

Abstract
Histone deacetylases (HDACs) regulate chromatin status and gene expression, and their inhibition is of significant therapeutic interest. To date, no biological substrate for class IIa HDACs has been identified, and only low activity on acetylated lysines has been demonstrated. Here, we describe inhibitor-bound and inhibitor-free structures of the histone deacetylase-4 catalytic domain (HDAC4cd) and of an HDAC4cd active site mutant with enhanced enzymatic activity toward acetylated lysines. The structures presented, coupled with activity data, provide the molecular basis for the intrinsically low enzymatic activity of class IIa HDACs toward acetylated lysines and reveal active site features that may guide the design of class-specific inhibitors. In addition, these structures reveal a conformationally flexible structural zinc-binding domain conserved in all class IIa enzymes. Importantly, either the mutation of residues coordinating the structural zinc ion or the binding of a class IIa selective inhibitor prevented the association of HDAC4 with the N-CoR·HDAC3 repressor complex. Together, these data suggest a key role of the structural zinc-binding domain in the regulation of class IIa HDAC functions.
Eukaryotic histone deacetylases (HDACs)3 were initially discovered in transcriptional repressor complexes, where they deacetylate acetylated lysines in histone N termini (1). Histone deacetylation is thought to promote the tight histone-DNA interactions characteristic of nucleosomes in condensed chromatin, where access to DNA for processes including transcription, DNA repair, recombination, or replication is limited. More recently, HDACs have also been shown to deacetylate nonhistone proteins, predominantly transcription factors (2-5). Due to their critical role in regulation of chromatin structure and gene expression, HDACs have become major drug targets. Indeed, several HDAC inhibitors have entered clinical trials (6, 7), and one compound, suberoylanilide hydroxamic acid, was recently approved for the treatment of cutaneous T-cell lymphomas.
There are currently 18 known mammalian HDACs, grouped into four classes based on sequence similarity (8). The class I, II, and IV HDACs are zinc-dependent enzymes, whereas class III HDACs (SIRT1-SIRT7) require NAD+ for activity. The class I HDACs (HDAC1, -2, -3, and -8) are ∼400 residues long and are generally nuclear, whereas the class II HDACs (HDAC4, -5, -6, -7, -9, and -10) exhibit nucleocytoplasmic shuttling. In turn, class II HDACs are subdivided into class IIa (HDAC4, -5, -7, and -9) and IIb (HDAC6 and -10). Class IIa enzymes are characterized by the presence of an N-terminal extension of ∼600 residues with distinct regulatory and functional properties, whereas the class IIb enzymes contain two catalytic domains (9-11). Class IV currently includes only HDAC11, which shares greatest sequence similarity to the class I enzymes.
HDACs have been extensively studied, but the function of the class IIa enzymes is only partially understood. The most well characterized roles of class IIa HDACs are as transcriptional repressors, often resulting in the suppression of cellular hypertrophy (reviewed recently in Refs. 12 and 13). For example, HDAC4 was shown to regulate chondrocyte hypertrophy and skeletogenesis, an activity at least partially exerted through a repressive interaction with the transcription factor RUNX2 (14, 15). Although RUNX2 has been implicated as a substrate for deacetylation by HDAC4 and -5 (16), the involvement of a class IIa deacetylase activity has not been demonstrated. Further, HDAC4 was shown to form a complex with HDAC3 via N-CoR, but therein the catalytic domain of HDAC4 did not contribute to the enzymatic activity toward acetylated histone peptides (17). Consistently, we recently reported that class IIa HDACs possess only weak, but measurable, lysine deacetylation activity in vitro (18).
For a long time, structural information on zinc-dependent HDAC catalytic domains has been limited to a bacterial HDAC-like protein (HDLP) resembling a class I enzyme (19), a eukaryotic class I enzyme (HDAC8) (20, 21), and a bacterial HDAC-like amidohydrolase (HDAH) resembling a class IIb enzyme (22, 23). However, recent progress includes the structures of a catalytically dead HDAC8 active site mutant bound to an acetyl-lysine peptidic substrate (24) and the class IIa HDAC7 catalytic domain (25).
Here we report the crystal structures of the HDAC4 catalytic domain (HDAC4cd) and of an active site mutant (H976Y) with enhanced activity toward acetylated lysines, both in complexes with two different inhibitors. Moreover, we present the structure of an inhibitor-free HDAC4cd. These structures reveal a flexible structural zinc-binding domain conserved in all class IIa enzymes. Mutagenesis, activity, and binding data suggest a key role of this domain in substrate recognition and in the association of HDAC4 with the HDAC3·N-CoR co-repressor complex. In addition, the structures and accompanying activity data provide the molecular basis for the intrinsically low enzymatic activity of class IIa HDACs toward acetylated lysines and for the design of class-selective HDAC inhibitors.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification—The human HDAC4 catalytic domain (residues Thr648-Thr1057) was produced in Escherichia coli BL21 cells using the pETM-11 vector (EMBL Heidelberg) and was purified via a His6 tag later removed by a solubility-enhanced TEV protease (26). The complete expression and purification protocol is given in the supplemental materials.
Deacetylation Assays—Deacetylation assays were performed as reported previously using either the “Fluor de Lys” acetamide substrate (BIOMOL International) or the trifluoroacetamide substrate (18), which both comprise an acetylated lysine side chain and a methyl-coumarin group. Deacetylation sensitizes the substrate so that treatment with the “Fluor de Lys” developer produces a fluorophore.
Crystallization of the HDAC4 Catalytic Domain—Prior to crystallization screening of protein-inhibitor complexes, the HDAC4cd was incubated for 30 min at 4 °C with a 3-fold molar excess of inhibitor dissolved in DMSO. The synthesis of inhibitors used has been reported elsewhere (27). Crystals of protein-inhibitor complexes were obtained by the hanging drop vapor diffusion technique. Drops were mixed to contain 1 μl each of protein-inhibitor complex at 6-8 mg/ml and reservoir solution containing 1.6 m ammonium sulfate, 0.1 m MES buffer, pH 6.5, 10% dioxane, and 1 mm dithiothreitol. Crystals of HDAC4cd-inhibitor complexes grew after 2 days of equilibration at 18 °C. The wild type (WT) crystals belonged to the C2221 space group with one molecule per asymmetric unit, whereas the cysteine-mutant crystals belonged to the P21 space group with two molecules per asymmetric unit. The two crystal forms have an almost identical crystal packing; the noncrystallographic 2-fold axis relating two HDAC4cd molecules in the P21 space group becomes crystallographic in the C2221 space group (Table S1).
After preincubating the protein with a molar excess of substrate for 30 min at 4 °C, crystals of the inhibitor-free “gain of function mutant” (GOF) HDAC4cd protein were obtained in 6-8 h. Crystals were not obtained if substrate was omitted. The inhibitor-free crystals grew in 0.1 m Hepes, pH 7.5, 18% polyethylene glycol 10,000, 1 mm dithiothreitol.
Data Collection, Structure Determination, and Refinement—X-ray diffraction data were collected at the ID14, BM30, and ID29 beam lines at ESRF (Grenoble, France) from single crystals flash-frozen in liquid nitrogen. Data were indexed and integrated with MOSFLM (28). The CCP4 program suite (29) was used for further data processing and analysis.
