

Abstract
Bax is activated and translocated onto mitochondria to mediate cytochrome c release and apoptosis. The molecular mechanisms of Bax activation during apoptosis remain a subject of debate. We addressed the question of whether reactive oxygen species could directly activate Bax for its subsequent translocation and apoptosis. Using the SW480 human colon adenocarcinoma cell line stably expressing Bax fused to GFP, we showed that H2O2 induces Bax conformational change, mitochondrial translocation, and subsequent oligomerization at mitochondria. We found that H2O2-induced Bax activation is dependent on the conserved cysteine residue 62 of Bax. Mutation of cysteine 62, but not cysteine 126, to serine or alanine abolished its activation by H2O2 but not other death stimuli, both in SW480 and Bax-deficient HCT116 cells, whereas wild type Bax sensitizes these cells to apoptosis. Cysteines of Bax could chemically react with H2O2. Mutation of Bax BH3 domain in the presence of cysteine 62 also abolished Bax proapoptotic activity. We conclude that reactive oxygen species could be a direct signal for Bax activation by reacting with cysteine residues. Our results identify a critical role of cysteine 62 in oxidative stress-induced Bax activation and subsequent apoptosis.
Bcl-2 family proteins constitute a central checkpoint for apoptotic pathways. Upon death stimulation, the cytosolic proapoptotic proteins, such as Bax, translocate onto mitochondria, where it interacts with and neutralizes the antiapoptotic proteins Bcl-2/Bcl-xL, which serve as a gateway for cytochrome c (cyt c)5 release and the apoptotic cascade (1). Bax contains three functional domains (BH1, BH2, and BH3) in addition to a transmembrane domain. Despite the presence of membrane localization domain, Bax resides primarily in the cytosol and requires an initial activation step to promote its translocation to the mitochondrial outer membrane (MOM) (2-4). The mechanisms of Bax activation and translocation remain a subject of debate. Mutational studies with Bax suggest that Bax translocation to the MOM during apoptosis involves the unfolding of the hydrophobic C-terminal end (i.e. helix α9, αH-9) from its hydrophobic groove and a change in the conformation in the N terminus of the protein (2). Thus, Bax appears to undergo one or more conformational changes during its activation, leading to its anchoring in the MOM and its oligomerization (5). An additional step, the disruption of an electrostatic bond between Asp-33 (Hα1), Lys-64, and Leu-70 (within the BH3 domain), results in Bax being addressed to the mitochondria (6). These studies are important for understanding how Bax is activated. However, it remains to be determined if this type of activation is physiologically relevant, since some of these mutations may result in the irreversible alteration of the conformation of the protein and constitutive activation.
Critical questions remain regarding how apoptotic stimuli and the intracellular environmental cues trigger Bax conformational change and subsequent activation during the onset of apoptosis. A high pH has been reported to promote conformational change (7). However, other studies, including ours, show a decrease in pH during apoptosis (8, 9). Also, an NMR study did not support pH being involved in the structural rearrangement or change of the aggregation state of Bax in solution (10). BH3-containing proapoptotic molecules, such as Bim and Bid, could bind to and activate Bax. However, in some systems, the direct interaction between these proapoptotic molecules could not be detected, at least in cytosol. Instead, Bim and Bid could interact and neutralize Bcl-2, Bcl-xL, and/or Mcl-1, leading to Bax release from its antiapoptotic partners and activation (11).
Reactive oxygen species (ROS) could signal for apoptosis (3). Most studies point out that ROS exert their nonspecific damaging effects on DNA and proteins, which activate multiple signaling pathways for Bax transcription and activation (12). Intracellular ROS or perturbation of the intracellular redox could be the upstream factors for Bax translocation and homooligomerization in the apoptotic pathway (8, 13-15); Bax translocation was found to be associated with the superoxide anion production linked to respiratory chain inhibition and uncoupling of mitochondrial respiration (and H2O2 as a secondary product) (16). H2O2 is a strong activator of c-Jun N-terminal protein kinase, which can phosphorylate Bax prior to its translocation to mitochondria and apoptosis (17). It remains to be determined if intracellular H2O2 could directly activate Bax. We here tested whether ROS is a direct signal for Bax activation during H2O2-induced apoptosis. Specifically, we sought to understand whether the two conserved cysteines (Cys-62 and Cys-126) mediate Bax conformation change, mitochondrial translocation, and oligomerization in response to H2O2. Experiments in a colon cancer cell line and Bax knockout HCT116 cells showed that oxidation of cysteine 62 could lead to the conformational change and redox-dependent proapoptotic activity of Bax.
EXPERIMENTAL PROCEDURES
Materials—H2O2, xanthine (X), xanthine oxidase (XO), N-acetyl-l-cysteine (NAC), campothecin, S-nitroso-N-acetylpenicillamine anti-actin (A-5411), and anti-Bax 6A7 monoclonal antibody were purchased from Sigma. Manganese (III) tetrakis (N-methyl-2-pyridyl) porphyrin was purchased from Alexis Biochemicals (San Diego, CA). Disuccinimidyl suberate was obtained from Pierce. Anti-GFP (B-2, sc-9996) monoclonal and anti-Bax (N-20, sc-493) were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Anti-caspase 3 polyclonal antibody was obtained from Cell Signaling Technology, Inc. Anti-cyt c and polyclonal Bcl-xL antibody were from BD Biosciences PharMingen (San Jose, CA). DAPI and Mito-Tracker Red CMXRos were purchased from Molecular Probes, Inc. (Eugene, OR).
Plasmids—The mammalian expression vector encoding human Bax (pEGFP-C3-Bax) constructs was obtained from R. Youle (National Institutes of Health, Bethesda, MD). Bcl-xL was inserted into pCDNA 4-TO/B. The Bax site-directed mutant constructs were made using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). The following primers were used to generate the site-directed mutant constructs used in this study: Cys/Ser-62 (C62/S) sense, 5′-CACCAAGAAGCTGAGCGAGTCTCTCAAGCGCATCGGGGACG-3′; C126S forward, 5′-GGCCCTGTGCACCAAGGTCCCGGAACTGATCAGAACC-3′.
