-2 bands (marked by arrows) in Fig. 4B (second panel), it is likely that the upper nonspecific bands are not sumoylated SREBP-2 bands because the COS-1 cell lysates were prepared without NEM, a desumoylation inhibitor, which was essential for detection of sumoylated SREBP-2 in HepG2 cells (Fig. 3C). To further confirm the IGF-1-mediated SREBP activation, we employed a heterologous Gal4 system using expression plasmids encoding a nuclear form of SREBP-2 with or without a mutation coupled to the DNA-binding domain of yeast Gal4. This assay system allows direct evaluation of the effect of IGF-1 on the transcriptional activity of SREBP-2, independent of the possible effect on SREBP processing. Consistent with our previous finding (8), mutation of the sumoylation site (K464R) stimulated the transcriptional activity of SREBP-2 by >3-fold, but mutation of the phosphorylation site (S455A) had no effect (Fig. 4C). IGF-1 treatment for longer than 6 h significantly transactivated wild-type SREBP-2, but not mutant SREBP-2 missing the sumoylation (K464R) or phosphorylation (S455A) site. These results suggest that phosphorylation at Ser455 induced by IGF-1 treatment enhances the transcriptional activity through blocking sumoylation at Lys464. Supplemental Fig. S2 also shows that IGF-1 stimulated the SREBP-1a transcriptional activity as long as one of the sumoylation sites (Lys123) and the phosphorylation site (Ser117) were intact, suggesting that the phosphorylation at Ser117 induced by IGF-1 treatment is crucial for regulating the transcriptional activity of SREBP-1a.
FIGURE 4.
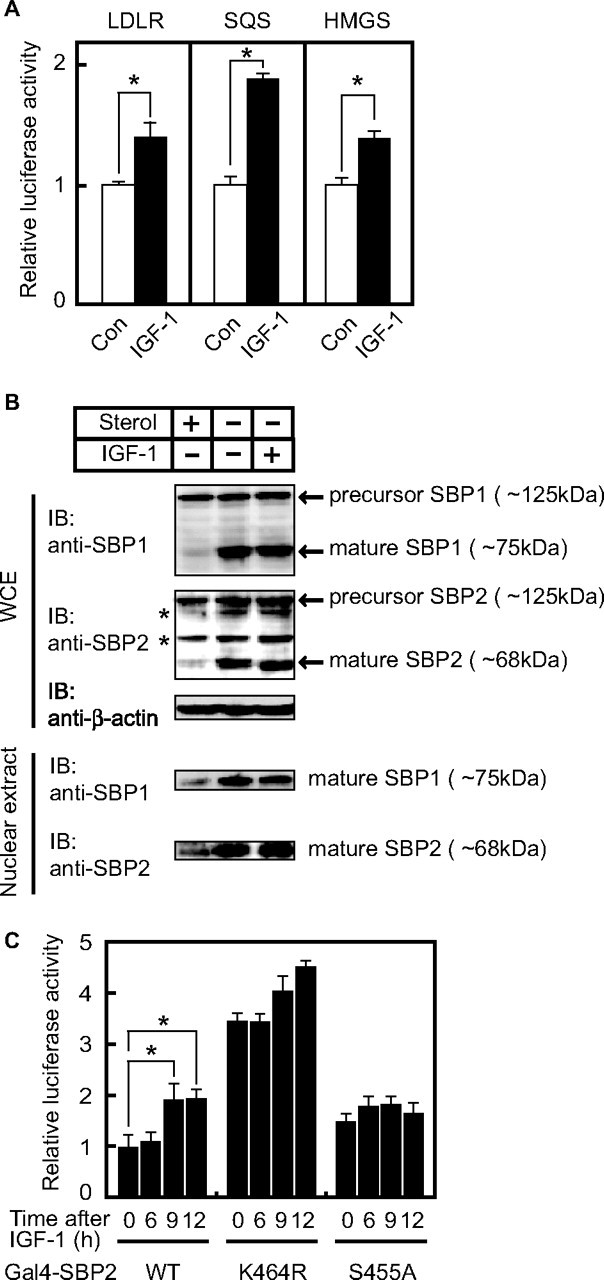
IGF-1 stimulates the expression of SREBP target genes through inhibition of sumoylation. A, COS-1 cells were transfected with 100 ng of reporter plasmid containing the human LDLR, squalene synthase (SQS), or HMG-CoA synthase (HMGS) promoter and 10 ng of pRL-CMV. The cells were cultured under cholesterol-depleted conditions for 24 h to increase the amount of endogenous active SREBPs in the nucleus. After the cells were further incubated with or without IGF-1 for 12 h, luciferase assays were performed. The promoter activities in the absence of IGF-1 were set as 1. All data are presented as the means ± S.D. of three independent experiments performed in triplicate. *, p < 0.05. Con, control. B, COS-1 cells were cultured under cholesterol-depleted or cholesterol-loaded conditions for 12 h and further incubated with 30 nm IGF-1 for 6 h. All cells were incubated with 50 μm N-acetyl-Leu-Leu-norleucinal for the