

Abstract
In alphaviruses, here represented by Semliki Forest virus, infection requires an acid-responsive spike configuration to facilitate membrane fusion. The creation of this relies on the chaperon function of glycoprotein E2 precursor (p62) and its maturation cleavage into the small external E3 and the membrane-anchored E2 glycoproteins. To reveal how the E3 domain of p62 exerts its control of spike functions, we determine the structure of a p62 cleavage-impaired mutant virus particle (SQL) by electron cryomicroscopy. A comparison with the earlier solved wild type virus structure reveals that the E3 domain of p62SQL forms a bulky side protrusion in the spike head region. This establishes a gripper over part of domain II of the fusion protein, with a cotter-like connection downward to a hydrophobic cluster in its central β-sheet. This finding reevaluates the role of the precursor from being only a provider of a shield over the fusion loop to a structural playmate in formation of the fusogenic architecture.
When an enveloped virus infects its target cell, the mechanism usually involves a step with hairpin refolding of the viral fusion protein to promote merging of virus and target membranes. Opposite to the class I fusion proteins, which are trimers from the start and in which the fusion-related refolding involves formation and backfolding of α-helical bundles, the class II fusion proteins elaborate on homotrimer formation to mediate membrane fusion with target membrane, as discussed by Kielian (1, 2) and others (3–5). In the alphaviruses, here represented by Semliki Forest virus (SFV),3 the fusion proteins are of class II and essentially lack helical motifs. Homotrimers of the fusion protein E1, formed in relation to the membrane fusion process, are stable associations (6–13). The strong interaction would be the driving force for completion of fusion, after low pH and membrane contact trigger. However, it would be suicidal for virus transmission if the fusion protein were allowed to create such a configuration prematurely. The SFV assembly pathway handles this problem by providing a chaperon protein, the precursor of glycoprotein E2, in SFV named p62. The p62 precedes the E1 in the proprecursor sequence (p62–6K-E1; see Scheme 1) and waits in the ER to form dimers with the nascent E1, thereby allowing transport to the Golgi compartment and forestalling E1 self-aggregation (10, 15). The E2 itself, if translocated into ER by a cleavable signal sequence, is not sufficient for the purpose. This was shown with an E3 deletion mutant, where the N-terminal E3 domain of the precursor was exchanged for a cleavable artificial signal sequence to preserve the membrane topology of authentic E2. In this E3 deletion mutant, expressed via a recombinant vaccinia virus, the heterodimerization of the spike proteins was abolished, and the E1 was completely retained in the ER (14). In the early Golgi compartment, the p62-E1 heterodimers may form trimers of dimers (16) that in the trans-Golgi undergo furin-dependent maturation cleavage (9, 17, 18), transport to the plasma membrane, and assembly with nucleocapsid components into infectious virus particles. The furin cleavage of p62 into the small external glycoprotein E3 and the membrane-anchored spike glycoprotein E2 is not a prerequisite for transport to the plasma membrane and virus assembly, since virions are formed in furin-deficient cells (19), as well as with cleavage-impaired p62 mutants (18–22). Such “nonmature” particles, or virion equivalents, are less sensitive to pH trigger for fusion than the maturation-cleaved wild type (WT) particles with E2 and are noninfectious under normal cell conditions. It is therefore assumed that the p62 protects the prespike structure against acid-induced reorganization with fusion loop exposure during the low pH passage through Golgi compartments. By the maturation cleavage in late trans-Golgi, the pH-sensitive virus shell architecture, prompted for merging interaction with a target membrane, is established.
SCHEME 1.

Pathway for the formation of acid sensitive spike configuration in SFV.
By what means does the E3 domain of the precursor constitute such major control? A first essential feature of the p62 would be to establish control over the nascent E1 to prevent E1 homotrimer formation during spike assembly. Although it would be expected that it exerts this function by a tighter E1 binding, or even an extra contact, compared with the E2 itself, it has never been shown that such a contact exists. The sequence of the about 66-residue-long E3 domain is relatively well conserved within the alphaviruses and implies conserved structural elements (Fig. 1A); The N-terminal domain is the signal sequence that was directly glycosylated after translocation into ER and retained as the N terminus of the precursor (23). This sequence is followed by a region dominated by a highly conserved pattern of Pro and Cys. In the C-terminal half of the molecule, the sequence indicates two α-helices with amphipathic patterns that would allow hairpin back-folding along each other's. After the helical domain follows a conserved Cys, making an even total number of cysteines, and at the C terminus a cluster of basic residues form the furin cleavage site, of which the outermost two may be removed after cleavage (24). In summary, the E3 domain that is the first part of the p62 entering into ER may comprise the retained signal sequence with its carbohydrate moiety, a cross-linked domain with a protruding hairpin pair of α-helices, and the furin cleavage region linking to the E2 sequence.
FIGURE 1.

A, structural elements in the E3 sequence. A Kyte-Doolittle hydropathy plot is shown under a schematic diagram depicting conserved structural elements of the E3 sequence in a set of alphaviruses. The sequences (accession numbers) are drawn from the Swiss Protein Data Bank (UniProtKB/Swiss-Prot): SFV (Q87051); SIN (P03316); BFV (P89946); AV (Q86925); WEEV (P13897); NNV (P22056); EEEV (P08768); RRV (P13890); SDV (Q8QL52); SPDV (Q8JJX0). SIN, Sindbis virus; BFV, Barmah Forest virus; AV, Aura virus; WEEV, Western equine encephalomyelitis virus; NNV, O'Nyong-Nyong virus; EEEV, Eastern equine encephalomyelitis virus; RRV, Ross River virus; SDV, sleeping disease virus rainbow trout; SPDV, Salmon pancreas disease virus. The conserved N-linked sugar site (CHO) in the signal peptide is indicated, as is the one close to the cleavage site occurring in SFV and a few other alphaviruses. B, pattern of fusion loop exposure relative pH in SFV WT and SQL particles. The availability of the fusion loop for external interaction was assayed using the fusion loop-specific monoclonal antibody E1f. In this ELISA, the plates were coated with equal protein amounts of purified SFV WT (SFV) or mutant (SQL) particles and blocked with bovine serum albumin. The E1f was introduced to the particles at different pH for 1 h, and the wells were then washed with a neutral buffer. Bound monoclonal antibody was quantified with the aid of a horseradish peroxidase-conjugated goat anti-mouse antibody. Maximum reading with the SQL sample (pH 5.0) was about 40% of that of the SFV WT (pH 6.2). To demonstrate the difference in pH profiles between the particles, the diagram shows the maximum-normalized readings. C, E3 content in the WT virus preparation at different stages of purification. The E3 content is measured as the amount of deglycosylated E3 relative the total spike protein content in the crude pellet, the pooled virion peak after sucrose or tartrate density gradient centrifugation, and the thereafter pelleted virions. As seen, the E3 is almost totally lost in the postgradient pellet, leaving 3% or less. D, a one-dimensional plot that shows the radial density distribution in the solved structures of SFV WT and mutant SQL. Locations of major regions of the particles are indicated above the graph.
