

Abstract
MicroRNAs are small non-coding RNA molecules that can regulate gene expression by interacting with multiple mRNAs and inducing either translation suppression or degradation of mRNA. Recently, several miRNAs were identified as either promoters or suppressors of metastasis. However, it is unclear in which step(s) of the multistep metastatic cascade these miRNAs play a defined functional role. To study the functional importance of miRNAs in epithelial-mesenchymal transition (EMT), a process thought to initiate metastasis by enhancing the motility of tumor cells, we used a well established in vitro EMT assay: transforming growth factor-β-induced EMT in NMuMG murine mammary epithelial cells. We found that members of the miR-200 family, organized as two clusters in the genome, were repressed during EMT. Overexpression of each miRNA individually or as clusters in NMuMG cells hindered EMT by enhancing E-cadherin expression through direct targeting of ZEB1 and ZEB2, which encode transcriptional repressors of E-cadherin. In the 4TO7 mouse carcinoma cell line, which expresses low levels of endogenous E-cadherin and displays a mesenchymal phenotype, ectopic expression of the miR-200 family miRNAs significantly increased E-cadherin expression and altered cell morphology to an epithelial phenotype. Furthermore, ectopic expression of each miR-200 miRNA cluster significantly reduced the in vitro motility of 4TO7 cells in migration assays. These results suggested that loss of expression of the miR-200 family members may play a critical role in the repression of E-cadherin by ZEB1 and ZEB2 during EMT, thereby enhancing migration and invasion during cancer progression.
MicroRNAs are a large family of small (21–23-nt)4 RNAs that exhibit a high degree of structural and functional conservation throughout metazoan species. miRNAs are initially synthesized by polymerase II as long primary transcripts, which are subsequently processed into ∼70-nt stem-loop pre-microRNAs by Drosha RNase III endonuclease (1) and are transported out of the nucleus by exportin 5 (2). Pre-microRNAs are further processed in the cytoplasm by Dicer to yield the final ∼22-nt mature miRNAs (3). Binding of miRNA to target mRNAs with perfect or near perfect complementarity induces mRNA degradation, whereas imperfect complementarity often induces translational repression. It is believed that 7–8 nt in the 5′ end of miRNAs, referred to as the seed sequence, are critical for efficient targeting.
miRNAs have been implicated in regulating complex physiological processes such as embryogenesis (4), organ development (5), and oncogenesis (6, 7). However, the functional roles of a vast majority of miRNAs remain unknown. Recently, several groups have used a variety of model systems to identify different miRNAs as promoters or suppressors of metastasis (8–12). Although these studies clearly implicate these miRNAs in metastasis, it is unclear which step(s) in the multistep metastatic progression these miRNAs regulate. In the present study, we sought to define a role for miRNAs in regulating the initiating step in metastasis, epithelial-mesenchymal transition (EMT). Applying a classical model system of inducing EMT in NMuMG cells (normal murine mammary epithelial cells), we found that members of the miR-200 family, existing as two clusters in the genome, are significantly repressed during EMT, suggesting a role as suppressors of EMT. We further discovered that members of the miR-200 family hinder EMT by positively regulating E-cadherin expression through direct targeting of ZEB1 and ZEB2. Furthermore, ectopic expression of the miR-200 family in 4TO7 mammary carcinoma cells, which express low endogenous levels of these miRNAs, induced mesenchymal-epithelial transition by up-regulating E-cadherin expression and inhibited migration of these tumor cells. These results strongly suggested an important role of the miR-200 family miRNAs in repressing epithelial-mesenchymal transition and cancer progression.
EXPERIMENTAL PROCEDURES
Cell Lines and Cell Culture—NMuMG, 4TO7, and HeLa cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. NMuMG medium was further supplemented with 10 μg/ml insulin (Sigma).
RNA Extraction and Quantitative Real-time PCR—Total RNA was extracted using the miRVana miRNA isolation kit (Ambion). For miRNA analysis, mature miRNAs were reverse-transcribed, and real-time PCR was performed using TaqMan microRNA assays (Applied Biosystems). All data were normalized to U6 expression. For mRNA analysis, real-time PCR was performed using Power SYBR® green PCR master mix (Applied Biosystems) on an ABI 7900HT series PCR machine Applied Biosystems, and data were normalized to GAPDH expression and further normalized to the negative control unless otherwise indicated.
