

Abstract
The mitogen-activated protein (MAP) kinases are essential signaling molecules that mediate many cellular effects of growth factors, cytokines, and stress stimuli. Full activation of the MAP kinases requires dual phosphorylation of the Thr and Tyr residues in the TXY motif of the activation loop by MAP kinase kinases. Down-regulation of MAP kinase activity can be initiated by multiple serine/threonine phosphatases, tyrosine-specific phosphatases, and dual specificity phosphatases (MAP kinase phosphatases). This would inevitably lead to the formation of monophosphorylated MAP kinases. However, the biological functions of these monophosphorylated MAP kinases are currently not clear. In this study, we have prepared MAP kinase p38α, a member of the MAP kinase family, in all phosphorylated forms and characterized their biochemical properties. Our results indicated the following: (i) p38α phosphorylated at both Thr-180 and Tyr-182 was 10–20-fold more active than p38α phosphorylated at Thr-180 only, whereas p38α phosphorylated at Tyr-182 alone was inactive; (ii) the dual-specific MKP5, the tyrosine-specific hematopoietic protein-tyrosine phosphatase, and the serine/threonine-specific PP2Cα are all highly specific for the dephosphorylation of p38α, and the dephosphorylation rates were significantly affected by different phosphorylated states of p38α; (iii) the N-terminal domain of MPK5 has no effect on enzyme catalysis, whereas deletion of the MAP kinase-binding domain in MKP5 leads to a 370-fold decrease in kcat/Km for the dephosphorylation of p38α. This study has thus revealed the quantitative contributions of phosphorylation of Thr, Tyr, or both to the activation of p38α and to the substrate specificity for various phosphatases.
Mitogen-activated protein kinases (MAPKs)3 play a pivotal role in controlling numerous cellular processes, including differentiation, mitogenesis, oncogenesis, and apoptosis (1–6). A typical MAPK cascade consists of three tiers of sequentially activating protein kinases, which commonly are referred to as MAPK, MAPK kinase (MAPKK), and MAPK kinase kinase (MAPKKK). An activated MAPKKK phosphorylates and activates a specific MAPKK, which then activates a specific MAPK. The three best characterized MAPK cascades are the extracellular signal-regulated kinase (ERK) pathway, the c-Jun N-terminal kinase (JNK) pathway, and the p38 kinase pathway. ERKs are activated by a range of stimuli, including growth factors, cell adhesion, tumor-promoting phorbol esters, and oncogenes, whereas JNK and p38 are preferentially activated by proinflammatory cytokines, and a variety of environmental stresses such as UV and osmotic stress. After activation, each MAPK phosphorylates a distinct spectrum of substrates, which include key regulatory enzymes, cytoskeletal proteins, nuclear receptors, regulators of apoptosis, and many transcription factors.
Like many protein kinases, the activity of MAPKs is regulated by phosphorylation in an activation loop located near their active sites (7). The hallmark of the MAPKs is their unique requirement for dual phosphorylation at a conserved threonine and tyrosine residue belonging to the consensus sequence TXY for catalytic activation (where X is Glu in ERKs, Pro in JNKs, and Gly in p38 kinases) (8–13). Because both threonine and tyrosine residues must be phosphorylated for full kinase activity, dephosphorylation on either site is sufficient to inactivate MAPK. Increasing evidence suggests that multiple phosphatases are involved in MAPK inactivation, thus forming a negative feedback mechanism. Several serine/threonine phosphatases, such as PP2A and PP2Cα, and tyrosine phosphatases, such as PTP-SL and HePTP, have been shown to shut off MAPK activity (14–17). In addition, there are several MAPK phosphatases that show dual specificity and could inactivate MAPKs (18).
The MAPK phosphatases (MKPs) are dual specificity phosphatases that inactivate MAPKs through dephosphorylation of phosphothreonine and phosphotyrosine residues in the activation loop of MAPKs (18, 19). MKPs belong to the protein-tyrosine phosphatases (PTPase) superfamily, which is defined by the PTPase signature motif (H/V)C(X)5R(S/T). Mechanistic studies with small molecule aryl phosphates have shown that the tyrosine-specific PTPases and the dual specificity phosphatases share a common catalytic mechanism (20). In mammalian cells, at least 13 MKPs have been identified, and they are grouped into four categories on the basis of their structural and functional characteristics (21). Type I MKPs contain only an ∼200-residue dual specificity phosphatase (DSP) domain. Type II MKPs consist of an N-terminal MAP kinase-binding (MKB) domain and a DSP domain. Type III subgroup of MKPs contains only one member, MKP5, which possesses an N-terminal domain of unknown function in addition to the MKB and DSP domains. Like type II subgroup of MKPs, type IV MKPs contain both the MKB and DSP domains. Their unique feature, however, is that they contain a sequence of about 300 residues C terminus to the DSP domain, which is rich in prolines, glutamates, serines, and threonines.
Many MKPs exhibit distinct substrate specificity toward three major classes of MAPKs. For instance, PAC1 and MKP3 are highly selective in inactivating ERKs, whereas MKP5 selectively dephosphorylate p38 and JNK MAPKs (22–26). This substrate selectivity of MKPs is in part due to their ability to recognize distinctively different dual phosphorylation sites containing the pTXpY motif in the three different classes of MAPKs. Although the activation of MAPKs has been extensively studied, the equally important process of quenching MAPK activity is poorly understood. Most previous mechanistic studies of protein phosphatases used small molecule of substrates, and there are few detailed investigations of protein phosphatases with physiological substrates. The most commonly used approaches for studying the substrate specificity of protein phosphatases involve overexpression experiments followed by either monitoring a target reporter gene expression or in vitro assay of phosphorylation level of protein substrate in the immunoprecipitate of cellular extracts. These kinds of experiments do not provide definitive information about the identity of protein phosphatases involved in certain cellular functions. Indeed, one would almost always observe substrate dephosphorylation if a sufficient amount of a protein phosphatase is provided to the system. Therefore, further understanding of the specific functional role of MKPs in cellular signaling requires quantitative study of protein phosphatases with physiological substrates.
In this study, we prepared milligram quantities of p38α in its various phosphorylation states. This has allowed us to fully characterize the biochemical properties of p38α in different phosphorylation states. Our results indicate the following. (i) p38α phosphorylated at both Thr-180 and Tyr-182 was 10–20-fold more active than p38α phosphorylated at Thr-180 only, whereas p38α phosphorylated at Tyr-182 alone was inactive. (ii) The dual-specific MKP5, the tyrosine-specific HePTP, and the Ser/Thr-specific PP2Cα are all highly specific for the dephosphorylation of p38α, and the dephosphorylation rates were significantly affected by different phosphorylated states of p38α. (iii) The N-terminal domain of MPK5 has no effect on enzyme catalysis, whereas deletion of the MKB domain in MKP5 leads to a 370-fold decrease in kcat/Km for the dephosphorylation of p38α/pTpY. This study provides a quantitative understanding of the roles of phosphorylation at each site with respect to the activation mechanism of p38α.
EXPERIMENTAL PROCEDURES
Materials—ATP, phosphoenolpyruvate (PEP), NADH, lactate dehydrogenase (LDH), pyruvate kinase, p-nitrophenyl phosphate (pNPP), bacterial purine nucleotide phosphorylase, and the ingredients to generate the purine nucleotide phosphorylase substrate 7-methyl-6-thioguanosine (MESG) were purchased from Sigma. MOPS was purchased from Amresco. EGF receptor peptide (KRELVEPLTPSGEAPNQALLR) was synthesized using standard protocol, purified by reverse-phase preparative high pressure liquid chromatography, and characterized by matrix-assisted laser desorption ionization time-of-flight mass spectrometry by Scilight Biotechnology Ltd. (Beijing, China). MESG were synthesized as described previously (27). All other chemicals were local products of analytical grade used without further purification. Double-deionized water was used throughout.