In the whole virus structure, revealed by electron cryomicroscopy (cryo-EM), the trimeric spikes protrude above a protein shell layer that surrounds the virus at some distance above its membrane (25–31). Modeling of the solved crystal structure of the ectodomain of fusion protein E1 (32, 33) into cryo-EM-derived whole virion structures implies that E1 constitutes the protein shell by intermolecular E1-E1 interactions and internal interactions between its domain III and I (31–34). E1 is part of the spikes by heterodimeric interactions with E2. The fusion loop is thereby hidden by E2 under the rim of the spike wing (31–33). A similar organization is deduced from studies on sugar deletion mutants of Sindbis virus (34). There seems to be no direct contact between the E1 molecules within the spike, which is held together only by pairwise E2-E2 contacts (31). Thus, the E1 protein in the virion is inter- and intramolecularly self-associated but in an alternative fashion to the fusion-related configuration of stable trimers for which the atomic structure is determined (12, 13). The further domains of E1 that are required for homotrimerization are controlled by E2 (31, 33). Then, while the rearrangement of the virus envelope to form fusion-related E1 homotrimers seems complex enough as it is, what are the location and crucial interactions of the E3 domain that hold back the fusion-related refolding? Venien-Bryan and Fuller (35) early found that the E3 was primarily interacting with the E2 in the spike structure, and Paredes et al. (36), who studied a cleavage-impaired Sindbis virus mutant, proposed that the E3 domain in the nonmature virion would “protrude midway between the center of the spike complex and the tips.” In that constellation, the E3 domain would not be able to shield the fusion loop, at least not in the same spike.
We approach the question of how the p62 exerts its action by searching the location of the E3 domain in the whole particle structure of an SFV mutant, here referred to as SQL. In SQL, the furin cleavage site of p62 is impaired by amino acid substitutions (21, 22) and thereby gives rise to an acid-resistant virion. This is noninfectious at around pH 6, where the WT virus is prone to fuse. However, it may turn fusion-competent at a lower pH, indicating that the structure of the functional machinery remains in working order (37). The location of the E3 domain is then deduced from comparison of the SQL structure with our recently reported SFV WT structure (31). Our analyses give details on the mutant virion structure and essentially confirm the location of the E3 domain suggested by the Paredes studies on Sindbis virus (36). A similar location would also be deduced from the structure of the same SFV mutant studied by Ferlenghi et al. (27). Furthermore, a direct contact of the p62 with a hydrophobic cluster in the E1 molecule is demonstrated. The properties of this contact and implications for how the functional spike architecture is created are discussed.
EXPERIMENTAL PROCEDURES
Wild-type and Mutant Virus—The SFV WT used in this study was generated from the plasmid pSFV4 (38), and the mutant SQL was generated from a corresponding plasmid where the sequence RHRR in the p62 cleavage site is replaced with SHQL (21). The mutant virus is noninfectious, but cleavage with chymotrypsin removes the block and results in wild type-like infectivity (21).
Virus Purification by Tartrate Gradient Centrifugation and Quality Control—The SFV4 was propagated and purified, essentially as described previously (29, 39). The SFV SQL was activated for infection by chymotrypsin treatment but otherwise handled in the same way as the wild type virus. Briefly, monolayers of BHK-21 cells were grown in 225-cm2 T-flasks using Glasgow's modified Eagle's minimal essential medium (Invitrogen), supplemented with 5% fetal calf serum, 10% tryptose phosphate broth, 2 mm glutamine, 20 mm HEPES (Sigma), and 20 μg/ml cholesterol. The SQL stock was activated on ice by α-chymotrypsin for 2 h, when the reaction was quenched with aprotinin (21). At 90% confluence, the cells were infected with SFV WT or the chymotrypsin-activated SQL and further incubated at 37 °C. Virus was harvested from the cell supernatant at 18 h postinfection. The virus-containing supernatant was cleared from cell debris, and the virus was pelleted at 17,000 × g at 4 °C for 18 h. The virus pellet was soaked in TNM buffer (50 mm Tris-HCl, 50 mm NaCl, 10 mm MgCl2, pH 7.4) and applied to isopycnic centrifugation using a 10–30% (w/w) potassium tartrate density gradient in 0.1 m MOPS, pH 7.4, run at 100,000 × g for 5 h at 4 °C. The virus was eluted from the gradient, diluted with TNM buffer, and pelleted by centrifugation at 100,000 × g for 5 h at 4 °C. The final virus pellet was soaked in TNM buffer and kept at 4 °C to avoid structural damage. For comparison of E3 content, a parallel experiment was done by the traditional scheme in the literature (i.e. using sucrose to create the density gradient). The quality of the virus was controlled by SDS-PAGE, ELISA, and negative stain EM. For structural analyses, the samples were applied on cryogrids, plunge-frozen into liquid ethane, and transferred to liquid nitrogen and subsequently to liquid helium before microscopy imaging.
