

Abstract
Altered inositol metabolism is implicated in a number of diabetic complications. The first committed step in mammalian inositol catabolism is performed by myo-inositol oxygenase (MIOX), which catalyzes a unique four-electron dioxygen-dependent ring cleavage of myo-inositol to d-glucuronate. Here, we present the crystal structure of human MIOX in complex with myo-inosose-1 bound in a terminal mode to the MIOX diiron cluster site. Furthermore, from biochemical and biophysical results from N-terminal deletion mutagenesis we show that the N terminus is important, through coordination of a set of loops covering the active site, in shielding the active site during catalysis. EPR spectroscopy of the unliganded enzyme displays a two-component spectrum that we can relate to an open and a closed active site conformation. Furthermore, based on site-directed mutagenesis in combination with biochemical and biophysical data, we propose a novel role for Lys127 in governing access to the diiron cluster.
myo-Inositol is a cyclitol that plays a crucial role in all eukaryotic cells by serving as a backbone for the most important second messengers: inositol phosphates and their lipid derivatives, phosphoinositides. Although inositol phosphates and phosphoinositides have been and continue to be the focus of intense study, the mechanisms for maintenance of total cellular inositol levels and the medical implications of deranged intracellular inositol levels are less studied. It is known that inositol homeostasis is often disturbed in diabetes (1) and that intracellular depletion of myo-inositol is associated with common diabetic complications, such as cataracts, nephropathies, retinopathies, and neuropathies (2). In mammalian cells, the only pathway for inositol breakdown utilizes myo-inositol oxygenase (MIOX)3 to catalyze the first committed step by a dioxygen-dependent cleavage between C1 and C6 of the inositol ring to form d-glucuronate, which can then enter the glucuronate-xylulose pathway (2). Therapeutic intervention aimed at inhibiting MIOX activity may be a future cure for diabetic complications caused by inositol depletion. 18O labeling studies have shown that MIOX incorporates only a single oxygen into the product and that oxygen is found exclusively in the d-glucuronate carboxylate group. MIOX has a pH optimum of 9.5 (3) with myo-inositol (Km = 5.9 mm; kcat = 11 min-1 (2)) and its epimer d-chiro-inositol (Km = 33 mm; kcat = 2.3 min-1 (2)) as its only known substrates. Moreover, the only known reasonably potent inhibitor is myo-inosose-1 (Ki = 62 μm (3)). It has been noted that an N-terminally truncated fragment starting at Thr32 is formed upon storage of recombinant protein (4). Interestingly, this uncharacterized N-terminal fragment is also observed in vivo (5).
It was recently shown, using EPR and Mössbauer spectroscopy, that MIOX is a new member of the nonheme diiron-dependent dioxygen-activating family (6). Interestingly, the sequence indicated strong dissimilarity to other members of this family (6) that all feature a core four-helix bundle, as shown by the structures of, for example, protein R2 of ribonucleotide reductase (7), soluble methane monoxygenase (8), and stearoylacyl carrier protein Δ9-desaturase (9, 10). Furthermore, MIOX uses an antiferromagnetically coupled high spin mixed valent diiron (II/III) cluster for catalysis, and the mixed valent state is regenerated after each cycle of catalysis (6).
In studies of MIOX from mice (Mm-MIOX) it was found that myo-inositol perturbs the diiron cluster's Mössbauer and EPR spectra, presumably by binding directly to the cluster (11). A tentative (superoxo) diiron (III/III) complex (“G-complex”) can be trapped using a deuterated substrate (12). The formation of the G-complex is reversible (40-s), thus disfavoring a scenario involving O–O cleavage and the utilization of higher iron oxidation states than the ferric (12). Furthermore, EPR hyperfine interaction data support a coordination of the dioxygen species, where it binds to only one of the irons in the diiron cluster. Based on spectroscopic data, a mechanism was proposed in which the G-complex, after dioxygen species-mediated aliphatic hydrogen abstraction from the myo-inositol C1, decomposes to product via either a 1-hydroperoxy or a gem-diolate intermediate (12). Furthermore, the substrate was proposed to bridge the two irons with C1-O as an alkoxide (12). The presence of an iron-substrate alkoxide bond could be confirmed with 2H ENDOR spectroscopy (13).
The three-dimensional structure of Mm-MIOX determined at 2.0 Å resolution has recently been published (14). Mm-MIOX was shown to have a novel overall topology reminiscent of the HD domain superfamily of metal-binding phosphohydrolases and to possess a novel diiron cluster first coordination sphere with four histidines and two aspartates, and, contrary to what had been previously proposed (6, 11–13), substrate was found to bind terminally to the diiron cluster (14). The structure in combination with site-directed mutagenesis, in essence, confirmed a mechanism previously proposed from spectroscopic data (12). Furthermore, in the Mm-MIOX structure, a hairpin loop (residues 83–93) was found lining the entrance to the active site and was proposed to be important for substrate access to the active site through the formation of a lid covering the substrate-binding pocket (14). Additionally, in the mechanism based on the Mm-MIOX structure, a lysine (Lys127) was given a critical role in promoting ionization of the C1 hydroxyl and in activation of the bond between C1 and its aliphatic hydrogen (14). The Mm-MIOX K127A mutant was inactive.