Preparation of Unphosphorylated p38α and Bisphosphorylated p38α/pTpY—The cDNA encoding mouse p38α (a generous gift from Dr. Zhenguo Wu) was subcloned into pET15b (Novagen). The N-terminally His6-tagged unphosphorylated p38α was expressed in Escherichia coli BL21(DE3) and purified by Ni-NTA column (Qiagen), followed by an anion exchange Source 15Q HR 10/10 column (GE Healthcare). The cDNAs for human wild-type MKK6 and MKK6CA, the constitutively active MKK6 mutant (S207E/T211E) in pET15b (Novagen), were kindly provided by Dr. Zhenguo Wu. Bisphosphorylated p38α/pTpY was obtained by coexpressing p38α and MKK6CA in vivo. Both p38α and MKK6CA were subcloned into the same plasmid pETDuet-1 (Novagen) used to transform competent E. coli BL21(DE3) cells. Four liters of culture medium was inoculated with an overnight culture of Luria Broth (LB, 100 ml) containing 100 μg/ml ampicillin and grown at 37 °C to an A600 of 0.8. Expression was induced with 0.2 mm isopropyl 1-thio-β-d-galactopyranoside for 12–16 h at 25 °C. The cell pellets were resuspended immediately in buffer (50 mm Tris, pH 8.0, and 300 mm NaCl). Cells were lysed in 1 mg/ml lysozyme and 0.1 mm phenylmethylsulfonyl fluoride by sonication at 4 °C. The cell lysate was clarified by centrifugation (13,500 rpm, 4 °C, 40 min). Ni-NTA column was used to separate N-terminal His6-tagged p38α/pTpY from other cell components. The fraction of eluted protein from Ni-NTA column were pooled and applied directly to an ion exchange Source 15Q column equilibrated with 50 mm Tris, pH 8.0, 2 mm dithiothreitol. The protein eluted using a 100-ml gradient of 0–500 mm NaCl. The protein purity was over 98% as judged by SDS-PAGE. High performance liquid chromatography coupled with mass spectrometry analysis confirmed that the purified p38α/pTpY was effectively homogeneous and was phosphorylated stoichiometrically to a ratio of 2 mol of phosphate per mol of p38α. The protein was stored in buffer (20 mm HEPES, pH 7.5, 100 mm NaCl, 2 mm dithiothreitol, 20% glycerol) at -80 °C. The concentration of p38α was determined using the molar extinction coefficient (A280) of 52,501 cm-1 m-1, following the method of Gill and von Hippel (28).
Expression and Purification of Protein Phosphatases and ATF2-(1–109)—Bacterial expression plasmid for the N-terminal His6-tagged HePTP was kindly provided by Dr. Tomas Mustelin. The human N-terminal His6-tagged PP2Cα expression plasmid pET28a-His6-PP2Cα was a generous gift from Dr. Mark Solomon. The human MKP5 cDNA was a generous gift from Dr. Eisuke Nishida and subcloned into pET15b vector (Novagen). The catalytically inactive mutant HePTPC270S, PP2CαD239N, and MKP5C408S were generated by PCR oligonucleotide site-directed mutagenesis, and all mutations were confirmed by DNA sequencing. The N-terminally His6-tagged HePTP, PP2Cα, and MKP5 and the inactive mutants were expressed in E. coli BL21(DE3) and purified using standard procedures of Ni-NTA column, followed by an anion exchange source 15Q column. Recombinant GST fusion human HePTP and PP2Cα (in pGEX-4T vector; GE Healthcare) were expressed in E. coli BL21(DE3) and purified according to standard procedures using the affinity matrix glutathione-Sepharose 4B (GE Healthcare), followed by an anion exchange Source 15Q column. The GST fusion ATF2-(1–109) expression plasmid pGEX-4T-1-ATF2(1–109) was kindly provided by Dr. Eui-Ju Choi. The GST-ATF2(1–109) construct was expressed in E. coli BL21(DE3) and purified according to standard procedures with an additional high resolution Source 15Q anion exchange chromatography step. The GST tag was removed by incubation with thrombin at 4 °C for 6 h. The untagged ATF2-(1–109) was concentrated to 200 μm in buffer (20 mm HEPES, pH 7.5, 100 mm NaCl, 2 mm dithiothreitol, 20% glycerol). All proteins prepared were examined by SDS-PAGE analysis, and the contents of each were judged at least 98% pure. The purified protein was made to 20% glycerol and stored at -80 °C. Protein concentrations were determined spectrophotometrically using theoretical molar extinction coefficients at 280 nm.
Enzyme Assays for p38α—The enzymatic activity of p38α was measured spectrophotometrically using EGF receptor peptide and ATF2Δ109 as substrates (29). This assay couples the production of ADP with the oxidation of NADH by pyruvate kinase and LDH. The standard assay was carried out at 25 °C in 1.8-ml reaction mixture containing 50 mm MOPS, pH 7.0, 100 mm NaCl, 0.1 mm EDTA, 10 mm MgCl2, 0.2 mm NADH, 1.0 mm PEP, 20 units/ml LDH, and 15 units/ml pyruvate kinase and varying amounts of ATP, EGF receptor peptide, and enzyme. Based on the theoretical calculation and control experiments, the amount of the coupling enzymes used in our assays are more than sufficient to determine the initial velocity of the protein kinase reaction (30) (supplemental Fig. 1). Reactions were initiated by the addition of p38α to the reaction mixture. Progress of the reaction was monitored continuously by following the formation of NAD+ at 340 nm, on a Lambda 45 spectrophotometer (PerkinElmer Life Sciences) equipped with a magnetic stirrer in the cuvette holder. The concentrations of ADP formed in p38α-catalyzed reaction were determined using an extinction coefficient for NADH of 6220 cm-1 m-1 at 340 nm. The concentration of EGF receptor peptide was determined by turnover with the enzyme under conditions of limiting peptide at a fixed concentration of ATP.
Enzyme Assays for Protein Phosphatases—With pNPP as a substrate, the reaction was initiated by the addition of the enzyme in a reaction mixture, and the initial rates for hydrolysis of pNPP by phosphatase were measured at 25 °C in 1.8 ml of reaction mixture containing 50 mm MOPS, pH 7.0, 100 mm NaCl, 0.1 mm EDTA, in the presence of either 10 mm MgCl2 for MKP5, HePTP or 2 mm MnCl2 for PP2Cα. The nonenzymatic hydrolysis of the substrate was corrected by measuring the control without the addition of enzyme. The amount of product p-nitrophenol was determined from the absorbance at 405 nm using a molar extinction coefficient of 18,000 m-1 cm-1 (31). Kinetic parameters for the dephosphorylation of the phosphorylated p38α was determined using a continuous spectrophotometric assay (17, 32). This assay incorporates a coupled enzyme system, which uses purine nucleoside phosphorylase and its chromogenic substrate 7-methyl-6-thioguanosine (MESG) for the quantification of inorganic phosphate produced in the phosphatase reaction (33). All experiments were carried out at 25 °C in 1.8 ml of reaction mixture containing 50 mm MOPS, pH 7.0, 100 mm NaCl, 0.1 mm EDTA, 10 mm MgCl2, 100 μm MESG, 0.1 mg/ml purine nucleoside phosphorylase. The reactions were initiated by the addition of phosphatase unless indicated otherwise. The time courses of absorbance change at 360 nm were recorded on a Lambda 45 spectrophotometer (PerkinElmer Life Sciences) equipped with a magnetic stirrer in the cuvette holder. Initial rates were determined from the linear slope of progress curves obtained, and the experimental data were analyzed using a nonlinear regression analysis program. Quantitation of phosphate release was determined using the extinction coefficient of 11,200 m-1cm-1 for the phosphate-dependent reaction at 360 nm at pH 7.0 (34). The concentration of MESG was determined at 331 nm, using a molar extinction coefficient of 32,000 m-1cm-1.
Western Blotting—The phosphorylation states of p38α were assessed by Western blot analysis using specific antibodies. The same amount of p38α in various phosphorylation states were resolved by SDS-PAGE and electrotransferred onto polyvinylidene difluoride membranes (Bio-Rad). After incubation of the membranes with anti-p38 (sc-7149, Santa Cruz Biotechnology, Inc.), anti-bisphosphorylated p38 (catalog number 9211, Cell Signaling Technology, Inc.), and anti-phospho-Tyr antibodies (catalog number 9411, Cell Signaling Technology, Inc.), specific immunocomplexes were detected by chemiluminescence using ECL reagents. The membrane was finally exposed to x-ray film (Eastman Kodak Co.).