Cell Culture and Transfection—SW480 and HCT116 (Bax-/-) cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (Hyclone) and 1% penicillin-streptomycin at 37 °C under 5% CO2. To establish Bax-stable transfectants, SW480 cells were transfected with pEGFP-C3-Bax and Bax mutant plasmid DNA, and positive clones overexpressing Bax were selected with 1 mg/ml G418 as described previously (13, 18). HCT116 (Bax-/-) cells were placed on 6-well tissue plates and then transfected with the appropriate plasmid DNA using the protocol described by the manufacturer. Typically, 1 μg of plasmid DNA and 4 μl of DMRIE-C reagent (Invitrogen) were used per coverslip. The cells were incubated for 4 h in the transfection mixture, which was then replaced with fresh culture medium. Positive clones overexpressing Bax were selected with 1 mg/ml G418 for 2 weeks. The transfected cells were visualized by fluorescence microscopy.
Apoptosis Assays and Fluorescence Microscopy—Apoptosis was induced by incubation for the indicated times with H2O2, 500 μm X, or 0.1 units of XO in culture medium. The percentage of living cells with high mitochondrial membrane potential was determined by 3,3′-dihexyloxacarbocyanine iodide and propidium iodide staining, followed by flow cytometry. Apoptotic cells were identified as Annexin V+ cells. Nuclear condensation was assessed by staining with DAPI. Cells were grown to 70% confluence on a coverslip. Mitochondria was stained with 50 nm MitoTracker Red CMXRos at 37 °C for 20 min, and the cells were washed twice with phosphate-buffered saline and then fixed with freshly prepared 3.7% formaldehyde at 37 °C for another 20 min. Cell images were captured with a LSM 510 Zeiss confocal microscope.
Cell Fractionation—Cells were fractionated by differential centrifugation as previously described (3, 19). Briefly, cells were harvested and resuspended in 3 volumes of hypotonic buffer (210 mm sucrose, 70 mm mannitol, 10 mm Hepes, pH 7.4, 1 mm EDTA) containing 1 mm phenylmethylsulfonyl fluoride, 50 mg/ml trypsin inhibitor, 10 mg/ml leupeptin, 5 mg/ml aprotinin, and 10 mg/ml pepstatin. After gentle homogenization with a Dounce homogenizer, cell lysates were centrifuged at 1,000 × g for 5 min to remove unbroken cells and nuclei. The supernatant was collected and centrifuged at 10,000 × g to pellet the mitochondria-enriched heavy membrane fraction. The supernatant was further centrifuged at 100,000 × g to obtain a cytosolic fraction. To determine the mitochondrial membrane integral Bax, the mitochondrial pellets (1 mg of protein/ml) were resuspended in freshly prepared 0.1 m Na2CO3 (pH 11.5) and incubated for 20 min on ice. The membranes were then collected by centrifugation.
SDS-PAGE and Immunoblotting—SDS-PAGE and immunoblotting were performed as described elsewhere (3, 20). Briefly, the cells or the membrane fractions were resuspended in lysis buffer containing Nonidet P-40 (10 mm Hepes, pH 7.4, 2 mm EGTA, 0.5% Nonidet P-40, 1 mm NaF, 1 mm NaVO4, 1 mm phenylmethylsulfonyl fluoride, 1 mm dithiothreitol, 50 μg/ml trypsin inhibitor, 10 μg/ml aprotinin, and leupeptin) and placed on ice for 30 min. The lysates were centrifuged at 12,000 × g for 12 min at 4 °C, and the protein concentration was determined. Equivalent samples (30 μg of protein) were subjected to SDS-PAGE on 12% gels. The proteins were then transferred onto nitrocellulose membranes and probed with the indicated antibodies followed by the appropriate secondary antibodies conjugated to horseradish peroxidase (KPL, Gaithersburg, MD). Immunoreactive bands were visualized using enhanced chemiluminescence (Pierce). The molecular sizes of the proteins detected were determined by comparison with prestained protein markers (Invitrogen).
Detection of Bax Conformational Change—This assay was performed as described previously (3). Cytosolic or mitochondrial fraction was lysed with Chaps lysis buffer (10 mm Hepes, pH 7.4, 150 mm NaCl, 1% Chaps) containing protease inhibitors as described. The cell lysates were normalized for protein content, and 250 μg of total protein was incubated with 1 μg of anti-Bax 6A7 monoclonal antibody in 500 μl of Chaps lysis buffer at 4 °C for 3 h or overnight. Then 25 μl of protein G-agarose was added, and the reactions were incubated at 4 °C for an additional 2 h. The beads were washed three times in Chaps lysis buffer, boiled in loading buffer, and the conformationally changed Bax protein in the immunoprecipitates was subjected to SDS-PAGE and immunoblot analysis with anti-Bax polyclonal antibody as described above.
Cross-linking of Bax Protein—Following H2O2 treatment, cells were washed with conjugating buffer containing 150 mm NaCl, 20 mm Hepes (pH 7.2), 1.5 mm MgCl2, and 10 mm glucose. Disuccinimidyl suberate in DMSO was added from a 10-fold stock solution to a final concentration of 2 mm (3, 21). The samples were incubated at room temperature for 30 min with no reduced buffer, and the cross-linker was then quenched by the addition of 1 m Tris-HCl (pH 7.5) to a final concentration of 20 mm and incubation at room temperature for 15 min. The samples were then solubilized in 0.5% Nonidet P-40 lysis buffer without a reducing agent and centrifuged at 12,000 × g for 10 min. Bax was detected by Western blotting with anti-Bax polyclonal antibody.
Determination of Bax Redox Status—HCT116 (Bax-/-) cells overexpressing GFP-Bax and Bax mutants were treated with H2O2 for indicated times and lysed with no reduced buffer. Aliquots of the cells were mixed with one-tenth volume of 100% trichloroacetic acid. Precipitated cells were collected by centrifugation, and the pellet was dissolved in 40 μl of reaction buffer (0.1% SDS, 0.67 m Tris-HCl, pH 7.5) supplemented with a mixture of protease inhibitors with 15 mm freshly prepared 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid (AMS; Molecular Probes). Samples were incubated for 30 min at room temperature and for 10 min at 37 °C and then loaded on SDS-PAGE. Bax was detected by Western blotting with anti-Bax polyclonal antibody (22). Alkylation with AMS adds ∼500 Da of molecular mass to every sulfhydryl group. When run on nonreducing SDS-PAGE, proteins in their reduced (alkylated) and oxidized (nonalkylated) forms can be resolved by the increase in molecular weight due to the addition of AMS.