ZEB1 and ZEB2 3′-UTR Luciferase Reporter Assays—The 3′-UTRs for both ZEB1 and ZEB2 were PCR-amplified from genomic DNA extracted from NMuMG cells. PCR primers used to amplify the Zeb1 3′-UTR include 5′-AAAAATCCGGGTGTGCCTGA-3′ (forward) and 5′-AACTGCTTTCTACTGCTCTG-3′ (reverse), whereas the primers used to amplify the Zeb2 3′-UTR include 5′-CAGTTCAGCCAAGACAGAGT-3′ (forward) and 5′-TTCGAGCATGGTCATTTTC-3′ (reverse). Amplified 3′-UTRs were cloned downstream of the firefly luciferase coding region in the pMIR-REPORT™ (Ambion). HeLa or 4TO7 cells were seeded in 24-well plates 24 h prior to transfection. The following day, 200 ng of reporter plasmid along with 200 ng of control Renilla-luciferase plasmid were co-transfected using Lipofectamine 2000 (Invitrogen). Cells were collected 24 h after transfection and assayed for luciferase activity using the Glomax 96 luminometer (Promega). To assess the effect of miRNAs on reporter activity, 50 pm of synthetic precursor miRNAs (pre-miRs) (Ambion) were co-transfected. All experiments were performed in triplicates.
Immunoblot Analysis—For EMT assays, NMuMG cells were seeded in 6-well plates, and the next day, cells were transfected with synthetic miRNAs individually or as clusters. 24 h after transfection, cells were treated with 200 pm TGFβ1 (R&D systems) for 48 h. Cells were lysed in prechilled lysis buffer, and 10 μg of protein from the supernatant was loaded per lane and resolved by SDS-polyacrylamide electrophoresis. Protein was transferred onto nitrocellulose membranes, blocked, and probed with mouse anti-E-cadherin (BD Biosciences), mouse anti-N-cadherin (BD Biosciences), or mouse anti-β-actin (AbCam).
Immunofluorescence—NMuMG cells were transfected in 6-well plates with synthetic pre-microRNAs (pre-miRs). 4–6 h after transfection, the cells were dissociated and seeded onto gelatin-coated glass coverslips placed in 24-well plates and stimulated with recombinant TGFβ for an additional 48 h. 4TO7 cells were similarly treated except that stimulation with TGFβ did not take place. 72 h after transfection, media were aspirated, and cells were fixed with ice-cold methanol for 10 min, permeabilized with 0.2% Triton for 3 min, and blocked in 10% goat serum for 1 h at room temperature. E-cadherin was probed with mouse-anti-E-cadherin for 1 h at room temperature followed by detection with a rhodamine-conjugated goat anti-mouse secondary antibody for 1 h at room temperature. Hoechst dye (1 μg/ml) was subsequently used to stain nuclei. Cells were observed on a Zeiss microscope, and pictures were taken using an Axiocam Icc3 camera.
Transwell Migration Assays—4TO7 cells were transfected with pre-miRs for 48 h and subsequently dissociated. 1 × 105 cells were resuspended in serum-free media and placed in inserts containing 8-μm pores. These inserts were placed in wells with serum-containing media. 12 h after seeding, serum-containing media were aspirated, and trypsin was placed into the wells to trypsinize the cells that had passed through the pores, which were counted using a hemocytometer.
RESULTS
Down-regulation of miR-200 Family miRNAs during EMT—To study EMT, we used a classical model system: TGFβ1-induced EMT in the NMuMG mouse mammary epithelial cell line. Within 72 h of TGFβ treatment, NMuMG cells undergo a dramatic morphological change, from compact, cobblestone-like epithelial structures to fibroblastoid spindle-shaped cells, together with significant disintegration of cell-cell adhesions (Fig. 1A). This morphological transition is accompanied by E-cadherin down-regulation with reduced membrane localization (Fig. 1, A and B) and N-cadherin up-regulation (Fig. 1B) with increased localization to the membrane (Fig. 1A). These hallmark shifts at the morphological and molecular levels indicate a successful EMT program in NMuMG cells.
FIGURE 1.