RESULTS
Preparation of Bis- and Monophosphorylated p38α—The bisphosphorylated p38α, p38α/pTpY (p38α phosphorylated on both Thr-180 and Tyr-182), was prepared by coexpression of N-terminal His6-tagged p38α and constitutively active MKK6 encoded on a single plasmid in E. coli and followed by purification using Ni-NTA column and Source 15Q anion exchange chromatography. To characterize the phosphorylated states of the p38α preparation, we employed a continuous spectrophotometric enzyme-coupled assay that measures the inorganic phosphate produced in a phosphatase reaction (17, 32, 33). A summation of the two enzyme activities involved is as shown in Reactions 1 and 2.
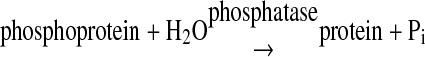 |
REACTION 1 |
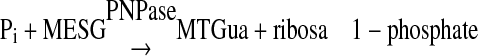 |
REACTION 2 |
In this coupled enzyme system, the coupling enzyme, purine nucleoside phosphorylase (PNPase), uses the inorganic phosphate, generated by the action of the phosphatase, to convert 7-methyl-6-thioguanosine to 7-methyl-6-thioguanine (MTGua) and ribose 1-phosphate, resulting in an increase in absorbance at 360 nm. Fig. 1 shows time course of MKP5-catalyzed p38α/pTpY dephosphorylation. The continuous assay for phosphatases was carried out at 25 °C in a 1.8-ml reaction mixture. After 5 min of equilibration at 25 °C, the reaction was initiated by adding catalytic amount of MKP5 (186 nm), and absorbance at 360 nm was recorded. The change in absorbance was because of the conversion of MESG to 7-methyl-6-thioguanine in the presence of inorganic phosphate released from the dephosphorylation of p38α by MKP5. The MKP5-catalyzed dephosphorylation was near 100% complete in 3 min. Quantitation of phosphate release was determined using the extinction coefficient of 11,200 m-1cm-1 for the phosphate-dependent reaction at 360 nm at pH 7.0. As shown in Fig. 1, the phosphorylation stoichiometry of p38α was determined to be close to 2 mol of phosphate/mol of p38α. Thus the change in absorbance at 360 nm is a direct measure of the stoichiometric, phosphorylated p38α concentration.
FIGURE 1.

The time course of MKP5-catalyzed p38α/pTpY dephosphorylation reaction. The reaction mixture contained the standard phosphatase assay buffer (50 mm MOPS, pH 7.0, 100 mm NaCl, 0.1 mm EDTA, 10 mm MgCl2, 100 μm MESG, 0.1 mg/ml purine nucleotide phosphorylase), and 1.7 μm bisphosphorylated p38α. The absorption at 360 nm was recorded following the addition of 186 nm MKP5 at 25 °C.
MKP5 is a dual specificity phosphatase capable of dephosphorylating both Tyr(P) and Thr(P) in the activation loop of p38α. In addition to MKP5, bisphosphorylated p38α can also be inactivated through the action of serine/threonine protein phosphatase PP2Cα (14) and tyrosine-specific PTP-SL and HePTP (35–37). Fig. 2A shows the time dependence of HePTP-catalyzed reaction. Upon the addition of HePTP (470 nm), the dephosphorylation reaction of bisphosphorylated p38α was initiated as detected by monitoring the real time phosphate release. At the end of the HePTP-catalyzed dephosphorylation reaction, all bisphosphorylated p38α molecules were converted to the threonine 180 phosphorylated form of p38α; 186 nm MKP5 was added to the reaction mixture, and the threonine 180-phosphorylated p38α underwent MKP5-catalyzed dephosphorylation, resulting in a further increase of phosphate release. Similarly, Fig. 2, B, C, and D, shows the time-dependent absorption profiles (at 360 nm) for sequential additions of different phosphatases into the coupled reaction system. It can be seen from these figures that HePTP and PP2Cα specifically dephosphorylated the phosphotyrosine and phosphothreonine of p38α, respectively, and the ratio of phosphothreonine and phosphotyrosine in this p38α preparation is close to 1:1. These results also indicate that the continuous spectrophotometric enzyme-coupled assay is suitable for measuring the inorganic phosphate production in the protein phosphatase-catalyzed MAPK dephosphorylation reaction.
FIGURE 2.

The time dependence absorption profiles for sequential additions of different phosphatases to the MESG/phosphorylase coupled enzyme assay system at 25 °C. The initial mixture contains the standard phosphatase assay buffer, and 1.9 μm bisphosphorylated p38α. The absorption at 360 nm was recorded following the addition of individual phosphatase (as indicated) to the reaction mixture: A, 470 nm HePTP and 186 nm MKP5; B, 670 nm PP2Cα and 186 nm MKP5; C, 470 nm HePTP and 670 nm PP2Cα; D, 670 nm PP2Cα and 470 nm HePTP.
Because tyrosine-specific phosphatase HePTP and serine/threonine-specific phosphatase PP2Cα can effectively remove the phosphate from Thr(P)-180 or Tyr(P)-182 in bisphosphorylated p38α, we can generate monophosphorylated forms of p38α through the action of these protein phosphatases on bisphosphorylated p38α. To prepare monophosphorylated p38α/pT (p38α phosphorylated on Thr-180) and p38α/pY (p38α phosphorylated on Tyr-182), the N-terminal His6-tagged bisphosphorylated p38α/pTpY (0.5 mg in 500 μl) was treated with either GST-HePTP (final concentration 1 μm) or GST-PP2Cα (final concentration 1 μm) in standard buffer (50 mm MOPS, pH 7.0, 100 mm NaCl, 10 mm Mg2+) at 25 °C for 60 min, at which time dephosphorylation was near 100% complete as examined by the phosphatase assay. The monophosphorylated p38α proteins were separated from GST-tagged phosphatases by Ni-NTA column. The homogeneity of the p38α preparations were verified by SDS-PAGE. The bisphosphorylated and monophosphorylated p38α comigrated on SDS-PAGE and could be resolved from the unphosphorylated p38α because of gel mobility retardation. As shown in Fig. 3, all forms of p38α appeared to migrate as single bands (greater than 98% purity as judged by SDS-PAGE). To directly and quantitatively measure the phosphorylated states of the p38α preparation, the stoichiometry of p38α phosphorylation was determined by the amount of inorganic phosphate released from p38α upon treatment by various phosphatases using the coupled enzyme procedure. Incubation of the monophosphorylated p38α/pT with HePTP did not produce any inorganic phosphate, which is consistent with no p38α/pY in the sample, whereas stoichiometric amounts of phosphate were released from p38α/pT upon the addition of MKP5 (Fig. 4A). Similarly, incubation of the monophosphorylated p38α/pY with PP2Cα did not produce any inorganic phosphate, indicating that there were no p38α/pT in the sample. Further treatment with MKP5 yields stoichiometric amounts of phosphate released from p38α/pY (Fig. 4B). Finally, to confirm that the increased absorbance at 360 nm is dependent on phosphatase activities of MKP5, HePTP, and PP2Cα and that these phosphatases per se do not have any effect on MTGua production, we have demonstrated that the absorbance remained unchanged when the catalytically inactive mutants MKP5C408S, HePTPC270S, and PP2CαD239N were used (supplemental Fig. 2).
FIGURE 3.

SDS-PAGE of p38α, p38α/pT, p38α/pY, and p38α/pTpY. Lane 1, molecular mass standard; lane 2, 1 μg of purified p38α; lane 3, 1 μg of purified p38α/pTpY; lane 4, 1 μg of purified p38α/pT; lane 5, 1 μg of purified p38α/pY.
FIGURE 4.

Phosphorylation stoichiometry of various phosphorylated forms of p38α determined by an enzyme-coupled assay that measures the amount of phosphate released from p38α upon treatment with an appropriate phosphatase. The phosphate release from p38α was monitored by the increase in absorbance at 360 nm, 25 °C. A, 0.9 μm p38α/pT was first treated with 50 nm HePTP and then with 43 nm MKP5. B, 0.9 μm p38α/pY was first treated with 50 nm PP2Cα and then with 43 nm MKP5.