RESULTS
H2O2-induced Bax Translocation and Nuclear Condensation in SW480 Colon Cancer Cells—We established the GFP-Bax SW480 colon cancer cell line stably expressing wild-type human Bax fused to GFP at its N terminus (GFP-Bax) and cells carrying the empty vector (GFP-SW480 cells). We then studied Bax translocation and nuclear condensation in response to H2O2 treatment. GFP was diffuse in the cytosol of both cell lines before treatment and became punctate and co-localized with the mitochondria in 40-50% of GFP-Bax cells but not in GFP-SW480 cells 6 h after treatment. Thus, GFP-Bax, but not GFP alone, could sensitize the cells toward apoptosis. The punctate Bax fluorescence overlapped with Mitotracker staining. Nuclear condensation occurs in the Bax translocated cells (Fig. 1, A and B), suggesting that H2O2-induced Bax translocation causes typical apoptosis. Bax translocation and H2O2-induced cell death were blocked by pretreatment of the cells with a commonly used antioxidant, NAC (Fig. 1, A and B), indicating that ROS-induced apoptosis is associated with Bax translocation. Under the high dose of H2O2 (100 μm) and long duration of the treatment (10 h) SW480 cells expressing GFP only or vector control are sensitive to H2O2-induced apoptosis, whereas under the physiological dose of H2O2 (25 μm) and short time (6 h) the endogenous Bax proapoptotic activity was not detected (supplemental Fig. 1A). These data indicated that only under this condition endogenous Bax is not activated.
FIGURE 1.

Translocation of Bax in the SW480 colon cancer cell line during H2O2-induced cell death. A, confocal microscopy analysis of Bax translocation to mitochondria. GFP-vector and GFP-Bax cells were cultured in the presence or absence of 25 μm H2O2 for 6 h, and mitochondria were fluorescently stained with 50 nm MitoTracker. Nuclei were counterstained with DAPI. B, quantitative analyses are represented in the graphs. Cell death was assessed by measuring cells with condensed nuclear. At least 200 cells were counted in each experiment (n = 3, mean ± S.D.). C, GFP-Bax cells were stably transfected with the control vector (Ctrl vector) and Bcl-xL constructs. The isolated cell clones were analyzed. Left, cell lysates were subjected to SDS-PAGE, blotted, and probed with the indicated antibodies. Right, representative cells analyzed by confocal microscopy for cell death and mitochondrial morphology. Bax translocation and apoptotic cells were characterized as indicated in A and B.
We also investigated whether H2O2-induced Bax translocation and apoptosis were inhibited by Bcl-xL overexpression. Wild-type Bcl-xL was expressed in GFP-Bax-overexpressing cells. As expected, we found that Bax translocation and cell death were clearly inhibited by Bcl-xL overexpression in GFP-Bax cells (Fig. 1C). However, mitochondria become fragmented even in cells overexpressing Bcl-xL, which prevents H2O2 induced Bax translocation, cyt c release, and apoptosis (data not shown).
Cysteine 62 of Bax Is Critical for Bax Translocation to Mitochondria in H2O2-induced Apoptosis—We analyzed the degree of conservation of two cysteine residues at positions 62 and 126 of Bax (supplemental Table 1) and found that exempted isoform δ cysteine 62 is conserved for all the different isoforms. In addition, we found that this cysteine is conserved from Epinephelus coioides to humans.
We reasoned whether these two highly conserved residues may be responsible for H2O2-induced Bax activation. Thus, we established SW480 cell clones stably expressing recombinant GFP-Bax plasmids carrying mutations corresponding to cysteine to serine substitutions at position 62 (GFP-Bax C62S) or 126 (GFP-Bax C126S) or both (GFP-Bax C62S/126S). These different cells expressed equivalent amounts of Bax, and the level is similar to endogenous Bax expression (Fig. 2A). We used a physiologically relevant concentration (25 μm) of H2O2 to treat the stably transfected cells for various times. H2O2 induced apoptosis in a time-dependent manner in GFP-Bax and GFP-Bax C126S. However, H2O2-induced apoptosis in cells expressing GFP-Bax C62S, and a double mutant is significantly reduced (Fig. 2B). The mitochondrial membrane potential was maintained in the cells expressing GFP-Bax C62S and the double mutation, but not in cells expressing GFP-Bax C126S, after H2O2 treatment (Fig. 2B). High doses of H2O2 (above 100 μm) induced cell lysis in all different cell types.
FIGURE 2.
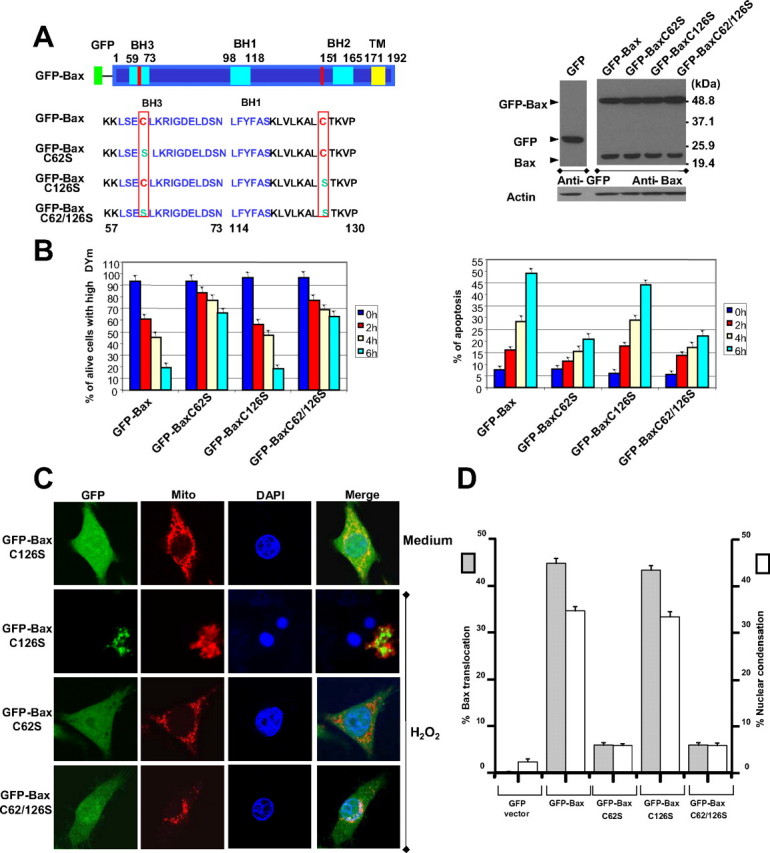
Cysteine 62 to serine substitution prevents Bax translocation and apoptosis. A, left, schematic overview of GFP-Bax and GFP-Bax mutant constructs. Right, expression levels of GFP-Bax and its mutants in SW480 cells. Cell lysates were subjected to SDS-PAGE, blotted, and probed with the indicated antibodies. B, cells were exposed to H2O2 (25 μm) for indicated times. Left, the percentage of living cells with high mitochondrial membrane potential was determined by 3,3′-dihexyloxacarbocyanine iodide and propidium iodide staining, followed by flow cytometry. Right, apoptotic cells were identified as Annexin V+ cells. C, confocal microscopy analysis of translocation of GFP-Bax mutants to mitochondria. Cells were cultured in the presence or absence of 25 μm H2O2 for 6 h and then subjected to mitochondrial fluorescence staining with 50 nm MitoTracker. Nuclei were counterstained with DAPI. D, graphs showing results of quantitative analyses. Apoptotic cells were characterized by nuclear condensation. At least 200 cells were counted in each experiment (n = 3, mean ± S.D.).