ELISA for pH-dependent Epitope Exposure—ELISA experiments were done using Protein A (GE Healthcare) affinity-purified mAb E1f against the E1 glycoprotein (39). The SFV WT and mutant SQL were purified using tartrate gradient ultracentrifugation, as described above (29). Equal amounts (∼85 ng/well) of purified SFV WT and SQL were coated overnight at 4 °C onto 96-well ELISA plates (high binding enzyme immunoassay/radioimmunoassay plate; Corning Glass). After blocking with TBS-B (10 mm Tris-HCl, 150 mm NaCl, 5% bovine serum albumin, pH 7.4) for 1 h, the mAb E1f, suspended in a series of buffers of different pH, was added (∼50 ng/well). The pH of these buffers was obtained by mixing appropriate volumes of 50 mm Tris and 50 mm MES, both containing 50 mm NaCl. After a 1-h incubation, the excess of antibody was removed by washes with TBS-b (10 mm Tris-HCl, 150 mm NaCl supplemented with 0.5% bovine serum albumin, pH 7.4) before a 1-h incubation with an Fc-specific, peroxidase conjugated, anti-mouse reporter antibody (A-2554; Sigma). Finally, the wells were washed with TBS-b, and bound reporter antibody was developed using ortho-phenylenediamine dihydrochloride (Dako A/S) according to the manufacturers' recommendations. The reaction was quenched when adequate color intensity was reached or after ∼15 min, and the light absorbance at 490 nm was measured in a PowerwaveX spectrophotometer (Bio-Tek Instruments Inc., Winooski, VT).
Cryo-EM and Image Processing—The SFV WT and SFV SQL specimens were prepared for electron cryomicroscopy and recorded under low dose conditions in a JEM3200F field emission gun transmission electron microscope equipped with the top entry type liquid helium cold stage at a magnification of 40,000 and an accelerating voltage of 300 kV. Micrographs were digitized and further bin-averaged to give a step size of 14 μm, corresponding to 3.5 Å/pixel at the specimen. Particle images were manually boxed out using the RobEM package. The parameters of orientation and origin were determined by model-based polar Fourier transform routines (40, 41). The resolution of the final reconstruction was determined using a 0.5 cutoff of the Fourier shell correlation coefficient between the “odd” and “even” image reconstructions. The numbers of particles included in the reconstructions are 834 (SFV WT) and 1,580 (SQL). Image reconstruction procedures were carried out essentially as described previously (31), using the initial SFV model provided by Dr. Bomu Wu. We used the Iris explorer software (NAG, Inc., Downers Grove, IL), supplemented with custom-made modules4 for the three-dimensional visualization, along with Pymol™ (DeLano Scientific LLC; available on the World Wide Web).
RESULTS
The pH Profile of Fusion Loop Exposure—A functional difference of the SFV WT virus and the SQL is the threshold for fusion activation. Here, we tested the exposure of the fusion loop relative pH as the capacity to bind mAb E1f, an antibody that recognizes the fusion loop sequence (39). Thus, coated in an ELISA plate at the same protein load, the particles were exposed to mAb E1f at different pH, and the bound antibody, reflecting the accessible sequence, was measured. The pH profiles obtained showed an optimum for the WT at pH 6.2, similar to a previously reported value (39), whereas the peak for the mutant appeared first at below pH 5 (Fig. 1B). The maximal response was lower with the mutant than with the WT virus, reaching its optimum at about 40% of the WT value. This may partly reflect that the antibody is less stable at the lower pH and/or that the mutant particle is too rigid to allow access to the same number of sites as the WT particle. Therefore, we present the result normalized to optimal mAb E1f binding value, simply to demonstrate the difference in the pH response profile.
Spike Homogeneity—A problem with structure determination by cryo-EM is the purity and homogeneity of the material studied. Therefore, the virus material was extensively purified, and loosely attached components were removed. In SFV, the E3 may partly stay associated to the WT virions after maturation cleavage. This varied with the purification efficiency (Fig. 1C). At the final stages of the extensive purification of SFV WT particles for cryo-EM, the glycoprotein E3 was essentially removed. Such E3-depleted particles have been the source for our recently presented structure of SFV (29, 31, 42). As medium for the density gradient centrifugation, tartrate has been preferred to sucrose, since it is easier to remove and therefore allows a better situation for the imaging.
A similar homogeneity problem is imposed by the incomplete maturation cleavage of the glycoprotein E2 precursor, p62, in the WT virus. This cleavage is not essential for the transport of the envelope proteins to the plasma membrane and assembly of virions (18, 21), which is why traces usually remain in the WT virus preparations. This may also vary with the condition of the producer cells. Under the conditions used in this study, we found that 5 ± 3%, n = 12 (mean ± S.D.), of the heterodimers contained p62. It is uncertain whether this inhomogeneity in spike composition was arbitrary distributed. However, its impact on the final reconstruction of the WT structure, after averaging and refinement, would be minor. In the mutant SQL, the p62 equivalent, p62SQL, remained uncleaved (no E2 was present), and the composition of the spikes would be homogeneous.
Three-dimensional Reconstruction of WT and Mutant of Semliki Forest Virus—The structure of the cleavage-impaired SFV mutant SQL determined in this work was achieved at a resolution computed to 17 Å. The WT particle structure at neutral pH was earlier obtained at a resolution computed to 11 Å (31). For an accurate comparison of the two particles, the reconstruction of SFV WT was here band pass-filtered to 17 Å resolution. With a diameter of 703 Å, the SQL particles are only slightly larger than the WT ones, reaching a diameter of 700 Å. As judged from the one-dimensional radial density distribution plot (Fig. 1D) and whole particle renderings (not shown), the general organization of the two icosahedral particles is essentially similar, except for the details in the spike morphology described below.
Revealing the Location of the E3 Domain—Common and discriminating details between the SFV WT and SQL particles are revealed by superimposing three-dimensional reconstructions of the two structures. They overlap well in the limb and shell domains of the envelope above the membrane and in the center of the spikes. The part of the head domain of the spike, known to cover the distal part of E1 domain II with its fusion loop, is also overlapping, as demonstrated in the close-up top and side views of the 3-fold spike in Fig. 2A (σ = 1 rendering). However, the SQL particle structure (shown as a white net) presents an extra density at the side of the spike head facing the quasi-2-fold axis. Most remarkably, this density forms a sidewise protrusion covering the small thumblike pointer of the WT structure. This extra volume would be constituted by a configuration imposed by the lack of the maturation cleavage and most probably represents the E3 domain of the p62SQL.