To further examine the various forms of p38α, we performed Western blot analysis using anti-p38α, anti-bisphosphorylated p38α, and anti-phosphotyrosine antibody (Fig. 5). It can be seen from this figure that when probed with an anti-p38α antibody, all forms of p38α showed immunoreactivity (Fig. 5, top panel), and their gel mobility was consistent with the Coomassie staining in Fig. 3. As expected when probed with an anti-bisphosphorylated p38α antibody, only p38α/pTpY and p38α/pT displayed immunoreactivity (Fig. 5, middle panel), and when probed with an anti-Tyr(P) antibody, only p38α/pTpY and p38α/pY showed immunoreactivity (Fig. 5, bottom panel). No measurable immunoreactivity was apparent with the p38α/pT sample toward anti-Tyr(P) antibody and with p38α/pY sample toward anti-bisphosphorylated p38α antibody, indicating that the dephosphorylation was complete, and there was no p38α/pTpY present in the p38α/pT and p38α/pY samples. In addition, high performance liquid chromatography coupled with mass spectrometry analysis also confirmed that the purified p38α/pT and p38α/pY were phosphorylated stoichiometrically to a ratio of 1 mol of phosphate per mol of p38α.
FIGURE 5.

Western blot analysis of various forms of p38α. Approximately 100 ng of various forms of p38α were transferred to polyvinylidene difluoride membrane from 10% SDS gel and probed with anti-p38α, anti-bisphosphorylated p38α, and anti-Tyr(P) antibodies. The immunoactivity was detected by chemiluminescence.
Regulation of p38α Activity by Differential Phosphorylation in the Activation Loop—To characterize the biochemical properties of various forms of p38α, we first measured the ATPase and kinase activities of p38α in various phosphorylation states using an enzyme-coupled spectrophotometric assay (29, 30). All steady-state kinetic studies were performed at pH 7.0 and 25 °C. As reported previously, the activated p38α displays a significant ATPase activity in the absence of any added protein substrate (38). Thus, we were able to obtain the kinetic parameters for the ATP hydrolyzing activity of p38α from a catalytic amount of the activated p38α. A typical set of initial velocities versus ATP concentration is shown in Fig. 6A. Direct curve fitting of the data to the Michaelis-Menten equation yielded kcat and Km values, which were 0.562 ± 0.007 s-1 and 212 ± 12.6 μm, respectively, for the p38α/pTpY-catalyzed ATP hydrolysis. The kcat and Km values for the hydrolysis of ATP by p38α/pTpY (prepared by in vitro phosphorylation using constitutive active MKK6) are comparable with those determined at pH 7.6 and 30 °C (38). Similarly, we also examined the ATPase activity for the unphosphorylated and monophosphorylated p38α (Table 1). The kcat and Km values for p38α/pT were determined to be 0.062 ± 0.002 s-1 and 1669 ± 16 μm, respectively. No measurable ATPase activity was observed for p38α and p38α/pY.
FIGURE 6.
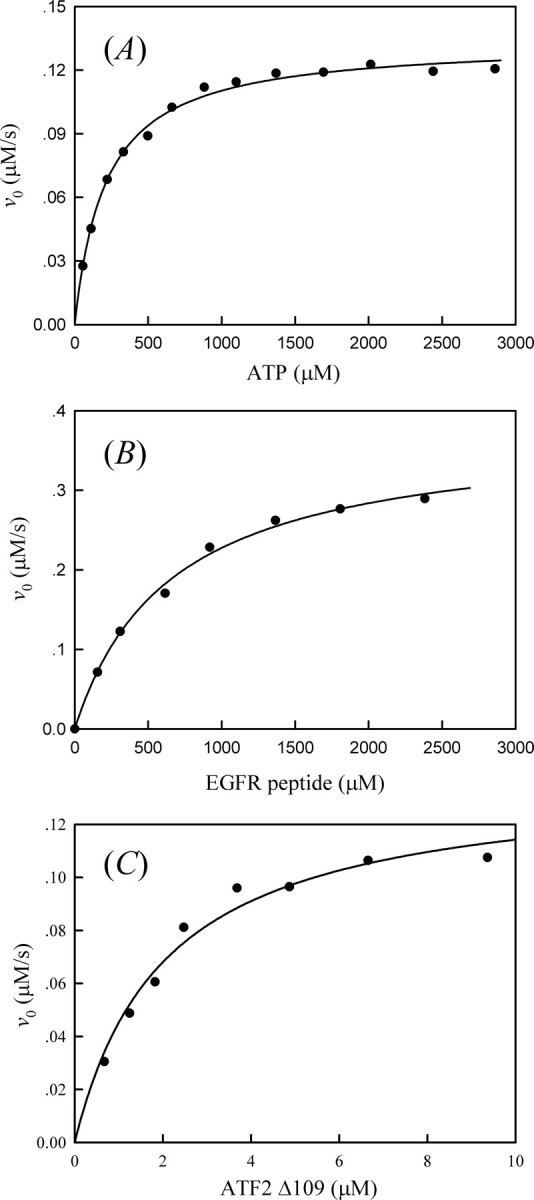
A, plot of initial rate of p38α/pTpY-catalyzed ATP hydrolysis versus the ATP concentration. Reaction mixtures contained the standard kinase assay buffer (50 mm MOPS, pH 7.0, 100 mm NaCl, 0.1 mm EDTA, 10 mm MgCl2, 0.2 mm NADH, 1.0 mm PEP, 20 units/ml LDH, and 15 units/ml pyruvate kinase) 238 nm p38α/pTpY and indicated concentrations of ATP, at 25 °C. The solid line is the best fitting result according to the Michaelis-Menten equation with kcat = 0.562 ± 0.007 s-1 and Km = 212 ± 12.6 μm. B, plot of the initial rate of p38α/pTpY-catalyzed EGF receptor peptide phosphorylation versus the peptide concentration. Reaction mixtures contained the standard kinase assay buffer, 1 mm ATP, 11 nm p38α/pTpY, and the indicated concentrations of the peptide, at 25 °C. The solid line is the best fitting result according to the Michaelis-Menten equation with kcat = 31.6 ± 1.1 s-1 and Km = 656 ± 61.7 μm. C, plot of the initial rate of p38α/pTpY-catalyzed ATF2Δ109 phosphorylation versus the ATF2Δ109 concentration. Reaction mixtures contained the standard kinase assay buffer, 1 mm ATP, 29 nm p38α/pTpY, and the indicated concentrations of ATF2Δ109, at 25 °C. The solid line is the best fitting result according to the Michaelis-Menten equation with kcat = 4.7 ± 0.3 s-1 and Km = 2.03 ± 0.34 μm.
TABLE 1.
ATPase activity of p38α in different phosphorylation states (pH 7.0, 10 mm Mg2+, 25°C)
| kcat | Km | kcat/Km | |
|---|---|---|---|
| s–1 | μm | m–1 s–1 | |
| p38α/pTpY | 0.562 ± 0.007 | 212 ± 13 | (2.65 ± 0.19) × 103 |
| p38α/pT | 0.062 ± 0.002 | 1669 ± 16 | 37.1 ± 1.7 |
| p38α/pY | No activity | ||
| p38α | No activity |
To determine the kinetic parameters of p38α kinase activity, we selected a synthetic peptide derived from the EGF receptor (residues 661–681; EGFR peptide) and the transcription factor ATF2Δ109 (residues 1–109) as p38α substrates. Fig. 6B shows the dependence of the initial rate of the p38α/pTpY-catalyzed phosphorylation on the peptide concentration. It can be seen from this figure that the phosphorylation of EGFR peptide catalyzed by p38α/pTpY obeyed classical Michaelis-Menten kinetics. By fitting the Michaelis-Menten equation to the experimental data, the values of kcat and Km were determined to be 31.6 ± 1.1 s-1 and 656 ± 62 μm, respectively. These results were similar to those obtained from earlier studies (measured at pH 7.6 and 30 °C) in which the kcat and Km values for the p38α/pTpY-catalyzed peptide phosphorylation at 1 mm ATP were found to be 22.6 ± 0.8 s-1 and 840 ± 80 μm, respectively (38). Under the same conditions, the kcat /Km value for the kinase activity of unphosphorylated p38α was estimated as 3.2 m-1s-1, which is 15,000-fold lower than that for p38α/pTpY.
Raingeaud et al. (39) showed the transcription factor ATF2 was a good in vitro substrate for p38 and suggested ATF2 as a potential in vivo substrate for p38 as well. Fig. 6C shows the dependence of the initial rate of the p38α/pTpY-catalyzed ATF2Δ109 phosphorylation on the substrate concentration. With ATF2Δ109 as a substrate, we obtained a kcat of 4.7 ± 0.3 s-1 and Km of 2.03 ± 0.34 μm for p38α/pTpY. These compared with a kcat of 1.1 ± 0.03 s-1 and Km of 1.4 ± 0.1 μm for ATF2Δ115 obtained by a radioisotope assay in which the rate of incorporation of 32P from [γ-32P]ATP into a substrate was directly measured (40). Similarly, the unphosphorylated p38α showed very low kinase activity toward ATF2Δ109 substrate, and the kcat/Km value was determined to be 691 ± 34 m-1 s-1.
