

Abstract
Disulfide bonding contributes to the function and antigenicity of many viral envelope glycoproteins. We assessed here its significance for the hepatitis C virus E2 envelope protein and a counterpart deleted for hypervariable region-1 (HVR1). All 18 cysteine residues of the antigens were involved in disulfides. Chemical reduction of up to half of these disulfides was compatible with anti-E2 monoclonal antibody reaction, CD81 receptor binding, and viral entry, whereas complete reduction abrogated these properties. The addition of 5,5′-dithiobis-2-nitrobenzoic acid had no effect on viral entry. Thus, E2 function is only weakly dependent on its redox status, and cell entry does not require redox catalysts, in contrast to a number of enveloped viruses. Because E2 is a major neutralizing antibody target, we examined the effect of disulfide bonding on E2 antigenicity. We show that reduction of three disulfides, as well as deletion of HVR1, improved antibody binding for half of the patient sera tested, whereas it had no effect on the remainder. Small scale immunization of mice with reduced E2 antigens greatly improved serum reactivity with reduced forms of E2 when compared with immunization using native E2, whereas deletion of HVR1 only marginally affected the ability of the serum to bind the redox intermediates. Immunization with reduced E2 also showed an improved neutralizing antibody response, suggesting that potential epitopes are masked on the disulfide-bonded antigen and that mild reduction may increase the breadth of the antibody response. Although E2 function is surprisingly independent of its redox status, its disulfide bonds mask antigenic domains. E2 redox manipulation may contribute to improved vaccine design.
Disulfide bond formation is mediated during biosynthesis by thiol oxidoreductases in the endoplasmic reticulum (1, 2). Some of these catalysts are also present at the cell surface (3).
The primary function of disulfides is in folding of nascent proteins (1). Secondary functions include stabilizing mature proteins for bioactivity and protection against degradation or immune response (4, 5). A strict conservation of Cys residues highlights their key role for exofacial proteins, and manipulation of their redox state using redox reagents often abrogates their function. Disulfide bonding of mature proteins has therefore long been considered a fixed, inert feature.
However, several exofacial proteins exhibit plasticity in their disulfide-bonding post-biosynthesis, and their function may be triggered by redox changes at the cell surface (6, 7). For instance, viral envelopes, which exhibit receptor-mediated conformational change to activate their fusogenicity, are, in some cases, subject to a redox reaction controlled by cell surface or autocatalytic oxidoreductase activities that trigger such structural changes (7).
Infectivity of the pestivirus bovine viral diarrhea virus depends on the redox status of its surface glycoprotein, and reactive thiols appear to induce structural destabilization to become fusogenic at endosomal acidic pH (8). Because the bovine viral diarrhea virus envelope is functionally similar to the related flavivirus HCV3 (9), yet the redox dependence of viral envelopes is quite diverse (7), we have studied the role of the disulfides of mature HCV envelope on its function during virus entry.
The HCV envelope complex consists of two membrane-glycoproteins, E1 (HCV envelope glycoprotein 1) and E2 (HCV envelope glycoprotein 2), which form a heterodimer (10–12). E2 was the focus of the present study because: (i) E2 is the larger of the two envelope proteins, a ≈370 residue polypeptide including 18 Cys in its outer membrane region; (ii) E2 mediates virus attachment to cell receptor(s) such as CD81 (10–12), and by analogy with other viral subunits responsible for receptor binding that also exhibit a high Cys content (7), E2 is a candidate-substrate for redox reactions during entry; and (iii) E2 is the target of a neutralizing immune response, anti-E2 antibodies protecting from infection and making the protein a candidate vaccine component (13–16). The E2 sequence is variable, however, which facilitates escape from the host immune response. Two hypervariable regions (HVR) are present, with HVR1 at the extreme N terminus of E2 the most prominent (17, 18).
We report here the capacity of E2 in a range of redox states to trigger virus entry as well as the consequence of E2 disulfide network manipulation on antigenicity.
EXPERIMENTAL PROCEDURES
Materials—Expression of the proteins using the baculovirus system is described in Ref. 19. Expression of E2 (genotype 1a; accession number M62321) (20) as a secreted protein fused to the human Fc tag (E2 thereafter) was done as in (21). An antigen lacking HVR1 was similarly expressed (see Fig. 1). Virus-infected supernatants (2–5 liters) were harvested 2 days post-infection, and Fc-tagged fusion proteins recovered on lentillectin-Sepharose and protein-A prior to concentration (1–2 mg/ml; >98% pure). Soluble-CD81, the complete large extracellular loop fused to maltose-binding protein and previously shown to bind E2 with same characteristics as cell surface CD81, was produced as in Ref. 19.
FIGURE 1.

Production of envelope antigens. A, sequence. The HVR1 sequence deleted in E2 HVR1–is in bold characters. The Cys residues are underlined. The gray and white boxes correspond to the H47 and H52 epitopes, respectively. B, purity. The antigens were submitted to reducing SDS-PAGE and Western blotting. The filter was stained using Amido Black (protein labeling), the MPB/streptavidin-peroxidase/diaminobenzidine system (thiol labeling) or an anti-human Fc coupled to peroxidase.
