

Abstract
HspA, a member of the GroES chaperonin family, is a small protein found in Helicobacter pylori with a unique histidine- and cysteine-rich domain at the C terminus. In this work, we overexpressed, purified, and characterized this protein both in vitro and in vivo. The apo form of the protein binds 2.10 ± 0.07 Ni2+ or 1.98 ± 0.08 Bi3+ ions/monomer with a dissociation constant (Kd) of 1.1 or 5.9 × 10-19 μm, respectively. Importantly, Ni2+ can reversibly bind to the protein, as the bound nickel can be released either in the presence of a chelating ligand, e.g. EDTA, or at an acidic pH (pH½ 3.8 ± 0.2). In contrast, Bi3+ binds almost irreversibly to the protein. Both gel filtration chromatography and native electrophoresis demonstrated that apo-HspA exists as a heptamer in solution. Unexpectedly, binding of Bi3+ to the protein altered its quaternary structure from a heptamer to a dimer, indicating that bismuth may interfere with the biological functions of HspA. When cultured in Ni2+-supplemented M9 minimal medium, Escherichia coli BL21(DE3) cells expressing wild-type HspA or the C-terminal deletion mutant clearly indicated that the C terminus might protect cells from high concentrations of external Ni2+. However, an opposite phenomenon was observed when the same E. coli hosts were grown in Bi3+-supplemented medium. HspA may therefore play a dual role: to facilitate nickel acquisition by donating Ni2+ to appropriate proteins in a nickel-deficient environment and to carry out detoxification via sequestration of excess nickel. Meanwhile, HspA can be a potential target of the bismuth antiulcer drug against H. pylori.
Helicobacter pylori infects nearly half of the population worldwide (1). It is a Gram-negative, microaerophilic, spiral organism and the only discovered species that can survive through the highly acidic environment produced by the gastric epithelium. Colonization of H. pylori in the human gastric mucosa leads to chronic gastritis, peptic ulcer, and even gastric carcinogenesis (2–4). Survival of this bacterium under acidic conditions (pH ∼ 3) heavily relies on its capability of producing two nickel-containing enzymes, urease and hydrogenase. Urease, which accounts for up to 10% of the total cellular proteins, is essential for colonization and virulence (5). It catalyzes the hydrolysis of urea to yield carbamate and ammonia and thus neutralizes its immediate environments (6), whereas hydrogenase utilizes hydrogen to provide the bacterium a high-energy substrate for respiratory-based energy generation (7, 8).
Similar to other essential elements, nickel is accumulated in certain kinds of cells, but its effects are double-edged: it operates as a cofactor for many metalloenzymes. However, it also exhibits toxic effects as a result of interfering with normal metal-protein binding and generating reactive oxygen. Therefore, regulation of nickel ions is crucial in nickel-desiring species such as H. pylori. For example, nickel-responsive gene regulation in H. pylori can be mediated by the nickel uptake regulator NikR. Working more sophisticatedly than other homologues such as the Escherichia coli NikR repressor (9), H. pylori NikR not only inhibits the transcription of the nickel importer gene, e.g. nixA, but also directly activates genes relevant to nickel utilization, such as ureA-ureB, hpn, and hpn-like (10).
It has been demonstrated that nickel can be imported into H. pylori primarily via the nickel-cobalt transporter NixA (11), which is required for effective H. pylori colonization (12). An ABC-type transporter has also been proposed as an alternative for H. pylori, although direct evidence of nickel import through this transporter has not been reported (13). Once nickel enters into the cells, it is likely to be delivered to apoenzymes such as urease and/or hydrogenase through certain accessory proteins (14). For example, the insertion of nickel ions into urease in H. pylori may require UreE, UreF, UreG, and UreH. The metalloprotein UreE has been proposed to be responsible for the delivery of Ni2+ to the UreDFG-urease apoprotein complex in Klebsiella aerogenes, probably through the histidine-rich region at the C terminus (15). Surprisingly, it has been found that the H. pylori UreE does not have a histidine-rich C-terminal region like most other homologues. A very recent study on H. pylori demonstrated that interaction between nickel-binding accessory proteins HypA and UreE would allow nickel to be transferred eventually from HypA to the apo-urease (16). Besides, two histidine-rich proteins that were reported only in H. pylori, Hpn and Hpn-like, were also suggested to be relevant to nickel utilization (17). However, it is still far from clear in terms of nickel homeostasis in H. pylori even if a considerable amount of research has been carried out.
On the other hand, the growth of H. pylori can be greatly inhibited by bismuth drugs, which have been widely used to treat various gastrointestinal diseases and H. pylori infection together with antibiotics (18). Although the antimicrobial activity of bismuth against H. pylori has been demonstrated (19), the mechanism of the action is not well understood. A number of proteins and enzymes have been demonstrated previously as intracellular targets of bismuth (20). Using a metalloproteomic approach, it was found that intracellular levels of four proteins (HspA, HspB, NapA, and TsaA) were significantly up- or down-regulated after bismuth treatment (21), and thus bismuth-binding proteins in H. pylori were identified. Moreover, HspA was found to be one of the Bi3+-binding proteins by immobilized bismuth affinity chromatography. Binding of bismuth to these proteins is likely to disrupt their functions and may also interfere with nickel homeostasis in H. pylori.
HspA, a homologue of the GroES chaperonin family, has been discovered previously to be essential for urease activation by gene complementary experiments (22). It is capable of binding nickel to the His- and Cys-rich C terminus, which consists of 27 amino acid residues and is absent in most of the other GroES members (23). In this study, we cloned the hspA gene from H. pylori strain 26695, overexpressed the protein in E. coli, and biophysically characterized the protein, in particular, for its interaction with Ni2+ and Bi3+. We also constructed an HspA mutant with the C-terminal deletion and compared nickel/bismuth resistance between the wild-type and mutant expression hosts.
EXPERIMENTAL PROCEDURES
Sequence Analyses
The amino acid sequence of H. pylori HspA was submitted to the well known BLAST program (24) against the data base of non-redundant protein sequences. The homologues belonging to either human disease-related microorganisms or evolutionary representatives were retrieved for sequence analyses (www.ncbi.nlm.nih.gov). As a result, the alignments were divided into two groups, one for microbial pathogens and the other for model species. Multiple alignments were performed by ClustalW (version 1.83) (25), and similarities to H. pylori HspA were calculated by the Needle program of the EMBOSS package (26). In addition, a pattern designated as the chaperonin Cpn10 signature was identified within the HspA sequence using ScanProsite (27, 32) against the ExPASy PROSITE Database (28).
Bacterial Strains and Growth Conditions
H. pylori strain 26695 was obtained from the American Type Culture Collection (ATCC 700392) and cultured on Brucella agar plates (Oxoid) supplemented with 10% defibrinated horse blood and Campylobacter Selective Supplement® at 37 °C under microaerophilic conditions (5% CO2, 4% O2, and 91% N2) produced by AnaeroGen® (Oxoid). All plasmids used were maintained or amplified in E. coli XL1-Blue (Stratagene). Both wild-type and mutant HspA were overexpressed in E. coli BL21(DE3) (Stratagene). All E. coli strains were cultured in LB medium (U. S. Biochemical Corp.) or M9 minimal medium. Ampicillin (100 μg/ml) was added to the culture medium when necessary.
DNA Manipulation
The genomic DNA was extracted from H. pylori culture using a QIAamp® DNA mini kit (Qiagen), from which the hspA gene was cloned and ligated to the pGEM-T easy vector (Promega). The expression vector pET-32a (Novagen) was double-digested by NdeI/SacI to remove all expression tags and then used to construct pET-32-hspAwt (expressing wild-type hspA) and pET-32-hspAmt (expressing the C-terminal deletion mutant). All DNA sequences were confirmed to have no mutation by DNA sequencing.