We next determined the kinase activity for the two monophosphorylated forms of p38α, p38α/pT, and p38α/pY. Similarly, with EGFR peptide and ATF2Δ109 as substrates, the phosphorylation reactions by p38α/pT and p38α/pY also followed Michaelis-Menten kinetics. Tables 2 and 3 summarize the kinetic parameters of kinase activity of p38α in different phosphorylation states. As shown in Tables 2 and 3, p38α/pT exhibited dramatically higher kinase activity than the unphosphorylated p38α. The overall catalytic efficiencies kcat/Km, also known as substrate specificity constant, are about 300–800-fold higher than those of the unphosphorylated p38α, and only 1 order of magnitude lower than those of the bisphosphorylated p38α. These results were similar to those obtained from an earlier study for ERK2 (41). It should be noted that if the observed activity in the monophosphorylated sample was because of the more active p38α/pTpY, then the Km value should reflect that of p38α/pTpY. As can be seen in Tables 2 and 3, the Km values of p38α/pT are substantially different from those of p38α/pTpY, suggesting that the kinase activities of the monophosphorylated p38α/pT are intrinsic to the protein, not from contaminating p38α/pTpY. In contrast, p38α/pY exhibited much lower kinase activities. The kcat/Km value for the p38α/pY kinase activity is 1000–2000-fold lower than that of p38α/pTpY and only severalfold higher than that of p38α. Taken together, our results suggest that unlike ERK2, phosphorylation of Tyr-183 in the activation loop of p38α alone does not have significant effect on both kinase and ATPase activities.
TABLE 2.
Kinase activity of p38α in different phosphorylation states with EGFR peptide as a substrate (pH 7.0, 1 mm ATP, 10 mm Mg2+, 25°C)
| kcat | Km | kcat/Km | |
|---|---|---|---|
| s–1 | μm | m–1 s–1 | |
| p38α/pTpY | 31.6 ± 1.1 | 656 ± 62 | (4.8 ± 0.6) × 104 |
| p38α/pT | 6.99 ± 1.09 | 2800 ± 691 | (2.5 ± 1.0) × 103 |
| p38α/pY | 22.4 ± 3.1 | ||
| p38α | 3.20 ± 0.48 |
TABLE 3.
Kinase activity of p38α in different phosphorylation states with ATF2Δ109 as a substrate (pH 7.0, 1 mm ATP, 10 mm Mg2+, 25°C)
| kcat | Km | kcat/Km | |
|---|---|---|---|
| s–1 | μm | m–1 s–1 | |
| p38α/pTpY | 4.7 ± 0.3 | 2.03 ± 0.34 | (2.3 ± 0.5) × 106 |
| p38α/pT | 3.8 ± 0.3 | 20.1 ± 3.2 | (1.9 ± 0.5) × 105 |
| p38α/pY | 2497 ± 382 | ||
| p38α | 691 ± 34 |
Specificity of p38α Dephosphorylation by MAP Kinase Phosphatases—To establish the utility of the continuous spectrophotometric enzyme-coupled assay to the study of protein phosphatase-catalyzed MAPK dephosphorylation, we first applied this assay to analyze the dephosphorylation of p38α/pTpY by MKP5. Fig. 7 shows typical progress curves of the MKP5-catalyzed reaction at several p38α/pTpY concentrations. Assays were carried out by using the indicated concentrations of bisphosphorylated p38α. It can be seen from the inset of Fig. 7 that there is a nice linear correlation between the final absorbance at 360 nm and the amount of p38α/pTpY present in the reaction. The initial rates of the dephosphorylation reaction were determined from the slopes of the initial linear portion of the progress curves. Fig. 8 shows the variation of initial rates of the MKP5-catalyzed reaction with p38α concentrations. In the case of the MKP5-catalyzed p38α dephosphorylation reaction, a quantitative description of kinetic behavior cannot be based on the Michaelis-Menten equation, because the assumption that the free substrate concentration is equal to the total substrate concentration is not valid. A general initial velocity equation, taking substrate depletion into account, is given by Equation 1 (42),
 |
(Eq. 1) |
By fitting Equation 1 to the experimental data, the values of kcat and Km were determined to be 0.144 ± 0.012 μm and 0.547 ± 0.012 s-1, respectively. The substrate specificity constant kcat/Km for the MKP5-catalyzed p38α/pTpY dephosphorylation (4 × 106 m-1 s-1) was more than 5 orders of magnitude higher than that of MKP5-catalyzed pNPP hydrolysis (12.4 m-1 s-1), indicating that p38α/pTpY is a very specific substrate for MKP5. By using the same procedures, we also determined the kinetic parameters for the dephosphorylation of p38α/pT and p38α/pY by MKP5. As can be seen from Table 4, the dephosphorylation rate of p38α/pY is slightly faster than that of p38α/pT, and the kinetic constants for the MKP5-catalyzed dephosphorylation of p38α/pT and p38α/pY are comparable with those of the bisphosphorylated p38α. Thus, both forms of the monophosphorylated p38α can serve as effective MKP5 substrates, independently.
FIGURE 7.

The time courses of MKP5-catalyzed reaction in the presence of different concentrations of p38α/pTpY. The reaction mixture contained the standard phosphatase assay buffer, 43 nm MKP5, and the various concentrations of p38α/pTpY. The absorption at 360 nm was recorded following the addition of 43 nm MKP5 at 25 °C. The p38α/pTpY concentrations were as follows: curve 1, 0.12 μm; curve 2, 0.24 μm; curve 3, 0.36 μm; curve 4, 0.60 μm; curve 5, 0.95 μm; curve 6, 1.2 μm.
FIGURE 8.

Dependence of the initial rate of MKP5-catalyzed reaction on the concentration of bisphosphorylated p38α. The reaction mixture contained the standard phosphatase assay buffer, 43 nm MKP5, and the various concentrations of bisphosphorylated p38α at 25 °C. The solid line is the best fitting result according to Equation 1 with kcat = 0.547 ± 0.012 s-1 and Km = 0.144 ± 0.012 μm.
TABLE 4.
Kinetic parameters of MKP5, HePTP, and PP2Cα (pH 7.0, 10 mm Mg2+, 25 °C)
| Phosphatase | Substrate | kcat | Km | kcat/Km |
|---|---|---|---|---|
| s–1 | μm | m–1 s–1 | ||
| MKP5 | p38α/pTpY | 0.55 ± 0.01 | 0.14 ± 0.01 | (4.01 ± 0.42) × 106 |
| p38α/pT | 0.42 ± 0.02 | 0.33 ± 0.04 | (1.28 ± 0.21) × 106 | |
| p38α/pY | 0.78 ± 0.02 | 0.46 ± 0.032 | (1.69 ± 0.16) × 106 | |
| pNPP | 0.22 ± 0.01 | 17680 ± 755 | 12.4 ± 0.8 | |
| HePTP | p38α/pTpY | 0.86 ± 0.03 | 2.86 ± 0.27 | (3.0 ± 0.4) × 105 |
| p38α/pY | 0.27 ± 0.01 | 2.37 ± 0.33 | (1.14 ± 0.22) × 105 | |
| pNPP | 3.52 ± 0.12 | 8675 ± 74 | 406 ± 17 | |
| PP2Cα | p38α/pTpY | ≫12 | (6.55 ± 0.17) × 104 | |
| p38α/pT | 2.75 ± 0.31 | 15.5 ± 2.4 | (1.77 ± 0.47) × 105 | |
| pNPP (Mn2+) | 0.75 ± 0.02 | 585 ± 75 | (1.28 ± 0.20) × 103 |
Among the serine/threonine-specific and tyrosine-specific protein phosphatases, PP2Cα and HePTP have been suggested as negative regulators of p38 MAP kinase involved in stress responses. However, it is not known how efficiently HePTP and PP2Cα can dephosphorylate and inactivate p38α. Using the continuous spectrophotometric assay, we examined the purified and phosphorylated p38α as a substrate of HePTP. Fig. 9 shows the dependence of the initial rate of HePTP-catalyzed reaction on the concentration of p38α. The initial velocity gives rise to the hyperbolic dependence on the substrate concentration. The experimental data fit the Michaelis-Menten equation with a Km of 2.86 ± 0.27 μm and kcat of 0.86 ± 0.034 s-1. The kcat/Km value for HePTP-catalyzed p38α/pTpY dephosphorylation (with 10 mm Mg2+) was 3.0 × 105 m-1 s-1, indicating that p38α/pTpY is an efficient substrate for HePTP. With p38α/pY as a substrate, the kinetic constants, kcat and Km, for the HePTP-catalyzed dephosphorylation were only slightly lower than those for the bisphosphorylated p38α (Table 4). These results suggest that dephosphorylation of the Tyr(P) residue by HePTP does not require the presence of Thr(P) in the p38α substrate. Similarly, we also examined the ability of PP2Cα to dephosphorylate both p38α/pTpY and p38α/pT. With p38α/pTpY as a substrate, initial rate analysis indicated that the Km for the PP2Cα-catalyzed reaction was much greater than 12 μm. Thus, we were only able to obtain a kcat/Km value of 6.55 × 104 ± 0.17 × 104 m-1 s-1 for the PP2Cα-catalyzed dephosphorylation of Thr(P) in p38α/pTpY. Interestingly, when the monophosphorylated p38α/pT was used as a substrate, the kcat/Km value for the PP2Cα-catalyzed reaction was 1.77 × 105 ± 0.47 × 105 m-1 s-1, which is 3-fold higher than that for p38α/pTpY, indicating that the hydrolysis of p38α/pT by PP2Cα is favored over p38α/pTpY.