Microscopic analysis indicated that wild type GFP Bax and Bax C126S became punctate and redistributed to mitochondria following H2O2 treatment for 6 h (Fig. 2C). In contrast, Bax carrying the cysteine 62 to serine substitution in GFP-Bax C62S cells and GFP-Bax C62S/C126S cells was diffuse in the cytosol and did not translocate to mitochondria following H2O2 treatments. However, mitochondria become fragmented in those cells expressing the C62S mutation. As expected, no nuclear condensation was observed in GFP-Bax C62S and double mutant cells. When cysteine 62 of Bax was mutated, only background levels of Bax translocation and apoptotic cells were obtained following H2O2 treatment (Fig. 2D). These results show that H2O2-induced mitochondrial translocation of Bax requires cysteine 62.
Cysteine 62 of Bax Is Necessary for the Bax Conformational Change and Its Oligomerization during H2O2-induced Apoptosis—Conformational change of Bax represents a key step for its activation and subsequent apoptosis. We thus analyzed the conformation change of our stably expressed GFP-Bax mutants during apoptosis using anti-Bax 6A7 monoclonal antibody that specifically recognizes the conformationally changed Bax protein (2, 3). Following H2O2 treatment, Bax conformational change was detected only in cells expressing GFP-Bax and GFP-Bax C126S (Fig. 3A). As expected, this Bax conformational change was inhibited by NAC (Fig. 3A) and SOD mimetic (data not shown). This experiment shows that the ROS-induced Bax conformational change requires cysteine at position 62. Bax conformational change started after 2 h of H2O2 treatment, concomitant with Bax translocation in GFP-Bax cells (Fig. 3B). The substitution of cysteine 62 to serine mutants itself does not confer the conformational changes (Fig. 3A), although this occurs when cells were treated with staurosporine (STS) (data not shown). The oligomerization of Bax has previously been reported to occur only in apoptotic cells and possibly mediates cyt c release (3, 23). Following H2O2 treatment, GFP-Bax and GFP-Bax mutant cells were exposed to the membrane-permeable cross-linking agent disuccinimidyl suberate and subjected to SDS-PAGE and immunoblotting (Fig. 3C). A GFP-Bax-immunoreactive band of ∼48 kDa was detected in GFP-Bax and GFP-Bax mutant SW480 cells. Following H2O2 treatment, two Bax-immunoreactive bands of ∼90 and 140 kDa were detected with GFP-Bax and GFP-Bax C126S cells but not with GFP-Bax C62S or double mutant cells. As expected, Bax oligomerization was inhibited by NAC (Fig. 3C) and by Bcl-xL (not shown).
FIGURE 3.

Cysteine 62 of Bax is important for Bax conformational change and its oligomerization in H2O2-induced apoptosis. A, detection of the conformational change of Bax. GFP-Bax and Bax mutant cells were cultured in the presence or absence of 25 μm H2O2 for 6 h and then lysed in Chaps buffer and subjected to immunoprecipitation with anti-Bax 6A7 antibody. A cell lysate obtained by Nonidet P-40 was used as positive control. GFP-Bax total lysates were also applied directly onto SDS-PAGE, transferred to blots, and probed with the indicated antibodies, including a specific anti-Bax polyclonal antibody. B, GFP-Bax cells were immunostained with mouse monoclonal anti-Bax 6A7 antibody, and immunoreactivity was visualized by laser-scanning confocal microscopy. Nuclei were counterstained with DAPI. C, cells were treated with H2O2 (25 μm) for 6 h, and then the oligomerization of Bax was assessed by cross-linking with disuccinimidyl suberate. The samples were then solubilized in Nonidet P-40 lysis buffer, and Bax was detected by Western blotting with anti-Bax polyclonal antibody. β-Actin was used as a protein loading control. D, GFP-Bax and GFP-Bax C126S cells were treated with H2O2 (25 μm) for the indicated times and then subjected to subcellular fractionation and detection of Bax oligomerization in heavy membrane.
We next examined the distribution of the oligomerized Bax. Cells were treated with H2O2 for the indicated time and fractionated for analysis by Western blotting. It was found that oligomerized Bax only existed in mitochondria (Fig. 3D). Bax oligomerization started after 3 h of treatment and peaked after 6 h in isolated mitochondria. It appears that this event is later than the occurrence of Bax conformational change (see Fig. 3B, 2 h) and its translocation. In the same conditions, we did not observe oligomerized Bax or conformationally changed Bax in the cytosol (data not shown). The cysteine 62 to serine substitution prevents Bax oligomerization in H2O2-induced apoptosis. These data showed that cysteine 62 is important for H2O2-induced Bax conformational change and subsequent oligomerization at mitochondria.
The endogenous Bax conformational change, its oligomerization, and translocation to the mitochondria during H2O2-induced apoptosis could be detected only 10 h after H2O2 treatment in SW480 cells. Under the conditions used in this study, a physiological dose of H2O2 (25 μm) and short time (6 h), the endogenous Bax conformational activation is not detected (supplemental Fig. 1, B-D). This is in sharp contrast to the data obtained with wild type GFP-Bax and Bax C126S that could be oligomerized and redistributed to mitochondria and subsequent cell death following H2O2 (25 μm) treatments for 6 h (Figs. 2B and 3, A and D). Cysteine 62 to Serine Substitution Prevents cyt c Release and Caspase-3 Processing Associated with H2O2-induced Apoptosis—The release of cyt c and activation of caspase-3 are causally linked to Bax activation during the onset of apoptosis. As expected, following H2O2 treatment, cyt c is released from mitochondria into cytosol in GFP-Bax and GFP-Bax C126S cells but not in GFP-Bax C62S and GFP-Bax C62S/C126S cells (Fig. 4, A and B). In contrast to the mitochondrial localization in untreated cells, cyt c staining became, in part, diffuse 3 h after H2O2 treatment in GFP-Bax cells and GFP-Bax C126S cells. Bax translocation from the cytosol to mitochondria started 2 h and peaked 3 h after the treatment. Furthermore, caspase 3 was processed only in GFP-Bax and GFP-Bax C126S cells (Fig. 4B). These results suggest that cysteine 62 of Bax by H2O2 is required for cyt c release and caspase activation.