FIGURE 2.
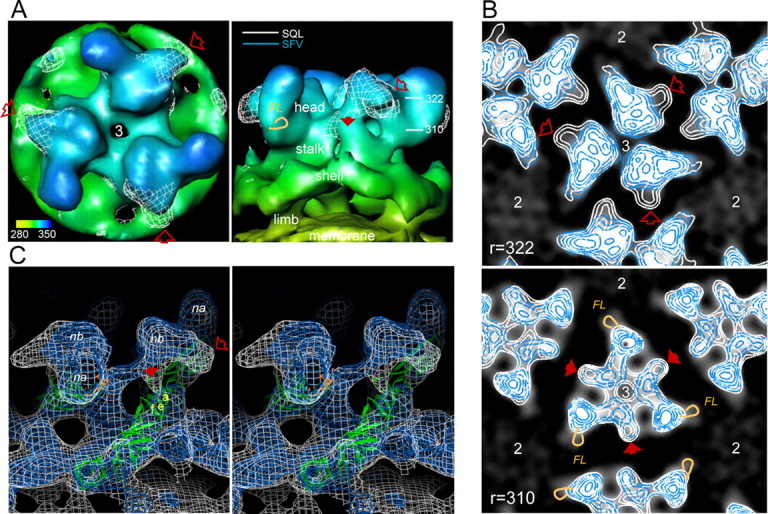
A, top and side views of the 3-fold spike structure, rendered at a stringency of σ = 1. The WT structure is shown as a continuous surface with a radial color code from yellow at the top of the membrane, through green at the shell layer, to dark blue at the top of the spike. The side view shows the side toward the neighbor spike. The structure of the SQL is superimposed in a white net rendering. The two structures are essentially in good agreement in large domains of the envelope and in part of the spike structure. However, a prominent extra density in the mutant particle is protruding at the side of the spike head (open red arrow) with a trace downward along the stalk region (solid red arrow). Radial distances in Å, marked to the right in the side view panel, refer to the position of the radial cuts shown in B. The location of the fusion loop, FL, is indicated by an orange loop at the side of the spike wing. B, superimposed radial cuts through the spike head (same orientation as in A, top view) of SFV WT and SQL to reveal mutant specific configurations. A serial of virtual 6-Å radial sections through the spike head domain, from the top and downward, were made with inlaid density contours for structural stringency (σ = 1 to σ = 5). Here the ones at radii 322 and 310 Å are shown with SFV WT and SQL density contours colored in blue and white, respectively. The background is created from the WT structure, with the high density within the WT spike in opaque white to make the blue lines visible. The arrows refer to the positions marked in A and point to SQL-specific substructures (high stringency in white contours only). In the 310-Å section, the fusion loop position is indicated by orange loops in the 3-fold spike and one of the neighbor spikes. The positions of the 2- and 3-fold axes of the particle are indicated. C, high stringency renderings in stereo view. To show details of the difference between the SFV WT and SQL structures they are both rendered as transparent net surfaces at a stringency of σ = 3 and superimposed. The WT is represented by a blue net, and the SQL is shown in white. The side protrusion of the mutant is pointing out from the position of the small pointer seen in the WT structure. Compared with the side view in A, the spike is tilted and rotated to demonstrate the vertical cotter structure (solid red arrow) observed as a node among the radial sections (310 Å in B). The cotter structure goes vertically under the spike head connecting down to a narrow location in the common structure. This location is deduced from molecular fitting to be occupied by glycoprotein E1, here shown as a secondary structure schematic diagram in green, with the fusion loop in orange. The strand a, strand e, and strand f of the central β-sheet in E1 domain II are indicated in yellow. The common high stringency node a and node b of the spike structure are indicated in white as na and nb, respectively.
To reveal to what extent the differences seen in the σ = 1 surface rendering represent extra volume or variations in the WT structure due to the maturation state, a serial of radial sections through the spike head domain with inlaid contour curves for structural stringency from σ = 1 to σ = 5 were made, as shown in the Fig. 2B. Here, the WT structure is presented as blue contour curves, and SQL structure is shown as white contour curves on a black background (with the high density within the WT spike in opaque white to make the lines visible). This demonstrates the locations of stable domains in the structure as centers of high accuracy. In an earlier paper, we identified a number of such “nodes” in the WT envelope (31). These are retained with minor variation in their outlines in the SQL structure, as seen from the domains with a coinciding blue and white pattern. Structures unique to the SQL are recognized as white contours lacking a corresponding outfit of blue curves. The SQL-specific extra volume appearing in Fig. 2A as a white net that reaches slightly above the WT structure and forms a side bulk down to a radius of 318 Å can be followed as the outer white contour in Fig. 2B (open arrow). The fusion loop would locate at a radius of 310 Å (FL; the location is marked by an orange loop). At higher radii, the E3 bulk has an anticlockwise direction that may possibly produce a shield for access between the spikes down to the fusion loop in the neighbor spike wing.
One can also find a structure that is partly within the σ = 1 rendering of the WT but forms a unique configuration of high stringency in the SQL structure. This starts from the bottom of the bulk protrusion, where it only slightly deviates from the WT (solid arrow). It appears as a high stringency density, or node, reaching above σ = 5. This node continues downwards from a radius of 316 Å and finally connects to the WT-SQL common density at radius 304 Å. It is indicated by a solid arrow in Fig. 2B, r = 310. This vertical structure connects the inner bottom of the bulky side protrusion down to the shell domain in the particle where the E1 glycoprotein resides (see stereo view in Fig. 2C).