The structure of WT HDAC4cd-trifluoromethylketone (TFMK) complex was solved by the single anomalous dispersion method. A 2.5 Å resolution data set was collected at the absorption peak for zinc as determined by x-ray fluorescence. The position of the zinc ion was located in an anomalous Patterson map. A combination of single anomalous dispersion phasing and electron density modification as implemented in CNX (Accelrys Software Inc.) yielded an interpretable electron density map. Several cycles of model building in Coot (30) and positional restrained refinement in REFMAC (29) allowed building of the entire model, excepting the N- and C-terminal regions Thr648-Pro650 and Glu1051-Thr1057.
The structures of the complex with the HA and of the other HDAC4cd mutants were solved by molecular replacement with Phaser (29), using as a search model the refined WT HDAC4cd-TFMK complex after removal of the inhibitor, side chains of active site residues, and loops containing residues involved in binding the structural zinc ion. When the data resolution was better than 2.5 Å, the electron density maps were improved using Arp/Warp in Molrep mode (31). All of the structures were refined using restrained positional refinement, isotropic B-factor refinement, and bulk solvent correction with a maximum likelihood target as implemented in REFMAC. Structure quality and Ramachandran statistics were assessed using Molprobity (32); images were prepared with PyMol (DeLano Scientific LLC).
Iodoacetamide Alkylation Protection Experiments—Alkylation reaction experiments were performed by incubating 20 μm WT HDAC4cd in buffer containing 25 mm Hepes, pH 7.5, 150 mm KCl, 0.1% n-octyl-β-glucopyranoside with 200 μm iodoacetamide in the dark for 1 h at room temperature. Following the reaction, excess iodoacetamide was removed by dialysis. Prior to analysis by mass spectrometry, 10-μg aliquots of the alkylated HDAC4 samples and nonalkylated HDAC4 controls were incubated overnight at 37 °C with 0.4 μg of trypsin in 100 mm ammonium bicarbonate, pH 8.0, in 30 μl. Subsequently, 1 μl of each sample was mixed with 1 μl of matrix solution (cyano-4-hydroxycinnamic acid in CH3CN plus 0.1% trifluoroacetic acid (10 mg/ml)) and loaded directly onto the matrix for analysis by matrix-assisted laser desorption ionization time-of-flight spectrometry.
In the alkylated and digested WT HDAC4cd sample, we observed that the peptide containing both Cys667 and Cys669 had reacted with two molecules of iodoacetamide. Likewise, a peptide containing Cys751 had reacted with one molecule of iodoacetamide. We did not identify a Cys667/Cys669-containing peptide that had reacted with only one alkylated group. In addition, we found that cysteines buried in the structure were not alkylated. These results suggest that in solution, Cys667 and Cys669 are both exposed to the alkylating agent, and thus neither is protected by binding to the zinc ion.
Mammalian Cell Expression of Full-length HDAC4-FLAG—HEK293 cells, maintained in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum, were transfected using Lipofectamine 2000 (Invitrogen) with mammalian pcDNA3 expression plasmids encoding full-length, C-terminal FLAG-tagged, wild type or doubly mutated (C669A/H675A) HDAC4 proteins. Cells were harvested after 24 h, as described previously (18). The double mutation was introduced using the QuikChange mutagenesis kit (Stratagene).
To investigate the effects of a cell-permeable HDAC4 inhibitor, HEK293 cells were stably transfected to enable the doxycyclin-inducible expression of full-length HDAC4-FLAG, as described previously (33). HEK293 cells transfected with an empty vector and induced with doxycyclin were used as a control. Cells were maintained at 37 °C, 5% CO2 in DMEM supplemented with 10% fetal bovine serum and, when 90% confluent, were induced with 1 μg/ml doxycyclin 20 h prior to the addition of inhibitor. A cell-permeable trifluoromethylketone inhibitor closely resembling the co-crystallized TFMK was added to the medium at 10 μm final concentration for 4 h, after which cells were harvested.
Immunoprecipitation of HDAC4-FLAG and Analysis of Its Association with N-CoR·HDAC3—All cells were harvested in ice-cold PBS, homogenized, and sonicated in lysis buffer containing 20 mm Hepes, pH 7.9, 0.25 mm EDTA, 10% glycerol, 250 mm KCl, 0.5% Nonidet P-40, 1 mm phenylmethylsulfonyl fluoride, and a protease inhibitor mixture (Roche Applied Science), followed by a 1-h incubation at 4 °C. Soluble whole-cell extracts were obtained by centrifugation. HDAC4-F protein complexes were immunoprecipitated on anti-FLAG M2 affinity gel (Sigma) and eluted in buffer containing 50 mm Hepes, pH 7.4, 100 mm KCl, 5% glycerol, and 0.01% Triton X-100 in the presence of 100 mg/ml 3× FLAG peptide (Sigma). Purified HDAC4-F wild type and mutant protein concentrations were normalized via a Western blot developed using an anti-FLAG horseradish peroxidase-conjugated antibody. Finally, the co-immunoprecipitation of endogenous HDAC3 with HDAC4-FLAG was evaluated by Western blotting using a rabbit anti-HDAC3 antibody and developed using an anti-rabbit horseradish peroxidase-conjugated antibody (Upstate Biotechnology).
RESULTS
Identification of Novel HDAC4 Inhibitors—We previously found that HDAC4 had only marginal activity against acetylated lysine-containing peptides, whereas an active site mutant (H976Y), which we refer to as the GOF HDAC4, was active on these molecules (18). Moreover, we discovered one molecule, a trifluoroacetamide, on which both full-length and the catalytic domain of HDAC4 were highly active when tested in the same deacetylation assay previously reported. In contrast, this compound was a poor substrate for class I and IIb HDACs, including HDAC1, -3, and -6 (18) and the GOF HDAC4cd (Fig. 1, A and B). Within a medicinal chemistry effort, we have now identified two compounds that inhibit HDAC4 in the hydrolysis of the trifluoroacetamide substrate with IC50 values in the nanomolar range (Fig. 1, B and C). The two inhibitors have identical scaffolds but differ in their zinc-chelating groups: a hydroxamic acid (HA) or a TFMK. The inhibitory potency of these molecules on the GOF HDAC4 mutant was also evaluated using an acetamide version of the same substrate (Fig. 1, B and C).
FIGURE 1.

Catalytic activity and inhibition of HDAC4 wild type and mutants. A, the full-length WT HDAC4 is highly active on the trifluoroacetamide substrate, whereas the H976Y mutant is inactive. RFU, relative fluorescence units. B, top, the substrate, comprising an acetylated lysine side chain (where R represents CH3) or the trifluoroacetamide version (where R represents CF3). Bottom, the inhibitor; R represents CF3 for the trifluoromethylketone (TFMK) or NH-OH for the hydroxamic acid (HA). C, potency of TFMK and HA inhibitors for WT and GOF HDAC4cd proteins.
Structure Determination of Inhibitor-bound HDAC4 Catalytic Domains—To gain structural insights into class IIa HDACs and the mechanism of their inhibition, we crystallized the WT and GOF HDAC4cds in complex with the TFMK and HA inhibitors. The four complexes crystallized in the same conditions and gave similar crystals (see Table S1). We initially solved the structure of the WT HDAC4cd-TFMK complex by the single anomalous dispersion method. This structure was then used as the starting model to solve and refine all the structures reported here (see “Experimental Procedures”).