Expression and Purification
BL21(DE3) cells expressing wild-type HspA were collected and resuspended in a running buffer containing 20 mm Tris-HCl, 500 mm NaCl, and 5 mm imidazole at pH 7.9. Cells were disrupted by sonication and ultracentrifuged to remove cell debris. The supernatant was loaded onto a 5-ml NiSO4-impregnated HiTrap® chelating HP column (Amersham Biosciences). Unspecifically bound proteins were removed by the running buffer with 80 mm imidazole, and HspA was eluted with 1 m imidazole. To avoid oxidization and remove trace amounts of metal ions, the eluted protein was supplemented with 5 mm dithiothreitol and 25 mm EDTA overnight. Prior to biophysical characterization, the solution was buffer-exchanged into 50 mm Tris-HCl and 100 mm NaCl, pH 6.8, by a HiTrap desalting column (Amersham Biosciences). All of the above procedures were carried out at room temperature at a flow rate of 2 ml/min.
Protein Analyses
Protein concentrations were determined using a BCA assay kit (Novagen) with concentration-known bovine serum albumin as a standard. The protein purity was examined by high-resolution gel filtration and anion-exchange chromatography. UV absorption at 280 nm was recorded as the protein indicator when purified HspA was eluted from a Superdex 75 100/300 GL column (Amersham Biosciences) or a MonoQ anion-exchange column (GE Healthcare). The purity of the protein was judged from the elute profiles. To examine the fidelity of the expression, the expressed protein was digested in gel by trypsin and then subjected to MALDI-TOF3 mass spectrometry analysis with an m/z scan range from 800 to 3,500. The free thiolate contents of HspA were determined using the improved Ellman's test (29).
Electronic Absorption Spectroscopy
All electronic absorption spectra were recorded on a Varian Cary 3E UV-visible spectrophotometer with a 1-cm cuvette at room temperature. For titration experiments, typically, a 30–50 μm HspA solution in 20 mm Hepes at pH 7.2 was used, and prior to addition of metal ions, an aliquot of concentrated solution of tris(2-carboxyethyl)phosphine hydrochloride (0.1 m) was added to give rise to a concentration of 6-fold HspA. The spectrum of HspA was recorded after equilibration for 30 min. Aliquots of stock solutions of Ni2+ (as 1 mm NiSO4) or Bi3+ (as 1 mm Bi-NTA) were then added to the cuvette, and UV-visible absorption spectra were recorded after at least 30 min of equilibration for each titration. The binding and release of metal ions were monitored by changes in absorbance at 316, 425, and 554 nm for Ni2+ and 364 nm for Bi3+. A large excess of EDTA (∼40-fold metal) was added to Ni2+/Bi3+-saturated HspA solutions at pH 7.2 (or at pH 3.9). Absorbance at 316 nm for Ni2+ or 364 nm for Bi3+ was used to examine the kinetics of nickel or bismuth release. The data were fitted by nonlinear least square best fits based on an exponential function. To investigate the stability of Ni2+/Bi3+-HspA complexes at different pH values, HspA was presaturated with Ni2+ or Bi3+ in 20 mm Hepes at pH 7.2, aliquots of concentrated HCl were subsequently added to the solution, and UV-visible spectra were recorded after equilibrium for about 30 min for each pH value.
Equilibrium Dialysis
The dissociation constants of metal-protein complexes were determined by equilibrium dialysis using an 8-kDa cut-off mini dialysis kit (Amersham Biosciences). A known amount of the protein (300 μl, 20 μm) was loaded into the dialysis tube and placed into 100 ml of dialysis buffer (50 mm Tris-HCl and 100 mm NaCl, pH 6.9) supplemented with appropriate amounts of either Ni2+ (as NiSO4) or Bi3+ (as Bi-NTA) from 0.5 to 8 μm for overnight incubation at room temperature. The metal concentrations inside and outside the dialysis tubes were determined by inductively coupled plasma mass spectrometry. The data were analyzed by a Hill model (Origin®, version 7.5) using the one-site binding equation: Y = Bmax*X/(Kd + X), where Bmax is the maximal binding capacity, and Kd is the concentration of metal ions required to reach half-maximal binding, i.e. the dissociation constant. For Bi3+, the dissociation constant from HspA was derived by Kd = Kd′/Ka, where Kd′ is the dissociation constant of Bi-NTA determined from current equilibrium dialysis experiments, and Ka is the formation constant of Bi-NTA.
Oligomeric State of HspA
The native state of apo-HspA and the effects of Ni2+/Bi3+ on protein oligomeric states were examined by gel filtration chromatography on a Superdex 75 100/300 GL column, which was precalibrated using the standard proteins of the LMW calibration kit (Amersham Biosciences). A protein solution (250-μl volume) in its apo- or metal-bound form was sequentially injected into the column and eluted with a buffer containing 50 mm Tris-HCl and 100 mm NaCl, pH 6.8, at a flow rate of 0.6 ml/min. The metal-saturated protein was prepared by addition of ∼3 molar eq of Ni2+ (as NiSO4) or Bi3+ (as CBS) relative to the protein. To further verify the results obtained from gel filtration chromatography, protein electrophoresis using 13% polyacrylamide gel was employed in nondenaturing buffer (25 mm Tris and 192 mm glycine). The protein was visualized by Coomassie Brilliant Blue staining, and the molecular masses were estimated with reference to standard proteins (GE Healthcare).
Metal Susceptibility
To investigate the potential physiological function of the unique C terminus of HspA, an expression vector carrying the C-terminal deletion hspA gene (pET-32-hspAmt) was constructed and transformed into E. coli BL21(DE3). Using a procedure similar to that used for wild-type HspA, the mutant was overexpressed by isopropyl β-d-thiogalactopyranoside induction, and the expressed protein was identified via biological mass data.
Dose-dependent Metal Resistance—To compare the metal capacity of the wild type and mutant, two E. coli cultures hosting wild-type hspA (pET-32-hspAwt) and mutant hspA (pET-32-hspAmt), respectively, were grown in M9 minimal medium supplemented with gradient metal concentrations. The metal resistance of host cells has been tested according to the following procedure. After isopropyl β-d-thiogalactopyranoside induction, E. coli BL21(DE3) cells were harvested by spin-down and washed three times with M9 minimal medium to completely remove nutrient-rich LB medium. The collected cells were diluted to a density of 0.2 (A550) with M9 minimal medium as starter cultures. To rule out possible uncertainty caused by different quantities of expressed proteins, a fraction of starter cultures was subjected to SDS-PAGE analysis to ensure that the two proteins were expressed at a similar level. The starter cultures were then grown for ∼18 h at 37 °C in M9 minimal medium containing gradient concentration metals using 10 mm NiSO4 or 10 mm CBS, respectively. The absorbance values at 550 nm were recorded for each bacterial culture.
Metal Resistance of Cell Growth at Different Times—To investigate the effect of metal on the growth of E. coli cells, starter cultures (A550 = 0.5) expressing the wild type or mutant in M9 minimal medium were grown in the presence of either 5 μm NiSO4 or 500 μm CBS. Metal resistance was presented using A550 values of bacterial cultures at 0, 1, 2, 3, 4, 5, 6, 7, 19, and 24 h for nickel and 0, 2, 4, 7, 20, 22, 24, and 26 h for CBS. All A550 values were normalized by At = x/At = 0 to yield a growth curve against time.
Each set of data was independently recorded in triplicate, represented as the mean ± S.D., and the statistical difference between the wild type and mutant was analyzed by Student's t test.
RESULTS
Sequence Analyses—The sequence of H. pylori HspA was aligned with its GroES homologues from several pathogenic microorganisms (Fig. 1A) as well as from various model species (Fig. 1B). The results demonstrate that HspA is relatively conserved with >40% sequence identities compared with other GroES homologues. However, two distinctions between H. pylori HspA and all other members of the GroES family are noted. H. pylori HspA contains a unique His- and Cys-rich C terminus in its primary structure (supplemental Fig. S1). In addition, it features a histidine residue (His45) and two cysteine residues (Cys51 and Cys53) in the conserved N terminus that are not frequently found in other members of the GroES family. The alignment for the widely spread model species was also used to construct a phylogenetic tree (data not shown), where homologues from bacterium, fish, amphibian, insect, bird, and mammal form well separated groups as expected, in agreement with an early report (30). When using the ScanProsite program (27) to scan the amino acid sequence of HspA against the PROSITE Database (28), a fragment, FQPLGERVLVERLEEENKTSSGIII, hit the chaperonin Cpn10 signature (LIVMFY)-X-P-(ILT)-X-(DEN)-(KR)-(LIVMFA)3-(KREQS)-X8–9-(SG)-X-(LIVMFY)3 (supplemental Fig. S1), which taxonomically ranges from Archaebacteria prokaryotes to eukaryotes, suggesting a common thread among all GroES homologues during molecular evolution.