FIGURE 9.

Dependence of the initial rate of HePTP-catalyzed reaction on the concentration of p38α/pTpY. The reaction mixture contained the standard phosphatase assay buffer, 50 nm HePTP, and the various concentrations of p38α/pTpY at 25 °C. The solid line is the best fitting result according to according to the Michaelis-Menten equation with kcat = 0.86 ± 0.034 s-1 and Km = 2.86 ± 0.27 μm.
Types II–IV MKPs share two common structural features as follows: a conserved catalytic domain (DSP) that contains the PTPase active site signature motif (H/V)CX5R(S/T), and a MAP kinase-binding (MKB) domain. The MKB domain of MKP3 can physically associate with ERK2 and may play a role in controlling MKP3 substrate specificity (43). Compared with MKP3, MKP5 contains an N-terminal domain of unknown function in addition to the MKB and DSP domains (26). To quantitatively assess the contribution of the N-terminal domain and kinase-binding domain to the MKP5-catalyzed p38α dephosphorylation, we measured the kinetic parameters of MKP5ΔN121 (an N-terminally truncated MKP5, lacking the first 1–121 amino acids) and MKP5ΔN319 (an N-terminally truncated MKP5, lacking the first 1–319 amino acids) toward p38α/pTpY. The kcat and Km values for MKP5ΔN121-catalyzed reaction were determined to be 0.518 ± 0.0265 s-1and 0.217 ± 0.0375 μm, respectively (Table 5). The kcat/Km value was calculated as 2.4 × 106 m-1s-1, which was similar to that of wild-type MKP5, indicating that the N-terminal domain of MPK5 has no effect on enzyme catalysis. The kcat and Km value of MKP5ΔN319-catalyzed p38α/pTpY dephosphorylation was 0.383 ± 0.04 s-1, which is comparable with that of the wild-type MKP5. However, its kcat/Km value was more than 2 orders of magnitude (600-fold) lower than that of the wild-type MKP5, which is due largely to a dramatic increase in Km. Thus, the ability of MKP5 to bind p38α through its kinase-binding domain contributes directly to efficient p38α dephosphorylation by MKP5. In addition, we also prepared a truncated form of HePTP, HePTPΔN43, in which the first 1–42 amino acids were deleted. Although the catalytic activity of HePTPΔN43 is higher than that of wild-type HePTP using pNPP as a substrate (Table 5), no measurable phosphate activity was observed for the HePTPΔN43-catalyzed dephosphorylation of p38α/pTpY.
TABLE 5.
Kinetic parameters of MKP5ΔN121, MKP5ΔN319, and HePTPΔN43 (pH 7.0, 10 mm Mg2+, 25 °C)
| Phosphatase | Substrate | kcat | Km | kcat/Km |
|---|---|---|---|---|
| s–1 | μm | m–1 s–1 | ||
| MKP5ΔN121 | p38α/pTpY | 0.518 ± 0.027 | 0.217 ± 0.038 | (2.40 ± 0.54) × 106 |
| pNPP | 0.132 ± 0.004 | 21100 ± 1364 | 6.26 ± 0.60 | |
| MKP5ΔN319 | p38α/pTpY | 0.383 ± 0.041 | 59.6 ± 8.3 | (6.4 ± 1.6) × 103 |
| pNPP | 0.070 ± 0.005 | 36400 ± 4332 | 1.90 ± 0.35 | |
| HePTPΔN43 | p38α/pTpY | No activity | ||
| pNPP | 5.1 ± 0.2 | 9500 ± 82 | 536.8 ± 23.6 |
DISCUSSION
Protein phosphorylation is the most important post-translational modification mechanism utilized by eukaryotic cells to regulate protein functions. The extent of protein phosphorylation inside the cell is determined by the integrated actions of both protein kinases and phosphatases. Consequently, protein kinases and phosphatases are key components of intracellular signal transduction pathways through which cells respond to extracellular signals, such as hormones and growth factor, as well as environmental and nutritional stresses. Thus, the determination of detailed kinetic and thermodynamic parameters of the individual enzymes in the cycles is important to construct and analyze the biological networks (44).
The MAP kinases are unique among the Ser/Thr protein kinases in that they are activated by dual phosphorylation of the Thr and Tyr residues in the TXY motif of the activation loop. Because phosphorylation of both Thr and Tyr in the activation loop is required to maintain optimal MAP kinase activity, MAPK inactivation may involve Ser/Thr protein phosphatases, PTPs, and dual specificity phosphatases. Consequently, it is expected that monophosphorylated MAP kinases would be generated through the combined action of MAPKKs and various MAP kinase phosphatases. Indeed, biochemical evidence indicates that both forms of the monophosphorylated ERK2 exist in the cell in addition to bisphosphorylated and unphosphorylated ERK2 (45, 46). In a study on the heat shock regulation of the fission yeast Spc1 stress-activated protein kinase, a homolog of human p38, the tyrosine-phosphorylated form of Spc1 was also observed in heat-shocked cells (47). It is widely accepted in the literature that only the bisphosphorylated MAP kinase is active, whereas the monophosphorylated MAP kinases are inactive (2). Thus, the biological roles for the monophosphorylated MAP kinases are currently not clear. Zhou and Zhang (41) have recently shown that a single phosphorylation in the activation loop of ERK2 produced an intermediate activity state. The catalytic efficiencies of monophosphorylated ERK2/pY and EKR2/pT are only 1–2 orders of magnitude lower than that of the fully active bisphosphorylated ERK2/pTpY. Phosphorylation in the ERK2 activation loop primarily increases kcat, with only a moderate decrease in Km values. The fact that ERK2/pY is only about 3-fold less active than ERK2/pT suggests that the unique phosphorylation site to the MAP kinases, Tyr-185, may also play a crucial role in substrate binding and/or catalysis. This raises the possibility that the monophosphorylated ERK2s may have distinct biological roles in vivo. Would the monophosphorylated forms of other MAP kinase display intermediate levels of activity as well? To address this question, we determined the kinetic parameters for kinase and ATPase activities of all forms of p38α. Our results revealed that unlike ERK2, both kcat and Km values for the p38α/pT-catalyzed peptide phosphorylation change dramatically compared with those of fully active p38α/pTpY, and the kinase activity of monophosphorylated p38α/pY is about 3 orders of magnitude lower than that of p38α/pTpY. Therefore, it seems likely that phosphorylation of Tyr-182 in p38α may have different biological functions. We have found that tyrosine phosphorylation of p38α by MKK6 precedes threonine phosphorylation in vitro.4 Because it is well known that both the magnitude and duration of MAP kinase activation are important in determining cell fate, the accumulations of the inactive, tyrosine-phosphorylated forms of p38α in cells are perhaps a reservoir of rapidly activable enzyme.