Here we present the three-dimensional structure of human MIOX (Hs-MIOX) determined using single wavelength anomalous dispersion methods. Furthermore, we provide biochemical and biophysical evidence for the importance of two additional proteinous lids that cover and shield the enzyme's active site. In addition, we propose a model for how Lys127 mutants render the enzyme inactive by modulating access to the active site of MIOX.
EXPERIMENTAL PROCEDURES
Cloning and Protein Production—For crystallization, DNA encoding residues 38–285 of the human MIOX gene (gi:49522836) was cloned by ligation-independent cloning into a pET-28-based expression vector incorporating an N-terminal His6 tag fusion (pNIC-Bsa4; gi:EF198016). After transformation and liquid culture growth using standard methods, recombinant expression of Hs-MIOX in Escherichia coli strain BL21(DE3) was induced at 291 K by the addition of 0.5 mm IPTG to minimal media for selenomethionine incorporation according to the methionine pathway inhibition method (15). Induction was maintained for 18 h before harvesting. MIOX was purified using IMAC (16) on a 1-ml HiTrap chelating HP column followed by gel filtration on a 120-ml Superdex 75 column. The N-terminal tag with the His tag fusion and residual cloning elements were excised through incubation with tobacco etch virus protease (1:20 protease/protein ratio) for 48 h at 277 K. After protease digestion, the IMAC purification was repeated to remove noncleaved protein and the His-tagged tobacco etch virus protease. After additional purification by ion exchange chromatography on a 1-l MonoQ column (in Tris, pH 8.0, 10% glycerol by varying NaCl concentration), MIOX was essentially pure as judged by SDS-PAGE analysis. All columns used were from GE-Healthcare. Denaturing electrospray ionization mass spectrometry verified protein integrity and indicated almost full selenomethionine incorporation. Iron content was determined colorimetrically (17) to be ∼1.8 irons/MIOX molecule.
Crystallization and Structure Solution—Crystals of selenomethionine-labeled Hs-MIOX were grown by hanging drop vapor diffusion at 293 K. MIOX (15 mg/ml in 20 mm Hepes, pH 7.5, 300 mm NaCl, 10% glycerol, 2 mm TCEP, 5 mm myo-inositol, 1 mm l-cysteine, and 1 mm FeCl3) was mixed with an equal amount (0.6 μl) of reservoir solution (100 mm Tris, pH 7.8, 1.8 m ammonium sulfate). Crystals grew initially as rods, but after 2 months of incubation, platelike crystals appeared. The rod-shaped crystals diffracted to 3 Å resolution at best and were not significantly improved even after extensive optimization efforts. The platelike crystals, on the other hand, diffracted up to 1.5 Å on a synchrotron (see below).
A platelike crystal was flash-frozen in liquid nitrogen after having been swept through a reservoir solution with 10% (2R, 3R)-(-)-2,3-butanediol, 5 mm myo-inositol, 1 mm l-cysteine, and 1 mm FeCl3. A single wavelength anomalous dispersion data set was collected on the K-edge of selenium (0.9798 Å) at the beam line ID29 (ESRF, Grenoble, France). The data were indexed in space group C2 with unit cell parameters a = 119.7, b = 55.86, c = 111.5 Å, and β = 116.7°. XDS/XSCALE (18) was used to process the data (Table 1). XPREP (Bruker AXS) was used to prepare FA values for substructure solution in SHELXD (19) that found 17 of the possible 20 sites. The identified sites were used for phase calculation in SHARP (20), followed by density modification in PIRATE (21). The resulting map could be partially autotraced using ARP/wARP (22).
TABLE 1.
Data collection statistics
| Se-Met SADa | |
|---|---|
| Wavelength (Å) | 0.9798 |
| Resolution (Å) | 30-1.5 (1.6-1.5) [30-1.8] |
| Unique/observed reflections | 140563/206908 |
| Completeness (%) | 76.4 (27.0) [96.9] |
| 〈I/σ (I)〉 | 12.2 (2.36) [12.2] |
| Rsym (%) | 5.9 (36.9) [5.4] |
| Wilson B-factor | 15 |
Selenomethionine single wavelength anomalous dispersion
The data are essentially complete to 1.8 Å (statistics shown in square brackets in Table 1), but using higher resolution but very incomplete data in refinement enhanced model geometry and the quality of 2FO - FC maps; therefore, they were used. The optical resolution (minimum distance of two resolved peaks in the electron density map) of all of the data to 1.5 Å resolution is 1.36 Å, whereas using only 1.8 Å data gives an optical resolution of 1.48 Å, according to SFCHECK (23). SFCHECK calculates the optical resolution from the width of a single Gaussian fitted to the Patterson origin peak (23).
The model from ARP/wARP was completed and improved through several rounds of model building and refinement in COOT (24) and REFMAC5 (25). Refinement results are summarized in Table 2. Mm-MIOX rerefinement is briefly described in the supplemental materials.
TABLE 2.
