

Abstract
Mechanisms regulating the activation and delivery of function of Lck and Fyn are central to the generation of the most proximal signaling events emanating from the T cell antigen receptor (TcR) complex. Recent results demonstrate that lipid rafts (LR) segregate Lck and Fyn and play a fundamental role in the temporal and spatial coordination of their activation. Specifically, TcR-CD4 co-aggregation-induced Lck activation outside LR results in Lck translocation to LR where the activation of LR-resident Fyn ensues. Here we report a structure-function analysis toward characterizing the mechanism supporting Lck partitioning to LR and its capacity to activate co-localized Fyn. Using NIH 3T3 cells ectopically expressing FynT, we demonstrate that only LR-associated, kinase-active Y505FLck reciprocally co-immunoprecipitates with and activates Fyn. Mutational analyses revealed a profound reduction in the formation of Lck-Fyn complexes and Fyn activation, using kinase domain mutants K273R and Y394F of Y505FLck, both of which have profoundly compromised kinase activity. The only kinase-active Lck mutants tested that revealed impaired physical and enzymatic engagement with Fyn were those involving truncation of the C-terminal sequence YQPQP. Remarkably, sequential truncation of YQPQP resulted in an increasing reduction of kinase-active Lck partitioning to LR, in both fibroblasts and T cells. This in turn correlated with an ablation of the capacity of these truncates to enhance TcR-mediated interleukin-2 production. Thus, Lck-dependent Fyn activation is predicated by proximity-mediated transphosphorylation of the Fyn kinase domain, and targeting kinase-active Lck to LR is dependent on the C-terminal sequence QPQP.
Two Src family tyrosine kinases, Lck and Fyn, provide critical functions that predicate the generation of the most proximal signals emanating from the antigen receptor complex in T cells (1, 2). Lck- and Fyn-dependent phosphorylation of many cellular substrates is readily detectable within seconds after T cell receptor engagement (3), and the provision of catalytic activity requires an intact molecular structure (4) and post-translational lipid modifications of these kinases (5–7).
Similar to other Src family kinases, Lck and Fyn contain a short N-terminal lipid-modified region, a unique domain, Src homology 3 (SH3)3 and SH2 domains, a linker region, a catalytic domain, and a C-terminal tail involved in negative regulation of function (8). Biochemical and crystallographic studies revealed that kinase activity is regulated through reversible phosphorylation of two key tyrosine residues. Specifically, the negative regulatory Tyr505 and Tyr528 on Lck and Fyn, respectively, and the positive regulatory Tyr394 and Tyr417 of Lck and Fyn, respectively, are positioned within the activation loops of their respective kinase domains (9–12). As Lck and Fyn can be phosphorylated on either of these two regulatory tyrosine residues, the activation of Src kinases is modeled as a sequential two-step mechanism, which allows transitions between three functionally different states (4, 13). The inactive, autoinhibitory conformation is supported in large part by 2-week intra-molecular interactions formed between the SH2 domain and the phosphorylated C-terminal tyrosine and the SH3 domain and the linker region, which cooperatively contribute to down-regulate the kinase activity. CD45-mediated dephosphorylation of the C-terminal phosphotyrosine results in an “open” structure and an active conformation of the kinase domain (14). This initial step of kinase activation can be counteracted by action of the C-terminal Src kinase (Csk) (15, 16). In the second step, full kinase activity is achieved upon phosphorylation of the positive regulatory tyrosine in the activation loop (17).
Although not accounting for all possible activation scenarios, this simplified model highlights the important difference between the two activation steps of Lck and Fyn kinases. The initial transition from “closed” to open conformation is controlled by extracellular signals, notably by peptide-MHC-mediated co-ligation of TcR and CD4, which alters the balance between the positive and negative regulating enzymatic activities of CD45 and Csk, respectively, toward the former. In contrast, the phosphorylation of positive regulatory tyrosine in the activation loop is accomplished by intrinsic catalytic activity of the kinases themselves, acting in an intra-molecular (18–20) or inter-molecular fashion (21), as a consequence of kinase co-clustering. Moreover, some data indicate that phosphorylation within the activation loop in trans by other Src or non-Src family members can also occur (22).
An additional mode of activation, which does not involve dephosphorylation of the C-terminal tyrosine, is conferred by high affinity interactions of the SH3 domain with its ligands, resulting in the competitive displacement of low affinity intramolecular interactions (19, 23–25). This results in an alignment of kinase domain residues critical for catalysis and enables productive binding of ATP and substrates (26).
Whether or how the respective functions of Lck and Fyn in generating the earliest signals emanating from the TcR are coordinated and integrated has remained enigmatic. Because of their abundance in T cells, their involvement in early TcR signaling, their partial ability to compensate for each other's deficiency in the context of TcR signaling, as well as their ability to phosphorylate an overlapping set of substrates, it has been suggested that their functions are, at least in part, redundant (2). However, the above commonalities contrast markedly with differences related to the physiology of these kinases. Based simply on expression patterns, Lck and Fyn belong to separate subfamilies of Src family tyrosine kinases. Whereas Fyn together with Src and Yes are ubiquitously expressed, the expression of Lck, Hck, Lyn, and Blk are largely restricted to hematopoietic cells (11). Interestingly, this physiological distinction is paralleled by a structural difference in the form of a short C-terminal truncation in Lck subfamily members (14). Furthermore, Lck and Fyn reconstitution experiments demonstrated their discrete physiological role in the maintenance and activation of peripheral T cells (27, 28). Finally, in T cells, although the majority of plasma membrane-bound Lck is complexed with co-receptor CD4 (29), only a small proportion of Fyn tethers to CD3 chains of the TcR complex (30, 31). Thus, the association of Lck and Fyn with discrete signaling structures at the plasma membrane, as well as their preference for specific targets in vivo (28, 32), suggests that their roles in the initiation of signals emanating from the TcR could be distinct, independent, and specific.
In this regard, recent findings demonstrating that lipid rafts function not only to segregate Lck and Fyn but also to couple the process of membrane translocation with temporal and spatial regulation of their enzymatic activities provides strong evidence for their nonredundant and interdependent functions (2). Specifically, although Fyn localizes to LR, the majority of Lck partitions to soluble membrane fractions. Co-aggregation of TcR and CD4 results in Lck activation outside LR, followed by its translocation into LR and the activation of co-localized Fyn (3). Because activation-induced (3) or genetically targeted enrichment (33) of Lck in LR results in the activation of co-localized Fyn, the prediction follows that activation of the latter is a consequence of either direct or indirect interaction between Lck and Fyn.
To address this question, we created a model system using fibroblasts enabling both biochemical and genetic analyses of the mechanisms underpinning Lck-dependent Fyn activation. Here we report that Lck-Fyn complexes are revealed in NIH 3T3 fibroblasts co-expressing WT FynT and constitutively active Y505F Lck. Complex formation was predicated by the kinase activity of ectopically expressed Lck, occurred exclusively in LR, and resulted in the phosphorylation of the positive regulatory Tyr417 in the activation loop of LR resident Fyn. Mutational analyses identified two domains of Lck critical for Lck-Fyn complex formation as follows: (i) the active kinase domain of Lck that mediates transphosphorylation of the Fyn kinase domain when co-localized in LR, and (ii) the unexpected characterization of the C-terminal sequence of Lck that functions as a cis-acting structural element required for partitioning of Lck to LR.
EXPERIMENTAL PROCEDURES
Cell Lines and T Cell Clones—Generation, selection, and growth of NIH 3T3 cells expressing WT FynT (3T3-FynT) were described elsewhere (34). The Src-, Yes-, Fyn-triple knock-out mouse embryonic fibroblast cell line (SYF) was obtained from the ATCC and maintained according to distributor's recommendations. The CD4+ T cell clone 2.5 infected with empty MigR1 vector or MigR1 containing various mutant constructs of Lck were selected and maintained as described elsewhere (35). BI-141 is a CD4–/CD8– MHC class II-restricted T cell hybridoma specific for bovine insulin (36) and was maintained as described previously (37).
Antibodies and Reagents—Polyclonal rabbit anti-mouse Lck-(38), Fyn-(34), and Tyr(P)394 Lck (39)-specific antisera have been described elsewhere. The anti-TcRCβ-specific mAb, H57.597 (40), was purified on protein A-conjugated Sepharose 4B. For use in immunoprecipitation, IgG fractions of antisera were affinity-purified and covalently coupled to CNBr-activated Sepharose 4B (Amersham Biosciences). Rabbit anti-mouse Tyr(P)418 Src that cross-reacts with Tyr(P)417 Fyn was purchased from BIOSOURCE. Phosphotyrosine-specific mAb, 4G10, and anti-actin antibodies were purchased from Upstate Biotechnology, Inc. (Lake Placid, NY) and Sigma, respectively. Goat anti-mouse IgG-horseradish peroxidase (HRP) and protein A-HRP were purchased from Bio-Rad. Cholera toxin B subunit-HRP (CT-HRP) was purchased from Sigma.