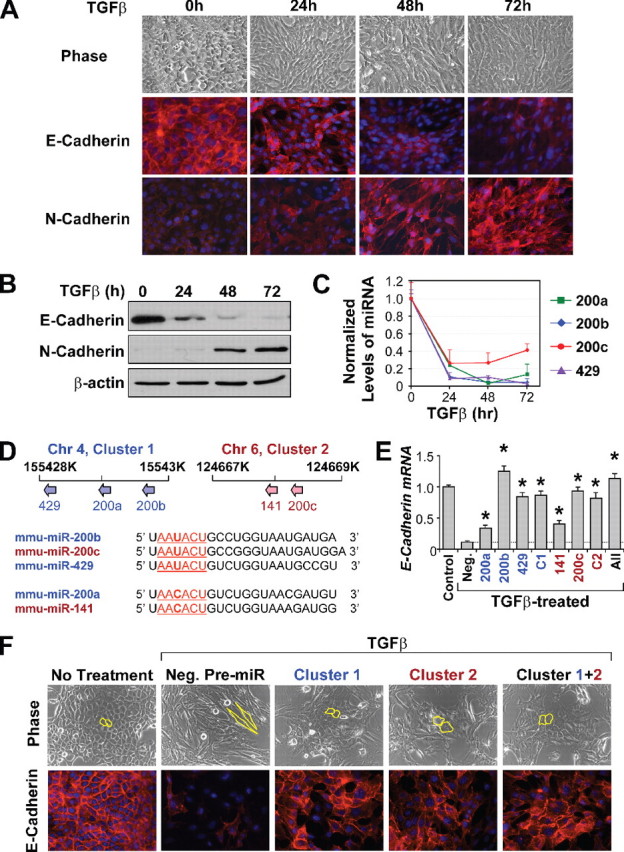
Overexpression of miR-200 family hinders EMT and up-regulates E-cadherin expression. A, phase contrast images and immunofluorescence staining of E-cadherin and N-cadherin in NMuMG cells undergoing EMT. B, Western blot analysis confirms down-regulation of E-cadherin and up-regulation of N-cadherin during EMT. C, changes in the miR-200 family levels in TGFβ-treated NMuMG cells, as measured by TaqMan qRT-PCR and normalized to U6 expression. The data are means from a representative time course experiment measured in triplicate and are presented as mean ± S.E. D, upper panel, schematic of chromosomal locations of the miR-200 family members in the mouse genome. Lower panel, sequence alignment of the miR-200 family members. Nucleotides 2–7, representing their seed sequences, are underlined. miR-200 members embedded within cluster 1 are in blue, whereas those embedded in cluster 2 are in red. Chr 4, chromosome 4. E, changes in expression of E-cadherin in NMuMG cells treated with TGFβ and transfected with miR-200 members individually, as clusters (C1 or C2), or altogether (All), as measured by real-time PCR. Expression levels are compared with cells untreated with TGFβ (control) or TGFβ-treated cells transfected with a negative control pre-miR (Neg.). * represents p < 0.05 as compared with control pre-miR. F, phase contrast microscopy and E-cadherin staining of NMuMG cells untreated or treated with TGFβ after being transfected with negative control pre-miR, cluster 1, cluster 2, or both clusters simultaneously (Cluster 1+2). Cell morphology is outlined in yellow.
Two miR-200 family miRNAs, miR-200b and miR-200c, have been previously correlated with the E-cadherin expression in epithelial cells (13, 14). Three additional miRNAs, miR-200a, miR-141, and miR-429, also belong to the miR-200 family. qRT-PCR analysis revealed that the expression of all miR-200 family miRNAs, except miR-141, were strongly down-regulated in NMuMG cells during TGFβ-induced EMT (Fig. 1C). The expression of each miRNA was reduced to less than 30% of the pretreatment level within 24 h of TGFβ exposure. We did not observe detectable expression of miR-141 in NMuMG cells.
The miR-200 family of miRNAs were mapped to two separate clusters of less than 2000 bp each in the mouse genome (Fig. 2D, upper panel). The first cluster (Cluster 1) contains miR-200a, miR-200b, and miR-429 and is located in mouse chromosome 4. Analysis of expression levels of miR-200a, miR-200b, and miR-429 in several cell lines revealed that the three members of this cluster are co-expressed (r = 0.97, data not shown), suggesting that a common promoter is likely to drive the expression of all three miRNAs. The second cluster (Cluster 2), consisting of miR-200c and miR-141, is located in a 500-bp region of chromosome 6. The five miR-200 family miRNAs contain very similar seed sequences (Fig. 1D, lower panel). The seed sequence of miR-200b, miR-200c, and miR-429, AAUACU, differs only by one nucleotide to the seed sequence of miR-200a and miR-141, AACACU.
FIGURE 2.