Global Structure Description—The HDAC4cd-inhibitor complex structures are essentially identical (Cα coordinate root mean square deviation ∼0.3 Å) except for differences in the interactions with inhibitors. The HDAC4cd core exhibits a layered α-β-α fold, with a central parallel β-sheet of eight β-strands (Fig. 2A) and overall is similar to HDAC8 (Cα coordinate root mean square deviation 2.2 Å). The catalytic zinc ion, located in the center of the catalytic domain, is bound by the inhibitors (see Fig. S1). In addition, as observed in HDAC8, two potassium ions are proximal to the active site, coordinated by residues largely conserved among HDACs (Figs. 2A and 3) (20, 21).
FIGURE 2.
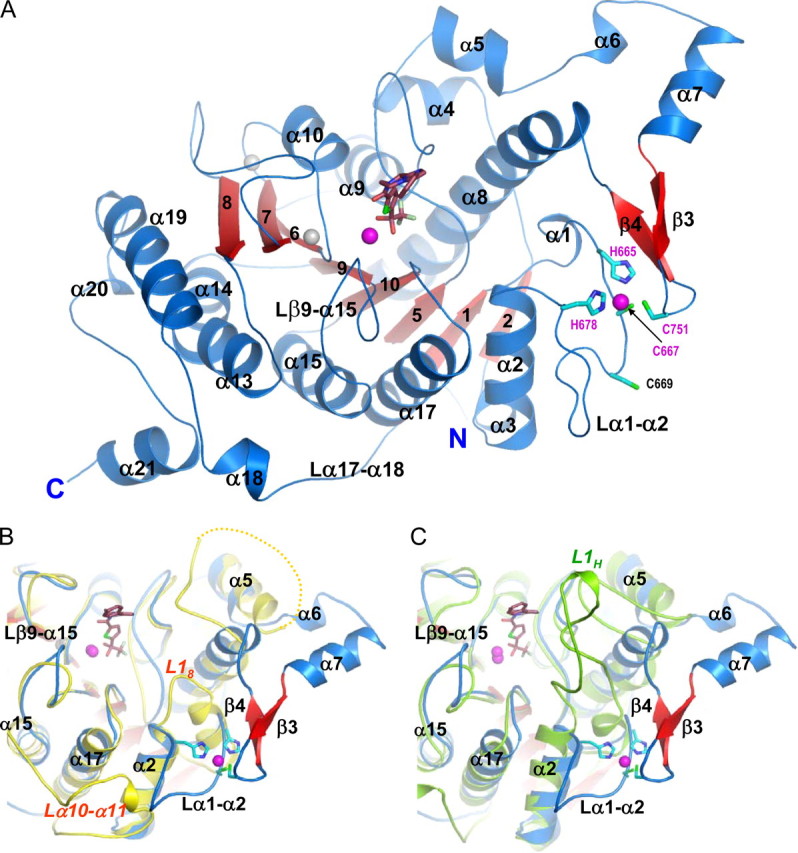
The inhibitor-bound HDAC4 catalytic domain. A, HDAC4cd bound to the TFMK inhibitor (sticks); Protein Data Bank code 2VQJ. Zinc and potassium ions are shown as magenta and gray spheres, respectively. The four residues coordinating the structural zinc ion are shown (sticks). B, a superposition of TFMK-bound HDAC4cd (blue/red) and HDAC8 (yellow). Each α-helix and β-strand of HDAC4 has a structural counterpart in HDAC8, except for α6, α7, β3, β4, and α18. The C terminus of HDAC8 coincides with the end of α19, such that α20 and α21 are also absent from HDAC8. Loop L18 of HDAC8 is much shorter than loop α1-α2 of HDAC4. The yellow dotted line indicates HDAC8 residues missing due to disorder. C, a superposition of TFMK-bound HDAC4cd (blue/red) and HDAH (green). Loop L1H of HDAH is much longer than loop α1-α2 of HDAC4. Both L1H and L18 loops turn inward toward the catalytic site. The major differences with HDAH occur in this loop and in the α6-α7-β3-β4 region and in the longer C terminus of HDAC4.
FIGURE 3.
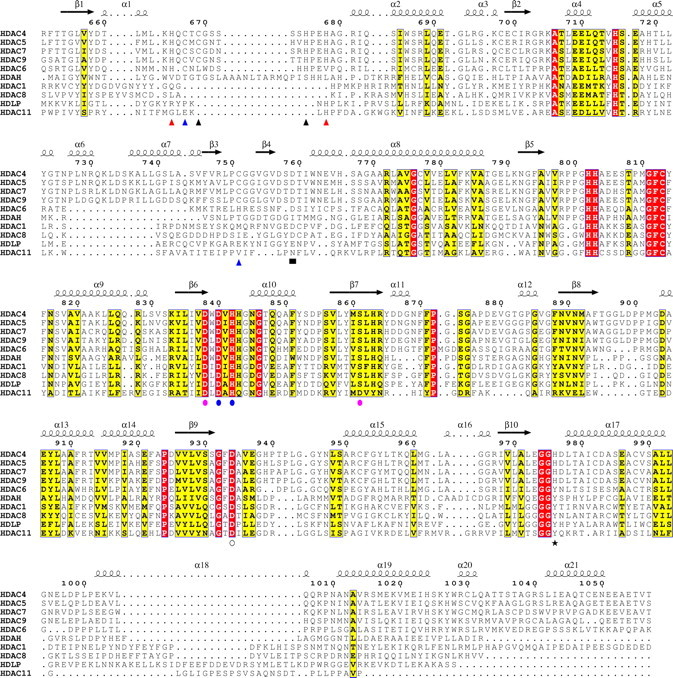
Multiple sequence alignment of HDAC catalytic domains. Shown is a structure-based multiple sequence alignment, made using the ESPript program, of HDACs from classes I, IIa, IIb, and IV and the bacterial HDAC-like proteins, HDLP and HDAH. Secondary structure elements are shown for the TFMK-bound HDAC4cd structure; numbering is for wild type HDAC4. Blue triangles, binds structural zinc ion in inhibited and inhibitor-free structures; red triangles, only binds structural zinc ion in the inhibited protein; black triangles, only binds structural zinc ion in the inhibitor-free protein; blue circles, binds both the catalytic zinc ion (via side chains) and potassium ion-1 (via backbone); open circle, binds the catalytic zinc ion (via side chain); pink circles, binds only potassium ion-1 via side chains (Asp838 and Ser861) or backbone (Leu862); black star, “gain-of-function” residue; black square, mutation to Ala reduces activity.
A Conserved Class IIa Structural Zinc-binding Domain—A novel feature distinguishing HDAC4cd from class I HDACs is the presence of a structural zinc ion holding together two protein segments that depart from the core of the molecule (Fig. 2). The first segment forms a 17-amino acid loop (Lα1-α2) and contributes three zinc ligands: His665, Cys667, and His678 (Fig. 2 and Table S2). The second segment is a 35-residue insertion (α6-α7-β3-β4) forming a helix-turn-helix motif followed by a β-hairpin containing at its tip a fourth zinc ligand, Cys751. Notably, these four zinc ligands are strictly conserved in all class IIa enzymes but are absent in other HDACs (Fig. 3). The presence of the structural zinc-binding domain and additional differences in loop structures result in HDAC4 having a more accessible active site than HDAC8 or HDAH (Fig. 2, A-C). Moreover, the water-filled tunnel proposed to enable release of the acetate reaction product in HDAC8, HDLP, and HDAH is not formed (19, 21, 22).