There are hundreds of protein phosphatases and thousands of potential protein substrates in the cell. However, it remains largely unknown as to how many phosphatases are involved in phosphorylation of a single substrate. Several years ago, Zhou et al. (17) performed a kinetic analysis of ERK2 dephosphorylation by protein phosphatases. Eleven different protein phosphatases were compared. They found that only the dual-specific MKP3, the tyrosine-specific HePTP, and the Ser/Thrspecific PP2A have the level of specificity and activity to support them being physiologically relevant regulators of ERK2 kinase activity. To compare the kinetic parameters for the phosphatase-catalyzed p38α dephosphorylation with those of the ERK2 reaction, we have performed kinetic analysis of three different protein phosphatases, MKP5, HePTP, and PP2Cα, which had been suggested previously to be involved in p38α regulation. Unlike MKP3, which displays a preference for ERK2, MKP5 has been shown to associate with p38 and JNK and block the enzymatic activation of MAPKs with the selectivity of p38 = JNK >> ERK2. Our results showed that indeed p38α/pTpY is highly efficient substrate for MKP5 with a kcat/Km second-order rate constant of 4 × 106 m-1 s-1, which is more than 100-fold higher than that of the MKP5-catalyzed ERK2/pTpY dephosphorylation. This result is consistent with the previous finding that MKP5 may selectively deactivate p38α signaling. As can be seen from Table 4, the rate of p38α/pY dephosphorylation by MKP5 is only slightly faster than that of p38α/pT. It is still unclear whether MKP5 uses the same catalytic mechanism to remove the phosphoryl group from threonine and tyrosine residues. HePTP is a cytosolic enzyme with a single catalytic domain and is most similar to the brain-specific PTPs, STEP and PTP-SL. There have been some controversial results regarding the specificity of HePTP toward p38α (35, 48, 49). To compare the ability of HePTP to dephosphorylate ERK2 and p38α, we also determined the kinetic parameters for the HePTP-catalyzed dephosphorylation of ERK2/pTpY (with Mg2+) and p38α/pTpY (without Mg2+) using the continuous spectrophotometric assay. In the presence of 10 mm Mg2+, the kcat and Km values for the ERK2/pTpY dephosphorylation by HePTP were found to be 1.23 ± 0.08 s-1 and 2.42 ± 0.34 μm, whereas those for the p38α/pTpY dephosphorylation were 1.28 ± 0.06 s-1 and 0.98 ± 0.078 μm in the absence of Mg2+, respectively. Thus, when HePTP activity was measured with Mg2+, the kcat/Km value was 4-fold lower than that obtained without Mg2+. The kcat/Km value for the HePTP-catalyzed ERK2/pTpY dephosphorylation is only 2-fold higher than that of the p38α/pTpY dephosphorylation under identical conditions, suggesting that p38α/pTpY is also a highly efficient substrate for HePTP. PP2Cα belongs to a large and widely distributed subfamily of Mg2+/Mn2+-dependent phosphatases of the PPM superfamily (50). Our kinetic analysis shows that PP2Cα is capable of dephosphorylating both p38α/pTpY and p38α/pT in the presence 10 mm Mg2+. The hydrolysis of p38α/pT by PP2Cα is favored by 3-fold over p38α/pTpY. Although PP2Cα can in vitro dephosphorylate Thr(P)-183 of ERK2 in the presence of Mn2+, no activity was detected with or without Mg2+ (data not shown). Because of its high concentration in the cell compared with other divalent metal ions, Mg2+ is considered the physiological activator of many enzymes (51). Thus, ERK2 may not be a physiological substrate for PP2Cα in vivo.
MKP5 is unique among the MKP family, which contains a novel N-terminal domain in addition to the MKB and DSP domains. Deletion of this domain from MPK5 had no effect on the MKP5-catalyzed dephosphorylation of p38α/pTpY. Thus, it is likely that the novel domain in MKP5 may be involved in interaction with other cellular proteins and thereby providing a mechanism for cross-talk between MAPK and other signaling pathways involved in regulating cellular functions (21). Like many other MKPs, the MKB domain of MKP5 contains a KIM motif that mediates phosphatase-substrate association using regions of p38α distant from the phosphorylation site. Deletion of the MKB domain in MKP5 leads to a 400-fold increase in Km, but it had only a modest effect (1.5-fold) on kcat. The results collectively demonstrated that substrate recognition by MKP5 may involve extensive protein-protein contacts in addition to the interactions that engage the phosphoamino acid residue. Thus, protein-protein interactions between the phosphatase and its substrate involving both the enzymatic active site and adjacent or remote noncatalytic site may be responsible for the observed high substrate specificity. Our data support the notation that the major function of the MKB domain in MKP5 is to increase the “effective concentration” of the p38α/pTpY motif in the vicinity of the active site of MKP5 for dephosphorylation. HePTP also contain an p38α kinase interaction motif (KIM, residues 16–31). Compared with MKP5, the KIM motif is more important for HePTP-catalyzed p38α/pTpY dephosphorylation. Deletion of this motif from HePTP completely abolished its phosphatase activity to p38α, although the catalytic activity of this mutant is similar to that of the wild-type HePTP using pNPP as a substrate. The results collectively demonstrated that protein phosphatases display exquisite substrate specificity toward this physiologically relevant protein substrate, the phosphorylated p38α. Interestingly, PP2Cα does not appear to have the canonical KIM sequence, which may explain why PP2Cα has a high Km value for p38α.
In summary, this study reveals that the kinetic properties for p38α/pY and p38α/pT are different from those measured for ERK2/pY and ERK2/pT. The kinase activities of p38α/pT and p38α/pY are about 10- and 1000-fold lower than that of the fully active p38α/pTpY, respectively. Therefore, it will be very informative to determine and compare the crystal structure of the bisphosphorylated p38α with that of ERK2/pTpY in a future study. Our results suggest that the monophosphorylated form of p38α/pY may have different biological functions in vivo compared with ERK2/pY. In addition, with different phosphorylated forms of p38α as substrates, our kinetic analysis shows that the dual-specific MKP5, the tyrosine-specific HePTP, and the Ser/Thr-specific PP2Cα are all highly specific for p38α dephosphorylation. Thus, our results highlight the importance of carrying out detailed kinetic analysis of dephosphorylation reaction in vitro to identify and validate physiological substrates for protein phosphatases.
Supplementary Material
This work was supported in part by Grants 2006CB503900-2007CB914400 from the Ministry of Science and Technology of China and Grants 30425005-30770476 from National Natural Science Foundation of China. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1 and 2.
Footnotes
The abbreviations used are: MAPK, mitogen-activated protein kinase; MAP, mitogen-activated protein; ERK, extracellular signal-regulated protein kinase; MKK6, mitogen-activated protein kinase kinase 6; MKP, mitogen-activated protein kinase phosphatase; HePTP, hematopoietic protein-tyrosine phosphatase; PTP, protein-tyrosine phosphatase; ATF2, activating transcription factor 2; MOPS, 3-(N-morpholino)propanesulfonic acid; MESG, 7-methyl-6-thioguanosine; pNPP, p-nitrophenyl phosphate; p38α/pT and p38α/pY, p38α phosphorylated on Thr-180 and Tyr-182; p38α/pTpY, p38α phosphorylated on both Thr-180 and Tyr-182; LDH, lactate dehydrogenase; KIM, kinase interaction motif; Ni-NTA, nickel-nitrilotriacetic acid; EGF, epidermal growth factor; EGFR, EGF receptor; JNK, c-Jun N-terminal kinase; GST, glutathione S-transferase; DSP, dual specificity phosphatase; MKB, MAP kinase-binding.
Y.-Y. Zhang, Z.-Q. Mei, J.-W. Wu, and Z.-X. Wang, unpublished data.