Site-directed Mutagenesis—Construction of WT and C20S/C23S (DC-S, double cysteine to serine mutation) (35) and Y505F (41) mutants of Lck were described previously. S59A, W97K, R154K, P249G, P252G, P249G/P252G (DP-G, double proline to glycine mutation), K273R, Y394F, ΔQP, ΔQPQP, and ΔYQPQP mutations were introduced on Y505F Lck template using “Genetailor” mutagenesis kit (Invitrogen). All mutants of Lck expressed in clone 2.5, except for those encoding the WT and DC-S Lck, were generated on a K273R + DC-S Lck template. Oligonucleotides carrying desired mutations were designed and used according the manufacturer's recommendation. All mutants were verified by sequencing. The wild type or mutant Lck constructs were inserted into the murine stem cell virus-based internal ribosome entry site-enhanced green fluorescent protein virus MIEV (35) or its analog MigR1 (42), both permitting the concurrent expression of a given gene and enhanced green fluorescent protein. Generation of retrovirus packaging cell lines and retrovirus stock as well as infection of 3T3-FynT were performed as outlined elsewhere (35, 43). The same protocol was applied to generate SYF, 2.5, and BI-141 cells expressing indicated Lck mutants.
Cell Lysis, Immunoprecipitations, Immunoblotting, and Densitometry—4G10 and anti-Lck immunoblots were performed on aliquots of SYF, BI-141, and 2.5 cell infectants directly lysed in Laemmli loading buffer and boiled for 10 min. For evaluation of Lck, Fyn, Tyr(P)394 Lck, and Tyr(P)417 Fyn levels in 3T3-FynT infectants, cells were lysed in TNE buffer (50 mm Tris, pH 8, 20 mm EDTA, 200 mm Na3VO4, 50 mm NaF, 1% Nonidet P-40, 20 μg/ml leupeptin, 20 μg/ml aprotinin) and immunoprecipitated, blotted, and probed as described in detail elsewhere (33). Densitometric analyses of immunoblots were performed on a GS800 densitometer (Bio-Rad) utilizing Quantity One quantification software (Bio-Rad). The signal quantifications were calculated using densitometric values obtained from nonsaturated signals.
Isolation of Lipids Rafts—Preparation of lipid rafts, the assessment of subcellular partitioning of Lck, Fyn, and the LR marker GM1, and the immunoprecipitation of Lck and Fyn from lipid raft fractions were performed as described elsewhere (33). For comparative analyses of LR solubility in selected detergents, TKM buffer containing 50 mm Tris, pH 7.4, 25 mm KCl, 5 mm MgCl2, 1 mm EDTA, 20 μg/ml leupeptin, 20 μg/ml aprotinin, and 100 μg/ml Pefabloc was supplemented with either 0.5% Brij-58, or 1% Nonidet P-40, or 1% n-dodecyl-β-d-maltoside (laurylmaltoside (LM), Sigma).
Immune Complex Kinase Assays—Lck immunoprecipitates from SYF infectants were subjected to in vitro kinase assay performed as described previously (33).
Immunofluorescence and Confocal Microscopy—The immunofluorescence protocol was obtained from Cell Signaling Technology web site. Briefly, 3T3-FynT infectants, grown on coverslips, were fixed in 4% paraformaldehyde and permeabilized with ice-cold 100% methanol. After washing, the samples were treated with a blocking solution containing 2.5% bovine serum albumin and 2.5% fetal cow serum. Immunostaining was performed with purified rabbit anti-Lck followed by Alexa-647-conjugated anti-rabbit (Invitrogen). In the second step, Alexa-555 directly conjugated to anti-Fyn was applied. Cells were mounted on slides using Vectashield mounting medium with 4′,6-diamidino-2-phenylindole (Vector Laboratories). Microscopy was performed on a confocal microscope Leica SP5, and images were acquired using an HCX PL APO 40×/1.25–0.75 Oil CS UV objective. Cropping and brightness/contrast adjustments were performed with Adobe Photoshop CS software.
IL-2 ELISA—Triplicate cultures containing 5 × 104 BI-141 infectants were stimulated or not with plate-bound TcRCβ-specific mAb, H57.597, at coating concentrations of 0.01, 0.03, or 0.1 μg/ml. Supernatants were harvested after 16 h, and IL-2 content was quantified using an IL-2-specific ELISA (eBiosciences), according to the manufacturer's recommendations.
RESULTS
Kinase-active Lck Forms Complexes with and Activates Fyn—To determine whether the mechanism underpinning Lck-dependent Fyn activation involves physical interaction of Lck and Fyn kinases, we generated an NIH 3T3 fibroblast cell line co-expressing wild type Fyn-T (WT Fyn) (3T3-FynT) with either wild type (WT) Lck or C20S/C23S (DC-S) Lck or constitutively active Y505F Lck. Because of the absence of CD4 expression in 3T3, the prediction is that partitioning of WT Lck, used as an internal control, would mimic that of DC-S Lck, unable to bind to intracellular portion of CD4 (44). As illustrated in Fig. 1A, Lck as well as Fyn expression levels were comparable (within 10%) among infectants. Of note is that NIH 3T3 cells do not express Lck (45), and the levels of endogenous Fyn-B are ∼30 times lower than those achieved through ectopic expression of Fyn-T (data not shown and see Ref. 43). Probing anti-Lck immunoprecipitates (left panels of Fig. 1, A–C) with antibodies specific for Fyn and anti-Fyn immunoprecipitates (right panels Fig. 1, A–C) with antibody specific for Lck revealed that only Y505F Lck reciprocally co-immunoprecipitates with Fyn (Fig. 1B). Redeveloping the blots using ECL+ significantly enhances the detection of these complexes and reveals low levels of co-immunoprecipitation of Fyn with both WT and DC-S Lck (Fig. 1B, bottom panel). This “background” co-immunoprecipitation is likely a reflection of basal kinase activity of WT and DC-S Lck (46).
FIGURE 1.

Kinase-active Lck complexes with and activates Fyn. NIH 3T3 cells stably expressing WT FynT (3T3-FynT) were further infected with either empty vector MIEV (0), or MIEV vector containing WT Lck or C20S/C23S Lck (DC-S Lck) or Y505F Lck cDNA. 105 cells were lysed in 1% Nonidet P-40 lysis buffer and immunoprecipitated (IP) with either anti-Lck-(left panels) or anti-Fyn-specific antibodies (right panels). The immunoprecipitates derived from each infectant were split into three equal portions and probed with immunoprecipitating antibody (A), with antibody against the alternative kinase (B), and with antibodies detecting Tyr(P)394 Lck and Tyr(P)417 Fyn (see text for details) (C). Histograms in B show the percentage of total Fyn and total Lck co-immunoprecipitated with the alternative kinase. Histograms in C show the relative kinase activity of Lck and Fyn as measured by the quantification of Tyr(P)394 Lck and Tyr(P)417 Fyn signals normalized to respective total kinase signals. Sample derived from the 3T3-FynT cells expressing WT Lck was given a reference value of 1. ECL, regular sensitivity reagent for chemiluminescent development of Western blots. ECL+, supersensitive reagent for chemiluminescent development of Western blots.
Quantification of the stoichiometry of Fyn-Lck interaction reveals that ∼1.8% of total Fyn and 4.3% of total Y505F Lck are complexed with the alternative kinase. As illustrated in Fig. 2A, this distinct stoichiometry is consistent with the differential partitioning of Fyn and Lck to LR in NIH 3T3 fibroblasts. Specifically >92% of Fyn is LR-associated, whereas up to ∼30% of Lck is not. Hence, anti-Lck immunoprecipitates are diluted with non-LR-associated Lck, which does not have access to Fyn. Using levels of Tyr(P)394 expression as a surrogate for Lck activation, Y505F Lck is ∼15 times more active compared with the other two variants of Lck (Fig. 1C). Importantly, as assessed by quantification of Tyr(P)417 levels, activation of WT Fyn immunoprecipitated from cells co-expressing Y505F Lck was increased ∼7-fold (Fig. 1C), compared with WT Fyn co-expressed with either WT or DC-S Lck. Thus, complex formation between kinase-active Lck and WT Fyn correlates with Fyn activation.
FIGURE 2.

Lck-Fyn complex formation is confined to lipid rafts. A, 106 3T3-FynT cells infected with empty vector MIEV (0), or MIEV vector containing WT Lck or S20C/S23C Lck (DC-S Lck) or Y505F Lck cDNA were lysed in TKM buffer containing 0.5% Brij 58 and subjected to sucrose gradient centrifugation. 10 μl of each fraction was probed with cholera toxin B-HRP recognizing lipid raft marker GM1 (top panels), anti-Lck (middle panel), or anti-Fyn (bottom panel). Numbers below the panels indicate the percentage distribution of Lck and Fyn to lipid rafts (R) and membrane-soluble (S) fractions. B, Lck and Fyn immunoprecipitates (IP) from LR (R) and soluble (S) fractions derived from each infectant highlighted in A were split into equal portions and immunoblotted with anti-Lck and anti-Fyn.
Of note is that although detection of Tyr(P)394 Lck was assessed using specific antibody (39), detection of Tyr(P)417 Fyn utilized a polyclonal antibody that also recognizes Tyr(P)394 Lck. As Fyn immunoprecipitates will contain complexed Lck in this system, the resulting Tyr(P)417 signal illustrated in Fig. 1C, right panel, is composed of the Tyr(P)394 Lck plus that of Tyr(P)417 Fyn. However, the contribution of Lck-derived signal is readily distinguishable from that of Fyn because of the difference in their molecular weights (Fig. 1C, right panel). Also, because of the low stoichiometry of the Y505F Lck-Fyn interaction, contribution of Tyr(P)394 Lck signal revealed by anti-Tyr(P)417 Fyn is negligible (Fig. 1C, right panel).