SDS-PAGE Analysis—E2 was submitted to 8% reducing SDS-PAGE analysis and Western blotting. The filter was then incubated with the biotin-coupled reagent 3-(N-maleimidylpropionyl)biocytin (MPB; Molecular-Probes), which reacts with sulfhydryls and is detected using streptavidin-coupled peroxidase, as detailed in Refs. 22 and 23.
Thiol Content Determination—Samples (250 ng of antigen/10 μl of phosphate-buffered saline) were dot-blotted onto a nitrocellulose membrane. After blocking with casein, the filters were incubated with MPB (0.1 mm; 30 min; 25 °C) (22, 23) and then with streptavidin-coupled peroxidase for 30 min before staining. Spot intensity was quantified by densitometry (22, 23), and its relationship with the thiol content was assessed as follows. Increasing amounts of thyroglobulin, previously reduced using β-mercaptoethanol, were dot-blotted and subsequently MPB-labeled as described above. Densitometry of the resulting thyroglobulin spots produced a linear dose-response curve between 1 and 100 ng. Taking into account its molecular mass (about 300 kDa) and cysteine content (122 Cys residues/molecule), we obtained a standard curve that enabled us to assign a thiol content to the measured densitometry value of the sample (range, 0.3–20 pmol; signal/background ratio, ≈50) (22, 23).
Chemical Reduction—As detailed in Refs. 22 and 23, the samples were treated with reducing agents (15 min; 25 °C) before dot-blotting and processing to assess the thiol content, as described above. For binding experiments, the samples were reduced (15 min; 25 °C) prior to incubation with iodoacetamide (≥3:1 iodoacetamide/β-mercaptoethanol ratio; 10 min; 25 °C). β-Mercaptoethanol, which interferes with the in vitro assay, was removed under vacuum.
CD81 Binding Assay—The plates were coated with soluble CD81 (500 ng/100 μl of 200 mm sodium bicarbonate, pH 8.2, for 24 h at 21 °C). E2 was added after serial dilution in 10 mm Tris-HCl, pH 7.4, 150 mm NaCl, 5% casein (25 °C; 60 min). Peroxidase-conjugated anti-human Igs (1:1000; 60 min) were used for detection.
HCVpp Infection—The pseudotyped particles were produced as in Ref. 24. HCVpps were generated with the H77 envelope (genotype 1a; AF011752) and produced in human embryonic kidney 293 cells. Huh7 cells were seeded and infected 24 h later with virus supernatant. The drugs or reagents were added during the infection step. Alternatively, the drugs were preincubated with virus or cells for 1–3 h prior to cell/virus co-incubation. After 5 h of co-incubation, the supernatants were removed, and the cells were incubated for 72 h at 37 °C. GFP expression was determined by flow cytometry. In some experiments, the cells were treated using Bafilomycin A1 (15 nm) prior to incubation with HCVpp (2 h; 4 °C) followed by washing and DTT reduction together with treatment using fusion buffer (130 mm NaCl, 15 mm sodium citrate, 10 mm 2-(N-morpholino)ethanesulfonic acid, 5 mm Hepes) adjusted to various acidic pH levels.
HCVcc Infection—The pFK-venus-Jc1 is a chimeric HCV genome consisting of codons 1–846 derived from J6CF (genotype 2a; AF177036) and codons 847–3033 derived from JFH1 (AB047639); it also encodes Venus, a variant of yellow fluorescent protein (YFP) (25). It was used to produce viral particle-containing supernatants (cell culture-derived HCV (HCVcc)) following electroporation of Huh7.5 cells; 24 h before harvesting viral supernatants, the cells were incubated in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum (Hyclone). For infection experiments, viral supernatants were incubated with Huh7.5 cells for 4 h prior to washing and overlay with medium. At 72 h post-infection YFP expression was determined by flow cytometry.
Immunization of Mice—BALB/C mice, housed and handled under statutory care regimes, were immunized subcutaneously with 10 μg of each antigen at 14-day intervals. The Fc domain acts as a molecular adjuvant by targeting immunogen entry via FcR, and no additional adjuvant was used (21). Three immunizations were done, and the animals were sacrificed 2 weeks after the final immunization.
ELISA—After coating (100 ng/100 μl of phosphate-buffered saline for 3 h at 21 °C) and washing with phosphate-buffered saline, 0.5% casein, 0.3% Tween 20, the sera were incubated (90 min at 21 °C). Peroxidase-coupled anti-human or anti-mouse Fab antibodies (1:2,000 for 60 min) were then added before staining (22 min; 21 °C). Background signal was determined using pre- or nonimmune sera and irrelevant antigens. Alternatively, the plates were coated with protein G (250 ng/100 μl). After saturation with casein, the antigens were added (1 μg for 90 min at 21 °C) before blocking the protein G-Fc binding domain with calf serum (10% in buffer). Anti-E2 antibody dilution in calf serum (5% in buffer) was used for labeling.