E3 Domain Contact with E1—By rendering the structures of the two particles at high stringency (σ = 3), details in the more stable domains can be compared, as here shown in stereo in Fig. 2C. The WT structure is represented as blue, and the superimposed SQL structure is shown as a white net surface. By this means, it is seen that there is a good agreement between large parts of the two structures and the domains of the WT envelope assigned to external E1 and E2 (31) are essentially contained within the SQL structure. Therefore, the location and configuration of E1 and of the E2 domain of the p62 molecule would be very similar in the WT and the mutant. In the SQL-specific sideways bulk, the high stringency core appears as a gripper at some distance above the common structure where the central β-sheet (Roussel terminology) (33) of domain II of E1 locates and reaching out over the empty cleft between the spikes. A notable feature is a narrow pillar or cotter-like structure (see also Fig. 2, A and B, solid red arrow) that connects the gripper down to the common structure at a radius of 304 Å. This is clearly separate from the common spike stalk structure but is overshadowed by the less stringent rendering at σ = 1 of the SFV WT (Fig. 2A, solid arrow). In the σ = 3 rendering (Fig. 2C) and at higher stringencies (Fig. 2B), it appears as a cotter-like node, unique for the SQL structure, which otherwise shows a good agreement with the WT in the nearby stalk region (Fig. 2C). The connecting cotter points down to a narrow site occupied by E1. By deduction from the fitted molecular structure of external E1 in the WT virus structure (31), this contact meets a hydrophobic cluster in E1 domain II (Fig. 3, hydrophobic surface in white). This hydrophobic cluster in E1 is formed by residues in strand a, strand e, and strand f of the antiparallel central β-sheet of domain II, which bridges between the elongated fusion loop-bearing β-sandwich and the domain I and II connecting β-sheet (Fig. 4). Part of the latter is often referred to as the hinge region (31–33, 43). The structure implies that the fusion loop-bearing β-sheet is covered by E2-assigned domains of the WT and SQL structures alike (Fig. 2).
FIGURE 3.

The SQL-specific high stringency cotter structure and its contact with E1 domain II. A detail of the spike is in focus to demonstrate the unique cotter node of the SQL structure. The fitted atomic structure of E1 is shown in a surface representation with surface-directed hydrophobic side chainsin white, histidyls in blue, Ser120 and Tyr122 in pale green, and Glu209 in light red. The two right panels highlight, from different angles, possible contacts between the cotter structure and E1 residues. The strands a, e, and f in the E1 central β-sheet are highlighted in yellow.
FIGURE 4.
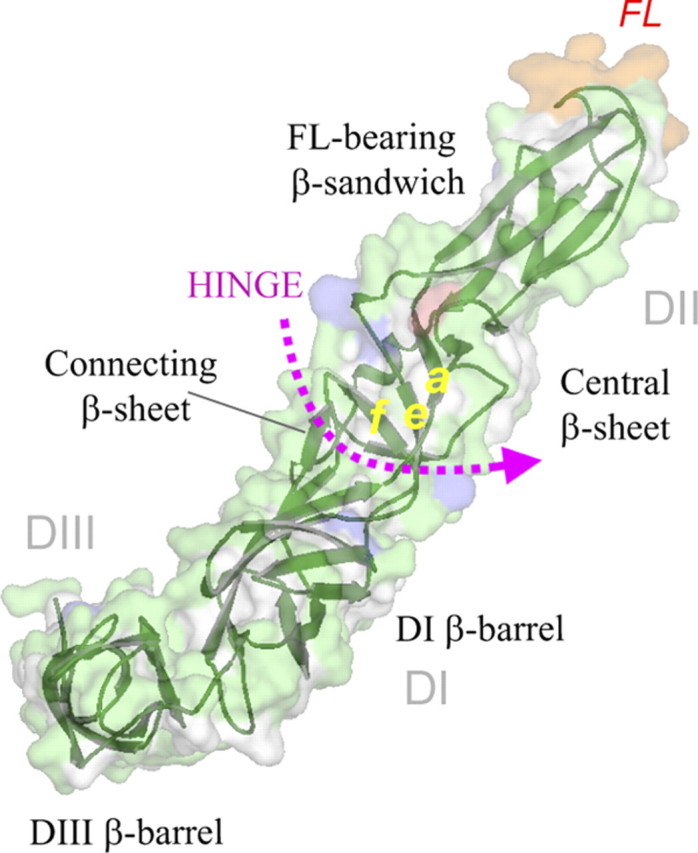
The nomenclature of the domains (DI–III) and β-sheets of the E1 structure. The E1 molecule (33) is viewed in the same perspective and color coding as in Fig. 3. The fusion loop is in orange.
According to the fitting of the atomic structure of external E1, the amino acid residues that would be very close or directly involved in the contact are Leu44 and Ile47 in strand a, and Tyr122 and Ser120 in strand e and, contacting the side of the cotter node, also the Val208 and, above that, Glu209 in the h-h′ loop (Fig. 3, insets). Furthermore, in close vicinity behind the cotter node toward the center of the spike, the His118 in strand e points toward the cotter domain, whereas the His116 of the short helix-η1, located above the central β-sheet, points away. Of the apparently contact-forming residues, all are common among the alphaviruses, but none is absolutely conserved. Residues of this domain that have been observed in mutation studies are Leu44 in strand a and Val178 in strand f, both related to lipid dependence (33). In the reported structure of the E1 homotrimer, this hydrophobic surface domain is made internal by a slight torsion of the region (44). In the homotrimer structure, the Val44 in one subunit is facing Pro382-Pro383 of domain III (the limb connector sequence) in the adjacent subunit. The limb region in the whole virus structure connects E1 and E2, in consort, to their transmembrane domains. In the fusion-related E1 homotrimer, the limb sequence of E1 instead follows the cleft between the E1 subunits toward the fusion loops, thus representing a past membrane-merging structure. We conclude that the contact domain of the cotter node is a functional center in the spike structure.