Lck-Fyn Complex Formation Is Confined to Lipid Rafts—To determine whether the interaction between Y505F Lck and WT Fyn is spatially confined or occurs ubiquitously on the plasma membrane, we prepared LR and soluble fractions from 3T3-FynT cells expressing either WT, DC-, or Y505F mutants of Lck. As illustrated in Fig. 2A, >92% of WT Fyn and 67–75% of Lck partitions to LR in each of the four infectants. Probing Lck and Fyn immunoprecipitates derived from LR and soluble fractions with antibodies specific for the alternative kinase demonstrates that Lck-Fyn interaction is detected exclusively in LR (Fig. 2B), consistent with the observation that virtually all WT Fyn is localized within LR. It was therefore critical to determine whether the co-immunoprecipitation of Y505F Lck and Fyn from LR reflects a bona fide interaction and is not simply an artifact of isolating LR using nonstringent conditions of solubilization that could promote nonspecific interactions.
Toward this end, we assessed the presence of Lck-Fyn complexes in circumstances in which LR integrity is disrupted. For this purpose we used LM, a member of the n-alkyl glycosidic type of detergents, frequently used for immunoprecipitation of LR-associated proteins (47–52) as it efficiently dissociates LR (53). We first compared the LR-solubilizing capacity of LM to those of Brij 58 and Nonidet P-40 by quantifying the relative amount of Fyn in LR and soluble fractions prepared from NIH 3T3 cells. For purposes of quantification in these experiments, we denote fractions 1–8 as LR, and 9 and 10 as soluble fractions. As illustrated in Fig. 3, A and B, three distinct patterns of Fyn distribution were observed in the three detergents. More than 90% of Fyn partitions to LR in Brij 58 lysates. In contrast, partitioning of Fyn in Nonidet P-40 lysates redistributed 45 and 55% to LR and soluble fractions, respectively. As expected, given the reported efficiency of LM to disaggregate LR, Fyn is virtually undetectable in low buoyant density fractions of LM lysates. Specifically, 100% of Fyn was detected in the soluble fractions derived from LM lysates (Fig. 3, A and B), demonstrating that LR harboring Fyn are efficiently solubilized in LM. Importantly, setting the Fyn signal detected in Brij 58 to 100%, >95% of the Fyn signal is rescued in both Nonidet P-40 and LM lysates, while partitioning to distinct fractions of the gradient. This was not the case for the parallel assessment of GM1 distribution in the three detergents.
FIGURE 3.

Co-immunoprecipitation of Lck-Fyn complexes is not dependent on integrity of lipid rafts. A and C, 5 × 105 3T3-FynT cells were lysed in buffers containing either Brij 58, Nonidet P-40, or laurylmaltoside (LM) and subjected to sucrose gradient centrifugation. An equal amount of each fraction was probed using anti-Fyn antibody (A) or cholera toxin B subunit-HRP detecting lipid raft marker GM1 (C). B and D, quantification of A and C, relative distribution of Fyn (B) and GM1 (D) signals in LR (fractions 1–8, open bars) and soluble fractions (fractions 9–10, SF, closed bars) in all three detergents are expressed in %. The relative total signal of Fyn (B) and GM1 (D) detected in each lysis condition is normalized to the total amount of Fyn or GM1 in Brij 58 lysate, which were assigned an arbitrary reference value of 100%. ND = not detectable. E, 3T3-FynT cells expressing either WT Lck or Y505F Lck cDNA were lysed in Nonidet P-40 or LM containing buffers and subjected to immunoprecipitation (IP) using anti-Lck antibody. Immunoprecipitates were split and probed using either anti-Lck (top panel) or anti-Fyn (bottom panel) antibodies.
The distribution of the classical LR resident ganglioside, GM1, was assessed in parallel to Fyn in each of the three detergents. As illustrated in Fig. 3, C and D, >99% of the GM1 signal in Brij 58 lysates localized to LR. The re-distribution of GM1 in Nonidet P-40 and LM lysates was qualitatively comparable with that observed for Fyn. Specifically, streaming of GM1 to higher buoyant density fractions was observed in Nonidet P-40 and virtually all detectable GM1 localized to SF in LM lysates (Fig. 3C). However, in striking contrast to quantitative analyses of Fyn redistribution in these three detergents, setting the value of the GM1 signal in Brij 58 to 100%, only ∼14 and 3.7% of the total GM1 signal was observed in Nonidet P-40 and LM lysates, respectively (Fig. 3D). Hence, after solubilization of LR, GM1 is largely undetectable as assessed by cholera toxin (CT) binding. Impaired CT binding to GM1 in Nonidet P-40 and LM lysates, in which LR are disaggregated, is consistent with previous reports suggesting that LR resident GM1 is clustered and therefore supports multivalent binding of CT (54). In contrast, disaggregation of LR in Nonidet P-40 and LM lysates would destroy the GM1 geometry predicating CT binding.
The key observation derived from this detergent analysis is illustrated in Fig. 3E. Notwithstanding undetectable LR derived from cells lysed in LM, the capacity to co-immunoprecipitate Y505F Lck-Fyn complexes is unimpaired (Fig. 3E). Thus, although LR play a fundamental role in supporting the co-localization of Lck and Fyn, the retention of these ordered microdomains upon solubilization does not predicate the stability of Y505F Lck-Fyn complexes once they are formed.
Structure-Function Analysis of Lck-Fyn Interaction—We next sought to define domain mutant(s) of Y505F Lck mediating its interaction with Fyn. Previous mutational and functional analyses suggest that several regions/residues on Lck could mediate either a direct Lck-Fyn interaction or interactions with intermediate molecule(s) as follows: the unique domain (55–57) and the SH2 (58) and SH3 (59) domains; the proline-mediated PPII conformation of the linker region (8); and the kinase domain (60, 61) or specific residues within the C terminus (17, 62). Toward determining which of these sequences might be involved in supporting Y505F Lck-Fyn complex formation, mutants with disabled function of each of these individual domains, and the last five amino acid truncated at the C terminus, were prepared on Y505F Lck cDNA template by site-directed mutagenesis. 3T3-FynT infectants, ectopically expressing matched levels of mutant forms of Lck (Fig. 4, top row) and WT Fyn (Fig. 4, 2nd row), were screened for their ability to support Lck-Fyn complex formation.
FIGURE 4.

Structure-function analysis of Lck-Fyn interaction. C20S/C23S, S59A, W97K, R154K, P249G, P252G, P249G+P252G, K273R, Y394F, and ΔFQPQP mutations on a Y505F Lck template, each inactivating a single functional domain/region of constitutively active Y505F Lck, were cloned into the retroviral vector Migr1 vector and expressed in 3T3-FynT cells. Anti-Lck or anti-Fyn immunoprecipitates (IP) were probed with anti-Lck (upper panels) and anti-Fyn (2nd row of panels), respectively, and illustrate the total amount of Lck and Fyn co-expressed in each mutant. To reveal which Lck domain mutant abrogates Lck-Fyn complex formation, anti-Fyn immunoprecipitates were probed with anti-Lck (3rd row of panels). Anti-Fyn immunoprecipitates from cells co-expressing WT Fyn with either WT Lck or Y505F Lck immunoblotted with anti-Lck served as negative and positive controls, respectively (3rd row, left panel). To assess the activation status of Fyn, anti-Fyn immunoprecipitates were also immunoblotted with anti-Tyr(P)417 Fyn (bottom panels).
Introduction of single point mutations ablating the function of either the SH2 or SH3 domain or substitution of the two proline residues in the linker region or various ablating substitutions in the unique domain, including the two cysteine residues involved in CD4 interaction and the serine residue known to impact the function of the SH3 domain, have no impact on Lck-Fyn co-immunoprecipitation (Fig. 4, 3rd row of panels). In contrast, a profound reduction in the formation of Lck-Fyn complexes is observed using K273R and Y394F mutations of Y505F Lck (Fig. 4, 3rd row of panels), both of which are unable to support the activation of WT Fyn as assessed by increased Tyr(P)417 levels (Fig. 4, bottom panels).
Sequential Truncation of FQPQP C-terminal Sequence Correlates with Increasing Loss of Lck-Fyn Complex Formation—The established role of C-terminal sequences in the regulation of Src family kinases and the Src-related kinase c-Abl (4, 14) prompted us to investigate the involvement of C-terminal residues unique to the Lck subfamily of Src family PTK in supporting Lck-Fyn interactions. Remarkably, truncation of the last five C-terminal residues (ΔFQPQP) profoundly reduces complex formation with Fyn in this fibroblast system (Fig. 4). Furthermore, this impaired complex formation correlates with decreased Tyr(P)417 Fyn levels (Fig. 4, bottom panels). This result was further analyzed by comparing the impact using sequential truncates of Y505FLck, ΔQP, ΔQPQP, and ΔFQPQP, and correlating levels of Tyr(P)394 Lck and Tyr(P)417 Fyn.
As illustrated in Fig. 5, A and B, left panels, these sequential truncations have no effect on levels of Tyr(P)394 Lck. In contrast, and as illustrated in the left panel of Fig. 5C, the ability to co-immunoprecipitate Fyn with anti-Lck is sequentially lost, such that the ΔFQPQP Lck co-immunoprecipitated ∼5–10% of the Fyn in comparison with that co-immunoprecipitated with Y505F Lck. This in turn is observed in the reciprocal circumstances in which anti-Fyn immunoprecipitates were probed with anti-Lck, as shown in the right panel of Fig. 5C. Furthermore, and consistent with the inability of these truncates to form stable complexes with Fyn, is the observed sequential loss of the Tyr(P)417 signal as illustrated in Fig. 5B, right panel.
FIGURE 5.