Analysis of the crystal packing revealed that an intermolecular disulfide bond had formed between Cys669, lying within the structural zinc-binding domain (Fig. 2), and Cys700 from a neighboring molecule. To exclude the possibility that disulfide formation might have affected the interaction of the inhibitors with active site residues, we also solved the structures of WT and GOF HDAC4cd containing either a C669A/C700A double mutation or a C700A single mutation in complex with the inhibitors. The structures of the mutants were identical to those of the corresponding nonmutated WT and GOF proteins except for the structural zinc-binding domain that was now disordered (i.e. no electron density was observed). Therefore, we conclude that in the HDAC4cd-inhibitor complexes, this domain is flexible and that the intermolecular disulfide bond had previously locked it in place. Hereafter, for each of the WT and GOF inhibitor complexes, only the higher resolution structures are described, independently of the presence of cysteine mutations (Table S1).
Analyses of the Inhibitor-bound HDAC4 Active Sites and Comparison with Class I and IIb HDACs—In the HDAC4cd structures, the inhibitors occupy the same position but establish different interactions with the zinc ion. In the complex with the TFMK, the inhibitor is in its hydrated form and employs both oxygens for coordinating the catalytic zinc ion (Fig. 4A and Table S2). Notably, NMR analyses showed that the TFMK is already 70-90% hydrated in solution (27). In contrast, in the WT HDAC4cd-HA complex, the inhibitor binds the catalytic zinc ion in a monodentate fashion, with its carbonyl oxygen replacing one of the TFMK-OH groups and a zinc-bound water molecule (W1 in Fig. 4A and Table S2) substituting the other.
FIGURE 4.
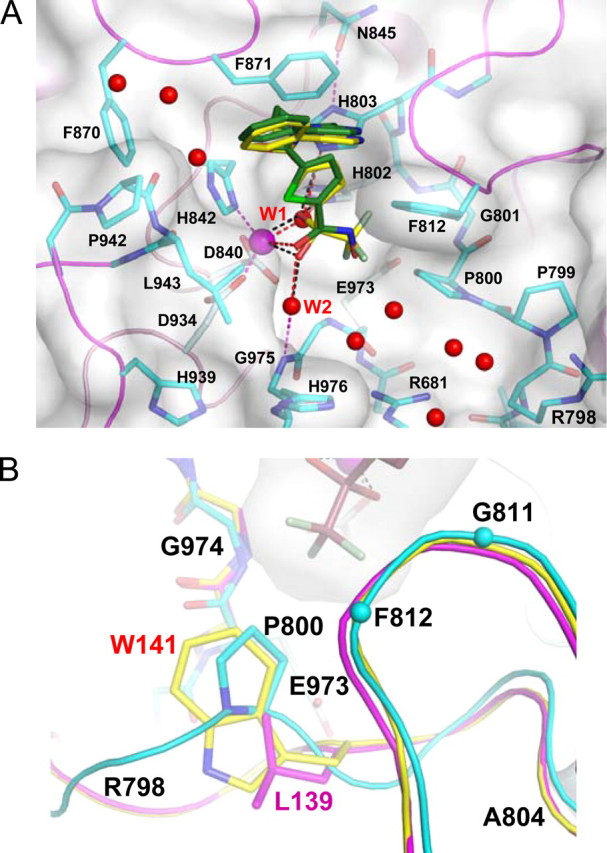
Interactions of HDAC4cd with inhibitors. A, interactions of the TFMK (yellow carbons) and HA (green carbons) with HDAC4; the complex structures have PDB codes 2VQJ and 2VQM, respectively. Red spheres, water molecules. The surface around the protein is shown for the TFMK-bound HDAC4cd. B, superposition of HDAC4cd (cyan) bound to TFMK (sticks and surface) with HDAC8 (yellow) and homology-modeled HDAC1 (magenta). Residues surrounding the trifluoro group are labeled. Cyan spheres, Cα atoms.
In the two complexes, the trifluoromethyl group of the TFMK and the hydroxyl group of the HA point toward a pocket formed by Pro800, Gly811, Phe812, Glu973, and Gly974. In particular, the trifluoromethyl group makes van der Waals contacts with the Pro800 pyrrolidine ring (Fig. 4). The residues composing this pocket are largely conserved among class II HDACs. However, a similar pocket is also formed in HDAC8 (class I) despite the sequence differences and lack of proline residues (Fig. 3). This is achieved through replacement of the HDAC4 Pro800 side chain with the indole ring of HDAC8 Trp141 and a different arrangement of the HDAC8 polypeptide chain in the region corresponding to HDAC4 residues Arg798-Gly801 (Fig. 4B). This region is relatively well conserved among class I HDACs, but Trp141 is replaced by Leu in HDAC1, HDAC2, and HDAC3, probably resulting in a larger pocket (Figs. 3 and 4B).
An additional difference between HDAC8, HDLP, and HDAH and the inhibitor-bound HDAC4cd structures is that in the latter, one side of the inhibitor is solvent-exposed. In particular, a pocket, only partially filled by a water molecule (W2 in Fig. 4A), is present between the zinc-bound carbonyl/-OH group of the inhibitors and the main chain of Gly975-His976. This space is entirely occupied by the side chain of Tyr306 (HDAC8 numbering) in HDAC8, HDLP, and HDAH, whereas the side chain of the corresponding residue in HDAC4, His976, points toward the solvent and away from the inhibitor (Figs. 4A and 5, A and B). These differences in the HDAC active sites could be exploited for the design of class-selective inhibitors.
FIGURE 5.

The active sites of HDAC4 and HDAC8. A, the active site of HDAC8 (yellow side chains) bound to a hydroxamic acid inhibitor (light brown) from Protein Data Bank entry 1W22. B, WT HDAC4cd with bound TFMK (yellow carbons) (Protein Data Bank code 2VQQ) and superposed HA (green carbons) (Protein Data Bank code 2VQM). The active site closely resembles HDAC8. C, the active site of GOF HDAC4cd with HA bound (Protein Data Bank code 2VQV); Tyr976 adopts the inward, class I-like conformation. D, the active site of GOF HDAC4cd with TFMK bound (Protein Data Bank code 2VQO); Tyr976 adopts an outward conformation.
Structural Basis for the Low Enzymatic Activity of Class IIa HDACs toward Acetylated Lysines—In class I and IIb HDACs, the hydroxyl group of Tyr306 has been proposed to stabilize the transition state by hydrogen bonding with the oxyanion intermediate (19). Instead, in HDAC4cd, the side chain of the corresponding residue, His976, is rotated away from the active site, and a water molecule, W2, forms hydrogen bonds with the backbone nitrogen of Gly975 and an oxygen atom of the inhibitor (2.43 Å to OH1 of the TFMK, 3.0 Å to the carbonyl of the HA) (Fig. 5, A and B). The latter interaction is reminiscent of the one established in HDAC8 by the Tyr306 hydroxyl group with the carbonyl oxygen of the HA inhibitor (20, 21, 24). Thus, in HDAC4, W2 could form a similar bond with the reaction intermediate and contribute, albeit less efficiently, to its stabilization and to catalysis.