FIGURE 1.
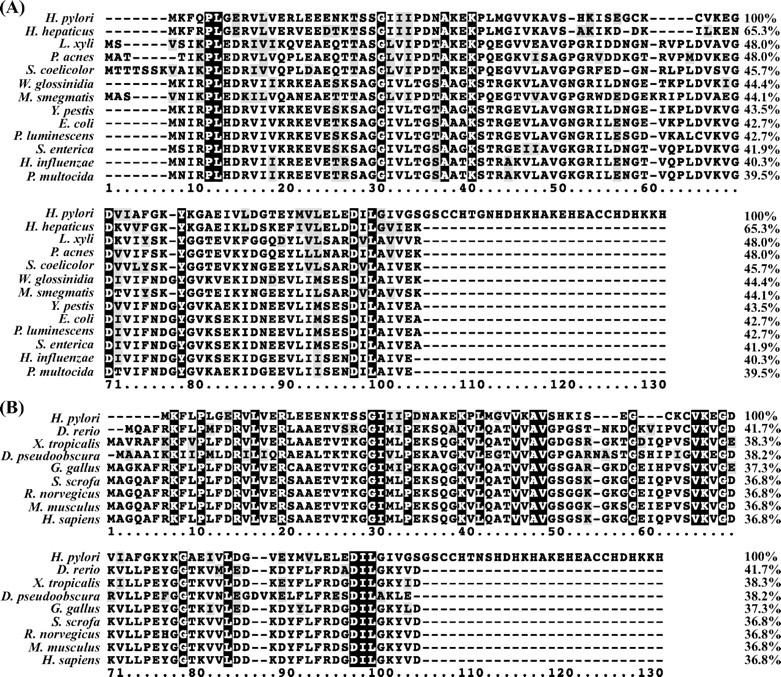
Sequence alignment of H. pylori HspA with homologues from other pathogenic microorganisms (A) or model species (B). Identical residues are marked with a black background, and conserved ones are shaded in light gray. The sequence alignment was done by ClustalW (version 1.83), and the sequence similarities were calculated by the Needle program from the EMBOSS package (version 3.0) using the H. pylori homologue as the template. The following sequences were retrieved from the GenBank™ Protein Databank. A, HspA (H. pylori; AAP51175.1), co-chaperonin GroES (Helicobacter hepaticus; NP_860731), co-chaperonin GroES (Leifsonia xyli xyli; YP_062808), 10-kDa chaperonin (Propionibacterium acnes; YP_056460), co-chaperonin GroES (Streptomyces coelicolor; NP_628919), GroES (Wigglesworthia glossinidia; AAK07426), chaperonin GroS (Mycobacterium smegmatis; YP_885961), co-chaperonin GroES (Yersinia pestis; NP_403998), co-chaperonin GroES (E. coli; NP_418566.1), co-chaperonin GroES (Photorhabdus luminescen; NP_931325), co-chaperonin GroES (Salmonella enterica; YP_219195), co-chaperonin GroES (Haemophilus influenzae; NP_438700), and co-chaperonin GroES (Pasteurella multocida; NP_246043). B, heat shock 10-kDa protein (Sus scrofa; AAP32465), heat shock 10-kDa protein (Homo sapiens; NP_002148), chaperonin 10 (Rattus norvegicus; AAC53361.1), chaperonin 10 (Mus musculus; AAF67345.1), chaperonin 10 (Gallus gallus; NP_990398.1), chaperonin 10 (Xenopus tropicalis; AAH77653.1), chaperonin 10 (Danio rerio; AAH71419.1), GA10877-PA (Drosophila pseudoobscura; EAL31011.1), and HspA (H. pylori; AAP51175.1).
Protein Expression, Purification, and Identification—Because the genomic sequence of H. pylori strain 26695 was the first to be determined among all H. pylori strains (31), the hspA gene was amplified from the genomic DNA of this strain through Taq PCR. The cloned DNA fragment was sequenced, and it was confirmed to be identical to the H. pylori hspA gene found in the GenBank™ Databank (accession number AAP51175.1) (supplemental Fig. S1). After PCR cloning and appropriate restriction digestion, the gene sequence was inserted into the pET-32a expression vector for high-level expression in E. coli BL21(DE3). The apparent molecular mass (15.5 kDa) of the recombinant protein was slightly higher than predicted (13 kDa), which occurred on many occasions for histidine-rich proteins (15, 22). To make our analyses more biologically relevant, HspA was expressed in its native form without any tag fused to the protein. Because of the histidine/cysteine-rich residues located on the C terminus, HspA exhibited a high affinity toward nickel-charged iminodiacetate-derivatized agarose, and thus the protein was purified effectively by a single-step purification protocol for histidine-tagged fusion proteins (see “Experimental Procedures”). The protein purity was examined by either size-exclusive gel filtration or anion-exchange chromatography, which showed the prepared protein was free of significant contaminants (supplemental Fig. S2). Data from MALDI-TOF mass spectra hit several peptide fragments belonging to H. pylori HspA at m/z 935.4434, 1000.5278, 1105.5508, 1215.6147, and 1957.9424, corresponding to EGDVIAFGK (calculated 935.4832), EKPLMGVVK (1000.5859), MKFQPLGER (1105.5823), TSSGIIIPDNAK (1215.658), and LEEENKTSSGIIIPDNAK (1958.0077).
Interaction of Ni2+/Bi3+ with HspA—Complexation of metal ions, e.g. Ni2+, to proteins often led to the production of new absorption bands in the UV-visible spectrum, which has often been used to monitor metal binding to proteins. When a 0.3 molar eq of Ni2+ was added to HspA solution, five new bands appeared in the UV difference spectra at ∼248, 290, 316, 423, and 547 nm, indicative of Ni2+ binding to HspA. The intensities of these bands increased upon further addition of Ni2+ and leveled off at a molar ratio of Ni2+ to HspA of 2.0, indicative of binding two Ni2+ ions/HspA monomer (Fig. 2A). Based on a previous report of Ni2+ binding to small molecules (33), the strong band at 248 nm is probably attributed to N(His) → Ni2+ ligand-to-metal charge transfer transition, and the bands at 290 and 316 nm are assigned to S(Cys) → Ni2+ ligand-to-metal charge transfer transition. The other two bands are possibly due to d → d′ transition of Ni2+. Similarly, titration of Bi3+ led to two new bands at ∼262 and 364 nm in the UV-visible difference spectra (Fig. 2B), which is characteristic for binding of bismuth to cysteine residues (34). By monitoring the absorbance change at 364 nm versus the Bi3+ amounts added, the stoichiometry of Bi3+ binding to HspA was determined to be 2, identical to that of Ni2+.
FIGURE 2.