DISCUSSION
Validity and interpretation of the Structural Information
Due to the reconstruction technique, using multiple particle projections and symmetry averaging (40, 41), the resolution and accuracy of the reconstruction is highly dependent on the homogeneity of the sampled particles, their random orientation, and their symmetrically positioned elements within each particle. Furthermore, secondary structure elements, such as β-sheets, give good signals both because the atoms are close, producing a high density of matter, and because they statistically provide a homogeneous configuration with little variation within and among the particles. In the reconstruction, such elements show up as high stringency centers or structural nodes (31). Therefore, what is usually referred to as full volume, or σ = 1 rendering, includes not only high to low density of matter but also averaged, nonsymmetrically distributed information. Here we elaborate on the structure of an enveloped, spherical virus with icosahedral symmetry. The information is strong and accurate at locations where there is a high density of matter and a homogeneous presence of that structure in the particle. Variable occurrence of a substructure is averaged out, resulting in a weak signal in the final picture.
When we are here comparing the mutant particle that contains the p62SQL equivalent of the precursor and E1 as the constituents of the spike, instead of E2 and E1 of the WT, the theoretical difference in matter is the E3 domain. However, the WT may contain minor amounts of p62 that, along with the E2, appear in complex with the E1 and traces of E3 (Fig. 1C). It is not known if the proportions of these are the same in all particles or how they may distribute within a particle. From our measures, we understand that (i) the remaining amount of E3 would be insignificant and (ii) the minor content of nonprocessed p62 that is always found to be there in the WT preparation would add a small volume to the E3 domain location. Thus, provided there is full agreement in other domains, a comparison of SQL and WT structures would give above 90% of the E3 domain volume in the mutant particle. This would be considered as trustworthy information regarding location and shape. The stringency in appearance is, as always dependent on consistency in configuration. In those domains where there is only a low stringency overlap of a high stringency structure in the superimposed reconstructions, it would be plausible that trace content of p62 in the WT is contributing. From this, we conclude that the location of the E3 domain is represented by the extra volume of the structure seen in the SQL sample compared with the WT but with a minor contribution to the real volume provided by the nonprocessed p62 in the WT itself. Thus, the E3 domain in SQL appears to protrude out from the node b in the top of the head region of the spike (Figs. 2C and 3 (nb)). It is tempting to assume that the thumblike pointer of the WT, toward the spike side, is the N-terminal of the E2, possibly enhanced by traces of the p62 that remain uncleaved in the WT particle. This would logically place the furin cleavage site in the precursor available for external processing at the top of the spike.
The Stringency in Rendering and What It May Reveal
The occurrence of a “unique” structure may represent a configuration of domains other than the E3 in the mutant particle. The E2 domain may occur in a different configuration in the p62 than as E2, and p62 may cause configurations of the envelope of the particles other than those found with the processed E2. However, in comparing the superimposed reconstructions, one has to conclude that the WT is well contained within the SQL structure, and the major structural nodes of the WT do essentially coincide with nodes in the SQL. The major nodes of the WT are described in a recent publication where their rearrangements during acidification could be followed (31). As deduced from the fitted E1 ectodomain, they would represent stable subdomains, like β-sheets. In the present study, where the WT structure is well retained within the SQL structure, the appearance of the extra cotter-like node would be a representation of a domain missing in the WT (i.e. be part of the E3 domain of the p62SQL molecule). This cotter node provides a direct and specific interaction with the E1 molecule that includes residuals involved in lipid responsiveness. It would control the bending and torsion in the hinge region of E1 that is part of the reconfiguration occurring during acidification and formation of homotrimers.
Cotter Bolt Theory and Implications
The maturation cleavage would allow the release of E3 and, like the removal of a cotter bolt, promote the further hold of the E1 top domain to be released by a fusion-related trigger. In the WT structure, the E1 is held by E2 at the transmembrane limb and shell regions of the envelope as well as by the heavy cover at the top of the spikes, with the rigid bodies that we earlier assigned node a and node b. The node a in the corner of the top triangle constitutes the control of the fusion loop-bearing β-sheet, whereas the node b connects node a to the E2-E2 pairwise spike connecting structure (31). In the mutant, the E3 domain appears to protrude out from node b into the space between the spikes and under it form a cotter, connecting to the central β-sheet of domain II in E1 (Figs. 2C and 3).
The Cleavage Site Location Would Be Available for External Processing—Assuming that the furin cleavage site, producing the C terminus of E3 and the N terminus of the E2, is located at the pointer location in node b, it is logical that the bulky side protrusion would be filled by the two predicted and relatively long C-terminal helices (Fig. 1A shows the sequence and assumed structural domains of E3) and that the N-terminal half of the sequence with the Pro-Cys domain forms the bottom of the gripper and the signal peptide forms the cotter, connecting down to the common structure. The connecting tools would be a salt bridge and a hydrogen bond, stabilized by hydrophobic effects created by the surrounding residues (Fig. 3). By such a configuration, the E3 domain would lock the release of E1 down to a pH providing carboxyl protonation, which is lower than required for WT fusion (the pH threshold of which implies histidyl protonation to be essential). If cleavage occurs, the E3 may well remain in E1 contact but would not seriously counteract its bending out from the stalk when E2 allows. And the contact point would not be shielded by the hydrophobic milieu to the same extent. Thus, after cleavage at the furin site, the particles would adopt to higher sensitivity to acidification. However, some mutants are described that prevent fusion without preventing cleavage; an example is the fus-1 mutant (45), with E2 T12I, an amino acid substitution that would locate close to the cleavage site. The much higher hydropathy of the isoleucine side chain, compared with that of threonine, would stabilize and retain the precursor configuration of the E2 domain, and possibly the cleaved E3 may retain in locking position in a configuration stable enough to withstand moderate pH variations. The pH suppression of fus-1 is reported to be less than in our cleavage-impaired mutant (45).