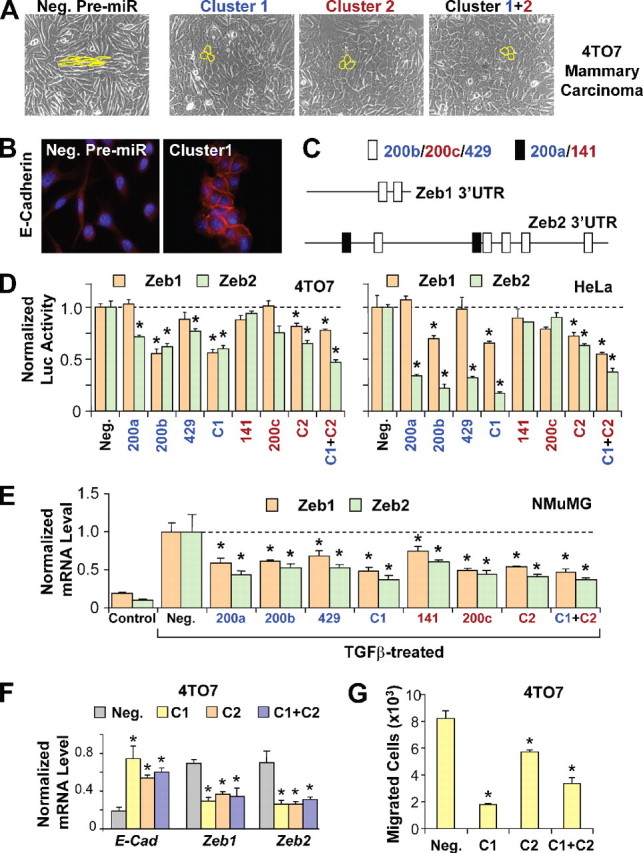
miR-200 family targets transcriptional repressors ZEB1 and ZEB2 to enhance E-cadherin expression and inhibit migration. A, phase contrast microscopy of 4TO7 cells transfected with negative control pre-miR (Neg.), cluster 1, cluster 2, or both clusters simultaneously (Cluster 1+2). Cell morphology is outlined in yellow. B, E-cadherin staining of 4TO7 cells transfected with negative control pre-miR or cluster 1. C, schematic of putative miR-200 target sites in the mouse ZEB1 and ZEB2 3′-UTRs. White boxes represent target sites for miR-200b/200c/429, whereas black boxes represent target sites for miR-200a/141. D, normalized activity of luciferase reporter with the ZEB1 (brown bars) or ZEB2 (green bars)3′-UTR in 4TO7 (left panel) or HeLa cells (right panel) in the presence of co-transfected negative control pre-miR or miR-200 members individually, as clusters (C1 or C2) or both clusters (C1 + C2). Luciferase activity was measured after 24 h. The data are mean ± S.E. of triplicates and are shown as the ratio of firefly to Renilla luciferase activity. E, expression levels of ZEB1 and ZEB2 in NMuMG cells untreated (Control) or treated with TGFβ, transfected with negative control pre-miR or miR-200 family members individually, as clusters (C1 or C2), or both clusters (C1 + C2). F, changes in expression of E-cadherin (E-Cad), ZEB1, and ZEB2 (normalized to GAPDH without further normalization to the negative control) in 4TO7 cells transfected with negative control pre-miR or with cluster 1, cluster 2, or both clusters. G, migration of 4TO7 cells transfected with negative control pre-miR, cluster 1, cluster 2, or both clusters, toward serum containing media. The data are the average number of cells that migrated in a representative experiment measured in triplicate and are presented as means + S.E. * represents p < 0.05.