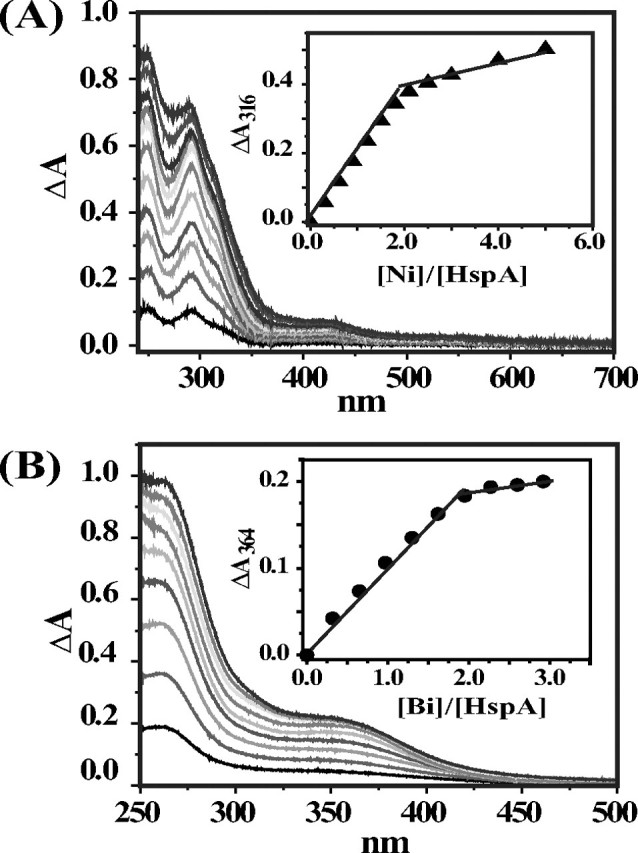
UV-visible difference spectra of apo-HspA (38 μm in 20 mm Hepes buffer, pH 7. 4, in the presence of 6-fold tris(2-carboxyethyl)phosphine hydrochloride at room temperature) after the addition of different molar equivalents of NiSO4 (A) or Bi-NTA (B). The insets are titration curves plotted at 316 nm for Ni2+ binding and 364 nm for Bi3+ binding, indicative of binding two metal ions/monomer.
To further study the stoichiometry of Ni2+/Bi3+ binding to HspA and to determine the binding affinities, equilibrium dialysis experiments were performed, and the results are shown in Fig. 3. The data were fitted by a one-site binding Hill plot, which gave rise to Bmax values of 2.10 ± 0.07 for Ni2+ and 1.98 ± 0.08 for Bi3+, indicating that two metal ions bound per monomer of HspA. The binding constant (Kd) was determined to be 1.09 ± 0.14 μm for Ni2+ by fitting the data. Because of a very high affinity of Bi3+ for HspA, NTA was used as a competing ligand in determination of the binding constant of Bi3+ and the protein. The binding constant (Kd′) of Bi-NTA and HspA was determined to be 0.21 ± 0.01 μm. Based on the formation constant of Bi-NTA of log Ka = 17.55 (35), the binding constant of Bi3+ and HspA was therefore calculated to be Kd′/Ka = 5.9 ± 0.03 × 10-19 μm.
FIGURE 3.
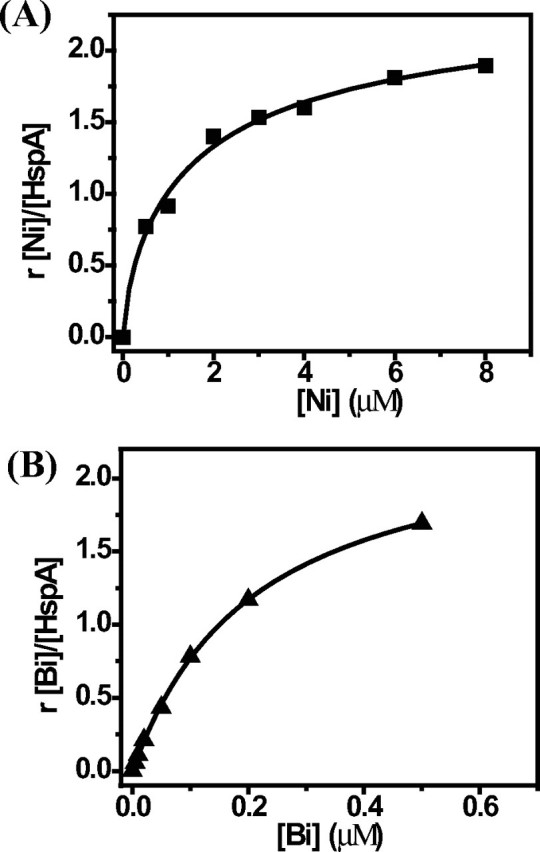
Binding profiles of HspA by equilibrium dialysis. HspA (20 μm in 300 μl of protein solution) was dialyzed against 100 ml of buffer containing 50 mm Tris-HCl and 100 mm NaCl at pH 7.2 with serial concentrations of NiSO4 (A) or Bi-NTA (B) at room temperature overnight. The graphs show Hill plots of the molar ratios of bound Ni2+/Bi3+ to HspA against concentrations of metals in the dialysis buffer.
To investigate whether metal binding to the protein is reversible, we monitored the intensities of the absorption bands (316 and 364 nm for Ni2+ and Bi3+, respectively) upon altering the pH of metal-saturated HspA solution. The absorbance was normalized to the percentages of bound metal. As shown in Fig. 4, the percentages of bound Ni2+ remained almost unchanged from pH 7 to 5 but decreased gradually upon further lowering of the pH and sharply decreased at pH <4.5. The bound Ni2+ was completely released at pH 2.5, and the profile was fitted with pH½ (a pH for half of the metal dissociated from the protein) ∼3.8 ± 0.2. In contrast, the percentage of bound Bi3+ only decreased slightly (∼10%) under identical conditions, and only 30% Bi3+ was released even when the pH was lowered to 1.5. This suggests that binding of Bi3+ to HspA is irreversible and hardly released from the protein, confirming its extremely strong binding. Note that within the pH range from 4.8 to 5.1, the absorbance cannot be detected precisely, presumably due to partial protein precipitation, as the predicated pI value is ∼6.1.
FIGURE 4.

pH-dependent profiles of Ni2+/Bi3+ release from metal-bound HspA. Metal-saturated HspA in 20 mm Hepes buffer was used for pH titration and monitored by the decrease in absorbance of the peaks at 316 nm for Ni2+-HspA (▪) and 364 nm for Bi3+-HspA (▴).
Similarly, a competition binding of Ni2+/Bi3+ between HspA and a chelating ligand (e.g. EDTA) was also carried out. A large excess of EDTA (40 molar eq to the metal concentration) was added to metal-saturated HspA solutions at pH 7.2, and absorbance at 316 and 364 nm was monitored for Ni2+ and Bi3+, respectively. The absorbance was again normalized to the percentages of bound metal for convenience. Ni2+ was released from the protein gradually (Fig. 5), and biphasic kinetics were observed with half-lives of 2.0 ± 0.3 and 92.8 ± 1.5 min for the fast and slow steps, respectively. In contrast, Bi3+ remained bound to HspA under identical conditions (Fig. 5). Again, it indicates that the binding of Ni2+ to the protein is reversible, whereas the binding of bismuth to HspA is almost irreversible at neutral pH, probably due to a higher affinity between Bi3+ and the protein. However, the majority of the bound Bi3+ could be extracted rapidly from HspA by excess EDTA in acidic solution (pH 3.9), as evidenced by decreases in absorbance at 364 nm (t½ = 0.11 ± 0.01 min), and the rest could be released relatively slowly (t½ = 5.35 ± 0.54 min) (supplemental Fig. S5).
FIGURE 5.

Kinetics of metal release from HspA. Time-dependent absorbance at 316 nm for Ni2+-HspA (▪) or 364 nm for Bi3+-HspA (▴) was recorded for Ni2+/Bi3+-saturated HspA upon the addition of 40 molar eq of EDTA, pH 7.4, at room temperature. The absorbance at each time point was normalized to percentages to represent the amount of bound metals.