FIGURE 4.

Cysteine 62 to serine substitution prevents cyt c release and caspase-3 processing associated with H2O2-induced apoptosis. A, analyses of cyt c release and Bax translocation. Cells were cultured on chamber slides in the presence or absence of 25 μm H2O2 for the times indicated, subjected to immunofluorescence staining with anti-cyt c antibody, and then analyzed by confocal microscopy. B, cells were cultured in the presence or absence of 25 μm H2O2 for 6 h and subjected to subcellular fractionation. The cytosolic fractions were immunoblotted (30 μg of protein/lane) with antibody specific for cyt c or caspase-3, which only detected the cleaved caspase 3. β-Actin was used as a protein loading control. Data are representative of at least three independent experiments.
Cysteine 62-dependent Bax Activation Requires the Function of the Bax BH3 Domain—The reaction of cysteine residue with ROS could result in the conformational change of Bax, leading to the exposure of the BH3 domain of Bax, which is essential for the proapoptotic activity of Bax (24). To directly prove the involvement of BH-3 in H2O2 induced apoptosis, we first removed the IGDE amino acids from the BH3 domain of Bax (Fig. 5A), which was reported to be critical for Bax proapoptotic activity (24). A polyclonal stable cell line that expresses mutated GFP-Bax C126S ΔIGDE and a monoclonal cell line expressing GFP-Bax C126S were generated (Fig. 5B). Following H2O2 treatment, GFP retained its diffuse cytosolic localization in GFP-Bax C126S ΔIGDE cells with no nuclear condensation. As expected, GFP-Bax C126S cells exhibited Bax translocation and apoptosis (35%). There was no conformational change in GFP-Bax C126S ΔIGDE cells, whereas the Bax conformation change was detected in GFP-Bax C126S cells (Fig. 5C). Thus, the cysteine 62-dependent activation of Bax by ROS requires a functional Bax BH3 domain.
FIGURE 5.

Deletion of the IGDE motif from the Bax BH3 domain inhibits Bax translocation and apoptosis. A, schematic overview of GFP-Bax and GFP-Bax truncation constructs. B, confocal microscopy analysis of GFP-Bax mutant translocation to mitochondria and apoptosis. GFP-Bax C126S and GFP-Bax C126SΔIGDE cells were cultured in the presence or absence of 25 μm H2O2 for 6 h and then subjected to mitochondrial fluorescence staining with 50 nm MitoTracker. Nuclei were counterstained with DAPI. Apoptotic cells, those with characteristic nuclear condensation, are shown on the graph. At least 200 cells were counted in each experiment (n = 3, mean ± S.D.). C, detection of the conformational change of Bax. GFP-Bax C126S and GFP-Bax C126SΔIGDE cells were cultured in the presence or absence of 25 μm H2O2 for 6 h and then were lysed in Chaps buffer and subjected to immunoprecipitation with anti-Bax 6A7 antibody. A cell lysate obtained by Nonidet P-40 lysis buffer was used as a positive control. GFP-Bax total lysates were also loaded directly in SDS-PAGE, transferred to membranes, and probed with the indicated antibodies, including a specific anti-Bax polyclonal antibody.
Cysteine 62 Is Necessary for H2O2-Bax Activation in HCT116 Bax-deficient Cells—Cells deficient in Bax are highly resistant to drug-induced apoptosis, and ectopic expression of wild-type Bax abolishes this resistance to apoptosis (25). To rule out the involvement of endogenous Bax in the cysteine 62-mediated Bax translocation and proapoptotic activity, we transiently expressed GFP-Bax and GFP-Bax mutants in HCT116 (Bax-/-) cells. Transfection of GFP-Bax and GFP-Bax mutant causes cell death when the expression levels were high (as judged by its higher fluorescence). Cells that survived in the presence of a certain level of Bax were then treated with H2O2. These different cells expressed an equivalent amount of Bax (supplemental Fig. 2, A and B).
HCT116 (Bax-/-) cells expressing wild type GFP-Bax and GFP-Bax C126S are sensitive to H2O2-induced cell death (Fig. 6A). In contrast, both GFP-Bax C62S and GFP-Bax C62S/C126S cells remained resistant to H2O2-induced apoptosis. Bax translocation was also observed in wild-type GFP-Bax and GFP-Bax C126S but not in GFP-Bax C62S or double mutant cells. Findings concerning the cysteine 62 to alanine substitution in H2O2-induced mitochondrial translocation of Bax were similar (GFP-Bax C62A, supplemental Fig. 2C). These findings confirm that H2O2-induced Bax translocation and the proapoptotic activity of Bax are dependent on the presence of cysteine 62 in an endogenous Bax-deficient background.
FIGURE 6.

The sensitivity of HCT116 (Bax-/-) cells carrying different mutant forms of Bax to different apoptotic stimuli. A, HCT116 (Bax-/-) cells were transfected with GFP-Bax and Bax mutants and subjected to 25 μm H2O2 for 6 h and then analyzed by confocal fluorescence microscopy. B, detection of the redox status of GFP-Bax and its mutants in HCT116 (Bax-/-) cells. GFP-Bax and its mutants were transfected into HCT116 (Bax-/-) cells and then treated with 25 μm H2O2 for 6 h. The protein was then precipitated with trichloroacetic acid and treated with AMS before being subjected to SDS-PAGE and probed with the anti-Bax polyclonal antibody. β-Actin was used as a protein loading control. C, HCT116 (Bax-/-) cells were transfected with GFP-Bax, and Bax mutants and subjected to 0.2 μm STS for 12 h and then detected as described in A. D, graphs showing results of quantitative analyses of apoptotic cells characterized by nuclear condensation or fragmentation. At least 200 cells were counted in each experiment. The graph shows the percentage of cells displaying Bax translocation and apoptosis (n = 3, mean ± S.D.). E, confocal microscopy analysis of Bax translocation and apoptosis following treatment with X, XO, both, or DEANO. Cells were exposed to X (500 μm), XO (0.1 units), DEANO (500 μm), or a combination of X and XO for 6 h and then subjected to mitochondrial fluorescence staining with 50 nm MitoTracker. Nuclei were counterstained with DAPI. F, Bax translocation and apoptotic cells were characterized by nuclear condensation. At least 200 cells were counted in each experiment. The graph shows the percentage of cells displaying Bax translocation and apoptosis (n = 3, mean ± S.D.).