The Cotter Node May Represent the Signal Sequence—The cotter node may represent the signal sequence that remains as the N-terminal sequence in the E2 precursor in alphaviruses after the capsid protein is folded and has cleaved itself off of the growing structural polyprotein (see Scheme 1). If so, the N-terminal amino group of the signal sequence could be stabilized in a salt bridge with the γ-carboxyl of E1 Glu209, located in the h-h′ loop above the central β-sheet in E1 domain II. Elaborating on this thinking, the side chain hydroxyl of the N-terminal serine (in some viruses threonine) of the signal peptide sequence would fit in position to interfere with E1 Tyr122 and/or Ser120 within a domain characterized by hydrophobic residues both in the signal sequence domain of the E3 and in the E1, where the cotter node seems to connect to Leu44, Ile47, and Val208 in strands e and a of the central β-sheet (Fig. 3). This hypothetical configuration is supported by a good fit (the narrow cotter node would accommodate a single chain but not a loop structure) and the conserved hydrophobic character of the contact in E1 domain, surrounded by polar structures. The highly conserved glycosylation site with its attached sugar moiety in the signal peptide would help to shield the contact in the same way. A weak point in this hypothesis is that the suggested contacts in E1 are not all highly conserved among the alphaviruses. However, the strength of the E3 domain involvement would also vary, as partly reflected in the variable ease of the E3 to leave the particle after cleavage, commented on in the literature (24, 35).
Hypothetical Order of Events to Produce Spike Metastability—The formation of the E3 contact would go back to the time when the p62-E1 heterodimers form in the ER (Scheme 1). During biosynthesis, the translocated signal sequence would be prone to find a hydrophobic surface, like the one present in domain II in the nascent E1 and, by associating there, guide the further orientation and heterodimer interactions. The E3 domain contact would be a central element in stabilizing the heteromeric association in the Golgi compartments and help to form the proper trimeric configuration that eventually will appear as spikes in the budding virion. In the virion, the protein shell domain is established by E1-E1 contacts, which are among the first to be released on experimental acidification (31). The somewhat acidic pH of the Golgi would there prevent shell formation, whereas at the plasma membrane the neutral conditions should promote it. In that sense, the shell association is a reversible zipper in the complex network, whereas the crucial metastability for fusion activation is created by the spike head associations, formed under the guidance of the E3 domain. This makes it resist the pH of the ER-Golgi compartment. At the mature state, the p62 scaffold is broken by the cleavage, and the E3 cotter bolt can be removed from its holder, so that it no longer locks the spike complex. This can then dissociate when exposed to endosomal low pH and a target membrane of the proper composition. The pH and lipid trigger of the E2 to release the remaining control of E1 and its drastic reshuffling in the envelope during membrane contact and merging remain to be understood.
In conclusion, we show that the E3 domain of the precursor of E2 locates in the head of the spike and fills part of the space between them like a bulky side protrusion. It creates a gripper over the central β-sheet in domain II of the fusion protein E1, with a cotter-like connection down to it. This domain of the fusion protein is central in its different configurations. The precursor contact would be established at an early stage in the biosynthetic pathway, possibly providing the crucial contact for p62-E1 oligomerization in the ER. We interpret the cotter-like connection to be formed by the hydrophobic signal sequence constituting the N-terminal region of the E3 domain.
Acknowledgments
We thank Drs. Li Xing (Davis), Kazuyoshi Murata (Tokyo) (SFV WT), and Kenji Iwasaki (Osaka) (SFV SQL) for skillful assistance with freezing and electron cryomicroscopy and Drs. R. H. Cheng (Davis) and Yoshinori Fujiyoshi (Tokyo) for kindly providing outstanding EM facilities and engagement in the SFV studies. We also thank Birgitta Lindqvist, Che-Yen Wang, and Leif Bergman for excellent technical support and encouragement.
This work has been supported by Swedish Medical Research Council, European Union 6th Framework Program (Project 512740) and by The Swedish Knowledge Foundation together with Crystal Research AB (Lund, Sweden), through the Industrial Ph.D. program in Medical Bioinformatics (to S.-R. W.) at the Karolinska Institutet, Sweden. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: SFV, Semliki Forest virus; EM, electron microscopy; cryo-EM, electron cryomicroscopy; MES, 2-(N-morpholino)ethanesulfonic acid; WT, wild type; ELISA, enzyme-linked immunosorbent assay; MOPS, 3-(N-morpholino)propanesulfonic acid; mAb, monoclonal antibody; ER, endoplasmic reticulum; C, E1, E2, and E3, structural proteins of Semliki Forest virus.
L. Bergman unpublished data.
References
- 1.Kielian, M. (2006) Virology 344 38–47 [DOI] [PubMed] [Google Scholar]
- 2.Kielian, M., and Rey, F. A. (2006) Nat. Rev. Microbiol. 4 67–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.White, J. M., Hoffman, L. R., Arevalo, J. H., and Wilson, I. A. (1997) in Structural Biology of Viruses (Chiu, W., Burnett, R. M., and Garcea, R. L., eds) pp. 80–104, Oxford University Press, Oxford