Refinement statistics and quality of structure
| Parameter | Value |
|---|---|
| Resolution (highest resolution shell) (Å) | 21.3-1.5 (1.6-1.5) |
| Rcrysta (%) (highest resolution shell) | 19.7 (34.2) |
|
Rfreea (%) (highest resolution shell) |
23.8 (41.6) |
| Root mean square deviation from ideal geometry | |
| Bond lengths (Å) | 0.020 |
| Bond angles (degrees) | 1.8 |
| Average B-factors (Å2) | 24.4 |
| Ramachandran plot (favored, allowed)b | 99.6, 0.4 |
| Ramachandran plot (most favored, additional allowed)c (%) | 91.1, 8.9 |
Mutagenesis and Biochemical Characterization—For biochemical and biophysical experiments, wild-type Hs-MIOX (wt-MIOX) and selected mutants were studied. Site-directed point mutations (Fig. 4) were obtained by a two-step PCR method. N-terminal deletions (Fig. 4) were performed by recloning consecutively shorter PCR fragments. Activity assays were performed as described (2) on purified Hs-MIOX. Relative errors were estimated from variation between several experiments.
FIGURE 4.

Relative enzymatic activity of mutations and N-terminal deletions compared with wild-type protein. Estimations of errors are denoted with error bars. ΔX denotes a construct entailing residues X + 1 to 285. Activity was measured using the standard orcinol-based assay (2).
Biophysical Characterization of Wild Type and Mutants—To obtain an EPR-observable mixed valent state of the diiron site in MIOX, 180 μl of 100 μm Hs-MIOX were dissolved in aerobic Tris buffer (pH 7.5, 50 mm) and reduced in the EPR tube by 5 mm ascorbate (prepared anaerobically) in the absence or presence of 50 mm myo-inositol. Samples that were reduced anaerobically and subsequently oxidized by molecular oxygen according to Ref. 6 resulted in comparatively lower yields of the mixed valent species.
9.5 GHz EPR spectra were recorded on a Bruker ESP 300 X-band spectrometer with an Oxford Instruments ESR9 helium cryostat in a temperature range of 5–20 K using a 0.5 millitesla modulation amplitude. The experimental spectra were corrected by subtracting a spectrum of the buffer alone and by a contaminant Cu2+ spectrum that in some cases was disturbing. Spin quantitation was performed by double integration of EPR spectra recorded at nonsaturating microwave power levels, comparing with a standard solution of 1 mm CuSO4 in 10 mm EDTA. Spectra simulations were performed with the Bruker SimFonia simulation package.
EPR saturation data were collected by measuring the EPR absorption derivative signal intensity as a function of microwave power at three different temperatures. The saturation data were fitted to the expression (26), S/P½ ∼ (1 + P/P½)—½, where S represents the EPR derivative signal intensity, P is the microwave power, and P½ is the half-saturation power. A nonlinear least squares fit to this equation yielded a P½ value for each particular temperature.
RESULTS
Overall Structure of Hs-MIOX—The three-dimensional structure of Hs-MIOX, residues 38–285, is shown in Fig. 1. The Hs-MIOX has an overall topology that consists of eight α-helices and a small two-stranded antiparallel β-sheet that act as a topological latch (Fig. 1). The overall structure of Hs-MIOX superimposes closely (Fig. 2A) with the Mm-MIOX structure with 0.5 Å root mean square deviation on Cα:s and with 86% sequence identity over the structural alignment. However, in the vicinity of the active site, there are significant differences relating to the lack of the 37 N-terminal amino acids in the Hs-MIOX construct used here for crystallization. Specifically, in the Mm-MIOX structure (Fig. 2B), Asp90 forms a strong interaction with the backbone NH of Arg29, and this governs the conformation of Asp88, through the rigid intermediate Pro89, to interact strongly with Lys257, which in turn forms a stacking interaction with Phe28 (not modeled in Protein Data Bank code 2HUO (14) but modeled in our redeposition, 3BXD). Hence, the three loops covering the active site form interdependent interactions in Mm-MIOX. The lack of one of the members (residues 28–32) in this group of three loops explains the observed disorder in these regions (Fig. 2B) in our crystallized construct of Hs-MIOX that lacks the first 37 amino acids (Δ37).
FIGURE 1.

Structure of human myo-inositol oxygenase. A ribbon diagram with numbered secondary structure elements is shown. Direct ligands to the diiron-cluster are shown in stick representation. In all panels, iron atoms are shown as orange spheres, and the oxygens are shown as small red spheres.
FIGURE 2.
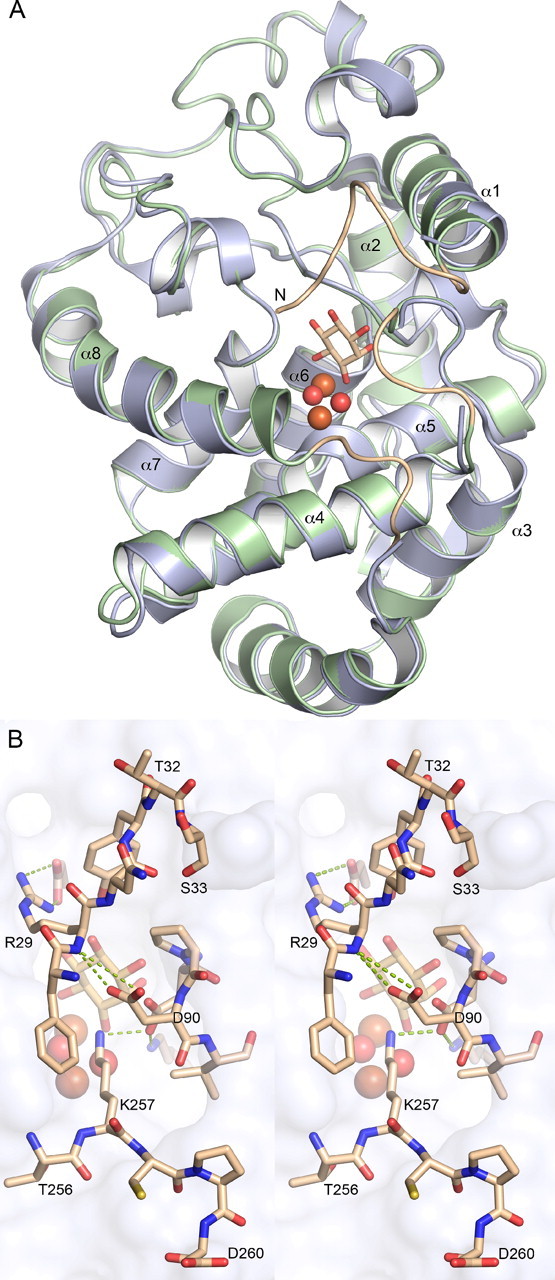
Comparison between human and mouse MIOX. A, overview of a structural imposition of mouse MIOX (green) and human MIOX (light blue). The three loops present in the model of mouse MIOX but not in the human MIOX model are shown in light brown. B, interactions, shown as green dotted lines, between residues in the three loops covering the active site.