Sequential truncation of FQPQP C-terminal sequence correlates with gradual decrease in Lck-Fyn complex formation without affecting kinase activity of Lck. 105 3T3-FynT cells co-expressing either WT, Y505F, Y505F+ΔQP (ΔQP505F), Y505F+ΔQPQP (ΔQPQP505F), or ΔFQPQP Lck were lysed in 1% Nonidet P-40 lysis buffer and immunoprecipitated (IP) with either anti-Lck (left panels) or anti-Fyn-specific antibodies (right panels). The immunoprecipitates derived from each infectant were split into three equal portions and probed with immunoprecipitating antibody-(A), Tyr(P)394 Lck-, and Tyr(P)417 Fyn-specific antibodies (B) and antibodies against the alternative kinase (C). Histograms in B show the relative kinase activity of Lck and Fyn measured by normalization of the Tyr(P)394 Lck and Tyr(P)417 Fyn signals to their respective total kinase signals. Histograms in C show the relative amount of Fyn and Lck co-immunoprecipitating with alternative kinase after normalization to their respective total kinase signals. Samples derived from the 3T3-FynT cells co-expressing Y505F Lck were given a reference value of “100%.” ECL+, supersensitive reagent for chemiluminescent development of Western blots.
Two criteria need be met to formally prove that this C-terminal sequence defines the structural domain of Lck predicating its interaction with Fyn. The first is that targeting of ectopically expressed Lck variants and Fyn to plasma membrane LR is not disrupted as a consequence of these truncations (64); and second, these truncates are indeed kinase-active. We first assessed an association of these mutants with plasma membrane and their partitioning to LR.
C Terminus of Lck Does Not Impact on Plasma Membrane Localization—As illustrated in Fig. 6, confocal images of 3T3-FynT cells infected with WT, Y505F, ΔQPY505F, ΔQPQPY505F, or ΔFQPQP Lck demonstrate comparable plasma membrane localization of all Lck variants, as well as that of Fyn. Thus, the absence of the QPQP sequence does not impair Lck tethering to the plasma membrane.
FIGURE 6.
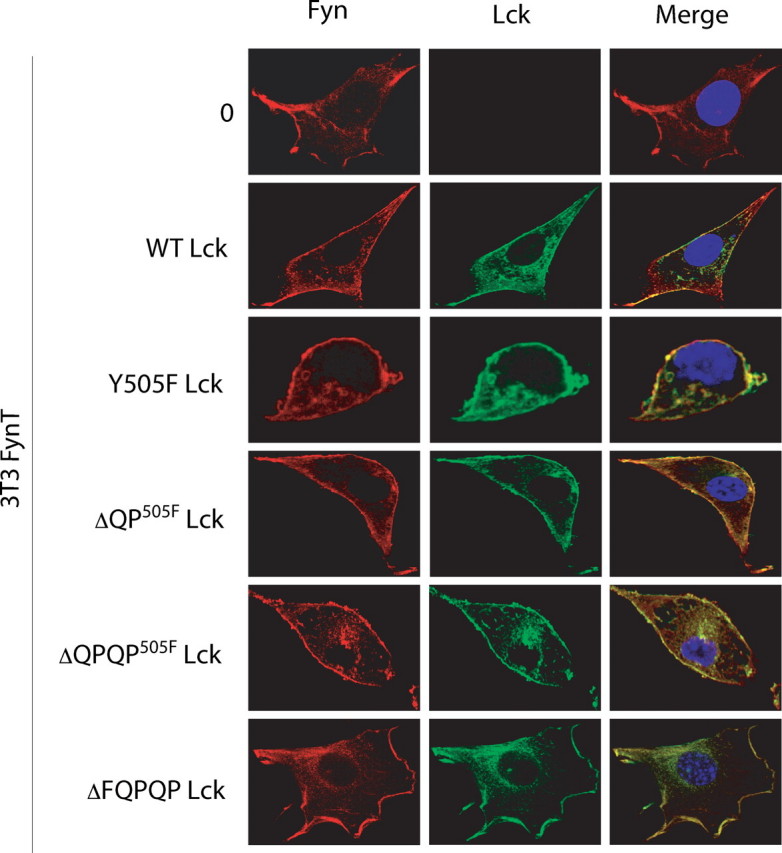
C-terminal truncation of Lck does not impair tethering to the plasma membrane. 3T3-FynT cells infected with either empty Migr1 vector (0) or vector containing either WT Lck, Y505F Lck, ΔQPY505F, ΔQPQPY505F, or ΔFQPQP were probed with anti-Lck (green) and anti-Fyn (red). One representative image of each infectant is shown. The nucleus was stained with 4′,6-diamidino-2-phenylindole (blue). (×400 zoom).
C Terminus of Lck Functions as a Lipid Raft Targeting Sequence—In marked contrast, sequential truncation of the C terminus of Lck has a profound impact on partitioning to LR. As control, the partitioning of WT Lck and Y505F Lck was reassessed. As illustrated in Fig. 7, 64 and 58% of WT Lck and Y505F Lck partition to LR, respectively. Sequential truncation of the five C-terminal residues of Lck exhibit increasing impact on LR localization (Fig. 7, bottom three rows of panels), Specifically, whereas a marginal (46%) to moderate (29%) reduction of ΔQP505FLck and ΔQPQP505FLck partitioning to LR was observed, respectively, only 10–15% of ΔFQPQP Lck partitioned to LR (Fig. 7, bottom panel). Of note, the sequential decrease in the proportions of these truncates partitioning to LR correlates well with the sequential loss of complex formation with FynT (Fig. 5). In contrast, Y394F and K273R kinase domain mutants of Y505F Lck (Y394F505F and K273R505F, respectively), which were also unable to form complexes with and activate Fyn (Fig. 4), exhibit Lck distribution similar to control samples (Fig. 7, 3rd and 4th row of panels).
FIGURE 7.

C terminus of Lck functions as a lipid raft targeting sequence. 106 3T3-FynT cells infected with Migr1 vector containing either WT Lck (WT), Y505F Lck (Y505F), Y505F+Y394F Lck (Y394F505F), Y505F+K273R Lck (K273R505F), Y505F+ΔQP Lck (ΔQP505F), Y505F+ΔQPQP Lck (ΔQPQP505F), or ΔFQPQP Lck cDNA were lysed in TKM buffer +0.5% Brij-58 and subjected to sucrose gradient centrifugation. 10 μl of each fraction was probed with cholera toxin B subunit-HRP detecting lipid raft marker GM1 (left panels), anti-Fyn (middle panels), and anti-Lck (right panels). Numbers below the panels indicate the percentage distribution of Lck and Fyn to lipid rafts (R) and membrane-soluble (S) fractions in each variant.
Two plausible scenarios could account for impaired partitioning of C-terminal truncates of Lck to LR: (i) the YQPQP sequence is required for Lck partitioning to LR, or (ii) its retention in LR. Although the former scenario discount the effect of LR-resident Fyn on Lck distribution, the latter could involve the role of LR-resident Fyn in retaining Lck in LR.
LR Resident Fyn Does Not Impact on Partitioning of Lck—To address the potential role of Fyn in retaining Lck in LR, the impact of the presence of WT Fyn in LR on partitioning of Y505F or ΔFQPQP Lck variants was assessed in stable infectants of the embryonic fibroblast cell line (SYF), genetically deficient in Src, Yes, Fyn, and Lck (not shown). The results presented in Fig. 8 demonstrate two points. The partitioning of ΔFQPQP Lck to LR in SYF cells is reduced ∼60–80% compared with that observed for Y505F Lck (Fig. 8), a reduction comparable with that observed in NIH 3T3 cells (Fig. 7); and that the extent to which either Y505F or ΔFQPQP Lck variants do partition to LR is independent of LR resident Fyn. Specifically, the partitioning of both Lck mutants was comparable in the presence or absence of WT Fyn as follows: 31 and 34% for Y505F Lck and 13 and 7% for ΔFQPQP Lck, respectively. Thus, if the C-terminal sequence of Lck is required for LR retention, the underlying molecular mechanism does not directly involve Fyn.
FIGURE 8.

Partitioning of Lck to lipid rafts is Fyn-independent. Lipid rafts from Fyn- and Lck-deficient SYF infectants ectopically expressing either WT FynT, or Y505F Lck, or ΔFQPQP Lck (single infectants) or a combination of WT Fyn and Y505F Lck or WT FynT and ΔFQPQP Lck (double infectants) were prepared and analyzed as in Fig. 7. Numbers below the panels indicate the percentage distribution of Lck and Fyn to lipid rafts (R) and membrane-soluble (S) fractions in each infectant.
Comparative Assessment of the Kinase Activity of C-terminal Truncates—The kinase activity of each of the Lck variants was assessed using stable infectants of SYF cells to ensure that activity measured was due exclusively to the Lck mutant. Two assays were utilized in this regard as follows: the induction of tyrosine phosphorylation of cellular substrates (Fig. 9A) and immune complex kinase assays, in which the phosphorylation of the exogenous substrate enolase was measured, as well as the transphosphorylation of immunoprecipitated Lck (Fig. 9B). As predicted and consistent with previously published data (65, 66), the kinase activities of both K273R505F Lck and Y394F505F Lck are profoundly impaired in comparison with the respective signals obtained with Y505F Lck (Fig. 9B). None of the other Lck mutants exhibit impaired global tyrosine phosphorylation with the unexpected exception of the ΔFQPQP C-terminal truncate.
FIGURE 9.