The “outward” orientation of the His976 side chain in the WT HDAC4cd structures and a limited stabilization of the transition state may explain the low activity of class IIa HDACs on acetylated lysines. This hypothesis is supported by the 3.3 Å resolution structure of the GOF HDAC4cd-HA complex showing repositioning of the Tyr976 side chain into the “inward” HDAC8-like orientation (Fig. 5C and Fig. S3). In this structure, the position of the Tyr976 hydroxyl group exhibits angles and distances (∼2.9 Å) enabling hydrogen bonding with the HA carbonyl oxygen, thus explaining the class I-like activity levels of this mutant.
The structure of the GOF HDAC4cd-TFMK complex (Table S1) provides further insight into our activity data and the observed inhibition profiles (Fig. 1). In this complex, the Tyr976 side chain is rotated away from the active site, like the corresponding His976 in the WT HDAC4cd structures, and the water molecule W2 forms a hydrogen bond with the inhibitor (Fig. 5, B and D). The different orientation of Tyr976 in the HA- and TFMK-bound structures reflects the different steric effects imposed by the two inhibitors. Indeed, a model based on the structure of the GOF HDAC4cd-TFMK complex suggests that the hydroxyl group of Tyr976 in the inward position would be too close (<2.0 Å) to the trifluoromethyl group of the inhibitor. A steric hindrance between the Tyr976 hydroxyl group and the trifluoromethyl group is in agreement with the low enzymatic activity of GOF HDAC4 and of class I HDACs toward the trifluoroacetamide substrate (Fig. 1) (18), despite these proteins being highly active on the canonical acetylated lysine substrate. Of note, in a recent structure of HDAH bound to a trifluoromethylketone inhibitor, the corresponding tyrosine is found in the inward position pointing toward the inhibitor, which occupies two alternative positions (23). Differences in length and flexibility of neighboring loops Lα1-α2 and Lβ9-α15 (Fig. 2, B and C) probably constrain this tyrosine rotamer in the HDAH enzyme but not in the GOF HDAC4cd.
Active Site Binding and the Conformation of the Structural Zinc-binding Domain—To obtain additional insight into the HDAC4 enzymatic mechanism, we attempted to crystallize WT and GOF HDAC4cd proteins in the presence of substrates (Fig. 1B). Only the GOF HDAC4cd incubated with the trifluoroacetamide substrate yielded crystals that diffracted to sufficient resolution (Table S1). Although the crystals grew only after preincubation with the substrate, the resulting structure showed no electron density for the substrate, suggesting that it had been hydrolyzed, thus yielding an inhibitor-free structure.
When compared with the inhibitor-bound structures, the inhibitor-free structure shows considerable differences in the conformation of the structural zinc-binding domain (Figs. 6, A and B, and S2). In particular, the two loops harboring the residues coordinating the structural zinc ion moved 10-20 Å toward the active site, resulting in a much greater similarity to the substrate-bound HDAC8 structure (Fig. 6C) (24). This “closed” conformation of the structural zinc-binding domain also results in the formation of the tunnel proposed to allow product release in other HDACs and not formed in the inhibitor-bound HDAC4cd structures.
FIGURE 6.
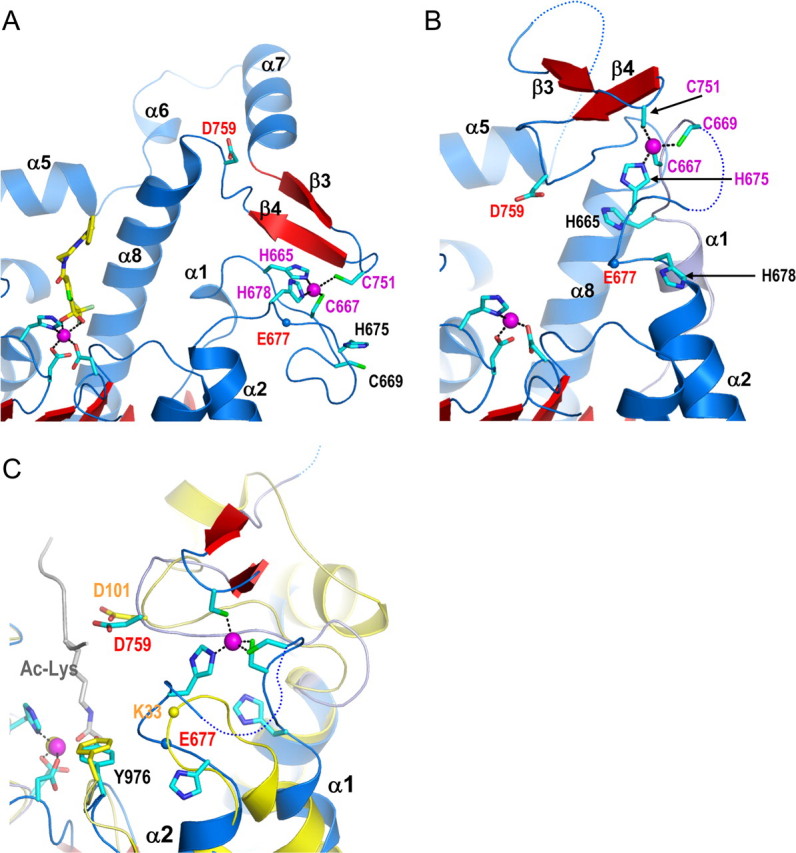
Conformational flexibility of the structural zinc-binding domain and role in substrate binding. A, the active site of TFMK-bound WT HDAC4cd showing the original structural zinc-binding scheme (cyan sticks). B, the new conformation of the zinc-binding region in the inhibitor-free GOF HDAC4cd structure (Protein Data Bank code 2VQW). C, a structural comparison of the active sites of the inhibitor-free GOF HDAC4cd structure (blue/red) and HDAC8 (yellow) bound to an acetylated lysine peptidic substrate (gray). The blue and yellow spheres representing the Cα atoms of HDAC4 Glu677 and HDAC8 Lys33, respectively, illustrate how this inhibitor-free HDAC4 structure has a more closed active site due to the new conformation of the α1-α2 loop.
The comparison of the inhibitor-free and inhibitor-bound HDAC4cd structures provides an explanation for the differences in the conformation of the structural zinc-binding domain (Fig. 6). In fact, superposition of the two structures reveals that in the “closed” conformation, the inhibitor would clash with the structural zinc binding domain, thus promoting the “open” conformation. In addition, the structural comparison reveals a switch in the ligation scheme of the structural zinc ion (Fig. 6, A and B, and Table S2) with His665 and His678 in the inhibitor-bound structure being replaced by Cys669 and His675 in the inhibitor-free structure (Fig. 3). Notably, the latter two residues are also conserved only in class IIa HDACs. This variability in the structural zinc ion coordination is supported by thiol alkylation experiments in which we observed that even in mild reaction conditions, all of the cysteines involved in zinc binding as well as those exposed to the solvent were alkylated, whereas the cysteines buried in the structure did not react (see “Experimental Procedures”). A loose coordination of the structural zinc ion may be required to allow conformational flexibility of the structural zinc-binding domain and might play a role in the regulation of HDAC4 function.