last 4 h to stabilize the nuclear form of SREBPs. The whole cell extracts (WCE) and nuclear extracts were subjected to immunoblotting (IB) with anti-SREBP-1, anti-SREBP-2, or anti-β-actin antibodies. The asterisks mark the nonspecific bands observed in all lanes. The same results were obtained in more than three separate experiments. C, COS-1 cells were transfected with expression plasmid for 300 ng of Gal4-SREBP-2 (either wild-type (WT) or mutant), 100 ng of pG5-Luc containing five copies of the Gal4-binding sites, and 10 ng of pRL-CMV and cultured under cholesterol-loaded conditions for 48 h to remove the possibility of unexpected effects of endogenous SREBPs in the nucleus. The cells were incubated with 30 nm IGF-1 for the indicated periods, and luciferase assays were then performed. The promoter activities driven by pGAL4-SREBP-2WT without IGF-1 treatment were set as 1. All data are presented as the means ± S.D. of three independent experiments performed in triplicate. *, p < 0.05.
HDAC3 Is Involved in the Sumoylation-mediated Suppression of the SREBP Transcriptional Activity—The above findings prompted us to examine what kinds of repressors are recruited to suppress the SREBP transcriptional activity in response to SUMO conjugation. To verify the involvement of histone deacetylases (HDACs) in the suppression, we first treated cells with TSA, an HDAC inhibitor. The SREBP transcriptional activity was enhanced when cells expressing Gal4-SREBP-2 (wild-type) were incubated with TSA, whereas there was no effect on the K464R mutant activity, suggesting that part of sumoylation-mediated reduction in SREBP activity is caused by the functions of HDACs (Fig. 5A). On the other hand, a previous study demonstrated that TSA attenuates the activity of ERK kinases (27). This effect should have down-regulated the SREBP transcriptional activity in this assay, but the result was just the opposite. To clarify which family member of HDAC can make a complex with SREBP-2, both GST-SREBP-2 and HA-HDAC were expressed in HEK293 cells, and GST pulldown experiments were performed. Immunoblotting with anti-HA antibody revealed that HDAC3 from among the family members was exclusively co-precipitable with GST-SREBP-2 (data not shown). When GST-SREBP-2(K464R), an SREBP-2 mutant lacking the sumoylation site, or GST was expressed, these proteins did not make a complex with HA-HDAC3 (Fig. 5B), suggesting that SUMO conjugation is required for formation of the complex. GST-SREBP-1 also formed a complex with HDAC3 (data not shown).
FIGURE 5.