Concerning the other mutants in BH3 (GFP-Bax C126S/K64A, GFP-Bax C126S/K57A/K58A, and GFP-Bax C126S/L70A), mutation K64A had little effect in altering the subcellular distribution of Bax, whereas mutation of L70 resulted in constitutive activation of Bax and subsequent cell death. Cells expressing all of the mutant forms of Bax are sensitive to H2O2 in Bax-deficient HCT116 cells (supplemental Fig. 2C). These results suggest that the mutation of these amino acids residues does not alter the H2O2-dependent activity of cysteine 62.
To confirm our hypothesis that cysteine residues indeed react with intracellular ROS, we investigated the redox status of Bax following H2O2 treatment. We used AMS to alkylate thiols of the protein cysteine residues in transfected HCT116 (Bax-/-) cells by GFP-Bax and GFP-Bax mutants, followed by the Western blot analysis. Alkylation with AMS adds ∼500 Da of molecular mass to every sulfhydryl group. When run on nonreducing SDS-PAGE, proteins in the reduced (alkylated) and oxidized (nonalkylated) forms can be resolved by the increase in molecular weight due to the addition of AMS. As expected, we observed two differently migrating forms of GFP-Bax and its mutants before and after treatment with hydrogen peroxide (Fig. 6B). In the absence of H2O2, GFP-Bax and its single mutants exist in the reduced form, but the double mutant cannot react with AMS and thus retains its molecular weight. Bax has only two cysteines, but in our conditions, AMS cannot distinguish the reaction on singular or double cysteine residues. Following the 25 μm H2O2 treatment, we observed the oxidized form for all of the samples, indicating that wild type and mutant forms containing cysteine were oxidized (Cys-126 in the C62S mutant; Cys-62 in the C126S mutant). As expected, the double mutant lacking cysteines cannot be oxidized and alkylated with AMS. These results show that both cysteines (Cys-62 and Cys-126) of Bax can be directly oxidized by H2O2.
Cysteine 62 Specifically Reacts with ROS, but Not with NO, for Promoting Bax Activation and Apoptosis—We then examined the effect of other inducers of apoptosis capable or not of generating ROS to demonstrate the specificity of Bax cysteine 62. We examined the effect of STS on Bax activation and apoptosis. Cells expressing all of the mutant forms of Bax are sensitive to STS-induced apoptosis both in SW480 cells (not shown) and in Bax-deficient HCT116 cells (Fig. 6, C and D). STS could induce Bax translocation into mitochondria in cells carrying different mutated Bax. These results suggest that the mutation of these cysteine residues does not alter Bax function and proapoptotic activity in STS-induced apoptosis. Findings with campothecin were similar (supplemental Fig. 3). We then used X and XO to generate 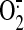 (14). GFP-Bax and GFP-Bax mutant cells were exposed to X (500 μm) and XO (0.1 units) or both for 6 h. Confocal microscopy analysis showed that neither X nor XO alone induced Bax translocation or significant apoptosis. The combination of X and XO induced Bax translocation and apoptosis in GFP-Bax and GFP-C126S cells (35% apoptotic cells) but not in the Bax C62S or double mutant cells (Fig. 6, E and F). Next, we examined the effect of NO on Bax activation and apoptosis. We used diethylamine NONDate and S-nitroso-N-acetylpenicillamine, two commonly used NO donors, to generate NO (26). GFP-Bax and GFP-Bax mutant cells were exposed to DEANO for 6 h. The GFP-Bax cells treated with DEANO showed only 2% apoptotic cells as assessed by confocal microscopy (Fig. 6, E and F). Findings with S-nitroso-N-acetylpenicillamine were similar (data not shown). These data reveal that cysteine 62 of Bax specifically reacts with H2O2 and
(14). GFP-Bax and GFP-Bax mutant cells were exposed to X (500 μm) and XO (0.1 units) or both for 6 h. Confocal microscopy analysis showed that neither X nor XO alone induced Bax translocation or significant apoptosis. The combination of X and XO induced Bax translocation and apoptosis in GFP-Bax and GFP-C126S cells (35% apoptotic cells) but not in the Bax C62S or double mutant cells (Fig. 6, E and F). Next, we examined the effect of NO on Bax activation and apoptosis. We used diethylamine NONDate and S-nitroso-N-acetylpenicillamine, two commonly used NO donors, to generate NO (26). GFP-Bax and GFP-Bax mutant cells were exposed to DEANO for 6 h. The GFP-Bax cells treated with DEANO showed only 2% apoptotic cells as assessed by confocal microscopy (Fig. 6, E and F). Findings with S-nitroso-N-acetylpenicillamine were similar (data not shown). These data reveal that cysteine 62 of Bax specifically reacts with H2O2 and 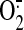 , but not with NO, to induce apoptosis.
, but not with NO, to induce apoptosis.
DISCUSSION
The principal finding of this paper concerns the molecular mechanisms by which Bax is activated and translocated to the mitochondria by intracellular ROS. We identified the conserved Cys-62 as a critical residue for Bax conformational change and its translocation to mitochondria during H2O2-induced apoptosis. Mutation of cysteine 62, but not cysteine 126, abolished the proapoptotic activity of Bax and its homo-oligomerization in response to H2O2 but not other death stimuli, such as STS and NO. This H2O2 activation of Bax appears to be specific, because NO donors did not activate Bax, and this effect was strongly inhibited by overexpression of Bcl-XL (Fig. 1C), by SOD mimetic (data not shown), and by the ROS scavenger NAC (Fig. 1A).