Comparative assessment of the kinase activity of C-terminal truncates. A, crude cell lysates derived from 3 × 105 uninfected (–) or SYF cells infected with empty Migr1 vector (0) or vector containing either WT Lck (WT) or Y505F Lck (Y505F), or Y505F+C20S/C23S (DC-S505F), or Y505F+S59A (S59A505F), Y505F+W97K (W97K505F), or Y505F+R154K (R154K505F), or Y505F+P249G+P252G (DP-G505F), or Y505F+K273R (K273R505F), or Y505F+Y394F (Y394F505F), or Y505F+ΔQP (ΔQP505F), or Y505F+ΔQPQP (ΔQPQP505F), or ΔFQPQP Lck cDNA were resolved on 12.5% SDS-PAGE and immunoblotted with anti-phosphotyrosine-specific mAb 4G10 (upper panel). Using the same lysates, 5 × 104 cell equivalents were resolved on 9% SDS-PAGE and immunoprobed using anti-Lck antibody. B, anti-Lck immunoprecipitates from 2 × 106 cells of selected SYF infectants shown in A were subjected to immune complex kinase assays. The relative Tyr(P) content of each infectant is expressed either as a relative level of phospho-Lck (pY-Lck, black bars) or phosphoenolase (pY-Eno, gray bars) signal (shown in the upper panel) after normalization to total Lck levels (bottom panel). Bars representing Tyr(P)-Lck and Tyr(P)-Eno from the same sample are grouped and aligned with their respective tracks.
As demonstrated in Fig. 9, A and B, the kinase activities of ΔQP505F Lck and ΔQPQP505F Lck assessed by both the induction of global tyrosine phosphorylation and immune complex kinase assays are comparable with that mediated by Y505F Lck. In striking contrast the capacity of ΔFQPQP Lck to induce global tyrosine phosphorylation is as compromised as that observed for both the K273R505F and Y394F505F mutants of Lck (Fig. 9A). This observation is recapitulated in immune complex kinase assays for ΔFQPQP Lck in which the enolase signal observed is less than that induced by WT Lck (Fig. 9B). In contrast, the ability of ΔFQPQP Lck to trans-phosphorylate other ΔFQPQP Lck molecules was ∼30% that observed with Y505F Lck (Fig. 9B). This apparent dichotomy of function is emphasized upon consideration of the “Tyr(P) index.” Specifically, the ratio of the Tyr(P)-Lck signal and the Tyr(P)-enolase signal calculated for each of the Lck mutants is approximately “1,” with the notable exception of ΔFQPQP Lck, which has a Tyr(P) index approaching 4 (Fig. 9B). Thus, with the exception of ΔFQPQP Lck, the efficacy with which each variant phosphorylates exogenous substrate, or itself, is comparable. Importantly, the unimpaired kinase activities associated with ΔQP505F Lck and ΔQPQP505F Lck prompted us to assess their partitioning to LR in more physiological circumstances.
C-terminal “QPQP” Sequence Targets Lck to Lipid Rafts in T Cells—To assess the function of C-terminal residues of Lck in partitioning to LR in T cells, we employed a T cell clonal system. Clone 2.5 is Lck-sufficient and CD4+ (44, 46). As controls, 2.5 cells were infected with WT Lck or DC-S Lck, which is unable to bind to CD4 (35). To obviate the potential role of kinase activity in the partitioning of DC-S Lck in clone 2.5, a kinase dominant negative mutant K273R of DC-S (DC-S/K273R) Lck was used as a template for the preparation of ΔQP, ΔQPQP, and ΔYQPQP Lck mutants. As illustrated in Fig. 10A, all of the Lck variants are expressed at levels ∼5–9-fold in excess of endogenous Lck in clone 2.5. The partitioning of Lck to LR and soluble fractions in each of these infectants was then assessed.
FIGURE 10.

C-terminal “QPQP” sequence targets Lck to lipid rafts in T cell clone 2.5. A, 105 2.5 T cells infected with the empty retroviral Migr1 vector (0), or Migr1 containing either WT Lck (WT), C20S/C23S Lck (DC-S), C20S/C23S+K273R Lck (DC-S/K273R), C20S/C23S+K273R+ΔQP Lck (DC-S/K273R/ΔQP), C20S/C23S+K273R+ΔQPQP Lck (DC-S/K273R/ΔQPQP), or C20S/C23S+K273R+ΔYQPQP Lck (DC-S/K273/ΔYQPQP) were resolved on SDS-PAGE and immunoblotted with anti-Lck. Histogram shows the relative cellular level of Lck in 2.5 infectants normalized to that in the empty Migr1 infectant, which was assigned an arbitrary value of 1. B, 106 2.5 infectants from A were lysed in TKM buffer +0.5% Brij 58 and subjected to sucrose gradient centrifugation. 15 μl of each fraction was subjected to SDS-PAGE and probed with anti-Lck. Numbers below the panels indicate the percentage distribution of Lck in lipid rafts (R) and membrane-soluble (S) fractions. Bar graph to the left represents the mean value of Lck amount (%) partitioning to LR. Values are derived from three independent experiments.
As reported previously (33) and illustrated in the top three panels of Fig. 10B, ectopically expressed WT Lck and DC-S Lck display distinct subcellular localization patterns. Specifically, although the overexpression of WT Lck achieved saturation of binding sites on endogenous CD4 with the excess Lck localizing to LR (∼20% of total Lck), the overexpressed DC-S Lck, unable to bind to CD4, is preferentially enriched in LR (∼50%). Furthermore, the basal kinase activity of DC-S Lck is dispensable for preferential targeting to LR, as DC-S/K273R Lck displays the same partitioning ratio (Fig. 10B). The prediction is that if the C-terminal sequence of Lck functions as an LR targeting sequence in T cells, sequential truncates of DC-S/K273R Lck will skew toward membrane-soluble fractions. As illustrated in Fig. 10B, DC-S/K273R truncates display increasingly impaired partitioning to LR, such that >90% of ΔYQPQP Lck was observed in the soluble fractions. Taken together these results define a novel role for the C-terminal residues in targeting Lck to LR in T cells.
LR Localization of Kinase-active Lck Predicates Enhanced TcR Signaling—Given that the indispensable role of Lck in TcR-mediated cellular activation has been established, and the role of LR in providing a signaling scaffold for TcR-mediated signaling is gaining acceptance (67), the results presented herein proffered the opportunity to assess whether LR localization of Lck predicates its function in TcR-mediated cellular activation. To address this question, we utilized the previously described system demonstrating that ectopic expression of Y505F Lck enhanced TcR-mediated IL-2 production in the T cell hybridoma BI-141 (37). Specifically, the prediction follows that if Lck needs be LR-associated to support enhanced TcR-mediated IL-2 production, the C-terminal truncates of LckY505F should not be as effective as LckY505F.
To test this prediction, BI-141 infectants expressing WT, Y505F, ΔQPY505F, ΔQPQPY505F, or ΔFQPQP Lck variants were prepared. As illustrated in Fig. 11A, these infectants expressed 5–7-fold higher levels of Lck than the empty vector control. As illustrated in Fig. 11B, Fyn and Lck distribution profiles in BI-141 recapitulated those observed in both 3T3 and SYF fibroblasts and the T cell clone 2.5. Specifically, the vast majority of Fyn localized to LR in all infectants. Furthermore, and as observed in both fibroblast lines and clone 2.5, the partitioning of ΔQPY505F, ΔQPQPY505F, or ΔFQPQP Lck to LR in BI-141 was significantly impaired. Specifically, 3–4 times more Y505F Lck was found associated with LR compared that observed in the C-terminal truncates (Fig. 11B). The skewed distribution of these truncates outside LR correlated with their inability to support enhanced TcR-mediated IL-2 production observed in LckY505F infectants. As illustrated in Fig. 11C, anti-TcR-mediated IL-2 production in ΔQPY505F, ΔQPQPY505F, or ΔFQPQP Lck infectants was 2–4 times less than that observed in LckY505F infectants and comparable with levels of IL-2 produced in WT Lck infectants (Fig. 11C). These results demonstrate that targeting of kinase-active Lck to LR predicates enhanced TcR-mediated IL-2 production and, furthermore, underscores the physiological and functional relevance of the C-terminal YQPQP sequence of Lck in targeting active Lck to LR in this CD4–/CD8– T cell line.
FIGURE 11.

C-terminal sequence-dependent enrichment of Lck in lipid rafts predicates enhanced TcR-mediated IL-2 production. A, BI-141 cells infected with either the empty Migr1 vector or the vector containing either WT, Y505F, ΔQPY505F, ΔQPQPY505F or ΔFQPQP Lck were analyzed by Western blot. Bar graph represents relative total levels of Lck expressed in each infectant after normalization to actin. B, GM1, Fyn, and Lck distribution to lipid rafts and soluble fractions in BI-141 infectants were performed as in Fig. 7C. Triplicate cultures of BI-141 infectants were stimulated with plate-bound anti-TcRCβ coated at the indicated concentrations, and IL-2 production was assessed by ELISA.
DISCUSSION
Modeling Lck activation and delivery of function in MHC class II-restricted T cells has been facilitated because of its high stoichiometric interaction with CD4 (68, 69). In contrast, no such functional analog has been described for Fyn. Furthermore, the similarity of functions, and overlapping arrays of targets of these two Src family PTKs, has led some to postulate their redundant function in TcR signaling. Our recent results provide evidence for nonredundant and essential functions of Lck and Fyn in generating the most proximal signals emanating from the TcR. The question of their redundancy is largely obviated with the demonstration that in primary resting CD4+ T cells, these two kinases are physically separated. Within seconds following TcR/CD4 engagement, Lck is activated outside of LR, translocates into LR, and the activation of LR-associated Fyn ensues (2, 3) Thus, although interdependent, the activation of Lck and Fyn is spatially and temporally uncoupled. The object of this study was to test the predication derived from the above observation that Lck-dependent Fyn activation may reflect the interaction of these two kinases. Toward this end, the characterization of the structural features of constitutively active Lck required for the activation of Fyn was assessed.