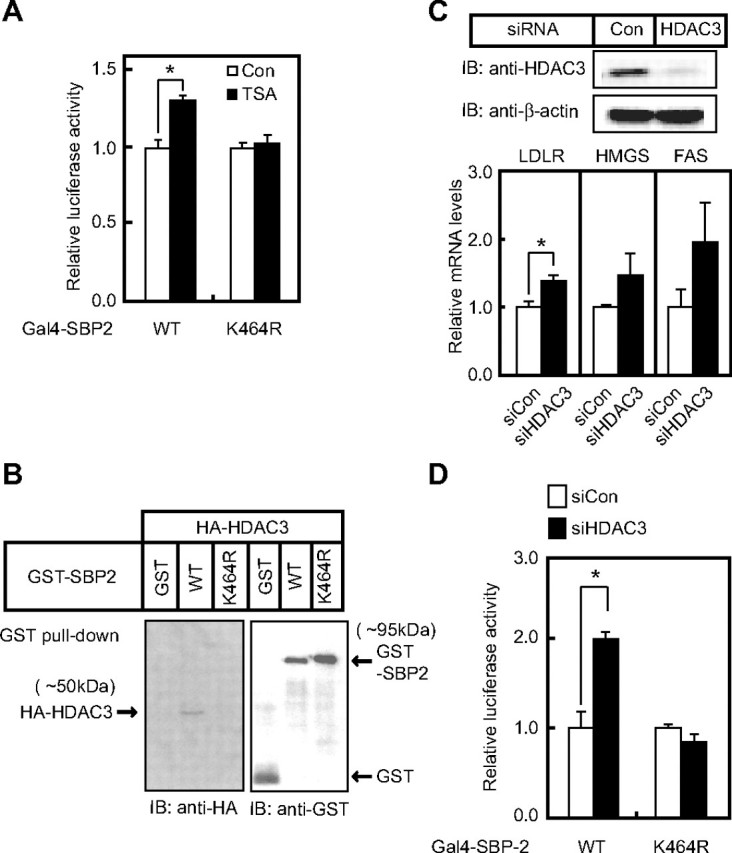
Involvement of HDAC3 in sumoylation-dependent suppression of SREBP transcriptional activity. A, HEK293 cells were transfected with expression plasmid for 300 ng of Gal4-SREBP-2 (either wild-type (WT) or mutant), 100 ng of pG5-Luc containing five copies of the Gal4-binding sites, and 10 ng of pRL-CMV and cultured under cholesterol-loaded conditions for 48 h. The cells were incubated with 330 nm TSA for the last 6 h. The promoter activities without TSA treatment were set as 1. All data are presented as the means ± S.D. of three independent experiments performed in triplicate. *, p < 0.05. B, HEK293 cells were transfected with expression plasmids for HA-HDAC3 and either GST-SREBP-2 (wild-type or K464R) or GST. The cells were analyzed in the same manner as described for Fig. 1C. The same results were obtained in more than three separate experiments. C, HepG2 cells were transfected with either control (Con) or HDAC3 siRNA and incubated for 72 h. The cell lysates were subjected to SDS-PAGE and immunoblotting (IB) with anti-HDAC3 or anti-β-actin antibodies, and total RNA was subjected to real-time PCR analysis. Relative mRNA levels were obtained after normalizing to the S17 rRNA protein transcript. All data are presented as the means ± S.D. of three independent experiments performed in triplicate. *, p < 0.05. HMGS, HMG-CoA synthase; FAS, fatty acid synthase. D, HepG2 cells were transfected with either control (siCon) or HDAC3 (siHDAC3) siRNA together with 300 ng of expression plasmid for Gal4-SREBP-2 (either wild-type or mutant K464R), 100 ng of pG5-Luc, and 10 ng of pRL-CMV. After a 4-day incubation, luciferase assays were performed. The promoter activities in the presence of the control siRNA were set as 1. All data are presented as the means ± S.D. of three independent experiments performed in triplicate. *, p < 0.05.
We next examined whether knockdown of endogenous HDAC3 with siRNA enhances the expression of SREBP target genes. When endogenous HDAC3 was significantly decreased by siRNA, the mRNA levels of the LDLR, HMG-CoA synthase, and fatty acid synthase were elevated (Fig. 5C). Moreover, the effect of HDAC3 knockdown on SREBP-2 transcriptional activity was confirmed using a heterologous Gal4 system. When cells were treated with siRNA for HDAC3, the luciferase activities driven by Gal4-SREBP-2, but not by the Gal4-SREBP-2 mutant, were significantly augmented (Fig. 5D). HDAC2 siRNA did not have any effect in the same assay system (data not shown). These results indicate that sumoylation of wild-type SREBP-2 suppresses its transcriptional activity through the recruitment of HDAC3.
We further examined whether HDAC3 knockdown leads to an increase in the LDLR protein. The uptake of DiI-labeled LDL in HepG2 cells was stimulated by HDAC3 siRNA treatment under cholesterol-depleted conditions, but not under cholesterol-loaded conditions, where only a negligible amount of active SREBPs exist in the nucleus (Fig. 6A). Moreover, chromatin immunoprecipitation assays revealed that wild-type SREBP-2 was capable of recruiting HDAC3 to the SRE sequence in the human LDLR promoter, whereas mutation of the sumoylation site (Lys464) abolished this recruitment (Fig. 6B). The same results were also obtained using a primer set covering an SRE in the human HMG-CoA synthase gene promoter (data not shown). These results indicate that the increased uptake of DiI-labeled LDL by HDAC3 knockdown is mediated by removal of an HDAC3-containing corepressor complex located on the SRE sequence in the LDLR promoter. During the entire course of in vitro GST pulldown experiments using recombinant HDAC3 and sumoylated SREBP-2 proteins under various conditions, no direct interaction between the two proteins was verified (data not shown). It seems likely that HDAC3 works as one of the components forming a corepressor complex that is recruited in response to sumoylation of SREBP-2 (see schematic diagram in Fig. 6B).