Role of the HDAC4 Structural Zinc-binding Domain in Catalytic Activity and Binding to the HDAC3·N-CoR Co-repressor Complex—A consequence of the refolding of the structural zinc binding domain, as observed in the absence of inhibitor, is the positioning of Asp759 at the border of the active site (Fig. 6B). Moreover, in this inhibitor-free structure, Asp759 forms a hydrogen bond with the backbone nitrogen of a neighboring crystallographically related molecule. This interaction is reminiscent of the one established between the structurally conserved HDAC8 residue Asp101 and the backbone nitrogens of a peptidic substrate (Fig. 6C) (24). Similarly to HDAC8, mutation of Asp759 to Ala in the GOF HDAC4cd resulted in a large decrease in deacetylase activity, indicating an involvement of this residue in substrate recognition (Fig. 7A).
FIGURE 7.

Role of the HDAC4 structural zinc-binding domain. A, activities on the acetamide substrate of the GOF HDAC4cd protein, the GOF HDAC4cd D759A mutant, and the GOF HDAC4cd C669A/H675A mutant. B, full-length HDAC4 proteins (WT and C669A/H675A mutant; top) were immunopurified and probed by Western blotting for HDAC3 (bottom). HDAC3 did not co-purify with the mutant protein. C, HDAC3 did not co-purify with full-length WT HDAC4 when expressed in the presence of a cell-permeable class IIa-specific trifluoromethylketone inhibitor, as shown by Western blotting.
To better define the role of the structural zinc-binding domain, we mutated into alanine Cys669 and His675, two ligands of the structural zinc ion in the inhibitor-free GOF HDAC4cd. These mutations are expected to affect the coordination of the structural zinc ion and ultimately destabilize the structure of the entire subdomain. The resulting double mutant protein was soluble but inactive (Fig. 7A). Likewise, the GOF HDAC4cd C669A/C700A double mutant showed reduced activity, whereas the C700A single mutant showed an activity equal to that of GOF HDAC4cd (data not shown). These results are in line with our structural studies. In fact, Cys700 is solvent-exposed and far away from both the active site and the structural zinc-binding domain in both the inhibitor-bound and inhibitor-free HDAC4cd structures, and therefore its mutation to alanine is expected not to have an affect on the HDAC4 catalytic activity. In contrast, Cys669 is involved in zinc chelation in the inhibitor-free form of the enzyme, and, similarly to the double mutant C669A/H675A, the single mutation C669A is likely to affect the coordination of the structural zinc ion. Together, these data support an involvement of the zinc-binding domain in substrate recognition and/or enzyme activity.
It has been previously shown that HDAC4 forms a multiprotein complex with the N-CoR·HDAC3 co-repressor complex (17). To explore the role of the structural zinc-binding domain on the association of HDAC4 with the HDAC3·N-CoR complex, we transfected HEK293 cells with either full-length FLAG-tagged wild type HDAC4 or with the C669A/H675A HDAC4 double mutant. The proteins were then immunoprecipitated, and Western blotting analysis was used to reveal additionally associated, co-purified proteins. In these experiments, we found that the endogenous HDAC3·N-CoR complex co-purified with wild type HDAC4 but not with the double mutant protein (Fig. 7B), indicating that a properly folded structural zinc-binding domain is required for the formation of the repressor complex.
The HDAC4cd structures reported here suggest that the zinc-binding domain is flexible and that binding of some active site inhibitors and/or substrates may affect its conformation. Thus, we also explored if, similarly to the C669A/H675A double mutant, an active site inhibitor may also interfere with the HDAC4·HDAC3·N-CoR interaction. Indeed, addition to cells of a cell-permeable class IIa selective structural analogue of the TFMK inhibitor prevented the formation of the complex (Fig. 7C). Therefore, some active site inhibitors may inhibit both the HDAC4 catalytic activity and interfere with the recruitment of the HDAC3·N-CoR co-repressor complex.
DISCUSSION
Class IIa HDACs have been implicated in several biological processes. To date, however, no histone or protein substrates have been identified for class IIa HDACs, and only a low catalytic activity toward acetylated lysines has been reported (18). Here we describe the inhibitor-bound and inhibitor-free structures of the class IIa HDAC4 catalytic domain and of an activesite mutant with enhanced catalytic activity toward acetylated lysine substrates. The structures and accompanying mutagenesis data provide insights into class IIa HDAC catalytic activity, design of class-selective HDAC inhibitors, and regulation of HDAC4 function.
The HDAC4cd structures reported here revealed the presence of an unexpected, class IIa-specific, structural zinc-binding domain. This domain adopted a different conformation in the inhibitor-free (“closed”) and in the inhibitor-bound (“open”) forms of the enzyme. In the initial structures of the HDAC4cd-inhibitor complexes, the zinc-binding domain was locked in the “open” position by an intermolecular disulfide bond between Cys669, in the zinc-binding domain, and Cys700 from a neighboring molecule. However, mutation of Cys700 to Ala resulted in a lack of electron density for the zinc-binding domain, presumably due to its increased mobility, demonstrating that this domain is intrinsically conformationally flexible.
The inhibitor-free structure resembles the substrate-bound HDAC8 and is likely to represent the active conformation of the enzyme. In particular, Asp759, which is located far from the active site in the HDAC4cd-inhibitor complex, is positioned at the rim of the active site in the inhibitor-free structure. The latter position of Asp759 is equivalent to that of Asp101 in HDAC8, a residue implicated in positioning the substrate (24). Moreover, similarly to the D101A mutant in HDAC8, mutation of Asp759 to Ala results in a strong decrease in HDAC4 enzymatic activity. Of note, mutation into alanine of two of the ligands of the structural zinc ion in the inhibitor-free structure, Cys669 and His675, also resulted in a catalytically inactive protein, further supporting a role of this domain in substrate binding.
An intriguing difference between the inhibitor-free and inhibitor-bound HDAC4cd structures is the exchange of two of the ligands of the structural zinc ion. In support of the crystallographic data, our alkylation experiments also suggest the zinc ion is only weakly bound. The concomitance of a loose zinc coordination scheme and the involvement of Cys669 in an intermolecular disulfide bond might have stabilized in the inhibitor-bound HDAC4cd structure a zinc ligation scheme that might otherwise only be transiently formed. It remains to be seen if such variability in zinc coordination plays a role in the regulation of HDAC4 function in vivo.
It has been shown that HDAC4 associates with the N-CoR·HDAC3 co-repressor complex (17). To assess the role of the structural zinc-binding domain in the association of HDAC4 with N-CoR·HDAC3, we evaluated the ability of full-length wild type and C669A/H675A mutant HDAC4-FLAG proteins to form the ternary complex. In these experiments, the wild type protein co-purified with the N-CoR·HDAC3 complex, whereas the double mutant did not. Thus, the HDAC4 structural zinc-binding domain has a key role for N-CoR·HDAC3 binding, and it has to be properly folded for the formation of a stable ternary complex.
Comparison of the inhibitor-bound and inhibitor-free HDAC4cd structures suggested that some inhibitors may affect the conformation of the structural zinc-binding domain by sterically interfering with the closed conformation of the zinc binding domain. In addition, our mutagenesis data point to a role of the structural zinc-binding domain for the association of HDAC4 with N-CoR·HDAC3. Thus, we investigated the effects of active site inhibitors on the formation of the HDAC4·N-CoR·HDAC3 complex. Strikingly, the same disruptive effect observed for the C669A/H675A double mutant was obtained by the addition to cells of a cell-permeable class IIa selective trifluoromethylketone inhibitor. Thus, inhibitor binding may destabilize the structural zinc-binding domain and consequently the association of HDAC4 with N-CoR·HDAC3 in cells. Together, these data point to a key role of the structural zinc-binding domain in the recruitment of HDAC4 partner proteins in repressor complexes. Importantly, our data suggest that some HDAC4 active site inhibitors may not only inhibit its enzymatic activity but may also interfere with the recruitment of the class I HDAC3 to the promoters of HDAC4-regulated genes.