Overexpression of miR-200 Members Individually or in Combination Represses EMT and Enhances E-cadherin Expression—Because miRNAs in the miR-200 family were similarly down-regulated during EMT, we tested the functional role of each miRNA in the regulation of E-cadherin expression and the control of EMT in NMuMG cells by transient overexpression of synthetic pre-miRs. Successful transfection was achieved in over 90% of the cell populations for up to 3 days as assessed by fluorescence-activated cell sorting analysis of a cy3-labeled control pre-miR (data not shown). NMuMG cells transfected with the negative control pre-miR responded to TGFβ treatment with more than 90% reduction of E-cadherin expression (Fig. 1E) and morphological changes characteristic of EMT (Fig. 1F). In contrast, overexpression of pre-miRs corresponding to each of the five miR-200 miRNAs led to significant resistance of E-cadherin to transcriptional repression during EMT (Fig. 1E). miR-200b, miR-200c, and miR-429 have the most robust effect in maintaining E-cadherin expression, whereas miR-200a and miR-141 have a relatively modest effect (Fig. 1E). Transfection of all five miRNAs, or miRNAs in two individual clusters, achieved a similar effect on E-cadherin expression as that of miR-200b, miR-200c, or miR-429 alone. Furthermore, expression of miRNAs as clusters (Fig. 1F) or individually (data not shown) significantly reduced the acquisition of mesenchymal characteristics during the TGFβ-induced EMT of NMuMG cells (Fig. 1F). In the miR-200-transfected cells, although the elongation of cells and the loss of compact cell-cell adhesion was apparent, the transition to fibroblastoid morphology was incomplete, with many clusters of cells maintaining the cobblestone-like epithelial characteristics (Fig. 1F). Immunofluorescence analysis of miR-200-transfected cells revealed a significant level of E-cadherin expression and the maintenance of adheren junctions. The activation of N-cadherin expression in TGFβ-treated cells, however, was not affected by miR-200 overexpression (data not shown). Overall, these results indicate that all five members of the miR-200 family are capable of functionally disrupting the epithelial-mesenchymal switch in NMuMG cells by maintaining high levels of E-cadherin expression.
Overexpression of the miR-200 Family Reverses the Mesenchymal Characteristics of the 4TO7 Mammary Carcinoma Cell Line—Next, we tested the ability of the miR-200 family of miRNAs to reverse the mesenchymal phenotype of metastatic breast cancer cells. 4TO7 is a mammary carcinoma cell line derived from a spontaneous mammary tumor in a wild-type BALB/c mouse (15). The 4TO7 cell line is highly invasive and is capable of seeding numerous micrometastases from orthotopic mammary gland tumors in BALB/c recipient animals (16). Microscopically, the 4TO7 cells display a typical fibroblastic morphology (Fig. 2A), consistent with a very low level of E-cadherin expression (Fig. 2B) and miR-200 family miRNA expression (data not shown). Overexpression of the two miR-200 clusters, individually or in combination, for 3 days in 4TO7 cells produced a dramatic shift in morphology, from a spindle-shaped mesenchymal population to a cobblestone-shaped epithelial population (Fig. 2A). The epithelial phenotype of miR-200-transfected 4TO7 cells was evident after 48 h, reaching a peak at 72 h and reverting back to mesenchymal phenotype at ∼84 h after transfection, possibly due to a degradation of transfected pre-miRs. Immunofluorescence analysis revealed a significant increase of E-cadherin expression. Importantly, E-cadherin is enriched on the plasma membrane in miR-200 overexpression cells and forms adherens junctions between neighboring cells, suggesting that E-cadherin can functionally contribute to the acquisition of epithelial phenotype in 4TO7 cells.
Direct Targeting of E-cadherin Transcriptional Repressors ZEB1 and ZEB2 by miR-200 Family miRNAs—To identify likely direct targets of miR-200 miRNAs, we searched the TargetScan data base (17) for miR-200 target sites in the mRNA sequences of known suppressors of E-cadherin expression, including Snail, Slug, Twist, Goosecoid, FoxC2, ZEB1, and ZEB2. E-box-binding zinc-finger transcription factor ZEB2 (SIP1/ZFXH1B) is predicted to be the most likely target gene of the miR-200 family since its 3′-UTR contains at least two sites for miR-200a/141 and five sites for miR200b/200c/429 (Fig. 2C). ZEB1 (TCF8/δEF1/Nil-2α), belonging to the same family as ZEB2, is also predicted to contain at least two target sites for miR-200b/200c/429. To test the direct targeting of ZEB1 and ZEB2 by miR-200, we cloned their 3′-UTR sequences downstream of a firefly luciferase reporter gene. Co-transfection of the reporter plasmid along with miR-200a, miR-200b, miR-429, as well as each or both of the clusters in 4TO7 cells, resulted in a significantly reduced ZEB2-3′-UTR-luciferase expression, suggesting that these miRNAs are likely to target ZEB2 directly (Fig. 2D, left panel). In reporter assays using the ZEB1 3′-UTR, only miR-200b and both clusters were able to significantly reduce luciferase reporter expression, possibly reflecting the lower abundance of miR-200 sites in the ZEB1 3′-UTR fragment cloned into the luciferase reporter plasmid. Similar reporter assay results were obtained from the HeLa cell line (Fig. 2D, right panel). Reporter assay results were less consistent in NMuMG cells (data not shown), possibly due to the high basal level of miR-200 expression in this epithelial cell line.