Oligomeric State of HspA—Gel filtration chromatography was used to estimate the molecular masses to unveil the oligomeric states of native HspA in its apo- or metal-bound form. In the presence of 24 molar eq of tris(2-carboxyethyl)phosphine hydrochloride, apo-HspA was eluted at ∼10 ml (Fig. 6A), which corresponded to a molecular mass of 95 kDa and thus indicated a heptamer (theoretically 91 kDa). The effect of metal ions, e.g. Ni2+ and Bi3+, on the oligomeric behavior of HspA was examined by addition of metal ions to the protein (∼4 molar eq) and overnight equilibration. The metal-saturated HspA solutions were loaded onto the column. The nickel-bound form migrated at an approximately similar elution volume (∼10 ml) to apo-HspA, suggesting a similar oligomeric state to its apo form (a heptamer). However, an elution peak for the bismuth-bound HspA was observed at ∼12.5 ml, corresponding to a molecular mass of ∼29 kDa. This indicated that the bismuth-bound form existed predominantly as a dimer (26 kDa), different from both the apo- and Ni2+-bound forms. The same protein samples were also subjected to polyacrylamide gel electrophoresis under nondenaturing conditions. The visualized protein clearly demonstrated a molecular mass migration from a larger size to a smaller one when HspA bound to bismuth, whereas Ni2+-bound protein resembled the apo form (Fig. 6B), in agreement with the gel filtration chromatography data.
FIGURE 6.
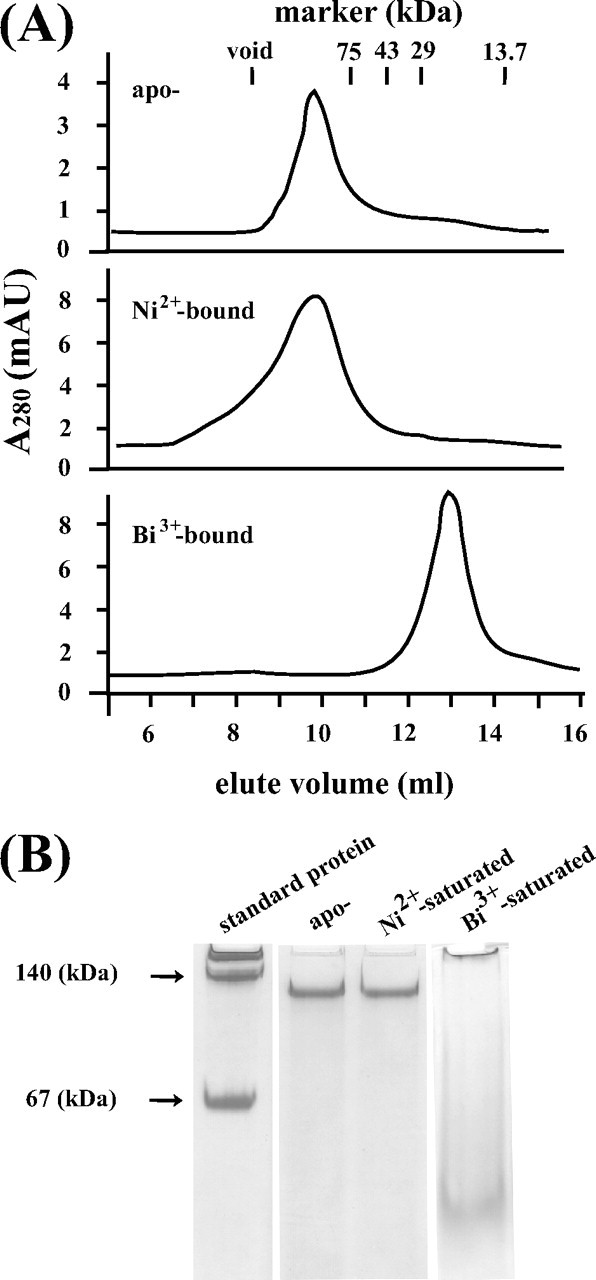
Analysis of the oligomeric states of apo- and metal-bound HspA by gel filtration chromatography and native electrophoresis. A, gel filtration profiles for apo-, nickel-, and bismuth-bound HspA on a Superdex 75 10/300 GL column eluted with a buffer containing 50 mm Tris-HCl and 100 mm NaCl, pH 7.0, at room temperature. Note that bismuth induced protein quaternary structure changes from a heptamer to a dimer. mAU, milli-absorbance units. B, native electrophoresis for apo-, nickel-, and bismuth-bound HspA. The electrophoresis was carried out in 50 mm Tris and 200 mm glycine, pH 8.8, at room temperature.
Metal Susceptibility of E. coli Cells Expressing Wild-type/Mutant HspA—To study the potential roles of the His- and Cys-rich terminus in vivo, the growth of E. coli BL21(DE3) cells expressing the wild type or C-terminal deletion mutant was examined in M9 minimal medium supplemented with metals in a concentration-dependent or time course manner. The absorbances were monitored at 550 nm as a marker of cell growth. To exclude the possible error caused by significantly different quantities of expressed proteins, the starter cultures of the wild type and mutant were induced deliberately to have comparable expression levels of the two proteins (supplemental Fig. S3). Using an A550 value of E. coli culture growing in Ni2+-free medium as a control (100%), the presence of wild-type HspA seemed to be able to partially protect the host from nickel toxicity in a certain range of metal concentrations, from 78% (of the cell number that was cultured without nickel) in 1 μm NiSO4 to 23% in 50 μm. In contrast, E. coli cells expressing the C-terminal deletion mutant were much more susceptible to increasing concentrations of nickel ions, concluded from the dramatically reduced growth rate, from 60% in 1 μm NiSO4 to 10% in 50 μm, compared with the nickel-free control (Fig. 7A). Hence, the addition of nickel to M9 medium inhibited the growth of mutant-expressing E. coli cells much more effectively compared with wild type-expressing E. coli cells. The effect of Bi3+ (as CBS) on E. coli cell growth was determined under similar conditions in M9 minimal medium. Unexpectedly, Bi3+ retarded the growth of the wild-type host more evidently than the mutant one, particularly at a bismuth concentration over 100 μm, where 76% of the mutant-hosting cells survived compared with 62% of the wild type-containing cells in the same medium. Even when 1000 μm CBS was supplemented, E. coli cells with the mutant hspA gene still reached 65% of the cell density cultured in the absence of bismuth; however, the wild-type gene host grew only to 40% cell survival (Fig. 7B).
FIGURE 7.
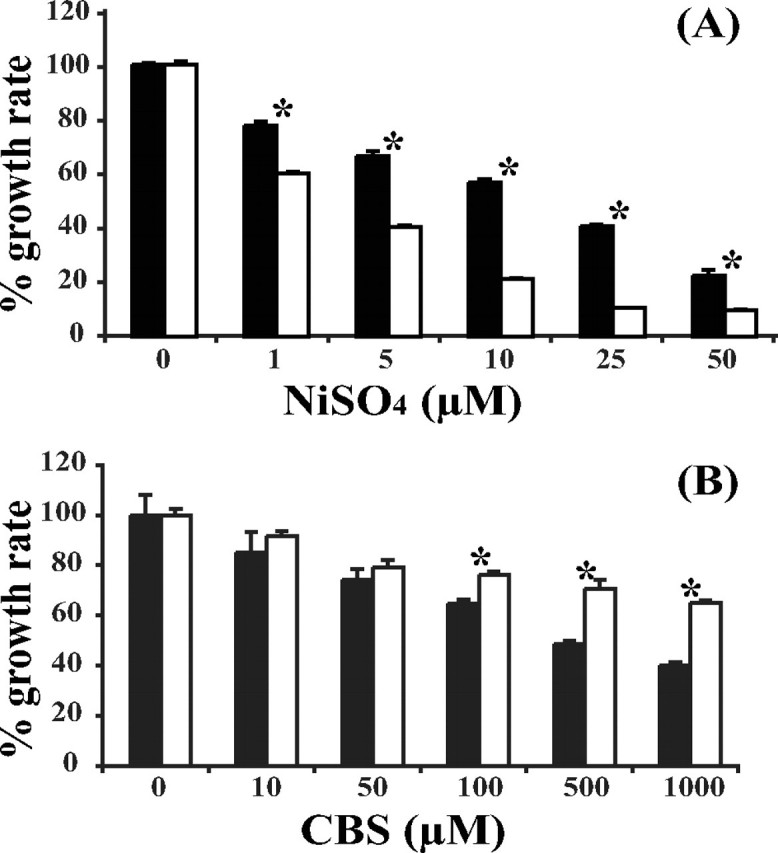
Effects of nickel and bismuth on the growth of E. coli cells expressing wild-type HspA (closed bars) or the C-terminal deletion HspA (open bars). Metal susceptibility is presented by bacterial growth in M9 minimal medium supplemented with NiSO4 (A) or CBS (B). The A550 of E. coli culture was measured as a growth indicator. The growth rate between the wild type and mutant was statistically compared by Student's t test, and asterisks indicate that the two sets of data are significantly different from each other. Each datum point was determined for independently cultured bacteria in triplicate, presented as the mean ± S.D.