- 4.Malashkevich, V. N., Singh, M., and Kim, P. S. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 8502–8506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lau, W. L., Ege, D. S., Lear, J. D., Hammer, D. A., and DeGrado, W. F. (2004) Biophys. J. 86 272–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wahlberg, J. M., Bron, R., Wilschut, J., and Garoff, H. (1992) J. Virol. 66 7309–7318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bron, R., Wahlberg, J. M., Garoff, H., and Wilschut, J. (1993) EMBO J. 12 693–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klimjack, M. R., Jeffrey, S., and Kielian, M. (1994) J. Virol. 68 6940–6946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garoff, H., Wilschut, J., Liljestrom, P., Wahlberg, J. M., Bron, R., Suomalainen, M., Smyth, J., Salminen, A., Barth, B. U., and Zhao, H. (1994) Arch. Virol. Suppl. 9 329–338 [DOI] [PubMed] [Google Scholar]
- 10.Andersson, H., Barth, B. U., Ekstrom, M., and Garoff, H. (1997) J. Virol. 71 9654–9663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gibbons, D., Ahn, A., Chatterjee, P., and Kielian, M. (2000) J. Virol. 74 7772–7780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gibbons, D., Erk, I., Reilly, B., Navaza, J., Kielian, M., Rey, F., and Lepault, J. (2003) Cell 114 573–584 [DOI] [PubMed] [Google Scholar]
- 13.Gibbons, D., Reilly, B., Ahn, A., Vaney, M., Vigouroux, A., Rey, F., and Kielian, M. (2004) J. Virol. 78 3514–3523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lobigs, M., Zhao, H. X., and Garoff, H. (1990) J. Virol. 6443 46–55 [Google Scholar]
- 15.Barth, B. U., Wahlberg, J. M., and Garoff, H. (1995) J. Cell Biol. 128 283–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mulvey, M., and Brown, D. T. (1996) Virology 219 125–132 [DOI] [PubMed] [Google Scholar]
- 17.Jain, S. K., DeCandido, S., and Kielian, M. (1991) J. Biol. Chem. 266 5756–5761 [PubMed] [Google Scholar]
- 18.Salminen, A., Wahlberg, J. M., Lobigs, M., Liljestrom, P., and Garoff, H. (1992) J. Cell Biol. 116 349–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang, X., Fugere, M., Day, R., and Kielian, M. (2003) J. Virol. 77 2981–2989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Glasgow, G. M., Sheahan, B. J., Atkins, G. J., Wahlberg, J. M., Salminen, A., and Liljestrom, P. (1991) Virology 185 741–748 [DOI] [PubMed] [Google Scholar]
- 21.Berglund, P., Sjoberg, M., Garoff, H., Atkins, G. J., Sheahan, B. J., and Liljestrom, P. (1993) Nat. Biotechnol. 11 916–920 [DOI] [PubMed] [Google Scholar]
- 22.Tubulekas, I., and Liljestrom, P. (1998) J. Virol. 72 2825–2831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garoff, H., Huylebroeck, D., Robinson, A., Tillman, U., and Liljestrom, P. (1990) J. Cell Biol. 111 867–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mayne, J. T., Rice, C. M., Strauss, E. G., Hunkapiller, M. W., and Strauss, J. H. (1984) Virology 134 338–357 [DOI] [PubMed] [Google Scholar]
- 25.Smith, T. J., Cheng, R. H., Olson, N. H., Peterson, P., Chase, E., Kuhn, R. J., and Baker, T. S. (1995) Proc. Natl. Acad. Sci. U. S. A. 92 10648–10652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fuller, S. D., Berriman, J. A., Butcher, S. J., and Gowen, B. E. (1995) Cell 81 715–725 [DOI] [PubMed] [Google Scholar]
- 27.Ferlenghi, I., Gowen, B., de Haas, F., Mancini, E. J., Garoff, H., Sjoberg, M., and Fuller, S. D. (1998) J. Mol. Biol. 283 71–81 [DOI] [PubMed] [Google Scholar]
- 28.Mancini, E. J., Clarke, M., Gowen, B. E., Rutten, T., and Fuller, S. D. (2000) Mol. Cell 5 255–266 [DOI] [PubMed] [Google Scholar]
- 29.Haag, L., Garoff, H., Xing, L., Hammar, L., Kan, S. T., and Cheng, R. H. (2002) EMBO J. 21 4402–4410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mukhopadhyay, S., Zhang, W., Gabler, S., Chipman, P. R., Strauss, E. G., Strauss, J. H., Baker, T. S., Kuhn, R. J., and Rossmann, M. G. (2006) Structure 14 63–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu, S. R., Haag, L., Hammar, L., Wu, B., Garoff, H., Xing, L., Murata, K., and Cheng, R. H. (2007) J. Biol. Chem. 282 6752–6762 [DOI] [PubMed] [Google Scholar]
- 32.Lescar, J., Roussel, A., Wien, M. W., Navaza, J., Fuller, S. D., Wengler, G., and Rey, F. A. (2001) Cell 105 137–148 [DOI] [PubMed] [Google Scholar]
- 33.Roussel, A., Lescar, J., Vaney, M. C., Wengler, G., Wengler, G., and Rey, F. A. (2006) Structure 14 75–86 [DOI] [PubMed] [Google Scholar]
- 34.Pletnev, S. V., Zhang, W., Mukhopadhyay, S., Fisher, B. R., Hernandez, R., Brown, D. T., Baker, T. S., Rossmann, M. G., and Kuhn, R. J. (2001) Cell 105 127–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Venien-Bryan, C., and Fuller, S. D. (1994) J. Mol. Biol. 236 572–583 [DOI] [PubMed] [Google Scholar]
- 36.Paredes, A. M., Heidner, H., Thuman-Commike, P., Prasad, B. V., Johnston, R. E., and Chiu, W. (1998) J. Virol. 72 1534–1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lobigs, M., Wahlberg, J. M., and Garoff, H. (1990) J. Virol. 64 5214–5218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liljestrom, P., Lusa, S., Huylebroeck, D., and Garoff, H. (1991) J. Virol. 65 4107–4113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hammar, L., Markarian, S., Haag, L., Lankinen, H., Salmi, A., and Cheng, R. H. (2003) J. Biol. Chem. 278 7189–7198 [DOI] [PubMed] [Google Scholar]
- 40.Baker, T. S., and Cheng, R. H. (1996) J. Struct. Biol. 116 120–130 [DOI] [PubMed] [Google Scholar]
- 41.Cheng, R. H., Kuhn, R. J., Olson, N. H., Rossmann, M. G., Choi, H. K., Smith, T. J., and Baker, T. S. (1995) Cell 80 621–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haag, L. (2006) The Dynamic Envelope of Semliki Forest Virus: Molecular Reorganizations during Pre-fusion Stages, Ph.D. thesis, Karolinska Institute, Huddinge, Sweden
- 43.Rey, F. A., Heinz, F. X., Mandl, C., Kunz, C., and Harrison, S. C. (1995) Nature 375 291–298 [DOI] [PubMed] [Google Scholar]
- 44.Gibbons, D., Vaney, M., Roussel, A., Vigouroux, A., Reilly, B., Lepault, J., Kielian, M., and Rey, F. A. (2004) Nature 427 320–325 [DOI] [PubMed] [Google Scholar]
- 45.Glomb-Reinmund, S., and Kielian, M. (1998) J. Virol. 72 4281–4287 [DOI] [PMC free article] [PubMed] [Google Scholar]