To test for the targeting of endogenous ZEB1 and ZEB2 transcripts by miR-200, we analyzed ZEB1 and ZEB2 mRNA levels in NMuMG cells transfected with the miR-200 family members individually, simultaneously, or in clusters. The transfected cells were then induced to undergo EMT with TGFβ1 treatment. Although ZEB1 and ZEB2 levels robustly increased during EMT in control cells, overexpression of each of the miR-200 family members significantly reduced the accumulation of both ZEB1 and ZEB2 transcripts. Thus, it is likely that the miR-200 family members enhance E-cadherin expression by directly reducing the expression of transcriptional suppressors of E-cadherin.
To test whether direct targeting of ZEB1 and ZEB2 is also involved in the up-regulation of E-cadherin in 4TO7 mammary tumor cells, we performed qRT-PCR analysis at 48 h following the transfection of miR-200 clusters. A dramatic increase in E-cadherin expression was accompanied by a significant decrease in ZEB1 and ZEB2 mRNA levels (Fig. 2F). Time course analysis revealed immediate targeting of ZEB1 and ZEB2 within 24 h in 4TO7 cells, which was maintained after 48 h. However, ZEB1 and ZEB2 targeting was lost after 72 h after transfection, consistent with the reversion back to the mesenchymal phenotype at ∼84 h after transfection.
miR-200 Family miRNAs Inhibit Migration of 4TO7 Mammary Tumor Cells—Loss of E-cadherin and acquisition of mesenchymal properties, including increased motility, are thought to be critical events during the transition of benign tumors to malignant carcinomas. Given the strong inhibition of E-cadherin and exhibition of epithelial characteristics in 4TO7 mammary carcinoma cells transfected with the miR-200 family miRNAs, we sought to determine the effect of these miRNAs in cell migration. Using an in vitro transwell migration assay, overexpression of each of the miR-200 clusters strongly reduced growth factor-induced directional migration of the 4TO7 cells (Fig. 2G).
DISCUSSION
During embryonic development, EMT is a crucial process in the formation of various tissues and organs such as the neural crest, heart, musculoskeletal systems, and peripheral nervous systems. However, during adult life, only a certain subset of cells retains the ability to undergo transient EMT, for example, keratinocytes during wound healing (18). Malignant breast tumor cells are known to reactivate embryonic programs such as EMT to obtain a selective advantage such as enhanced motility and invasiveness (16, 19, 20). As EMT is tightly linked to invasive carcinomas, molecular regulators of this process may serve as key points for therapeutic intervention.
Previous studies have linked the miR-200 family with the epithelial phenotype and the ZEB family. Expression of the miR-200 family was found to be enriched in epithelial tissues (21, 22) and negatively correlated with ZEB1 and ZEB2 expression during embryonic development (23). In addition, the ZEB family has been implicated in EMT, tumorigenesis, and metastasis (24–27). However, the functional involvement of miR-200 in EMT and tumor migration, as well as direct targeting of ZEB1 and ZEB2 by the entire miR-200 family, has not been investigated in these previous studies.
In our present study, we identified the miR-200 family as suppressors of EMT through direct targeting of ZEB1 and ZEB2, well known transcriptional repressors of E-cadherin. The miR-200 family is composed of five members arranged as two clusters, miR-200a/200b/429 and miR-200c/141. ZEB1 and ZEB2 were likely targets because both are EMT inducers and contain target sites for many of the miR-200 family members. ZEB2 had target sites for all five miRNAs, but the TargetScan data base indicated that ZEB1 had only two target sites for miR-200b, -200c, and -429. To the contrary, our results showed that overexpression of miR-200a and -141 also significantly reduced the expression of endogenous ZEB1 (Fig. 2D), leading us to believe that the 3′-UTR of ZEB1 may in fact harbor additional target sites for these miRNAs. Indeed, a recent report found that the Genbank™ Refseq entry for ZEB1 (NM_030751) was artificially truncated, and manual inspection revealed several target sites for miR-200a and miR-141 in the ZEB1 3′-UTR (28).