The Hs-MIOX Active Site—A diiron cluster is bound in a crevice formed between α4, α5, α6, α7, and α8 (Fig. 1). It represents a strictly conserved (supplemental Fig. S1) diiron cluster first coordination sphere with four histidines and two aspartates (Fig. 3, A and C), and, contrary to what has been previously proposed (6, 11–13) but similar to the Mm-MIOX structure, substrate binds terminally to the diiron cluster. In the cluster, the most solvent-exposed iron ion, FeA, is octahedrally coordinated by His98, His123, Asp253, a bridging Asp124, and one bridging and one directly bound oxygen species. The second, innermost iron, FeB, is coordinated by a distorted octahedron formed by His194, His220, the bridging Asp124, one bridging oxygen species, and the C1-O and C6-O oxygens from a terminally bound substrate-like molecule. In the active site, there are three major differences to the Mm-MIOX structure. First, the bridging oxygen species is asymmetrically bound (Fig. 1C) in Hs-MIOX while symmetrically bound in Mm-MIOX. Second, Asp124 bridges the diiron cluster asymmetrically in Hs-MIOX but symmetrically in Mm-MIOX. In addition, the outermost iron (FeA) is further away from Asp124 in the Hs-MIOX structure as compared with the Mm-MIOX structure. This is partly reflected in a 0.2-Å closer Fe-Fe distance in Mm-MIOX compared with Hs-MIOX. Third, the substrate, which ligates the innermost iron (FeB) in a strictly symmetrically bidental fashion (2.0 Å between FeB and both O1 and O6) in the Mm-MIOX structure (14), is highly asymmetrical in the Hs-MIOX structure (Fig. 1C). This can partly be explained by the different nature of the Hs-MIOX ligand (see below).
FIGURE 3.
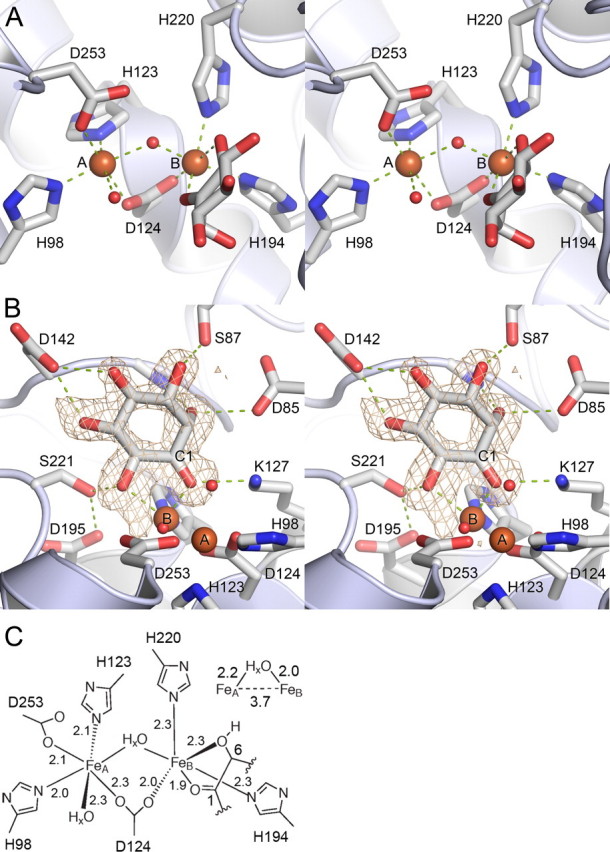
Close-up view on the active site. A, diiron cluster site (A and B, FeA and FeB, respectively). B, ligand-protein interactions. Omit Fo - Fc map calculated for myo-inosose-1 contoured at 3σ. C, diiron site coordination (distances in Å).