The fibroblast model described here enabled a biochemical analysis of Lck-Fyn complex formation. As observed for endogenous WT Fyn in T lymphocytes (3), 87–95% of the exogenous WT Fyn partitioned to LR in fibroblasts. However, in sharp contrast to primary CD4+ T cells, perhaps in part because of the absence of CD4 in NIH 3T3 fibroblasts, 53–75% of exogenous Lck also partitioned to LR. As described, this steady-state distribution of mutated forms of Lck to LR and their co-localization with WT Fyn enabled the analysis of Lck structures that predicate its partitioning to LR, its ability to complex with WT Fyn, and in turn the further characterization of the process(es) underpinning Lck-dependent Fyn activation.
The results presented demonstrate that Lck and Fyn physically interact with sufficient stability to enable co-immunoprecipitation. Thus, in the appropriate circumstances, Lck-Fyn complexes can be reciprocally immunoprecipitated with antibody specific for either Lck or Fyn. Revealing these complexes is predicated by co-localization of fully kinase-active Lck with Fyn in LR. The requirement for co-localization of kinase-active Lck with Fyn in LR in the fibroblast model reported here precisely parallels the sequence of events triggered by TcR/CD4 engagement in primary T cells (3). Specifically, in the latter system, Lck is activated within seconds outside of LR, and a fraction of kinase-active Lck translocates into LR. Hence the essential function of these lipid microdomains is to concentrate kinase-active Lck in a Fyn-rich environment. Consistent with this function is the observation that Lck-Fyn complexes and robust increases in Tyr(P)417 Fyn are observed exclusively in LR.
In this context, the demonstration that the integrity of LR is dispensable for detection of Y505F Lck-Fyn complexes contrasts with their critical role in initiation of this interaction by juxtaposing Lck and Fyn. However, several lines of evidence support this conclusion. First, Lck and Fyn complexes are detected exclusively in LR (Fig. 2B). Second, an argument that non-GM1 containing LR, which escape detection by cholera toxin, could harbor Fyn-Lck complexes is largely obviated by the demonstration that a full solubilization of GM1 containing LR with laurylmaltoside is concomitant with a complete shift of Fyn from LR to soluble fractions (Fig. 3). Third, despite the full solubilization of LR and displacement of Fyn to soluble fractions, the detection of Lck-Fyn complexes is unperturbed (Fig. 3E). These results strongly support the conclusion that interacting kinase-active Lck and Fyn in LR get transiently locked in a conformation resistant to LR solubilization.
The structure/function analysis reported herein revealed that stable complex formation between kinase-active Lck and Fyn was not dependent on any specific sequence(s) and/or modular domains of Lck, except those indispensable for its full kinase activity. These results support the conclusion that LR function to provide a constricted environment that supports proximity of the two kinases and increases the probability of Lck-mediated transphosphorylation of Fyn through kinase domain interaction. Importantly, whereas LR are required to mediate juxtaposition of kinase-active Lck with Fyn, they are dispensable for observing the two kinases in complex. The observed robust increase in LR-associated Tyr(P)417 Fyn levels, coupled with the observation that only a small fraction of kinase-active Lck and Fyn are found in complex at any given time, suggests that the transphosphorylation reaction is short lived with a high rate of turnover.
Furthermore, this study does not formally prove that the observed Lck-Fyn complexes are a result of the direct interaction between these two kinases. It is not implausible that the observed interaction is supported by an intermediate molecule(s). In this regard, protein analysis of Lck-Fyn complexes by silver staining did not reveal any bands other than those attributable to the two kinases (data not shown). However, the resolution of this assay may have precluded the visualization of a low stoichiometric interaction with a potential intermediate. Minimally, we can conclude that should an intermediate be involved, it would have to be one that is not lymphocyte-specific, and its function in this context is not dependent on Lck modular domains.
Two unexpected, although plausibly related results, are presented in this study. The C-terminal (Y/F)QPQP sequence is characterized as a novel cis-acting component essential for partitioning Lck to LR in at least two different fibroblast cell lines (3T3, Fig. 7, and SYF, Fig. 8) and, importantly, also in two unrelated T cell model systems (2.5, Fig. 10, and BI-141, Fig. 11). Several cis-acting protein sequences and domains supporting protein targeting to LR have been described in recent years. These sequences include the following: (i) serve as attachment sites for glycosylphosphatidylinositol anchor to proteins (39, 70); (ii) contain palmitoylated cysteine residues found at the N termini (71) or the C-terminal end of protein (72) or in the proximity of transmembrane domain (73); and (iii) can be post-translationally modified through acylation and/or addition of a cholesterol moiety (74, 75). In addition, it has been shown that the prohibitin homology domain (76) and the SoHo domain containing proteins (77) preferentially partition to LR. In comparison with the above modifications that impact on protein trafficking and subcellular partitioning, the (Y/F)QPQP sequence is unique. It does not contain residues permitting post-translational acylation nor does it exhibit any characteristics known to predispose to LR targeting. The working hypothesis is that the C-terminal sequence targets Lck to proteins of the cytoskeletal network, which in turn controls the re-distribution of Lck to LR. Although the ability of the C-terminal sequence of c-Src and Lck to interact with other proteins has been previously documented (62, 78, 79), the scope and nature of these interactions in terms of regulation of subcellular protein distribution remain uncharacterized. Thus, in aggregate, these results support a dual role for the C terminus of Lck as follows: (i) its previously established negative regulatory role through Tyr505, and (ii) its novel role in targeting to LR.
The C-terminal QPQP sequence was characterized as the minimal truncate that significantly affected Lck distribution to LR in the T cell clonal system 2.5 (Fig. 10B). Importantly, this trans-acting, Lck lipid-raft targeting sequence predicates the activation of T cells (Fig. 11, B and C). This is in agreement with our previously published data reporting a requirement for an enrichment of Lck in LR for the initiation of proximal TcRαβ signaling (33). Src family tyrosine kinases are generally considered as LR-associated molecules (6, 80, 81). The question remains whether QPQP-dependent LR targeting is a unique feature of Lck or represents a more universal trait among other Src family members. Analysis of the C-terminal sequences downstream of the negative regulatory tyrosine among Src family members reveals a striking conservation, one that discriminates among members of the Lck and Src subfamily members of Src family tyrosine kinases. Notably, the C terminus of Lck, Hck, Lyn, and Blk kinases invariably contain Gln and Pro residues in QQQP, QPQP, or LQP combinations, in striking contrast to the conserved QPGENL tail sequence found in Src, Fyn, Yes, and Fgr, the latter containing a slight modification of this sequence, QPGDQT (14). In this context, a recent report demonstrates that the intracellular C-terminal tail of the catfish gonadotropin-releasing and murine-luteinizing hormone receptors contribute to constitutive (82) and activation-induced (83) LR targeting, respectively. It was suggested that the underpinning mechanism involved palmitoylation of cysteine residues (63). However, analysis of their respective C-terminal sequences revealed the presence of QP-based motifs, specifically, QPSGQ and QQPIPP (63). These sequences may also contribute materially in targeting hormone receptors to LR. Thus, because of their sequence similarities, it is tempting to speculate that short, QP-based C-terminal motifs function as cis-acting elements guiding proteins to LR via their interaction with a postulated LR translocation/distribution machinery.
Occurrence of two types of QP-based motifs each associated with distinct subfamilies of Src family kinases could also provide the resolution for understanding their bias for differential distribution to LR. Specifically, although in the primary CD4+ T cells Fyn kinase resides in LR, preferential localization of Lck in membrane-soluble fractions is usually thought to reflect its high stoichiometric interaction with CD4 (68, 69). However, distinct LR distribution of Lck and Fyn is still observed in CD4-deficient 3T3-FynT cells (Fig. 2A and Fig. 6). These observations support the conclusion that additional structural factors contribute to this phenotype. In this regard, recent crystallographic data revealed that the dephosphorylated C-terminal tail of c-Src is folded back on the C-lobe of the kinase domain and further stabilized by the binding of the terminal leucine residue to a pocket (14). Thus, the structural constraints imposed by a shorter C-terminal tail of Lck subfamily members precludes interaction with the binding pocket, resulting in limited interaction with the surface of the C-lobe kinase domain. This in turn could result in increased availability for other protein interactions. We postulate that these interactions are precisely those involved in tethering Lck to cellular translocation/distribution machinery that could account for observed differences in partitioning to LR, and in turn dictate substrate specificity.
The second untoward observation reported herein relates to the impaired kinase activity associated with ΔFQPQP Lck (Fig. 9, A and B). Remarkably, the activity of this mutant in terms of its capacity to phosphorylate exogenous substrates is as low as that observed for Y394F and K273R kinase mutants of Y505F Lck. Of note, and in contrast to the latter two mutants, the Tyr(P)394 signal of ΔFQPQP Lck remains comparable with that observed in Y505FLck (Fig. 5B). Analysis of the structure of Lck does not provide any insight into the potential structural basis for the impaired kinase activity observed in this mutant. However, the results presented support the conclusion, as discussed above, that the C-terminal sequence of Lck may be involved in supporting/stabilizing its interactions with exogenous substrates.