The time course of the influence of NiSO4 (5 μm) or CBS (500 μm) was also studied for E. coli BL21 cells hosting the wild-type hspA or mutant gene in which the 3′-coding sequence of the His- and Cys-rich domain had been truncated. When the two E. coli hosts were cultured under identical conditions in M9 minimal medium with 5 μm Ni2+ or 500 μm Bi3+ at 37 °C and starting with similar cell density (A550 ∼ 0.5), they exhibited distinguishable growth curves. In nickel-supplemented medium, the bacteria containing wild-type HspA grew much faster than the bacteria containing the mutant after 3 h. When the two groups of bacteria reached their stationary phase of cell growth after incubation for 19 h, the cell density of E. coli cells expressing the wild type was nearly two times higher than that of the mutant host (Fig. 8A), indicative of a distinct ability of metal detoxification. However, in bismuth-supplemented medium, an opposite result was observed: after being cultured for ∼20 h, the cell densities of the E. coli cells with the mutant hspA gene increased by 4.7-fold of starter culture, whereas the wild-type host increased by merely 3.13-fold (Fig. 8B), suggesting that E. coli could be more resistant to bismuth drugs if the His- and Cys-rich C terminus was removed from HspA.
FIGURE 8.
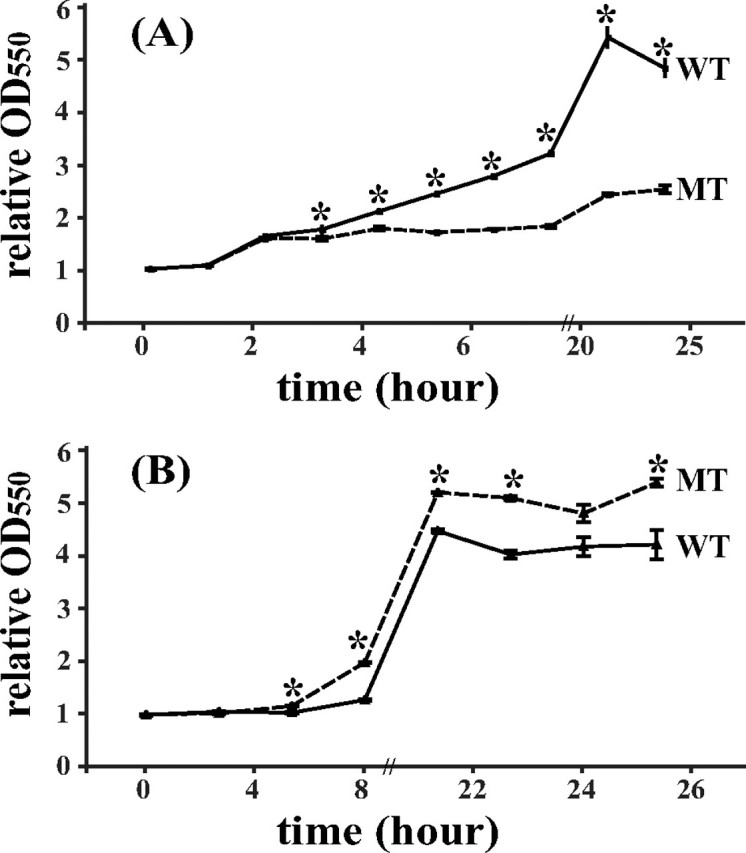
Growth curves of E. coli cells expressing wild-type HspA or C-terminal deletion mutant. E. coli cells were grown in M9 minimal medium supplemented with 5 μm NiSO4 (A) or 500 μm CBS (B). At each selected time point, the ratios of the A550 values at different times versus the value at starter culture (t = 0 h) were used to represent the relative growth rate. The statistical difference between the wild type (WT) and mutant (MT) was estimated by Student's t test, and asterisks indicated that the two sets of data are significantly different from each other. Each datum point was tested independently in triplicate, presented as the mean ± S.D.
DISCUSSION
GroES/GroEL, found in all discovered eubacteria and eukaryotic cells, is a highly conserved chaperone system during evolution (30). It is also named Hsp10/Hsp60 or Cpn10/Cpn60, and its major function is now considered to be recognition and refolding of denatured proteins (36) or induction of the rapid degradation of certain proteins (37). Unlike GroEL, which maintains an outstanding conservation in all kinds of species (38), it is not unusual that some GroES chaperonins vary in different species to meet specific functional requirements. For example, the T4 bacteriophage carries its own GroES copy (gp31) when infecting E. coli because the GroES homologue of the host cannot fold its major capsid protein (gp23) (39). The chloroplast Cpn20 chaperonin, a GroES homologue in plants, is believed to be essential in cellular protein folding, but it carries a unique version of the Cpn10 co-chaperonin that consists of two homologous Cpn10-like domains for full and efficient functioning (40).
It has long been known that metalloproteins are involved in numerous cellular events in prokaryotes or eukaryotes (41), such as ubiquitously found metallothionein (42); ZIP in eukaryotes (43); NikR in bacteria (44), NixA, Hpn, and Hpn-like in H. pylori (17, 45); and UreE in K. aerogenes (46). Many histidine-rich proteins including HspA have been speculated to serve the physiological function of metal homeostasis, e.g. storage and/or detoxification, in vivo (17, 47, 48). The H. pylori GroES homologue was identified previously as an essential factor for the urease maturation, and it could not be replaced by E. coli GroES, as deduced from functional complementation experiments (49). Moreover, HspA might cooperate with E. coli GroEL because H. pylori urease was still highly active even without the hspB gene, the H. pylori homologue of GroEL chaperonin (22). Given the fact that Ni2+ binds to HspA moderately and reversibly, if the binding constants (Kd) in vitro could be used to stand for the conditions in vivo, it is thermodynamically feasible for the histidine-rich proteins such as HspA (Kd of 1.1 μm) and HypB (2.3 μm) to sequester nickel ions from those with relatively lower affinities, e.g. Hpn (7.1 μm) and Hpn-like (5.1 μm), under nickel-deficient conditions. Because HspA was found to be essential for urease activation (49), our results raise the intriguing possibility that HspA subsequently releases bound Ni2+ ions and donates them to appropriate enzyme(s) when necessary.
The strong band at 248 nm of Ni2+-bound HspA is characteristic of N(His) → Ni2+, whereas the bands at 290 and 316 nm are assigned to S(Cys) → Ni2+ ligand-to-metal charge transfer absorption. A similar UV-visible absorption pattern was observed previously for another Ni2+-protein, e.g. E. coli NikR, a Ni2+ transcriptional regulator with known His and Cys coordination to the metal in a square planar geometry (50). This suggests that Ni2+ is probably coordinated to both nitrogen and thiolate centers, i.e. His and Cys residues, in HspA. To further confirm the coordination of Ni2+, a peptide containing the His- and Cys-rich domain (GSCCHTGNHDHKHAKEHEACCHDHKKH) was synthesized, and our UV-visible data showed that the peptide bound two Ni2+ ions (data not shown), in agreement with the intact protein, which also suggested that the C terminus should be the major nickel-binding domain if not the only one.