Binding of Substrate-like Ligand—The substrate-like molecule bound to FeB has suitable hydrogen bonding partners for all exocyclical hydroxyl groups (Fig. 3B), where it binds in a relatively tight groove. The hydrogen-bonding pattern is preorganized with special emphasis on the axial C2-O, which protrudes into a pocket, where it forms strong hydrogen bonds to nitrogen of Gly141 and Oδ2 of Asp85 from opposite sides. During refinement of the structure, it became apparent that C1 of the presumed myo-inositol molecule bound to FeB was, in fact, sp2-hybridized (supplemental Fig. S2). Therefore, the structure presented here represents myo-inosose-1 bound to Hs-MIOX. myo-Inosose-1 is a known inhibitor of the enzyme (3). It is, however, unclear whether it was formed during the 2-month crystallization period or if it is a minute contaminant in commercial preparations of myo-inositol.
Importance of the N Terminus; Structure and Activity—It has been noted that an N-terminally truncated fragment starting at Thr32 is formed upon storage of recombinant protein but also in mammalian cells expressing MIOX (5). The N terminus covers partly the active site, but, most importantly, it orchestrates the conformation of two other loops so that all three loops jointly seal off the active site from bulk solvent (see above and Fig. 2C). Therefore, we tested whether N-terminal truncations affected activity. Fig. 4 shows various protein constructs and their enzymatic activities compared with wt-MIOX. We find that the activity drops significantly only between cuts at residue Lys25 and Thr32 (Fig. 4). Mutating the first conserved residue in MIOX, Arg29 (supplemental Fig. S1), which is present in this region, shows intermediate activity compared with N-terminal deletions at residues Lys25 and Thr32 (Fig. 4). The Asp38–Trp285 fragment (called Δ37), corresponding to the crystallized molecule, has ∼18% of wild type activity. The K127S mutant is essentially inactive. We further investigated the effects of MIOX mutations by performing EPR spectroscopy on wt-MIOX and comparing the results with those from selected mutant proteins.
EPR Spectroscopy of Hs-MIOX—EPR spectroscopy was used to study wt-MIOX and the selected mutant proteins after ascorbate reduction under aerobic conditions, in the absence or presence of the substrate myo-inositol (Fig. 5, left and right, respectively). The studied mutants were Δ37 (containing residues 38–285), which lacks the residues necessary for forming the “dynamic lid” covering the substrate binding site (see above), the single mutant K127S probing the role of Lys127 near the substrate binding site, and the double mutant Δ37,K127S combining the two variants. The ascorbate/air reduction of all Hs-MIOX constructs results in formation of a mixed valent Fe(II)/Fe(III) species, which is paramagnetic and observable by EPR.
FIGURE 5.

EPR spectroscopy of wild type and mutants of human MIOX. A, EPR spectra of the mixed valent state of MIOX in the absence of substrate (left) and in the presence of the substrate myo-inositol (right). Spectra A and B, wild type protein; spectra C and D, Δ37; spectra E and F, K127S; spectra G and H, Δ37, K127S mutant protein. Shown is the sample preparation procedure according to “Experimental Procedures.” Spectra were taken at 7 K, at microwave power of 3 milliwatts (nonsaturating conditions for all paramagnetic mixed valent sites). The mixed valent state concentration was estimated by double integration of the spectra as 0.20(5) per polypeptide for spectra A and B and spectra E–H, whereas 0.02 perpolypeptide was found for spectra C and D. The g values indicated above spectra A and B give approximate descriptions of apparent spectral parameters (further discussed under “Experimental Procedures”).
Spectrum A in Fig. 5 shows the results of ascorbate reduction of wt-MIOX. This protein forms at least two types of mixed valent Fe(II)/Fe(III) states, resulting in a two-component EPR spectrum with axially symmetric signals. The overall intensity of the EPR spectrum corresponded to about 0.2 mixed valent sites per Hs-MIOX protein molecule. The two-component EPR signals are detectable in the same low temperature range and hardly saturated with microwave power even at low temperatures. Nevertheless, by changing the microwave power and temperature of the EPR recording, we could find conditions where one or the other component was predominant (Fig. 6). As is shown in Fig. 6, the experimental spectra could be simulated using the following parameters (g values (g) and line widths (L)) for the two components: Component I, g∥ = 1.96, g⊥ = 1.74, L∥ = 60 G, L⊥ = 220 G; Component II, g∥ = 1.94, g⊥ = 1.72, L∥ = 80 G, L⊥ = 250 G.
FIGURE 6.

EPR spectra and simulations of two components in the wild type human MIOX in the absence of substrate. The sample used in Fig. 5A was investigated under extreme conditions in order to emphasize the features of two distinct EPR spectral components. Upper trace, EPR at 20 K, 200 microwatts microwave power, showing dominating g∥ = 1.96 component I, assigned to the open lid conformation. The low signal-to-noise ratio and the spectral imperfections are due to the very low microwave power applied in order to single out this component. The line shape is mainly Lorentzian due to the relatively high recording temperature. Lower trace, EPR at 5 K, 200 milliwatt microwave power showing dominating g∥ = 1.94 component II, assigned to the closed lid conformation. The simulations (dashed lines superimposed on the experimental spectra) were made using the following parameters for g values and line widths. Upper trace, g∥ = 1.96, g⊥ = 1.74, L∥ = 60 G, L⊥ = 220 G and Lorentzian line shape; lower trace, g∥ = 1.94, g⊥ = 1.72, L∥ = 80 G, L⊥ = 250 G and Gaussian line shape.
The addition of the two-component spectra shows a quantitative ratio of 0.3/0.7 (supplemental Fig. S4), which fits well to the experimental spectrum of Fig. 5A. The g values of the simulated component spectra are the same as those determined for the components in Fig. 6, but the overall line widths are somewhat reduced to fit the less extreme conditions used for the Fig. 5 spectra.