While this manuscript was under review, it was reported that HDAC4 Cys669 and Cys667 can be alkylated in cells, form intramolecular disulfide bonds in response to reactive oxygen species, and be reduced by Trx1 (thioredoxin 1) (34). Importantly, the same authors found that formation of the disulfide bond causes shuttling of HDAC4 from the nucleus to the cytoplasm. These findings are in agreement with ours and further support our proposal that in HDAC4 the structural zinc ion is only loosely coordinated and has key regulatory roles. In addition, our structural data provide an explanation for the observations of Ago et al. (34). Under reducing conditions, Cys669 and Cys667 are involved in the coordination of the structural zinc ion, whereas under oxidative conditions, they form a disulfide bond probably resulting in impaired zinc binding and alteration of the conformation of the zinc-binding domain.
The structures reported here also provide insights into the catalytic activity of HDACs. Existing structural data on HDLP, HDAH, and HDAC8 enzymes have generated a plausible catalytic mechanism for lysine deacetylation (19). This mechanism is consistent with our HDAC4cd-inhibitor structures. Thus, the HDAC4cd-HA complex mimics the preintermediate state with a zinc-bound water molecule (W1) aptly positioned for nucleophilic attack on the carbonyl carbon of the substrate, whereas the complex with the hydrated TFMK resembles the reaction intermediate formed after nucleophilic attack by this water molecule. However, in WT HDAC4, there is no protein atom in a position corresponding to the Tyr306 hydroxyl group of HDAC8 that could stabilize the reaction intermediate. Instead, the side chain of the structurally equivalent His976 is rotated away from the active site, and a water molecule (W2) appears to functionally replace Tyr306. These structural differences may account for the strongly reduced activity of the class IIa HDACs toward acetylated lysines. The water molecule is probably a less efficient transition state stabilizer than the tyrosine hydroxyl group due to its faster exchange with the solvent and its lower hydrogen bond donating potential.
Our model is supported by the structure of the GOF HDAC4cd-HA complex, where the Tyr976 side chain points inwards, as do the equivalent tyrosines in class I and IIb HDACs. This tyrosine is likely to adopt a similar position in the presence of the reaction intermediate, thus resulting in its stabilization and the greatly enhanced catalytic activity of GOF HDAC4cd. Consistently, the HA inhibitor is 30-fold more potent on the GOF than on WT HDAC4, probably due to the favorable interactions of the inhibitor carbonyl and hydroxyl oxygens with the Tyr976 hydroxyl group in the GOF mutant. Together, these data provide a molecular framework for understanding the marginal activity of class IIa HDACs on acetylated lysines and suggest that these enzymes might act on intrinsically more reactive substrates. Interestingly, it has been shown recently that mutation of the corresponding active site Tyr to Phe in the bacterial class IIb homologue HDAH results in an enzyme with a preference for acetylester substrates rather than acetamides (35). However, a similar preference has not been reported for class IIa HDACs to date. Alternatively, a specific, yet unidentified, cofactor may be required for HDAC4 activation.
Our structures are consistent with the TFMK being a more potent inhibitor of WT HDAC4cd than is the HA (Fig. 1C). The TFMK is hydrated and binds the catalytic zinc ion in a bidentate fashion, whereas the HA inhibitor makes only one bond to the zinc ion (Fig. 4A). In the structures of HDAC8, hydroxamate compounds bind the zinc ion using a bidentate chelation (Fig. 5A). It is likely that the monodentate binding of the inhibitor in the HDAC4cd-HA complex was favored due to steric restraints and the lack of interaction with an active site tyrosine as found in class I or IIb-like HDACs. A second determinant of the difference in potency is that the trifluoro moiety fills a pocket in proximity of Pro800. Such interactions are greatly reduced in the complex with the HA. Similarly, sequence alignments and modeling suggest that the trifluoromethyl binding pocket would be larger in the HDAC1, -2, and -3 enzymes, thus forming fewer favorable contacts. Consistently, trifluoromethylketone compounds typically display 10-100-fold greater inhibition of class II HDACs than of class I HDACs (27). Thus, the differences in the size of the pocket around Pro800, together with the additional space generated in class IIa HDACs by the outward position of the His976 side chain, could be exploited for the design of improved class-selective inhibitors.
While this manuscript was being submitted, the structure of the HDAC7 catalytic domain alone and in complex with suberoylanilide hydroxamic acid and trichostatin A was published. These structures closely resemble the “closed” inhibitor-free HDAC4cd structure (Cα coordinate root mean square deviation ∼0.9 Å). Differently from the inhibitors used in our study, the more flexible suberoylanilide hydroxamic acid and trichostatin A capping groups do not clash with the structural zinc binding domain and therefore do not interfere with the “closed” conformation of the structural zinc-binding subdomain. In addition, in HDAC7cd, the His976 side chain adopts the same “outward” position observed in the WT HDAC4cd structure, further confirming that this is a general feature of class IIa HDACs.
In conclusion, we have shown that despite the overall similarity with class I HDACs, the class IIa enzymes present several novel features. Important differences were observed in the active sites that are relevant for the catalytic mechanism and inhibitor specificities. We have also identified an unexpected structural zinc-binding domain conserved only in class IIa enzymes. Our structural and mutagenesis data reveal a flexibility of this domain, with direct consequences on the boundaries of the enzyme active site. More importantly, we show that the structural zinc-binding domain has a key role for the association of HDAC4 with N-CoR and HDAC3 and therefore for the recruitment of these proteins to HDAC4 repressor complexes.
Supplementary Material
Acknowledgments
We thank S. Altamura, O. Cecchetti, and S. Serafini for performing activity assays, L. Orsatti and F. Talamo for mass spectrometry measurements, and the ESRF beamline scientists for assistance during data collection.
The atomic coordinates and structure factors (codes 2VQJ, 2VQQ, 2VQM, 2VQO, and 2VQV) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables S1 and S2 and Figs. S1-S3.
Footnotes
The abbreviations used are: HDAC, histone deacetylase; HDLP, HDAC-like protein; HDAH, HDAC-like amidohydrolase; MES, 4-morpholineethanesulfonic acid; WT, wild type; TFMK, trifluoromethylketone; HA, hydroxamic acid; GOF, gain of function; DMSO, dimethyl sulfoxide.