Previously, we characterized the structure of CBS, the recognition of Bi3+ by the iron and zinc storage proteins, and recently the histidine-rich protein Hpn (20, 34, 51–53). Using a metalloproteomic approach, we screened both Ni2+- and Bi3+-binding proteins in H. pylori. HspA was found to be not only a Ni2+-binding protein but also one of few bismuth-binding proteins (21). Although HspA also binds two Bi3+ ions, the binding is much stronger (Kd of 5.9 × 10-19 μm) than that of Ni2+, comparable with Bi3+ binding to the tripeptide glutathione (34). In combination with the observation of the characteristic UV-visible absorption at 364 nm upon addition of Bi3+ to the protein, we therefore speculate that Bi3+ binds thiolate groups of the protein, i.e. cysteine residues. Different from Ni2+, binding of Bi3+ to the protein is almost irreversible due to extremely tight binding, indicating that bismuth may disrupt the function of HspA in vivo once it binds to the protein. Unexpectedly, the binding of Bi3+ induces the protein dissociation from its native heptamer to a dimer. Therefore, bismuth may interfere with the biological functions of HspA by altering quaternary structures of the protein (Fig. 9). Such a phenomenon was observed previously for other drugs (e.g. Taxcol).
FIGURE 9.

Proposed scheme presenting the oligomeric state of HspA in the presence of nickel or bismuth. The front subunit is highlighted, and the rest are shaded in gray. The protein structure of H. pylori HspA was modeled based on the crystal structure of Mycobacterium tuberculosis GroES (Protein Data Bank entry 1hx5), except for the C-terminal domain due to lack of a modeling template. Ni2+ and Bi3+ presumably coordinate to different residual ligands. It is also worth mentioning that the C-terminal deletion mutant is unable to carry detectable Ni2+ (S. Cun, H. Li, and H. Sun, unpublished data), and it may be immune to the structural disruption induced by Bi3+ binding if the given speculation is real.
Some metalloenzymes from mucosal colonies and pathogens have evolved to have supplementary metal-binding domains that can help to maintain or reinforce the catalytic activity under specific conditions. For example, a histidine-rich domain at the N terminus (∼20 amino acids) in copper/zinc superoxide dismutase is found to be a metal-binding domain, which facilitates copper acquisition under copper deficiency and assists Cu+ transfer from the metal-binding domain to the copper/zinc superoxide dismutase active sites (54). To examine the role of the His- and Cys-rich domain of HspA in vivo, we induced the expression of wild-type HspA and its mutant (without the C-terminal His- and Cys-rich domain) in E. coli. Our data clearly show that in the presence of different concentrations of Ni2+ (0–50 μm), the cell growth in M9 minimal medium was much better for those E. coli BL21 cells with the wild-type hspA gene than those with the C-terminal deletion mutant. Such a difference was more pronounced when cells were grew over 20 h. This indicates that the C-terminal His- and Cys-rich domain plays a role in Ni2+ homeostasis. Unexpectedly, opposite results were observed for the bismuth antiulcer drug, indicating that Bi3+ binding to the C terminus of HspA may cause potential cytotoxicity in E. coli. Note that possibly because of a low efficiency of bismuth uptake (supplemental Fig. S4), E. coli cells exhibit very high resistance against bismuth among several tested Gram-negative bacteria (55). This could account for one of the reasons why the inhibitory effect of bismuth was not as significant on E. coli growth as was nickel. However, the result still provides direct evidence of the relationship between metal interaction and the C terminus in vivo. For a long time, the molecular mechanism that H. pylori benefits from nickel but suffers from bismuth has not been well established. Our in vitro and in vivo data indicated a double-edged role of the unique C terminus of HspA in H. pylori. On one hand, the C terminus can dynamically bind Ni2+ to assist nickel homeostasis, and it may donate Ni2+ to other metalloproteins with stronger affinity (e.g. UreE) under nickel-deficient conditions to help the maturation of Ni2+-incorporated metalloenzyme(s) such as urease. On the other hand, with a more efficient bismuth uptake system in H. pylori, Bi3+ may irreversibly interact with HspA, causing subsequent dysfunction of the protein and even cytotoxicity. Further work is required to investigate the differences in the effects of these two metals on microorganisms.
CONCLUSION
We have overexpressed and purified the H. pylori GroES chaperonin HspA and found that the protein bound two Ni2+ ions/monomer reversibly with moderate binding affinity. We have also established that Bi3+ bound to HspA much more strongly than did Ni2+, and the binding was almost irreversible at physiological pH. Importantly, Bi3+ induced the quaternary structural changes of HspA from its native form (heptamer) to a dimer. Bismuth may therefore interfere with the function of the protein. Our hypothesis of a double-edged role has been supported by in vivo data showing that the C-terminal His- and Cys-rich domain is beneficial in nickel resistance but harmful to cell growth when exposed to bismuth. Because histidine-rich proteins and motifs are commonly found in pathogenic microorganisms and in animals, this study may shed light on versatile roles of histidine-rich and other metal-binding residue-rich motifs.
Supplementary Material
Acknowledgments
We thank Dr. Xuesong Sun and Wei Xia for assistance with measurement of binding constants and helpful discussion.
This work was supported in part by Grants HKU7039/04P and HKU7042/07P from the Research Grants Council of Hong Kong, Grants HKUST4/03C and HKU1/07C from the Research Grants Council Central Allocation, the Area of Excellence Scheme of the University Grants Committee, and The University of Hong Kong. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S5.
Footnotes
The abbreviations used are: MALDI-TOF, matrix-assisted laser desorption ionization time-of-flight; Bi-NTA, bismuth(III)-nitrilotriacetate; CBS, colloidal bismuth subcitrate.
References
- 1.Covacci, A., Telford, J., Del Giudice, G., Parsonnet, J., and Rappuoli, R. (1999) Science 284 1328-1333 [DOI] [PubMed] [Google Scholar]
- 2.Marshall, B. J., and Warren, J. R. (1984) Lancet i 1311-1315 [DOI] [PubMed] [Google Scholar]
- 3.Dunn, B. E., Roop, R. M., Sung, C. C., Sharma, S. A., Perez-Perez, G. I., and Blaser, M. J. (1992) Infect. Immun. 60 1946-1951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parsonnet, J., Vandersteen, D., Goates, J., Sibley, R. K., Pritikin, J., and Chang, Y. (1991) J. Natl. Cancer Inst. 83 640-643 [DOI] [PubMed] [Google Scholar]
- 5.Bauerfeind, P., Garner, R., Dunn, B. E., and Mobley, H. L. (1997) Gut 40 25-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scott, D., Weeks, D., Hong, C., Postius, S., Melchers, K., and Sachs, G. (1998) Gastroenterology 114 58-70 [DOI] [PubMed] [Google Scholar]
- 7.Maier, R. J., Fu, C., Gilbert, J., Moshiri, F., Olson, J., and Plaut, A. G. (1996) FEMS Microbiol. Lett. 141 71-76 [DOI] [PubMed] [Google Scholar]