The appearance of two components in the EPR spectrum was much less prominent when the corresponding mixed valent state was induced in the Δ37 mutant. In this case, mainly one (g∥ = 1.96, g⊥ ∼ 1.77) type of mixed valent state was observed, and its relative yield was 10 times less compared with the wt-MIOX. (Fig. 5, spectrum C).
Binding of myo-inositol results in significant changes in the mixed valent state of wt-MIOX protein (Fig. 5, spectrum B). Although the EPR signal intensity remains approximately unchanged (0.2 spins/MIOX), both axially symmetric EPR components seen in the absence of substrate become replaced by a relatively sharp EPR signal with rhombic symmetry. From the simulation of the spectrum (supplemental Fig. S4), we obtained the following g value and line width parameters: gx = 1.96, gy = 1.82, gz = 1.79; Lx = 25 G, Ly = 30 G, Lz = 60 G. The spectrum appears free from any admixture of substrate-free fraction of enzyme that was seen in the initial mixed valent state of wt-MIOX (Fig. 5, spectrum A), and the anisotropy of the line width is smaller.
The microwave saturation behavior for the EPR signal of wt-MIOX as a function of temperature in the absence and presence of myo-inositol was investigated. The saturation was well described by an Orbach relaxation process at the temperatures below 12 K. The EPR half-saturation power (P½) as a function of absolute temperature (T) has been well fitted in other exchange-coupled systems by (27, 28) the equation, lnP½ = lnA -Δ/kT, where k is the Boltzmann constant and A is a constant characteristic of the particular system. The value of Δ is a measure of the energy separation between the ground and first excited states. Plots of P½, as a function of the reciprocal absolute temperature for the mixed valent Fe(II)/Fe(III) state of MIOX with and without inositol bound are shown in supplemental Fig. S5. At 7 K, both samples have similar P½ values of 8–10 milliwatts. The slopes for MIOX and MIOX-inositol were determined as -60 and -30 cm-1. For an anti-ferromagnetically coupled Fe(II)/Fe(III) system with ground spin state S = ½, the nearest excited energy level (Δ=-3J) is able to participate in spin lattice relaxation via the Orbach process. From this preliminary analysis, the exchange coupling constant, J (from Hex =-2J S1·S2), could be estimated from the slopes as -20 cm-1 and -10 cm-1. We conclude that binding of inositol weakens the exchange coupling. The results indicating that the substrate-free site has a stronger magnetic interaction between the two iron ions suggest that the filled ligation shell of the substrate-bound state gives rise to decreased coupling between the ions. A similar effect of ligand binding has been reported for uteroferrin. Also, this diiron enzyme has the Fe(II)/Fe(III) state as its active form, and its exchange coupling is reduced from |J| = 6 to 3 cm-1 upon binding of phosphate (29).
The K127S mutant can also form the mixed valent state under the same treatment of ascorbate reduction and with about the same yield as the wt-MIOX. The mixed valent site of the K127S mutant is characterized by an axially symmetric signal with g∥ = 1.94 and g⊥ ∼ 1.77. The EPR signal does not change in the presence of myo-inositol, suggesting that the substrate does not bind as in the wild type enzyme, if it binds at all (Fig. 5, spectra E and F). The EPR signal remains the same in the double mutant Δ37,K127S (Fig. 5, spectra G and H).
DISCUSSION
Mm- and Hs-MIOX are the first structurally characterized diiron-dependent dioxygen-activating proteins not belonging to class I of diiron carboxylate proteins. Clearly, MIOX has evolved from different ancestry, most probably from mammalian phosphodiesterases that belong to the HD-domain structural family. Furthermore, the MIOX family is the first of the diiron-dependent dioxygen-activating proteins where there is direct evidence for a terminal substrate-binding mode to a diiron cluster (14 and here). Clearly, by allowing selection on the exocyclical oxygens of the substrate in a pocket beside the diiron center (Fig. 3A) (14), the MIOX family, hereby proposed to form class V of diiron-carboxylate proteins, leaves the center of the diiron site open to iron-oxygen chemistry. The overall structures of Mm-MIOX and Hs-MIOX are quite similar, but distances differ significantly within the active site. Most importantly, there are clear differences close to the entrance of the active site. Clearly, the interdependence of the conformations of the three loops that shield the active site enable them to open and close access to the diiron site in concert. The three loops can therefore act as a single “dynamic lid” over the active site.
The activity studies (Fig. 4) emphasize the importance of the N terminus for enzymatic activity, most likely by contributing to cover the active site of MIOX. The complete loss of activity of K127S emphasizes the role of this residue for the active site integrity, in agreement with the observations on the K127A mutant in Mm-MIOX (14).