References
- 1.Cress, W. D., and Seto, E. (2000) J. Cell Physiol. 184 1-16 [DOI] [PubMed] [Google Scholar]
- 2.Ito, A., Kawaguchi, Y., Lai, C. H., Kovacs, J. J., Higashimoto, Y., Appella, E., and Yao, T. P. (2002) EMBO J. 21 6236-6245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Glozak, M. A., Sengupta, N., Zhang, X., and Seto, E. (2005) Gene (Amst.) 363 15-23 [DOI] [PubMed] [Google Scholar]
- 4.Chen, L., Fischle, W., Verdin, E., and Greene, W. C. (2001) Science 293 1653-1657 [DOI] [PubMed] [Google Scholar]
- 5.Yuan, Z. L., Guan, Y. J., Chatterjee, D., and Chin, Y. E. (2005) Science 307 269-273 [DOI] [PubMed] [Google Scholar]
- 6.Bolden, J. E., Peart, M. J., and Johnstone, R. W. (2006) Nat. Rev. Drug Discov. 5 769-784 [DOI] [PubMed] [Google Scholar]
- 7.Gallinari, P., Di Marco, S., Jones, P., Pallaoro, M., and Steinkuhler, C. (2007) Cell Res. 17 195-211 [DOI] [PubMed] [Google Scholar]
- 8.de Ruijter, A. J., van Gennip, A. H., Caron, H. N., Kemp, S., and van Kuilenburg, A. B. (2003) Biochem. J. 370 737-749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo, L., Han, A., Bates, D. L., Cao, J., and Chen, L. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 4297-4302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Verdin, E., Dequiedt, F., and Kasler, H. G. (2003) Trends Genet. 19 286-293 [DOI] [PubMed] [Google Scholar]
- 11.Yang, X. J., and Gregoire, S. (2005) Mol. Cell Biol. 25 2873-2884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McKinsey, T. A., and Kass, D. A. (2007) Nat. Rev. Drug Discov. 6 617-635 [DOI] [PubMed] [Google Scholar]
- 13.Martin, M., Kettmann, R., and Dequiedt, F. (2007) Oncogene 26 5450-5467 [DOI] [PubMed] [Google Scholar]
- 14.Vega, R. B., Matsuda, K., Oh, J., Barbosa, A. C., Yang, X., Meadows, E., McAnally, J., Pomajzl, C., Shelton, J. M., Richardson, J. A., Karsenty, G., and Olson, E. N. (2004) Cell 119 555-566 [DOI] [PubMed] [Google Scholar]
- 15.Arnold, M. A., Kim, Y., Czubryt, M. P., Phan, D., McAnally, J., Qi, X., Shelton, J. M., Richardson, J. A., Bassel-Duby, R., and Olson, E. N. (2007) Dev. Cell 12 377-389 [DOI] [PubMed] [Google Scholar]
- 16.Jeon, E. J., Lee, K. Y., Choi, N. S., Lee, M. H., Kim, H. N., Jin, Y. H., Ryoo, H. M., Choi, J. Y., Yoshida, M., Nishino, N., Oh, B. C., Lee, K. S., Lee, Y. H., and Bae, S. C. (2006) J. Biol. Chem. 281 16502-16511 [DOI] [PubMed] [Google Scholar]
- 17.Fischle, W., Dequiedt, F., Hendzel, M. J., Guenther, M. G., Lazar, M. A., Voelter, W., and Verdin, E. (2002) Mol. Cell 9 45-57 [DOI] [PubMed] [Google Scholar]
- 18.Lahm, A., Paolini, C., Pallaoro, M., Nardi, M. C., Jones, P., Neddermann, P., Sambucini, S., Bottomley, M. J., Lo Surdo, P., Carfi, A., Koch, U., De Francesco, R., Steinkuhler, C., and Gallinari, P. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 17335-17340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Finnin, M. S., Donigian, J. R., Cohen, A., Richon, V. M., Rifkind, R. A., Marks, P. A., Breslow, R., and Pavletich, N. P. (1999) Nature 401 188-193 [DOI] [PubMed] [Google Scholar]
- 20.Somoza, J. R., Skene, R. J., Katz, B. A., Mol, C., Ho, J. D., Jennings, A. J., Luong, C., Arvai, A., Buggy, J. J., Chi, E., Tang, J., Sang, B. C., Verner, E., Wynands, R., Leahy, E. M., Dougan, D. R., Snell, G., Navre, M., Knuth, M. W., Swanson, R. V., McRee, D. E., and Tari, L. W. (2004) Structure 12 1325-1334 [DOI] [PubMed] [Google Scholar]
- 21.Vannini, A., Volpari, C., Filocamo, G., Casavola, E. C., Brunetti, M., Renzoni, D., Chakravarty, P., Paolini, C., De Francesco, R., Gallinari, P., Steinkuhler, C., and Di Marco, S. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 15064-15069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nielsen, T. K., Hildmann, C., Dickmanns, A., Schwienhorst, A., and Ficner, R. (2005) J. Mol. Biol. 354 107-120 [DOI] [PubMed] [Google Scholar]
- 23.Nielsen, T. K., Hildmann, C., Riester, D., Wegener, D., Schwienhorst, A., and Ficner, R. (2007) Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 63 270-273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vannini, A., Volpari, C., Gallinari, P., Jones, P., Mattu, M., Carfi, A., De Francesco, R., Steinkuhler, C., and Di Marco, S. (2007) EMBO Rep. 8 879-884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schuetz, A., Min, J., Allali-Hassani, A., Schapira, M., Shuen, M., Loppnau, P., Mazitschek, R., Kwiatkowski, N. P., Lewis, T. A., Maglathin, R. L., McLean, T. H., Bochkarev, A., Plotnikov, A. N., Vedadi, M., and Arrowsmith, C. H. (2008) J. Biol. Chem. 283 11355-11363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van den Berg, S., Lofdahl, P. A., Hard, T., and Berglund, H. (2006) J. Biotechnol. 121 291-298 [DOI] [PubMed] [Google Scholar]
- 27.Jones, P., Bottomley, M. J., Carfi, A., Cecchetti, O., Ferrigno, F., Lo Surdo, P., Ontoria, J. M., Rowley, M., Scarpelli, R., Schultz-Fademrecht, C., and Steinkuhler, C. (2008) Bioorg. Med. Chem. Lett. 18 3456-3461 [DOI] [PubMed] [Google Scholar]
- 28.Leslie, A. G. W. (1992) Joint CCP4 + ESF-EAMCB Newsletter on Protein Crystallography 26
- 29.CCP4. (1994) Acta Crystallogr. Sect. D Biol. Crystallogr. 50 760-763 [DOI] [PubMed] [Google Scholar]
- 30.Emsley, P., and Cowtan, K. (2004) Acta Crystallogr. Sect. D Biol. Crystallogr. 60 2126-2132 [DOI] [PubMed] [Google Scholar]
- 31.Perrakis, A., Harkiolaki, M., Wilson, K. S., and Lamzin, V. S. (2001) Acta Crystallogr. Sect. D Biol. Crystallogr. 57 1445-1450 [DOI] [PubMed] [Google Scholar]
- 32.Davis, I. W., Murray, L. W., Richardson, J. S., and Richardson, D. C. (2004) Nucleic Acids Res. 32 W615-W619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Illi, B., Russo, C. D., Colussi, C., Rosati, J., Pallaoro, M., Spallotta, F., Rotili, D., Valente, S., Ragone, G., Martelli, F., Biglioli, P., Steinkhuler, C., Gallinari, P., Mai, A., Capogrossi, M. C., and Gaetano, C. (2007) Circ. Res. 102 51-58 [DOI] [PubMed] [Google Scholar]
- 34.Ago, T., Liu, T., Zhai, P., Chen, W., Li, H., Molkentin, J. D., Vatner, S.F., and Sadoshima, J. (2008) Cell 133 978-993 [DOI] [PubMed] [Google Scholar]
- 35.Moreth, K., Riester, D., Hildmann, C., Hempel, R., Wegener, D., Schober, A., and Schwienhorst, A. (2007) Biochem. J. 401 659-665 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.