References
- 1.Lewis, T. S., Shapiro, P. S., and Ahn, N. G. (1998) Adv. Cancer Res. 74 49-139 [DOI] [PubMed] [Google Scholar]
- 2.Cobb, M. H. (1999) Prog. Biophys. Mol. Biol. 71 479-500 [DOI] [PubMed] [Google Scholar]
- 3.Widmann, C., Gibson, S., Jarpe, M. B., and Johnson, G. L. (1999) Physiol. Rev. 79 143-180 [DOI] [PubMed] [Google Scholar]
- 4.Pearson, G., Robinson, F., Gibson, T. B., Xu, B., Karandikar, M., Berman, K., and Cobb, M. H. (2001) Endocr. Rev. 22 153-183 [DOI] [PubMed] [Google Scholar]
- 5.Kyriakis, J. M., and Avruch, J. (2001) Physiol. Rev. 81 807-869 [DOI] [PubMed] [Google Scholar]
- 6.Roux, P. P., and Blenis, J. (2004) Microbiol. Mol. Biol. Rev. 68 320-344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson, L. N., Noble, M. E. M., and Owen, D. J. (1996) Cell 85 149-158 [DOI] [PubMed] [Google Scholar]
- 8.Ahn, N. G., Seger, R., Bratlien, R. L., Diltz, C. D., Tonks, N. K., and Krebs, E. G. (1991) J. Biol. Chem. 266 4220-4227 [PubMed] [Google Scholar]
- 9.Payne, D. M., Rossomando, A. J., Martino, P., Erickson, A. K., Her, J. H., Shabanowitz, J., Hunt, D. F., Weber, M. J., and Sturgill, T. W. (1991) EMBO J. 10 885-892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zheng, C. F., and Guan, K. L. (1993) J. Biol. Chem. 268 11435-11439 [PubMed] [Google Scholar]
- 11.Seger, R., and Krebs, E. G. (1995) FASEB J. 9 726-735 [PubMed] [Google Scholar]
- 12.Zanke, B. W., Rubie, E. A., Winnett, E., Chan, J., Randall, S., Parsons, M., Boudreau, K., Mcinnis, M., Yan, M. H., Templeton, D. J., and Woodgett, J. R. (1996) J. Biol. Chem. 271 29876-29881 [DOI] [PubMed] [Google Scholar]
- 13.Canagarajah, B. J., Khokhlatchev, A., Cobb, M. H., and Goldsmith, E. J. (1997) Cell 90 59-869 [DOI] [PubMed] [Google Scholar]
- 14.Takekawa, M., Maeda, T., and Saito, H. (1998) EMBO J. 17 4744-4752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ogata, M., Oh-hora, M., Kosugi, A., and Hamaoka, T. (1999) Biochem. Biophys. Res. Commun. 256 52-56 [DOI] [PubMed] [Google Scholar]
- 16.Avdi, N. J., Malcolm, K. C., Nick, J. A., and Worthen, G. S. (2002) J. Biol. Chem. 277 40687-40696 [DOI] [PubMed] [Google Scholar]
- 17.Zhou, B., Wang, Z. X., Zhao, Y., Brautigan, D. L., and Zhang, Z. Y. (2002) J. Biol. Chem. 277 31818-31825 [DOI] [PubMed] [Google Scholar]
- 18.Camps, M., Nichols, A., and Arkinstall, S. (2000) FASEB J. 14 6-16 [PubMed] [Google Scholar]
- 19.Keyse, S. M. (2000) Curr. Opin. Cell Biol. 12 186-192 [DOI] [PubMed] [Google Scholar]
- 20.Zhang, Z. Y. (1999) Crit. Rev. Biochem. Mol. Biol. 33 1-52 [DOI] [PubMed] [Google Scholar]
- 21.Farooq, A., and Zhou, M. M. (2005) Cell. Signal. 16 769-779 [DOI] [PubMed] [Google Scholar]
- 22.Ward, Y., Gupta, S., Jensen, P., Wartmann, M., Davis, R. J., and Kelly, K. (1994) Nature 367 651-654 [DOI] [PubMed] [Google Scholar]
- 23.Muda, M., Boschert, U., Dickinson, R., Martinou, J.-C., Martinou, I., Camps, M., Schlegel, W., and Arkinstall, S. (1996) J. Biol. Chem. 271 4319-4326 [DOI] [PubMed] [Google Scholar]
- 24.Muda, M., Theodosiou, A., Rodrigues, N., Boschert, U., Camps, M., Gillieron, C., Davies, K., Ashworth, A., and Arkinstall, S. (1996) J. Biol. Chem. 271 27205-27208 [DOI] [PubMed] [Google Scholar]
- 25.Groom, L. A., Sneddon, A. A., Aleesi, D. R., Down, S., and Keyse, S. M. (1996) EMBO J. 15 3621-3632 [PMC free article] [PubMed] [Google Scholar]
- 26.Tanous, T., Moriguchi, T., and Nishida, E. (1999) J. Biol. Chem. 274 19949-19956 [DOI] [PubMed] [Google Scholar]
- 27.Killilea, S. D., Cheng, Q., and Wang, Z. X. (1998) Methods Mol. Biol. 93 23-33 [DOI] [PubMed] [Google Scholar]
- 28.Gill, S. C., and von Hippel, P. H. (1989) Anal. Biochem. 182 319-326 [DOI] [PubMed] [Google Scholar]
- 29.Roskoski, R., Jr. (1983) Methods Enzymol. 99 3-6 [DOI] [PubMed] [Google Scholar]
- 30.McClure, W. R. (1969) Biochemistry 7 2782-2787 [DOI] [PubMed] [Google Scholar]
- 31.Zhou, B., and Zhang, Z. Y. (1999) J. Biol. Chem. 274 35526-35534 [DOI] [PubMed] [Google Scholar]
- 32.Cheng, Q., Wang, Z. X., and Killilea, S. D. (1995) Anal. Biochem. 226 68-73 [DOI] [PubMed] [Google Scholar]
- 33.Webb, M. R. (1992) Proc. Natl. Acad. Sci. U. S. A. 89 4884-4887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sergienko, E. A., and Srivastava, D. K. (1994) Anal. Biochem. 221 348-355 [DOI] [PubMed] [Google Scholar]
- 35.Saxena, M., Williams, S., Brockdorff, J., Gilman, J., and Mustelin, T. (1999) J. Biol. Chem. 274 11693-11700 [DOI] [PubMed] [Google Scholar]
- 36.Saxena, M., Williams, S., Taskén, K., and Mustelin, T. (1999) Nat. Cell Biol. 1 305-311 [DOI] [PubMed] [Google Scholar]
- 37.Muñoz, J. J., Tárrega, C., Blanco-Aparicio, C., and Pulido, R. (2003) Biochem. J. 372 193-201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen, G., Porter, M. C., Bristol, J. R., Fitzgibbon, M. J., and Pazhanisamy, S. (2000) Biochemistry 39 2079-2087 [DOI] [PubMed] [Google Scholar]
- 39.Raingeaud, J., Gupta, S., Rogers, J. S., Dickens, M., Han, J., Ulevitch, R. J., and Davis, R. J. (1995) J. Biol. Chem. 270 7420-7426 [DOI] [PubMed] [Google Scholar]
- 40.Szafranska, A. E., Kevin, N., and Dalby, K. N. (2005) FEBS J. 272 4631-4645 [DOI] [PubMed] [Google Scholar]
- 41.Zhou, B., and Zhang, Z. Y. (2002) J. Biol. Chem. 277 13889-13899 [DOI] [PubMed] [Google Scholar]
- 42.Segel, I. H. (1975) Enzyme Kinetics, pp. 73-74, John Wiley & Sons, Inc., New York
- 43.Zhao, Y., and Zhang, Z. Y. (2001) J. Biol. Chem. 276 32382-32391 [DOI] [PubMed] [Google Scholar]
- 44.Graves, J. D., and Krebs, E. G. (1999) Pharmacol. Ther. 82 111-121 [DOI] [PubMed] [Google Scholar]
- 45.Cha, H., and Shapiro, P. (2001) J. Cell Biol. 153 1355-1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yao, Z., Dolginov, Y., Hanoch, T., Yung, Y., Ridner, G., Lando, Z., Zharhary, D., and Seger, R. (2000) FEBS Lett. 468 37-42 [DOI] [PubMed] [Google Scholar]
- 47.Nguyen, A. N., and Shiozaki, K. (1999) Genes Dev. 13 1653-1663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pettiford, S. M., and Herbst, R. (2000) Oncogene 19 858-869 [DOI] [PubMed] [Google Scholar]
- 49.Gronda, M., Arab, S., Iafrate, B., Suzuki, H., and Zanke, B. W. (2001) Mol. Cell. Biol. 21 6851-6858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barford, D. (1996) Trends Biochem. Sci. 21 407-412 [DOI] [PubMed] [Google Scholar]
- 51.Romani, A., and Scarpa, A. (1992) Arch. Biochem. Biophys. 298 1-12 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.