In the absence of substrate, the full-length wt-MIOX shows two components in its EPR spectrum of the mixed valent state (Figs. 5 (spectrum A) and 6). We propose that the two components represent contributions from two different structural states of the protein. The two components give rise to signals with distinct g∥ values: g∥ = 1.96 and g∥ = 1.94, respectively. Since the g∥ = 1.94 component is essentially absent in the Δ37 mutant, which has a deficient covering lid for the active site, we tentatively assign the g∥ = 1.94 component to a state where the lid is closed. Consequently, the g∥ = 1.96 component is assigned to an open state of the lid. Both of these components are transformed into one EPR spectrum of the wt-MIOX with bound substrate (Fig. 5, spectrum B). This shows that there should be a dynamic equilibrium between the open and closed states of the covering lid. We reason that binding of the substrate myo-inositol directly to the diiron site requires good accessibility for the rather bulky molecule, which may be provided only by an open conformation. As soon as substrate is bound, the lid closes to allow the catalytic reaction to proceed without influence of external reductants. Another conclusion may be drawn from the EPR observations; the decrease of the symmetry of the diiron site from axial to rhombic symmetry strongly supports the asymmetric binding of the substrate, as shown by the structure data.
The weak EPR signal of the Δ37-MIOX mutant without or with substrate (Fig. 5, spectra C and D) may be related to the deficient lid, whereby external reductants presumably obtain good access to the active site and give rise to extraneous reduction of the iron site, which accordingly loses enzymatic activity (Fig. 4). The results suggest that only the mixed valent Fe(II)/Fe(III) binds the substrate with productive results. The Δ37 mutant may also bind the substrate, but this complex is significantly weaker and less prevalent than the one that could be protected by the lid and is therefore not clearly observable in the EPR experiments (Fig. 5, spectrum D).
The two-state equilibrium does not seem to exist in the K127S mutant or in the double mutant Δ37,K127S. To explain these observations, we suggest that Lys127 governs access to the diiron site. A putative explanation for this is found by energy minimization of a model of the K127S mutant without substrate present. The removal of Lys127 enables access by Asp85 to the diiron cluster first coordination sphere, thus rendering the diiron site fully and strongly ligated but without possibilities of substrate access (supplemental Fig. S3). Indeed, the addition of substrate does not change the EPR spectra of either of these mutants, which would be a natural consequence of abolished access for the substrate to the active site. Interestingly, there is a large quantitative difference in the amount of mixed valent state induced by reduction in the single Δ37 and the double Δ37,K127S mutants (Fig. 5, spectra C and G. Presumably, the K127S mutation in the double mutant restricts access also to external reductants, and this explains why the mixed valent state is more prevalent in the double mutant.
Our EPR data on Hs-MIOX compared with the EPR studies on Mm-MIOX (6, 11, 12) demonstrate a general similarity of the spectra obtained in the absence and presence of substrate. However, there are several significant differences. The Hs-MIOX mixed valent state of the enzyme shown here has two distinct components, assigned to two clearly distinct conformations, both with the EPR signals significantly narrower than that reported for this state in Mm-MIOX with g values g∥ = 1.95, g⊥ ∼ 1.66 (4). The two different conformations of mixed valent Hs-MIOX are proposed to be open and closed relative to the active site. myo-Inositol binding to the metal site changes the EPR signal significantly. For Hs-MIOX, we find that substrate binding gives rise to one new site, with well resolved rhombic symmetry (in contrast to the axial symmetry reported for Mm-MIOX·MI (6, 11, 12)). It is important to note that when substrate has been added to Hs-MIOX, both components I and II with different g∥ values in the absence of substrate, apparently turn into one single species. This emphasizes that there is an equilibrium and dynamic interplay between the open and closed states before the substrate is bound.
The existence of open and closed active site conformations proposed here for Hs-MIOX may provide a regulating mechanism for enzyme activity. Furthermore, since proteolytic degradation of the N terminus also occurs in living cells (5), it is tempting to speculate that truncation of MIOX may play a role in the regulation of MIOX activity in vivo.
Supplementary Material
Acknowledgments
We are grateful to Laurent Terradot at ID29 (ESRF, Grenoble, France) for support during data collection.
The atomic coordinates and structure factors (codes 2IBN and 3BXD) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
The Structural Genomics Consortium is a registered charity (number 1097737) that receives funds from the Canadian Institutes for Health Research, the Canadian Foundation for Innovation, Genome Canada through the Ontario Genomics Institute, GlaxoSmithKline, Karolinska Institutet, the Knut and Alice Wallenberg Foundation, the Ontario Innovation Trust, the Ontario Ministry for Research and Innovation, Merck & Co. Inc., the Novartis Research Foundation, the Swedish Agency for Innovation Systems, the Swedish Foundation for Strategic Research, and the Wellcome Trust. This work was also supported by a grant from the Swedish Research Council (to A. G. and B. M. H.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S5 and Table S1.
Footnotes
The abbreviations used are: MIOX, myo-inositol oxygenase; Mm- and Hs-MIOX, mouse and human MIOX, respectively; wt-MIOX, wild-type human MIOX.