FIGURE 6.
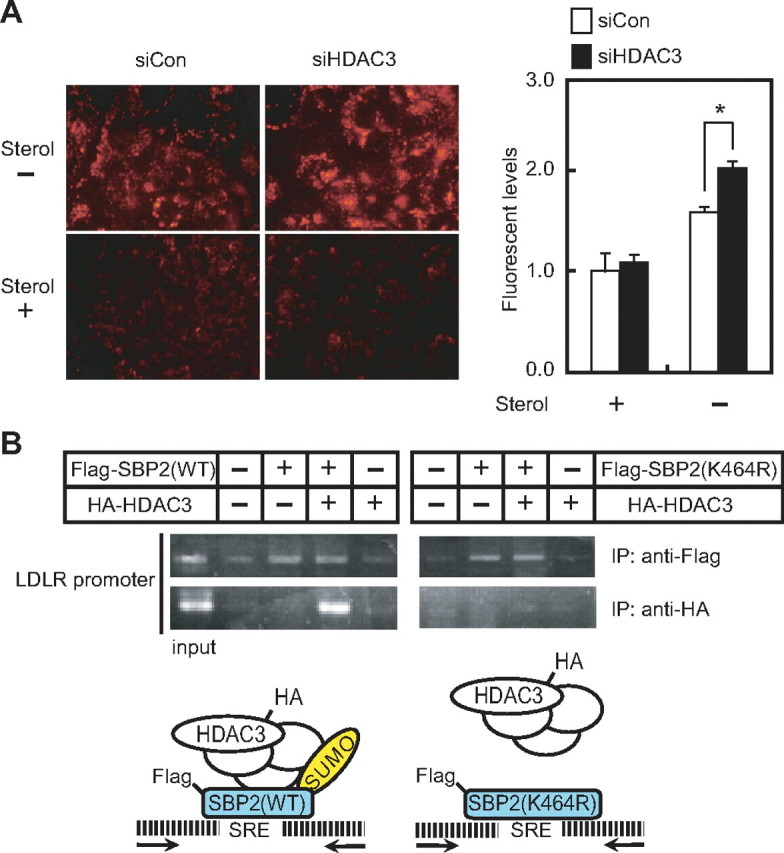
HDAC3 knockdown stimulates the uptake of DiI-labeled LDL in HepG2 cells. A, HepG2 cells were transfected with either the control (siCon) or HDAC3 (siHDAC3) siRNA and then cultured for 60 h. The cells were further cultured under cholesterol-loaded (Sterol+) or cholesterol-depleted (Sterol-) conditions for 12 h. The cells were placed in medium supplemented with 10 μg/ml DiI-labeled LDL for the last 4 h. The cells were washed, fixed, and examined by fluorescence microscopy (left). The fluorescence intensity was measured using a fluorometer (right). The fluorescence intensity in the cells cultured under cholesterol-loaded conditions with the control siRNA was set as 1. Data are represented as the means ± S.D. of three independent experiments performed in triplicate. *, p < 0.05. B, HEK293 cells were transfected with expression plasmids for HA-HDAC3 and FLAG-SREBP-2 (either wild-type (WT) or K464R) and processed for chromatin immunoprecipitation analyses as described under “Experimental Procedures.” After immunoprecipitation (IP) with anti-HA or anti-FLAG antibodies, PCR was performed with the primer set covering an SRE in the human LDLR promoter. 1% of the samples before immunoprecipitation were subjected to PCR as a control (input). The same results were obtained in more than three separate experiments. A schematic diagram of the SREBP·HDAC3 complex formation on the SRE sequence is shown.