We found that ectopic expression of the miR-200 family individually, as clusters, or altogether hindered EMT progression in TGFβ-treated NMuMG cells by keeping ZEB1 and ZEB2 expression levels low and E-cadherin expression levels high (Figs. 1E and 2D). Moreover, these cells overexpressing the miR-200 family maintained a cobblestone-like epithelial phenotype. Ectopic expression of each miR-200 miRNA was sufficient to hinder EMT progression, although miR-200a and miR-141 were consistently the weakest positive regulators of E-cadherin. Simultaneous overexpression of miRNAs from the same cluster or from both clusters did not appear to have a synergistic effect in regulating ZEB1, ZEB2, or E-cadherin, at least in the in vitro setting. Although we found that the miR-200 family can hinder EMT, their overexpression was not sufficient to completely block this process, suggesting that other miRNAs may also be involved in regulating EMT.
Ectopic expression of the miR-200 family in 4TO7 carcinoma cells caused a rapid and significant decrease in ZEB1 and ZEB2 levels. Overexpressing the miR-200 family in 4TO7 cells induced a dramatic morphological change from a spindle-like, mesenchymal phenotype toward a more pronounced epithelial phenotype with formation of adherens junctions. Furthermore, overexpression of miR-200 significantly inhibits growth factor-induced directional migration, a hallmark of metastatic cancer cells. This suggests that ectopic expression of the miR-200 family can promote the mesenchymal-epithelial transition and reduce tumor cell migration. Since ZEB1 and ZEB2 have been implicated in the progression of various tumor types, the miR-200 family may provide an important avenue for therapeutic targeting of metastatic carcinomas.
Acknowledgments
We thank members of the Kang laboratory for helpful suggestions and discussion. We are thankful to Drs. Hilary Coller and Aster Legesse-Miller for providing technical expertise and helpful discussions.
This work was supported by the U.S. Army Medical Research and Material Command (Grant W81XWH-06-1-0481) with additional support from the American Cancer Society (Grant RSG MGO-110765), and the Susan G. Komen Foundation (Grant BCTR0503765). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: nt, nucleotide; EMT, epithelial-mesenchymal transition; TGF, transforming growth factor; miRNA, microRNA; pre-miR, pre-microRNA; UTR, untranslated region; qRT-PCR, quantitative reverse transcription-PCR.
References
- 1.Lee, Y., Ahn, C., Han, J., Choi, H., Kim, J., Yim, J., Lee, J., Provost, P., Radmark, O., Kim, S., and Kim, V. N. (2003) Nature 425 415–419 [DOI] [PubMed] [Google Scholar]
- 2.Yi, R., Qin, Y., Macara, I. G., and Cullen, B. R. (2003) Genes Dev. 17 3011–3016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hutvagner, G., McLachlan, J., Pasquinelli, A. E., Balint, E., Tuschl, T., and Zamore, P. D. (2001) Science 293 834–838 [DOI] [PubMed] [Google Scholar]
- 4.Wienholds, E., Kloosterman, W. P., Miska, E., Alvarez-Saavedra, E., Berezikov, E., de Bruijn, E., Horvitz, H. R., Kauppinen, S., and Plasterk, R. H. (2005) Science 309 310–311 [DOI] [PubMed] [Google Scholar]
- 5.Yi, R., O'Carroll, D., Pasolli, H. A., Zhang, Z., Dietrich, F. S., Tarakhovsky, A., and Fuchs, E. (2006) Nat. Genet. 38 356–362 [DOI] [PubMed] [Google Scholar]
- 6.Esquela-Kerscher, A., and Slack, F. J. (2006) Nat. Rev. Cancer 6 259–269 [DOI] [PubMed] [Google Scholar]
- 7.Johnson, S. M., Grosshans, H., Shingara, J., Byrom, M., Jarvis, R., Cheng, A., Labourier, E., Reinert, K. L., Brown, D., and Slack, F. J. (2005) Cell 120 635–647 [DOI] [PubMed] [Google Scholar]