- 8.Olson, J. W., and Maier, R. J. (2002) Science 298 1788-1790 [DOI] [PubMed] [Google Scholar]
- 9.De Pina, K., Desjardin, V., Mandrand-Berthelot, M. A., Giordano, G., and Wu, L. F. (1999) J. Bacteriol. 181 670-674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Contreras, M., Thiberge, J. M., Mandrand-Berthelot, M. A., and Labigne, A. (2003) Mol. Microbiol. 49 947-963 [DOI] [PubMed] [Google Scholar]
- 11.Mobley, H. L., Garner, R. M., and Bauerfeind, P. (1995) Mol. Microbiol. 16 97-109 [DOI] [PubMed] [Google Scholar]
- 12.Maier, R. J., Benoit, S. L., and Seshadri, S. (2007) Biometals 20 655-664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hendricks, J., and Mobley, H. (1997) J. Bacteriol. 179 5892-5902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mehta, N., Olson, J. W., and Maier, R. J. (2003) J. Bacteriol. 185 726-734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brayman, T. G., and Hausinger, R. P. (1996) J. Bacteriol. 178 5410-5416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benoit, S., Mehta, N., Weinberg, M., Maier, C., and Maier, R. (2007) Microbiology (Read.) 153 1474-1482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seshadri, S., Benoit, S. L., and Maier, R. J. (2007) J. Bacteriol. 189 4120-4126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suerbaum, S., and Michetti, P. (2002) N. Engl. J. Med. 347 1175-1186 [DOI] [PubMed] [Google Scholar]
- 19.Phillips, R. H., Whitehead, M. W., Lacey, S., Champion, M., Thompson, R. P., and Powell, J. J. (2000) Helicobacter 5 176-182 [DOI] [PubMed] [Google Scholar]
- 20.Ge, R., and Sun, H. (2007) Acc. Chem. Res. 40 267-274 [DOI] [PubMed] [Google Scholar]
- 21.Ge, R., Sun, X., Gu, Q., Watt, R. M., Tanner, J. A., Wong, B. C., Xia, H. H., Huang, J. D., He, Q. Y., and Sun, H. (2007) J. Biol. Inorg. Chem. 12 831-842 [DOI] [PubMed] [Google Scholar]
- 22.Kansau, I., Guillain, F., Thiberge, J. M., and Labigne, A. (1996) Mol. Microbiol. 22 1013-1023 [DOI] [PubMed] [Google Scholar]
- 23.Kansau, I., and Labigne, A. (1996) Aliment. Pharmacol. Ther. 10 Suppl. 1, 51-56 [DOI] [PubMed] [Google Scholar]
- 24.Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990) J. Mol. Biol. 215 403-410 [DOI] [PubMed] [Google Scholar]
- 25.Thompson, J., Higgins, D., and Gibson, T. (1994) Nucleic Acids Res. 22 4673-4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rice, P., Longden, I., and Bleasby, A. (2000) Trends Genet. 16 276-277 [DOI] [PubMed] [Google Scholar]
- 27.De Castro, E., Sigrist, C., Gattiker, A., Bulliard, V., Langendijk-Genevaux, P., Gasteiger, E., Bairoch, A., and Hulo, N. (2006) Nucleic Acids Res. 34 W362-W365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hulo, N., Bairoch, A., Bulliard, V., Cerutti, L., De Castro, E., Langendijk-Genevaux, P., Pagni, M., and Sigrist, C. (2006) Nucleic Acids Res. 34 D227-D230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bulaj, G., Kortemme, T., and Goldenberg, D. (1998) Biochemistry 37 8965-8972 [DOI] [PubMed] [Google Scholar]
- 30.Gupta, R. S. (1995) Mol. Microbiol. 15 1-11 [DOI] [PubMed] [Google Scholar]
- 31.Tomb, J., White, O., Kerlavage, A., Clayton, R., Sutton, G., Fleischmann, R., Ketchum, K., Klenk, H., Gill, S., Dougherty, B., Nelson, K., Quackenbush, J., Zhou, L., Kirkness, E., Peterson, S., Loftus, B., Richardson, D., Dodson, R., Khalak, H., Glodek, A., McKenney, K., Fitzegerald, L., Lee, N., Adams, M., Hickey, E., Berg, D., Gocayne, J., Utterback, T., Peterson, J., Kelley, J., Cotton, M., Weidman, J., Fujii, C., Bowman, C., Watthey, L., Wallin, E., Hayes, W., Borodovsky, M., Karp, P., Smith, H., Fraser, C., and Venter, J. (1997) Nature 388 539-547 [DOI] [PubMed] [Google Scholar]
- 32.Wilkins, M., Gasteiger, E., Bairoch, A., Sanchez, J., Williams, K., Appel, R., and Hochstrasser, D. (1999) Methods Mol. Biol. 112 531-552 [DOI] [PubMed] [Google Scholar]
- 33.Stibrany, R. T., Fox, S., Bharadwaj, P. K., Schugar, H. J., and Potenza, J. A. (2005) Inorg. Chem. 44 8234-8242 [DOI] [PubMed] [Google Scholar]
- 34.Sun, H., Li, H., Harvey, I., and Sadler, P. J. (1999) J. Biol. Chem. 274 29094-29101 [DOI] [PubMed] [Google Scholar]
- 35.Petit, G., and Petit, L. D. (1997) IUPAC Stability Constant Database, International Union of Pure and Applied Chemistry Academic Software, Otley, UK
- 36.Tang, Y. C., Chang, H. C., Roeben, A., Wischnewski, D., Wischnewski, N., Kerner, M. J., Hartl, F. U., and Hayer-Hartl, M. (2006) Cell 125 903-914 [DOI] [PubMed] [Google Scholar]
- 37.Kandror, O., Busconi, L., Sherman, M. Y., and Goldberg, A. L. (1994) J. Biol. Chem. 269 23575-23582 [PubMed] [Google Scholar]
- 38.Gupta, R. S., and Golding, G. B. (1993) J. Mol. Evol. 37 573-582 [DOI] [PubMed] [Google Scholar]
- 39.Bakkes, P. J., Faber, B. W., van Heerikhuizen, H., and van der Vies, S. M. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 8144-8149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bonshtien, A. L., Weiss, C., Vitlin, A., Niv, A., Lorimer, G. H., and Azem, A. (2007) J. Biol. Chem. 282 4463-4469 [DOI] [PubMed] [Google Scholar]
- 41.Sarkar, B. (1987) Prog. Food Nutr. Sci. 11 363-400 [PubMed] [Google Scholar]
- 42.Henkel, G., and Krebs, B. (2004) Chem. Rev. 104 801-824 [DOI] [PubMed] [Google Scholar]
- 43.Guerinot, M. L. (2000) Biochim. Biophys. Acta 1465 190-198 [DOI] [PubMed] [Google Scholar]
- 44.Dosanjh, N. S., and Michel, S. L. (2006) Curr. Opin. Chem. Biol. 10 123-130 [DOI] [PubMed] [Google Scholar]
- 45.Mobley, H. L., Garner, R. M., Chippendale, G. R., Gilbert, J. V., Kane, A. V., and Plaut, A. G. (1999) Helicobacter 4 162-169 [DOI] [PubMed] [Google Scholar]
- 46.Lee, M. H., Pankratz, H. S., Wang, S., Scott, R. A., Finnegan, M. G., Johnson, M. K., Ippolito, J. A., Christianson, D. W., and Hausinger, R. P. (1993) Protein Sci. 2 1042-1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ge, R., Watt, R. M., Sun, X., Tanner, J. A., He, Q. Y., Huang, J. D., and Sun, H. (2006) Biochem. J. 393 285-293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Burk, R. F., and Hill, K. E. (2005) Annu. Rev. Nutr. 25 215-235 [DOI] [PubMed] [Google Scholar]
- 49.Suerbaum, S., Thiberge, J. M., Kansau, I., Ferrero, R. L., and Labigne, A. (1994) Mol. Microbiol. 14 959-974 [DOI] [PubMed] [Google Scholar]
- 50.Schreiter, E. R., Sintchak, M. D., Guo, Y., Chivers, P. T., Sauer, R. T., and Drennan, C. L. (2003) Nat. Struct. Biol. 10 794-799 [DOI] [PubMed] [Google Scholar]
- 51.Li, H., Sadler, P. J., and Sun, H. (1996) J. Biol. Chem. 271 9483-9489 [PubMed] [Google Scholar]
- 52.Sun, H., and Szeto, K. Y. (2003) J. Inorg. Biochem. 94 114-120 [DOI] [PubMed] [Google Scholar]
- 53.Li, W., Jin, L., Zhu, N., Hou, X., Deng, F., and Sun, H. (2003) J. Am. Chem. Soc. 125 12408-12409 [DOI] [PubMed] [Google Scholar]
- 54.Battistoni, A., Pacello, F., Mazzetti, A. P., Capo, C., Kroll, J. S., Langford, P. R., Sansone, A., Donnarumma, G., Valenti, P., and Rotilio, G. (2001) J. Biol. Chem. 276 30315-30325 [DOI] [PubMed] [Google Scholar]
- 55.Domenico, P., Reich, J., Madonia, W., and Cunha, B. A. (1996) J. Antimicrob. Chemother. 38 1031-1040 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.