Specifically, the assays used to directly measure the kinase activity of ΔFQPQP Lck are, with one notable exception, geared toward assessing phosphorylation of exogenous substrates: intracellular proteins when assessing global tyrosine phosphorylation, or enolase, in immune complex kinase assays. However, the observation that ΔFQPQP Lck itself is tyrosine-phosphorylated in immune complex kinase assays with efficacy ∼30% of that observed with Y505F Lck demonstrates that it is indeed kinase-active (Fig. 9B). However, the profoundly impaired global tyrosine phosphorylation (Fig. 9A) or phosphorylation of enolase (Fig. 9B) supported by this mutant strongly suggests its impaired capacity to interact with substrates with sufficient stability to support phosphotransfer.
Consideration of the “Tyr(P) Index” calculated for selected Lck variants as the ratio of the relative Tyr(P) content of the kinase itself, and that for enolase in immune complex kinase assays, underscores the point. Independent of relative kinase activity, the Tyr(P) index of all Lck variants tested approaches 1, except that for ΔFQPQP Lck, which approaches “4” (Fig. 9B). If the C terminus of Lck is indeed involved in supporting/stabilizing its interaction with potential substrates, then how could the transphosphorylation of Lck itself be 3–4 times more efficient than that of enolase? We posit that this mutant can function, albeit to a lesser degree, to transphosphorylate substrates held in stable proximity through immobilization on anti-Lck-Sepharose beads. In this way, both ΔFQPQP Lck phenotypes described in this study, the failure to distribute to LR, and the inability to transphosphorylate its in vivo and in vitro substrates could be uniformly explained by its impaired ability to engage substrates via an intact C-terminal tail sequence.
There is precedent for the role of the C-terminal sequence of Src kinase in either stabilizing and/or dictating substrate interactions. Specifically, whereas C-terminal truncates of chicken pp60Src, including the YQPGENL tail sequence, retained kinase activity, as assessed by their ability to transform NIH 3T3 cells (78, 79), they exhibited compromised activity in immune complex kinase assays (79). Furthermore, observed changes in the phosphorylation patterns of several intracellular targets support the involvement of the C terminus in targeting and/or stabilizing its substrate interactions (78). These observations are consistent with those reported here and further support the concept of the dual role of the C terminus of Src family kinases in both regulation of activity and substrate interaction.
In sum, the results presented are in agreement with and extend a model for Lck-dependent Fyn activation (2). They confirm the principal role of ordered lipid microdomains in the coordination of Lck and Fyn activation through the provision of an essential microenvironment that supports the proximity of the two kinases. This in turn enables the observed high turnover kinetics of the transphosphorylation reaction. The results also support the conclusion that the distribution and/or induced translocation of Lck to LR mediated upon TcR/CD4 co-aggregation (3, 33) functions as a critical regulatory step that limits kinase domain-mediated complex formation and predicates the activation of co-localized Fyn through kinase domainmediated transphosphorylation. Finally, the results demonstrate a previously uncharacterized function of the C-terminal sequence of Lck and establish its Fyn-independent role in partitioning of Lck to LR, and the physiological and functional relevance of targeting Lck to LR in the context of TcR-mediated IL-2 production in double negative T cells.
We posit that this sequence functions to tether Lck directly to cytoskeletal elements. Future studies will resolve the role of the QPQP sequence of Lck in a CD4+ system, and in the context of TcR/CD4-mediated initiation of the activation signaling sequelae induced upon TcR/CD4 engagement. However, even at this early juncture, the model proposed provides a biological framework based on structural distinctions, and we posit their related impact on subcellular distribution of Lck and Src subfamily members, that rationalizes the involvement and regulation of function of multiple Src family tyrosine kinases reported to be critically involved in a variety of receptor-mediated signaling pathways.
Acknowledgments
We gratefully acknowledge the expert technical assistance of Gisele Knowles, Director, Scanning Microscopy and Flow Cytometry Facility at the Sunnybrook Research Institute.
This work was supported by Grant FRN9735 from the Canadian Institutes of Health Research. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: SH3, Src homology 3; SH2, Src homology 2; TcR, T cell antigen receptor; LR, lipid raft; WT, wild type; IL, interleukin; ELISA, enzyme-linked immunosorbent assay; HRP, horseradish peroxidase; mAb, monoclonal antibody; MHC, major histocompatibility complex; LM, laurylmaltoside; CT, cholera toxin B.
References
- 1.Palacios, E. H., and Weiss, A. (2004) Oncogene 23 7990–8000 [DOI] [PubMed] [Google Scholar]
- 2.Filipp, D., and Julius, M. (2004) Mol. Immunol. 41 645–656 [DOI] [PubMed] [Google Scholar]
- 3.Filipp, D., Zhang, J., Leung, B. L., Shaw, A., Levin, S. D., Veillette, A., and Julius, M. (2003) J. Exp. Med. 197 1221–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harrison, S. C. (2003) Cell 112 737–740 [DOI] [PubMed] [Google Scholar]
- 5.Abraham, N., and Veillette, A. (1990) Mol. Cell. Biol. 10 5197–5206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Resh, M. D. (1999) Biochim. Biophys. Acta 1451 1–16 [DOI] [PubMed] [Google Scholar]
- 7.Webb, Y., Hermida-Matsumoto, L., and Resh, M. D. (2000) J. Biol. Chem. 275 261–270 [DOI] [PubMed] [Google Scholar]
- 8.Mayer, B. J. (1997) Curr. Biol. 7 R295–R298 [DOI] [PubMed] [Google Scholar]
- 9.Martin, G. S. (2001) Nat. Rev. Mol. Cell Biol. 2 467–475 [DOI] [PubMed] [Google Scholar]
- 10.Sicheri, F., Moarefi, I., and Kuriyan, J. (1997) Nature 385 602–609 [DOI] [PubMed] [Google Scholar]
- 11.Thomas, S. M., and Brugge, J. S. (1997) Annu. Rev. Cell Dev. Biol. 13 513–609 [DOI] [PubMed] [Google Scholar]
- 12.Xu, W., Harrison, S. C., and Eck, M. J. (1997) Nature 385 595–602 [DOI] [PubMed] [Google Scholar]
- 13.Hermiston, M. L., Xu, Z., Majeti, R., and Weiss, A. (2002) J. Clin. Investig. 109 9–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cowan-Jacob, S. W., Fendrich, G., Manley, P. W., Jahnke, W., Fabbro, D., Liebetanz, J., and Meyer, T. (2005) Structure (Lond.) 13 861–871 [DOI] [PubMed] [Google Scholar]
- 15.Chow, L. M., Fournel, M., Davidson, D., and Veillette, A. (1993) Nature 365 156–160 [DOI] [PubMed] [Google Scholar]
- 16.Schmedt, C., and Tarakhovsky, A. (2001) J. Exp. Med. 193 815–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamaguchi, H., and Hendrickson, W. A. (1996) Nature 384 484–489 [DOI] [PubMed] [Google Scholar]
- 18.Sugimoto, Y., Erikson, E., Graziani, Y., and Erikson, R. L. (1985) J. Biol. Chem. 260 13838–13843 [PubMed] [Google Scholar]
- 19.Moarefi, I., LaFevre-Bernt, M., Sicheri, F., Huse, M., Lee, C. H., Kuriyan, J., and Miller, W. T. (1997) Nature 385 650–653 [DOI] [PubMed] [Google Scholar]
- 20.Mendieta, J., and Gago, F. (2004) J. Mol. Graph. Model. 23 189–198 [DOI] [PubMed] [Google Scholar]
- 21.Cooper, J. A., and MacAuley, A. (1988) Proc. Natl. Acad. Sci. U. S. A. 85 4232–4236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brown, M. T., and Cooper, J. A. (1996) Biochim. Biophys. Acta 1287 121–149 [DOI] [PubMed] [Google Scholar]
- 23.Kjellen, P., Amdjadi, K., Lund, T. C., Medveczky, P. G., and Sefton, B. M. (2002) Virology 297 281–288 [DOI] [PubMed] [Google Scholar]
- 24.Heck, E., Friedrich, U., Gack, M. U., Lengenfelder, D., Schmidt, M., Muller-Fleckenstein, I., Fleckenstein, B., Ensser, A., and Biesinger, B. (2006) J. Virol. 80 9934–9942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mitchell, J. L., Trible, R. P., Emert-Sedlak, L. A., Weis, D. D., Lerner, E. C., Applen, J. J., Sefton, B. M., Smithgall, T. E., and Engen, J. R. (2007) J. Mol. Biol. 366 1282–1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hubbard, S. R., and Till, J. H. (2000) Annu. Rev. Biochem. 69 373–398 [DOI] [PubMed] [Google Scholar]