References
- 1.Winegrad, A. I. (1987) Diabetes 36 396-406 [DOI] [PubMed] [Google Scholar]
- 2.Arner, R. J., Prabhu, K. S., Thompson, J. T., Hildenbrandt, G. R., Liken, A. D., and Reddy, C. C. (2001) Biochem. J. 360 313-320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Naber, N. I., Swan, J. S., and Hamilton, G. A. (1986) Biochemistry 25 7201-7207 [DOI] [PubMed] [Google Scholar]
- 4.Arner, R. J., Prabhu, K. S., and Reddy, C. C. (2004) Biochem. Biophys. Res. Commun. 324 1386-1392 [DOI] [PubMed] [Google Scholar]
- 5.Arner, R. J., Prabhu, K. S., Krishnan, V., Johnson, M. C., and Reddy, C. C. (2006) Biochem. Biophys. Res. Commun. 339 816-820 [DOI] [PubMed] [Google Scholar]
- 6.Xing, G., Barr, E. W., Diao, Y., Hoffart, L. M., Prabhu, K. S., Arner, R. J., Reddy, C. C., Krebs, C., and Bollinger, J. M., Jr. (2006) Biochemistry 45 5402-5412 [DOI] [PubMed] [Google Scholar]
- 7.Nordlund, P., Sjöberg, B.-M., and Eklund, H. (1990) Nature 345 593-598 [DOI] [PubMed] [Google Scholar]
- 8.Rosenzweig, A. C., Frederick, C. A., Lippard, S. J., and Nordlund, P. (1993) Nature 366 537-543 [DOI] [PubMed] [Google Scholar]
- 9.Lindqvist, Y., Huang, W., Schneider, G., and Shanklin, J. (1996) EMBO J. 15 4081-4092 [PMC free article] [PubMed] [Google Scholar]
- 10.Nordlund, P., and Eklund, H. (1995) Curr. Opin. Struct. Biol. 5 758-766 [DOI] [PubMed] [Google Scholar]
- 11.Xing, G., Hoffart, L. M., Diao, Y., Prabhu, K. S., Arner, R. J., Reddy, C. C., Krebs, C., and Bollinger, J. M., Jr. (2006) Biochemistry 45 5393-5401 [DOI] [PubMed] [Google Scholar]
- 12.Xing, G., Diao, Y., Hoffart, L. M., Barr, E. W., Prabhu, K. S., Arner, R. J., Reddy, C. C., Krebs, C., and Bollinger, J. M., Jr. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 6130-6135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim, S. H., Xing, G., Bollinger, J. M., Jr., Krebs, C., and Hoffman, B. M. (2006) J. Am. Chem. Soc. 128 10374-10375 [DOI] [PubMed] [Google Scholar]
- 14.Brown, P. M., Caradoc-Davies, T. T., Dickson, J. M. J., Cooper, G. J. S., Loomes, K. M., and Baker, E. D. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 15032-15037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Duyne, G. D., Standaert, R. F., Karplus, P. A., Schreiber, S. L., and Clardy, J. (1993) J. Mol. Biol. 229 105-124 [DOI] [PubMed] [Google Scholar]
- 16.Porath, J., Carlsson, J., Olsson, I., and Belfrage, G. (1975) Nature 258 598-599 [DOI] [PubMed] [Google Scholar]
- 17.Atkin, C. L., Thelander, L., Reichard, P., and Lang, G. (1973) J. Biol. Chem. 248 7464-7472 [PubMed] [Google Scholar]
- 18.Kabsch, W. (1993) J. Appl. Crystallogr. 26 795-800 [Google Scholar]
- 19.Schneider, T. R., and Sheldrick, G. M. (2002) Acta Crystallogr. Sect. D 58 1772-1779 [DOI] [PubMed] [Google Scholar]
- 20.Bricogne, G., Vonrhein, C., Flensburg, C., Schiltz, M., and Paciorek, W. (2003) Acta Crystallogr. Sect. D 59 2023-2030 [DOI] [PubMed] [Google Scholar]
- 21.Cowtan, K. (2001) Acta Crystallogr. Sect. D 55 1555-1567 [DOI] [PubMed] [Google Scholar]
- 22.Perrakis, A., Harkiolaki, M., Wilson, K. S., and Lamzin, V. S. (2001) Acta Crystallogr. Sect. D 57 1445-1450 [DOI] [PubMed] [Google Scholar]
- 23.Vaguine, A. A., Richelle, J., and Wodak, S. J. (1999) Acta Crystallogr. Sect. D 55 191-205 [DOI] [PubMed] [Google Scholar]
- 24.Emsley, P., and Cowtan, K. (2004) Acta Crystallogr. Sect. D 60 2126-2132 [DOI] [PubMed] [Google Scholar]
- 25.Murshudov, G. N., Vagin, A. A., and Dodson, E. J. (1997) Acta Crystallogr. Sect. D 53 240-255 [DOI] [PubMed] [Google Scholar]
- 26.Rutter, R., Hager, L. P., Dhonau, H., Hendrich, M., Valentine, M., and Debrunner, P. (1984) Biochemistry 23 6809-6816 [DOI] [PubMed] [Google Scholar]
- 27.Pearce, L., Kurtz, D., Xia, Y.-M., and Debrunner, P. (1987) J. Am. Chem. Soc. 109 7286-7293 [Google Scholar]
- 28.Gayda, J.-P., Gibson, J., Cammack, R., Hall, D., and Mullinger, R. (1976) Biochim. Biophys. Acta 434 154-164 [DOI] [PubMed] [Google Scholar]
- 29.Yang, Y.-S., McCormick, J. M., and Solomon, E. I. (1997) J. Am. Chem. Soc. 119 11832-11842 [Google Scholar]
- 30.Lovell, S. C., Davis, I. W., Arendall, W. B., III, de Bakker, P. I., Word, J. M., Prisant, M. G., Richardson, J. S., and Richardson, D. C. (2003) Proteins 50 437-450 [DOI] [PubMed] [Google Scholar]
- 31.Laskowski, R. A., MacArthur, M. W., Moss, D. S., and Thornton, J. M. (1993) J. Appl. Crystallogr. 26 283-291 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.