We first show experimentally that ROS could be a direct signal for Bax activation and H2O2-induced cell death. The messenger function of H2O2 to activate Bax requires that its concentration increase above a certain threshold level. Mitochondria are a major source of superoxide anion that can be converted into H2O2; the latter can be released from mitochondria into cytosol. It is possible that the high levels of H2O2 released from mitochondria (8, 27, 28) could trigger Bax activation and its translocation to mitochondria for subsequent apoptosis under pathophysiological conditions. Even at mild oxidative burst, mitochondrial ROS formation may be comparable with that of NADPH oxidase, which could produce a high concentration of ROS (29). Since there is an equilibrium between the mitochondria-associated Bax (noninsertional) and cytosolic Bax, Bax could be primed (3, 8) or undergo a conformational activation when mitochondrial ROS increase to a high level. In this case, it is possible that Bax could be activated and targeted to mitochondria without the aid of other proapoptotic Bcl-2 family proteins, as previously suggested. Indeed, Bax does not interact with other BH3-containing proapoptotic molecules, at least in cytosol (11). Our results may explain how overexpressing Bcl-xL, which mainly localized at the outer membrane of mitochondria, effectively prevented the Bax conformational change and oligomerization, which only occur in the MOM in our system. Bcl-xL protects mitochondrial functions and thereby inhibits mitochondrial ROS. We did observe that mitochondrial is fragmented in cells overexpressing Bcl-xL, which are resistant to H2O2 treatment. This indicates that mitochondrial fragmentation is independent of H2O2-induced apoptosis. These results are in agreement with previous reports showing that Bcl-2 cannot prevent mitochondrial fragmentation (30, 31). It was proposed that mitochondrial fragmentation is a prerequisite for MOM permeabilization and cyt c release during apoptosis (32). However, other results suggest that mitochondrial fragmentation is not a prerequisite for mitochondrial cyt c release and apoptosis and that MOM permeabilization and cyt c release can occur without mitochondrial fragmentation (33), although there is extensive mitochondrial fragmentation during apoptosis. Further studies are under way to examine whether and how mitochondrial ROS activate Bax during apoptosis in the normal cellular context and how mitochondrial fragmentation is related to cyt c release and apoptosis.
The change of the redox status of cysteine residues could have a profound effect on the conformation of Bax (34). Computer simulation showed that both of the two cysteine residues of Bax (Cys-62 and Cys-126) may form disulfide bridges within or between Bax molecules following treatment with H2O2, although this dimerization appears to be independent of apoptosis (27). Cysteine residues can be oxidized by H2O2 to form a Cys-SOH, which then reacts with a neighboring cysteine to form a disulfide (18). This disulfide can then be reduced by GSH or by thioredoxin, two abundant antioxidant molecules. Thus, it is likely that intracellular ROS functions as a redox signal and is involved in a reversible reaction with Cys-SH. The specific requirement for cysteine 62 for Bax activation may be because it is located in the BH3 domain, whereas cysteine 126, which is not required, is located within α-helices 5 and 6. Oxidation of cysteine 62, but not cysteine 126, probably resulted in the exposure of N-terminal domain and C-terminal transmembrane domain, leading to its lethal attack on mitochondria. Indeed, the BH3 domain of Bax is critical for its apoptotic function, since deletion of the IGDE motif in the BH3 domain of Bax or removal of the entire BH3 domain prevented Bax protein homodimerization and cell death of mammalian cells (24). Previous work using NMR and analysis of mutations revealed that the Bax BH3 domain, in particular the side chains of the hydrophobic residues of Leu-59, Leu-63, Ile-66, and Leu-70, are critical for Bax conformational changes and dimerization (10). We confirmed that Leu-70 mutation, but not cysteine residues, resulted in the constitutive activation of Bax, as previously reported (35). Thus, we first demonstrated that the cysteine residue within the BH3 domain does not result in a constitutive activation, but it is required for Bax conformational change, subsequent homodimerization, and oligomerization at the mitochondria in response to H2O2. This may not be the only mechanism for Bax activation, since the mutant form of Bax at the 62-position is sensitive to STS (Fig. 6, C and D) and to campothecin (supplemental Fig. 3). Our results indicate that the modes of apoptosis caused by hydrogen peroxide and staurosporine are different, although both agents cause apoptosis through activating Bax prior to its translocation to mitochondria. It is worth noting that previous study showed that both STS and high doses of H2O2 promote Bax activation through phosphorylation by c-Jun N-terminal protein kinase (17). We also found that deletion of the αH5 and -6 domains inhibited Bax translocation to mitochondria and apoptosis (data not shown). This is in agreement with another report indicating that deletion of the pore-forming domain of αH5 and -6 prevented insertion of Bax into mitochondrial membranes and greatly reduced the cyt c-releasing activity of Bax (36).
In conclusion, we found that the highly conserved cysteine 62 of Bax is essential for the Bax conformational change, its translocation to mitochondria, and Bax oligomerization, which result in cyt c release and caspase-3 processing associated with H2O2-induced apoptosis. Our data showed that ROS could be a direct signal for activating Bax conformational changes by chemically reacting with cysteine residues. Mutation of Bax is closely related to many types of cancers, including colon cancers (20). Our results thus implicate that manipulation of Bax targeting to mitochondria by changing the redox status could be useful for cancer therapy.
Supplementary Material
Acknowledgments
We thank Stephen Manon (UMR5095 CNRS/Université Bordeaux 2, Bordeaux, France) for careful reading of the manuscript and useful comments.
The work was supported by CAS, INSERM, CNRS, the National Proprietary Basic Research Program, Natural Science Foundation of China (NSFC), Association pour la Recherche sur le Cancer (ARC), and Association Française contre les Myopathies (AFM). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Table 1 and Figs. 1-3.
Footnotes
The abbreviations used are: cyt c, cytochrome c; MOM, mitochondrial outer membrane; ROS, reactive oxygen species; X, xanthine; XO, xanthine oxidase; NAC, N-acetyl-l-cysteine; DAPI, 4′,6-diamidino-2-phenylindole; Chaps, 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid; AMS, 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid; GFP, green fluorescent protein; STS, staurosporine.