- 8.Huang, Q., Gumireddy, K., Schrier, M., le Sage, C., Nagel, R., Nair, S., Egan, D. A., Li, A., Huang, G., Klein-Szanto, A. J., Gimotty, P. A., Katsaros, D., Coukos, G., Zhang, L., Pure, E., and Agami, R. (2008) Nat. Cell Biol. 10 202–210 [DOI] [PubMed] [Google Scholar]
- 9.Ma, L., Teruya-Feldstein, J., and Weinberg, R. A. (2007) Nature 449 682–688 [DOI] [PubMed] [Google Scholar]
- 10.Tavazoie, S. F., Alarcon, C., Oskarsson, T., Padua, D., Wang, Q., Bos, P. D., Gerald, W. L., and Massague, J. (2008) Nature 451 147–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu, S., Wu, H., Wu, F., Nie, D., Sheng, S., and Mo, Y. Y. (2008) Cell Res. 18 350–359 [DOI] [PubMed] [Google Scholar]
- 12.Asangani, I. A., Rasheed, S. A., Nikolova, D. A., Leupold, J. H., Colburn, N. H., Post, S., and Allgayer, H. (2008) Oncogene 27 2128–2136 [DOI] [PubMed] [Google Scholar]
- 13.Christoffersen, N. R., Silahtaroglu, A., Orom, U. A., Kauppinen, S., and Lund, A. H. (2007) RNA (Cold Spring Harbor) 13 1172–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hurteau, G. J., Carlson, J. A., Spivack, S. D., and Brock, G. J. (2007) Cancer Res. 67 7972–7976 [DOI] [PubMed] [Google Scholar]
- 15.Aslakson, C. J., and Miller, F. R. (1992) Cancer Res. 52 1399–1405 [PubMed] [Google Scholar]
- 16.Yang, J., Mani, S. A., Donaher, J. L., Ramaswamy, S., Itzykson, R. A., Come, C., Savagner, P., Gitelman, I., Richardson, A., and Weinberg, R. A. (2004) Cell 117 927–939 [DOI] [PubMed] [Google Scholar]
- 17.Lewis, B. P., Burge, C. B., and Bartel, D. P. (2005) Cell 120 15–20 [DOI] [PubMed] [Google Scholar]
- 18.Thiery, J. P. (2003) Curr. Opin. Cell Biol. 15 740–746 [DOI] [PubMed] [Google Scholar]
- 19.Huber, M. A., Azoitei, N., Baumann, B., Grunert, S., Sommer, A., Pehamberger, H., Kraut, N., Beug, H., and Wirth, T. (2004) J. Clin. Investig. 114 569–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muraoka, R. S., Dumont, N., Ritter, C. A., Dugger, T. C., Brantley, D. M., Chen, J., Easterly, E., Roebuck, L. R., Ryan, S., Gotwals, P. J., Koteliansky, V., and Arteaga, C. L. (2002) J. Clin. Investig. 109 1551–1559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baskerville, S., and Bartel, D. P. (2005) RNA (Cold Spring Harbor) 11 241–247 [Google Scholar]
- 22.Lu, J., Getz, G., Miska, E. A., Alvarez-Saavedra, E., Lamb, J., Peck, D., Sweet-Cordero, A., Ebert, B. L., Mak, R. H., Ferrando, A. A., Downing, J. R., Jacks, T., Horvitz, H. R., and Golub, T. R. (2005) Nature 435 834–838 [DOI] [PubMed] [Google Scholar]
- 23.Miyoshi, T., Maruhashi, M., Van De Putte, T., Kondoh, H., Huylebroeck, D., and Higashi, Y. (2006) Dev. Dyn 235 1941–1952 [DOI] [PubMed] [Google Scholar]
- 24.Spoelstra, N. S., Manning, N. G., Higashi, Y., Darling, D., Singh, M., Shroyer, K. R., Broaddus, R. R., Horwitz, K. B., and Richer, J. K. (2006) Cancer Res. 66 3893–3902 [DOI] [PubMed] [Google Scholar]
- 25.Spaderna, S., Schmalhofer, O., Hlubek, F., Berx, G., Eger, A., Merkel, S., Jung, A., Kirchner, T., and Brabletz, T. (2006) Gastroenterology 131 830–840 [DOI] [PubMed] [Google Scholar]
- 26.Maeda, G., Chiba, T., Okazaki, M., Satoh, T., Taya, Y., Aoba, T., Kato, K., Kawashiri, S., and Imai, K. (2005) Int. J. Oncol. 27 1535–1541 [PubMed] [Google Scholar]
- 27.Imamichi, Y., Konig, A., Gress, T., and Menke, A. (2007) Oncogene 26 2381–2385 [DOI] [PubMed] [Google Scholar]
- 28.Gregory, P. A., Bert, A. G., Paterson, E. L., Barry, S. C., Tsykin, A., Farshid, G., Vadas, M. A., Khew-Goodall, Y., and Goodall, G. J. (2008) Nat. Cell Biol. 10.1038/ncb1722 [DOI] [PubMed]