DISCUSSION
In this study, we have shown that sumoylation of SREBPs constitutively attenuates their transcriptional activity through recruitment of a corepressor complex that includes HDAC3. This attenuation is reversed by activation of the MAPK pathway driven by IGF-1, which causes reduced sumoylation of SREBPs. Based on the fact that the phosphorylation sites of SREBP-1 and -2 are located within 10 amino acid residues of the sumoylation sites, it is likely that the phosphorylation of SREBPs interferes with sumoylation. Although in these experiments we focused only on SUMO-1, we have confirmed that SREBP-2 is modified by a single molecule of SUMO-2 or -3 at Lys464 (supplemental Fig. S3). Growth hormone stimuli appear to enhance the uptake and synthesis of cholesterol and fatty acid to provide a sufficiency of the membrane lipids required for the cell growth that takes place in a short period of time accompanied by a rapid increase in the SREBP transcriptional activities through an inhibition of SUMO conjugation. Growth factor-induced activation of SREBPs through the MAPK pathway has been proposed as one of the mechanisms responsible for the up-regulation of lipid synthesis in a subset of cancer cells (12). It was also demonstrated previously that growth factors, including platelet-derived growth factor and insulin, stimulate the proteolytic activation of SREBPs, thereby inducing membrane lipid synthesis in an SREBP-dependent manner (11). It is therefore conceivable that both the quantitative increase and qualitative activation of SREBPs are achieved by the sequential induction of signal transduction pathways in response to growth factor stimuli.
Previous studies demonstrated that phosphorylation of the transcription factor Elk-1 in response to activation of the MAPK pathway also results in a rapid loss of SUMO modification of Elk-1, leading to an increase in its transcriptional activity (13). Unlike SREBPs, Elk-1 is modified by SUMO-1 and -2 at both Lys230 and Lys249 at a distance from the phosphorylation site (Ser383) and thereafter recruits a corepressor complex containing HDAC2 (14). Another SUMO-conjugated transcription factor, p300, recruits HDAC6 through direct protein-protein interaction (15). These HDAC family members are thought to contain a SUMO-binding motif ((V/I)X(V/I)(V/I)) that exists in nearly all proteins known to be involved in SUMO-dependent processes (16). Based on the fact that HDAC3 also contains a couple of SUMO-binding motif-like sequences, we examined a direct interaction between SUMO-conjugated SREBP-2 and HDAC3. Using an Escherichia coli overexpression system in which substrate proteins are efficiently modified with SUMO by the action of SUMO-activating enzyme and SUMO carrier protein stably expressed in the bacteria (17), highly sumoylated GST-SREBP-2 protein was prepared, and GST pulldown experiments together with in vitro translation of 35S-labeled HDAC3 were carried out. There was not any detectable interaction observed in this assay (data not shown), suggesting that HDAC3 is likely to be recruited in a SUMO-dependent manner as a component of a corepressor complex. In this study, we found that SREBP-2(S455A), which lacks a phosphorylation site, is highly modified by SUMO-1 because of the absence of phosphorylation-dependent interference with sumoylation (Fig. 1C). Contrary to our expectation, this mutant form barely formed a complex with HDAC3 (supplemental Fig. S4), and its transcriptional activity was almost equal to that of wild-type SREBP-2 (Fig. 4C). This indicates that the recruitment of HDAC3, not SUMO conjugation itself, is critical for the SUMO-dependent repression of the transcriptional activity of SREBP-2. Furthermore, a chimeric protein composed of mutant SREBP-2 lacking a sumoylation site (K464R) and the SUMO-1 molecule (amino acids 1–95 lacking two Gly residues at the C terminus) fused to the C terminus of SREBP-2 also did not form a complex with HDAC3 (supplemental Fig. S4). These results indicate that both the SUMO-1 molecule binding to Lys464 and its neighboring amino acid sequence of SREBP-2, including Ser455, must be recognized by an as yet unidentified associated protein that is a component of the HDAC3-containing corepressor complex. The investigation to identify components of the complex other than HDAC3 and to elucidate the mechanism for the SUMO-dependent protein-protein interaction is now under way.