References
- 1.Wang, X. (2001) Genes Dev. 15 2922-2933 [PubMed] [Google Scholar]
- 2.Nechushtan, A., Smith, C. L., Hsu, Y. T., and Youle, R. J. (1999) EMBO J. 18 2330-2341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zheng, Y., Yamaguchi, H., Tian, C., Lee, M. W., Tang, H., Wang, H. G., and Chen, Q. (2005) Oncogene 24 3339-3347 [DOI] [PubMed] [Google Scholar]
- 4.Oltvai, Z. N., Milliman, C. L., and Korsmeyer, S. J. (1993) Cell 74 609-619 [DOI] [PubMed] [Google Scholar]
- 5.Cartron, P. F., Gallenne, T., Bougras, G., Gautier, F., Manero, F., Vusio, P., Meflah, K., Vallette, F. M., and Juin, P. (2004) Mol. Cell 16 807-818 [DOI] [PubMed] [Google Scholar]
- 6.Annis, M. G., Soucie, E. L., Dlugosz, P. J., Cruz-Aguado, J. A., Penn, L. Z., Leber, B., and Andrews, D. W. (2005) EMBO J. 24 2096-2103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khaled, A. R., Kim, K., Hofmeister, R., Muegge, K., and Durum, S. K. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 14476-14481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahmad, K. A., Iskandar, K. B., Hirpara, J. L., Clement, M. V., and Pervaiz, S. (2004) Cancer Res. 64 7867-7878 [DOI] [PubMed] [Google Scholar]
- 9.Chen, Q., Galleano, M., and Cederbaum, A. I. (1997) J. Biol. Chem. 272 14532-14541 [DOI] [PubMed] [Google Scholar]
- 10.Suzuki, M., Youle, R. J., and Tjandra, N. (2000) Cell 103 645-654 [DOI] [PubMed] [Google Scholar]
- 11.Willis, S. N., Fletcher, J. I., Kaufmann, T., van Delft, M. F., Chen, L., Czabotar, P. E., Ierino, H., Lee, E. F., Fairlie, W. D., Bouillet, P., Strasser, A., Kluck, R. M., Adams, J. M., and Huang, D. C. (2007) Science 315 856-859 [DOI] [PubMed] [Google Scholar]
- 12.Croteau, D. L., ap Rhys, C. M., Hudson, E. K., Dianov, G. L., Hansford, R. G., and Bohr, V. A. (1997) J. Biol. Chem. 272 27338-27344 [DOI] [PubMed] [Google Scholar]
- 13.Chen, Q., Gong, B., and Almasan, A. (2000) Cell Death Differ. 7 227-233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen, Q., Chai, Y. C., Mazumder, S., Jiang, C., Macklis, R. M., Chisolm, G. M., and Almasan, A. (2003) Cell Death Differ. 10 323-334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Orrenius, S., Gogvadze, V., and Zhivotovsky, B. (2007) Annu. Rev. Pharmacol. Toxicol. 47 143-183 [DOI] [PubMed] [Google Scholar]
- 16.Gonzalvez, F., Pariselli, F., Dupaigne, P., Budihardjo, I., Lutter, M., Antonsson, B., Diolez, P., Manon, S., Martinou, J. C., Goubern, M., Wang, X., Bernard, S., and Petit, P. X. (2005) Cell Death Differ. 12 614-626 [DOI] [PubMed] [Google Scholar]
- 17.Kim, B. J., Ryu, S. W., and Song, B. J. (2006) J. Biol. Chem. 281 21256-21265 [DOI] [PubMed] [Google Scholar]
- 18.Rhee, S. G. (2006) Science 312 1882-1883 [DOI] [PubMed] [Google Scholar]
- 19.Trojan, J., Brieger, A., Raedle, J., Weber, N., Kriener, S., Kronenberger, B., Caspary, W. F., and Zeuzem, S. (2004) Int. J. Colorectal Dis. 19 538-544 [DOI] [PubMed] [Google Scholar]
- 20.Zheng, Y., Shi, Y., Tian, C., Jiang, C., Jin, H., Chen, J., Almasan, A., Tang, H., and Chen, Q. (2004) Oncogene 23 1239-1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gross, A., Jockel, J., Wei, M. C., and Korsmeyer, S. J. (1998) EMBO J. 17 3878-3885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aslund, F., Zheng, M., Beckwith, J., and Storz, G. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 6161-6165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Antonsson, B., Montessuit, S., Sanchez, B., and Martinou, J. C. (2001) J. Biol. Chem. 276 11615-11623 [DOI] [PubMed] [Google Scholar]
- 24.Zha, H., Fisk, H. A., Yaffe, M. P., Mahajan, N., Herman, B., and Reed, J. C. (1996) Mol. Cell. Biol. 16 6494-6508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chandra, D., Choy, G., Daniel, P. T., and Tang, D. G. (2005) J. Biol. Chem. 280 19051-19061 [DOI] [PubMed] [Google Scholar]
- 26.Maragos, C. M., Morley, D., Wink, D. A., Dunams, T. M., Saavedra, J. E., Hoffman, A., Bove, A. A., Isaac, L., Hrabie, J. A., and Keefer, L. K. (1991) J. Med. Chem. 34 3242-3247 [DOI] [PubMed] [Google Scholar]
- 27.D'Alessio, M., De Nicola, M., Coppola, S., Gualandi, G., Pugliese, L., Cerella, C., Cristofanon, S., Civitareale, P., Ciriolo, M. R., Bergamaschi, A., Magrini, A., and Ghibelli, L. (2005) FASEB J. 19 1504-1506 [DOI] [PubMed] [Google Scholar]
- 28.Li, D., Ueta, E., Kimura, T., Yamamoto, T., and Osaki, T. (2004) Cancer Sci. 95 644-650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jezek, P., and Hlavata, L. (2005) Int. J. Biochem. Cell Biol. 37 2478-2503 [DOI] [PubMed] [Google Scholar]
- 30.Arnoult, D. (2007) Trends Cell Biol. 17 6-12 [DOI] [PubMed] [Google Scholar]
- 31.Sugioka, R., Shimizu, S., and Tsujimoto, Y. (2004) J. Biol. Chem. 279 52726-52734 [DOI] [PubMed] [Google Scholar]
- 32.Youle, R. J., and Karbowski, M. (2005) Nat. Rev. Mol. Cell. Biol. 6 657-663 [DOI] [PubMed] [Google Scholar]
- 33.Barsoum, M. J., Yuan, H., Gerencser, A. A., Liot, G., Kushnareva, Y., Graber, S., Kovacs, I., Lee, W. D., Waggoner, J., Cui, J., White, A. D., Bossy, B., Martinou, J. C., Youle, R. J., Lipton, S. A., Ellisman, M. H., Perkins, G. A., and Bossy-Wetzel, E. (2006) EMBO J. 25 3900-3911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu, H., Colavitti, R., Rovira, I. I., and Finkel, T. (2005) Circ. Res. 97 967-974 [DOI] [PubMed] [Google Scholar]
- 35.Zhou, H., Hou, Q., Hansen, J. L., and Hsu, Y. T. (2007) Oncogene 26 7092-7102 [DOI] [PubMed] [Google Scholar]
- 36.Heimlich, G., McKinnon, A. D., Bernardo, K., Brdiczka, D., Reed, J. C., Kain, R., Kronke, M., and Jurgensmeier, J. M. (2004) Biochem. J. 378 247-255 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.