- 27.Seddon, B., Legname, G., Tomlinson, P., and Zamoyska, R. (2000) Science 290 127–131 [DOI] [PubMed] [Google Scholar]
- 28.Zamoyska, R., Basson, A., Filby, A., Legname, G., Lovatt, M., and Seddon, B. (2003) Immunol. Rev. 191 107–118 [DOI] [PubMed] [Google Scholar]
- 29.Julius, M., Maroun, C. R., and Haughn, L. (1993) Immunol. Today 14 177–183 [DOI] [PubMed] [Google Scholar]
- 30.Samelson, L. E., Phillips, A. F., Luong, E. T., and Klausner, R. D. (1990) Proc. Natl. Acad. Sci. U. S. A. 87 4358–4362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Timson Gauen, L. K., Kong, A. N., Samelson, L. E., and Shaw, A. S. (1992) Mol. Cell. Biol. 12 5438–5446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsygankov, A. Y., Spana, C., Rowley, R. B., Penhallow, R. C., Burkhardt, A. L., and Bolen, J. B. (1994) J. Biol. Chem. 269 7792–7800 [PubMed] [Google Scholar]
- 33.Filipp, D., Leung, B. L., Zhang, J., Veillette, A., and Julius, M. (2004) J. Immunol. 172 4266–4274 [DOI] [PubMed] [Google Scholar]
- 34.Davidson, D., Chow, L. M., Fournel, M., and Veillette, A. (1992) J. Exp. Med. 175 1483–1492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leung, B. L., Haughn, L., Veillette, A., Hawley, R. G., Rottapel, R., and Julius, M. (1999) J. Immunol. 163 1334–1341 [PubMed] [Google Scholar]
- 36.Reske-Kunz, A. B., and Rude, E. (1985) Eur. J. Immunol. 15 1048–1054 [DOI] [PubMed] [Google Scholar]
- 37.Caron, L., Abraham, N., Pawson, T., and Veillette, A. (1992) Mol. Cell. Biol. 12 2720–2729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Veillette, A., Horak, I. D., and Bolen, J. B. (1988) Oncogene Res. 2 385–401 [PubMed] [Google Scholar]
- 39.Holdorf, A. D., Lee, K. H., Burack, W. R., Allen, P. M., and Shaw, A. S. (2002) Nat. Immunol. 3 259–264 [DOI] [PubMed] [Google Scholar]
- 40.Kubo, R. T., Born, W., Kappler, J. W., Marrack, P., and Pigeon, M. (1989) J. Immunol. 142 2736–2742 [PubMed] [Google Scholar]
- 41.Abraham, N., Miceli, M. C., Parnes, J. R., and Veillette, A. (1991) Nature 350 62–66 [DOI] [PubMed] [Google Scholar]
- 42.Pui, J. C., Allman, D., Xu, L., DeRocco, S., Karnell, F. G., Bakkour, S., Lee, J. Y., Kadesch, T., Hardy, R. R., Aster, J. C., and Pear, W. S. (1999) Immunity 11 299–308 [DOI] [PubMed] [Google Scholar]
- 43.Davidson, D., Fournel, M., and Veillette, A. (1994) J. Biol. Chem. 269 10956–10963 [PubMed] [Google Scholar]
- 44.Haughn, L., Gratton, S., Caron, L., Sekaly, R. P., Veillette, A., and Julius, M. (1992) Nature 358 328–331 [DOI] [PubMed] [Google Scholar]
- 45.Amrein, K. E., and Sefton, B. M. (1988) Proc. Natl. Acad. Sci. U. S. A. 85 4247–4251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Haughn, L., Leung, B., Boise, L., Veillette, A., Thompson, C., and Julius, M. (1998) J. Exp. Med. 188 1575–1586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Davidson, D., Bakinowski, M., Thomas, M. L., Horejsi, V., and Veillette, A. (2003) Mol. Cell. Biol. 23 2017–2028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brdickova, N., Brdicka, T., Angelisova, P., Horvath, O., Spicka, J., Hilgert, I., Paces, J., Simeoni, L., Kliche, S., Merten, C., Schraven, B., and Horejsi, V. (2003) J. Exp. Med. 198 1453–1462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Costa, M. J., Song, Y., Macours, P., Massart, C., Many, M. C., Costagliola, S., Dumont, J. E., Van Sande, J., and Vanvooren, V. (2004) Endocrinology 145 1464–1472 [DOI] [PubMed] [Google Scholar]
- 50.Irles, C., Symons, A., Michel, F., Bakker, T. R., van der Merwe, P. A., and Acuto, O. (2003) Nat. Immunol. 4 189–197 [DOI] [PubMed] [Google Scholar]
- 51.Molon, B., Gri, G., Bettella, M., Gomez-Mouton, C., Lanzavecchia, A., Martinez, A. C., Manes, S., and Viola, A. (2005) Nat. Immunol. 6 465–471 [DOI] [PubMed] [Google Scholar]
- 52.Falahati, R., and Leitenberg, D. (2007) J. Immunol. 178 2056–2064 [DOI] [PubMed] [Google Scholar]
- 53.Brdicka, T., Pavlistova, D., Leo, A., Bruyns, E., Korinek, V., Angelisova, P., Scherer, J., Shevchenko, A., Hilgert, I., Cerny, J., Drbal, K., Kuramitsu, Y., Kornacker, B., Horejsi, V., and Schraven, B. (2000) J. Exp. Med. 191 1591–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miller, C. E., Majewski, J., Faller, R., Satija, S., and Kuhl, T. L. (2004) Biophys. J. 86 3700–3708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kesavan, K. P., Isaacson, C. C., Ashendel, C. L., Geahlen, R. L., and Harrison, M. L. (2002) J. Biol. Chem. 277 14666–14673 [DOI] [PubMed] [Google Scholar]
- 56.Park, I., Chung, J., Walsh, C. T., Yun, Y., Strominger, J. L., and Shin, J. (1995) Proc. Natl. Acad. Sci. U. S. A. 92 12338–12342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Turner, J. M., Brodsky, M. H., Irving, B. A., Levin, S. D., Perlmutter, R. M., and Littman, D. R. (1990) Cell 60 755–765 [DOI] [PubMed] [Google Scholar]
- 58.Straus, D. B., Chan, A. C., Patai, B., and Weiss, A. (1996) J. Biol. Chem. 271 9976–9981 [DOI] [PubMed] [Google Scholar]
- 59.Hawash, I. Y., Kesavan, K. P., Magee, A. I., Geahlen, R. L., and Harrison, M. L. (2002) J. Biol. Chem. 277 5683–5691 [DOI] [PubMed] [Google Scholar]
- 60.Carrera, A. C., Alexandrov, K., and Roberts, T. M. (1993) Proc. Natl. Acad. Sci. U. S. A. 90 442–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bougeret, C., Delaunay, T., Romero, F., Jullien, P., Sabe, H., Hanafusa, H., Benarous, R., and Fischer, S. (1996) J. Biol. Chem. 271 7465–7472 [DOI] [PubMed] [Google Scholar]
- 62.Louie, R. R., King, C. S., MacAuley, A., Marth, J. D., Perlmutter, R. M., Eckhart, W., and Cooper, J. A. (1988) J. Virol. 62 4673–4679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Navratil, A. M., Farmerie, T. A., Bogerd, J., Nett, T. M., and Clay, C. M. (2006) Biol. Reprod. 74 788–797 [DOI] [PubMed] [Google Scholar]
- 64.Lingwood, D., and Simons, K. (2007) Nat. Protoc. 2 2159–2165 [DOI] [PubMed] [Google Scholar]
- 65.Kamps, M. P., and Sefton, B. M. (1986) Mol. Cell. Biol. 6 751–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.D'Oro, U., Sakaguchi, K., Appella, E., and Ashwell, J. D. (1996) Mol. Cell. Biol. 16 4996–5003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Viola, A., and Gupta, N. (2007) Nat. Rev. Immunol. 7 889–896 [DOI] [PubMed] [Google Scholar]
- 68.Maroun, C. R., and Julius, M. (1994) Eur. J. Immunol. 24 959–966 [DOI] [PubMed] [Google Scholar]
- 69.Bonnard, M., Maroun, C. R., and Julius, M. (1997) Cell. Immunol. 175 1–11 [DOI] [PubMed] [Google Scholar]
- 70.Yeh, E. T., Kamitani, T., and Chang, H. M. (1994) Semin. Immunol. 6 73–80 [DOI] [PubMed] [Google Scholar]
- 71.Shenoy-Scaria, A. M., Gauen, L. K., Kwong, J., Shaw, A. S., and Lublin, D. M. (1993) Mol. Cell. Biol. 13 6385–6392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lallemand-Breitenbach, V., Quesnoit, M., Braun, V., El Marjou, A., Pous, C., Goud, B., and Perez, F. (2004) J. Biol. Chem. 279 41168–41178 [DOI] [PubMed] [Google Scholar]
- 73.Resh, M. D. (2006) Sci. STKE 2006, RE14. [DOI] [PubMed]
- 74.Resh, M. D. (2004) Subcell. Biochem. 37 217–232 [DOI] [PubMed] [Google Scholar]
- 75.Porter, J. A., Young, K. E., and Beachy, P. A. (1996) Science 274 255–259 [DOI] [PubMed] [Google Scholar]
- 76.Morrow, I. C., and Parton, R. G. (2005) Traffic 6 725–740 [DOI] [PubMed] [Google Scholar]
- 77.Kimura, A., Baumann, C. A., Chiang, S. H., and Saltiel, A. R. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 9098–9103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Reynolds, A. B., Vila, J., Lansing, T. J., Potts, W. M., Weber, M. J., and Parsons, J. T. (1987) EMBO J. 6 2359–2364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cartwright, C. A., Eckhart, W., Simon, S., and Kaplan, P. L. (1987) Cell 49 83–91 [DOI] [PubMed] [Google Scholar]
- 80.Werlen, G., and Palmer, E. (2002) Curr. Opin. Immunol. 14 299–305 [DOI] [PubMed] [Google Scholar]
- 81.Horejsi, V. (2003) Immunol. Rev. 191 148–164 [DOI] [PubMed] [Google Scholar]
- 82.Pawson, A. J., Maudsley, S. R., Lopes, J., Katz, A. A., Sun, Y. M., Davidson, J. S., and Millar, R. P. (2003) Endocrinology 144 3860–3871 [DOI] [PubMed] [Google Scholar]
- 83.Roess, D. A., and Smith, S. M. (2003) Biol. Reprod. 69 1765–1770 [DOI] [PubMed] [Google Scholar]