With the increase in the number of proteins modified by SUMO, which have been identified over the past 10 years, it has become obvious that the effects of SUMO conjugation are diverse and largely dependent on the function of the protein targeted for sumoylation. A recent finding that a subunit of kainate receptors on the neuronal surface is conjugated with SUMO in response to kainate or glutamate, thereafter being rapidly internalized, highlights a novel role for this protein modification on the membrane (18), whereas sumoylation has been almost exclusively studied in the context of nuclear proteins. Most of these nuclear proteins are in some way involved in regulating transcription. For example, sumoylation of the transcription factors c-Myb and IRF-1, SREBPs, and several nuclear receptors results in down-regulation of their transcriptional activities (8, 19–21). However, the mechanism by which SUMO modification reduces their transcriptional capacity and how the switching between sumoylation and desumoylation of the substrate proteins is regulated remain unresolved. This study shows that sumoylation of SREBPs triggers recruitment of an HDAC3-containing corepressor complex, which reduces their transcriptional activity. In addition, phosphorylation of SREBPs through activation of the MAPKs by growth factors competes with sumoylation of the target proteins, thereby enhancing their transcriptional function of stimulating membrane lipid synthesis. Furthermore, it is still puzzling that IGF-1 treatment or HDAC3 knockdown brings about an almost 2-fold increase in the transcriptional activity of SREBPs (Figs. 4C and 5D), even though a small portion of SREBPs are detected in the SUMO-conjugated form upon immunoblotting (Fig. 1, C and D). It would appear that sumoylation is essential to recruiting an HDAC3-containing corepressor complex and that this SREBP/HDAC3-containing complex, once formed, does not require the SUMO moiety any longer, even though SUMO is rapidly separated from SREBPs through a sumoylation-and-desumoylation cycle. It is furthermore conceivable that the phosphorylation of SREBPs induced by growth factor stimuli, if this occurred prior to sumoylation, would strongly inhibit recruitment of the corepressor complex, resulting in an effective up-regulation of SREBP transcriptional activity. To identify the protein component(s) of the HDAC3-containing corepressor complex responsible for the direct interaction, mass spectrometry studies using recombinant SUMO-conjugated SREBP protein are now being undertaken.
Supplementary Material
Acknowledgments
We thank Dr. Michael S. Brown for insightful discussion and comments. We also thank Drs. Minoru Yoshida, Takayuki Ohshima, Tamotsu Nishida (Mie University), and Hisato Saito (Kumamoto University) for expression plasmids. We are grateful to Dr. Kevin Boru (Pacific Edit) for review of the manuscript.
This work was supported by research grants from the Ministry of Education, Science, Sports, and Culture of Japan and the Program for Promotion of Basic Research Activities for Innovative Biosciences. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S4.
Footnotes
The abbreviations used are: SREBPs, sterol regulatory element-binding proteins; LDL, low density lipoprotein; ER, endoplasmic reticulum; SCAP, SREBP cleavage-activating protein; INSIG, insulin-inducing gene; MAPK, mitogen-activated protein kinase; ERK, extracellular signal-regulated kinase; SUMO, small ubiquitin-like modifier; IGF-1, insulin-like growth factor-1; LDLR, LDL receptor; NEM, N-ethylmaleimide; TSA, trichostatin A; GST, glutathione S-transferase; HA, hemagglutinin; SRE, sterol regulatory element; siRNA, small interfering RNA; DiI, 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine; HMG, hydroxymethylglutaryl; HDACs, histone deacetylases.
References
- 1.Goldstein, J. L., DeBose-Boyd, R. A., and Brown, M. S. (2006) Cell 124 35-46 [DOI] [PubMed] [Google Scholar]
- 2.Bengoechea-Alonso, M. T., Punga, T., and Ericsson, J. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 11681-11686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kotzka, J., Müller-Wieland, D., Roth, G., Kremer, L., Munck, M., Schürmann, S., and Knebel, B. (2000) J. Lipid Res. 41 99-108 [PubMed] [Google Scholar]
- 4.Roth, G., Kotzka, J., Kremer, L., Lehr, S., Lohaus, C., Meyer, H. E., Krone, W., and Müller-Wieland, D. (2000) J. Biol. Chem. 275 33302-33307 [DOI] [PubMed] [Google Scholar]
- 5.Kotzka, J., Lehr, S., Roth, G., Avci, H., Knebel, B., and Müller-Wieland, D. (2004) J. Biol. Chem. 279 22404-22411 [DOI] [PubMed] [Google Scholar]
- 6.Punga, T., Bengoechea-Alonso, M. T., and Ericsson, J. (2006) J. Biol. Chem. 281 25278-25286 [DOI] [PubMed] [Google Scholar]
- 7.Sundqvist, A., Bengoechea-Alonso, M. T., Ye, X., Lukiyanchuk, V., Jin, J., Harper, J. W., and Ericsson, J. (2005) Cell Metab. 1 379-391 [DOI] [PubMed] [Google Scholar]
- 8.Hirano, Y., Murata, S., Tanaka, K., Shimizu, M., and Sato, R. (2003) J. Biol. Chem. 278 16809-1681912615929 [Google Scholar]
- 9.Hirano, Y., Yoshida, M., Shimizu, M., and Sato, R. (2001) J. Biol. Chem. 276 36431-36437 [DOI] [PubMed] [Google Scholar]
- 10.Hietakangas, V., Anckar, J., Blomster, H. A., Fujimoto, M., Palvimo, J. J., Nakai, A., and Sistonen, L. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 45-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Demoulin, J. B., Ericsson, J., Kallin, A., Rorsman, C., Rönnstrand, L., and Heldin, C. H. (2004) J. Biol. Chem. 279 35392-35402 [DOI] [PubMed] [Google Scholar]
- 12.Swinnen, J. V., Heemers, H., Deboel, L., Foufelle, F., Heyns, W., and Verhoeven, G. (2000) Oncogene 19 5173-5181 [DOI] [PubMed] [Google Scholar]
- 13.Yang, S. H., Jaffray, E., Hay, R. T., and Sharrocks, A. D. (2003) Mol. Cell 12 63-74 [DOI] [PubMed] [Google Scholar]
- 14.Yang, S. H., and Sharrocks, A. D. (2004) Mol. Cell 13 611-617 [DOI] [PubMed] [Google Scholar]
- 15.Girdwood, D., Bumpass, D., Vaughan, O. A., Thain, A., Anderson, L. A., Snowden, A. W., Garcia-Wilson, E., Perkins, N. D., and Hay, R. T. (2003) Mol. Cell 11 1043-1054 [DOI] [PubMed] [Google Scholar]
- 16.Song, J., Durrin, L. K., Wilkinson, T. A., Krontiris, T. G., and Chen, Y. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 14373-14378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Uchimura, Y., Nakamura, M., Sugasawa, K., Nakao, M., and Saitoh, H. (2004) Anal. Biochem. 331 204-206 [DOI] [PubMed] [Google Scholar]
- 18.Martin, S., Nishimune, A., Mellor, J. R., and Henley, J. M. (2007) Nature 447 321-327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Biesà, J., Markus, J., and Wolff, L. (2002) J. Biol. Chem. 277 8999-9009 [DOI] [PubMed] [Google Scholar]
- 20.Nishida, T., and Yasuda, H. (2002) J. Biol. Chem. 277 41311-41317 [DOI] [PubMed] [Google Scholar]
- 21.Park, J., Kim, K., Lee, E. J., Seo, Y. J., Lim, S. N., Park, K., Rho, S. B., Lee, S. H., and Lee, J. H. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 17028-17033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sato, R., Miyamoto, W., Inoue, J., Terada, T., Imanaka, T., and Maeda, M. (1999) J. Biol. Chem. 274 24714-24720 [DOI] [PubMed] [Google Scholar]
- 23.Ohshima, T., Koga, H., and Shimotohno, K. (2004) J. Biol. Chem. 279 29551-29557 [DOI] [PubMed] [Google Scholar]
- 24.Arimura, N., Horiba, T., Imagawa, M., Shimizu, M., and Sato, R. (2004) J. Biol. Chem. 279 10070-10076 [DOI] [PubMed] [Google Scholar]
- 25.Nakahara, M., Furuya, N., Takagaki, K., Sugaya, T., Hirota, K., Fukamizu, A., Kanda, T., Fujii, H., and Sato, R. (2005) J. Biol. Chem. 280 42283-42289 [DOI] [PubMed] [Google Scholar]
- 26.Kanayama, T., Arito, M., So, K., Hachimura, S., Inoue, J., and Sato, R. (2007) J. Biol. Chem. 282 10290-10298 [DOI] [PubMed] [Google Scholar]
- 27.Lee, H. J., Baek, K. H., Jeon, A. H., Kin, S. J., Jang, K. L., Sung, Y. C., Kim, C. M., and Lee, C. W. (2003) Oncogene 22 3853-3858 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.