RESULTS
Production of Envelope Antigens—The quantity of E2 necessary for the biochemical investigations performed here could not be purified from virus, so a recombinant HCV E2 protein (genotype 1a) was produced and examined by reducing SDS-PAGE analysis and Western blotting (Fig. 1). A single molecular species migrating as expected for the recombinant antigen was found following staining of the filter using Amido Black or anti-Fc antibodies. The possible presence of contaminants displaying free thiols was assessed following staining of the filter with the thiol reagent MPB. Only the species observed following protein staining also reacted with MPB, indicating that the molecular preparation did not contain detectable contaminants with Cys residues. Similarly, we produced an E2 antigen deleted of its HVR1 sequence (E2 HVR1–; Fig. 1). The slightly lower MW compared with the native recombinant E2 antigen is consistent with the removal of the HVR-containing 28-residue-long sequence. The sample was also essentially pure.
Cysteine Residues and Disulfide Bonding—The free sulfhydryl content of both native antigens was investigated. E2 ± HVR1 were dot-blotted and further incubated with MPB before staining. Spot intensity was measured by densitometry and the corresponding SH content, determined by comparison with a standard curve (see above and 22, 23), was found to be <0.5 SH/molecule (Fig. 2). A similar result was obtained when the antigen was incubated with MPB prior to dot-blotting (not shown). We conclude that the 18 Cys residues of the antigens are mainly involved in disulfides.
FIGURE 2.
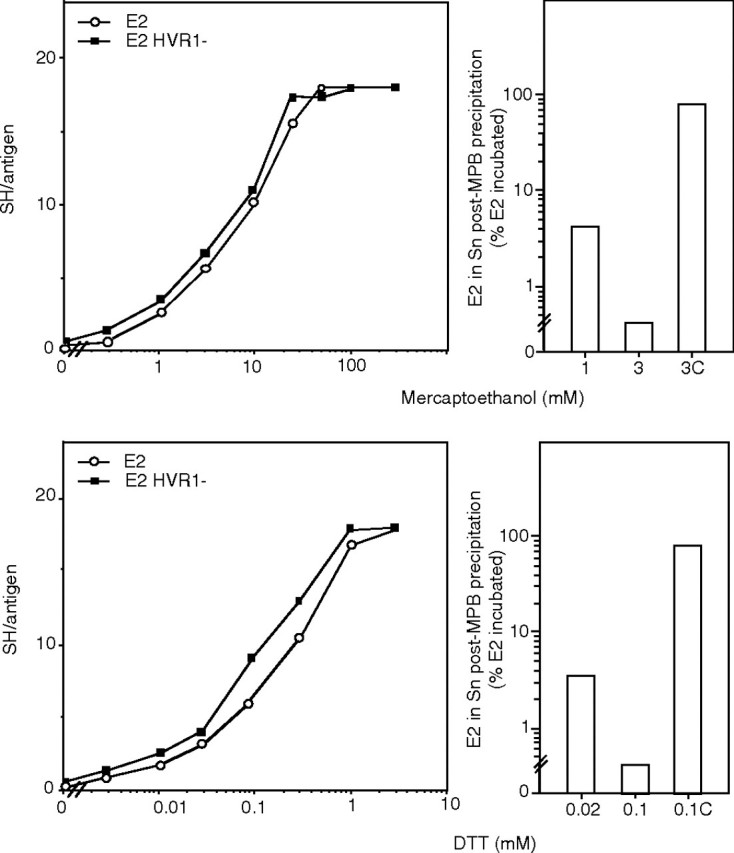
Sensitivity toward chemical reduction. The envelope-derived antigens were either mock treated (native; 0 mm reducer) or chemically reduced using β-mercaptoethanol or DTT. Left panels, after dot-blotting and staining using the thiol reagent MPB, the free sulfhydryl content/molecule (SH/antigen) was quantified by densitometry using a reference curve obtained as detailed under “Experimental Procedures” (n = 8; one experiment is shown). Right panels, the reduced samples were incubated with excess of MPB. MPB-labeled E2 was pulled down using streptavidin-coupled agarose beads, and the percentage of E2 remaining soluble, representing non reduced E2, was measured by quantitative dot-blot. Control condition: incubation of MPB with GSH prior to reaction with reduced E2 (0.1C; 3C) (n = 3; one experiment is shown).
Sensitivity Toward Chemical Reduction—The antigens were incubated with β-mercaptoethanol or DTT prior to dot-blotting, MPB labeling, and staining (Fig. 2) (22, 23). The free sulfhydryl content per molecule, reflecting the average number of reduced disulfide bonds/antigen (named ΔSS hereafter), was determined for each reducing condition as detailed above, establishing a relationship between reducer-concentration and a defined ΔSS (maximum relative standard-deviation (RSD): 8%). Because native, nonreduced species within the preparations produced using low reductor concentrations could have interfered with functional assays, we tested for their residual presence. To do this, the samples were treated using DTT or β-mercaptoethanol and then with MPB (a 3-fold molar excess of MPB to reducing agent). The reagent was then blocked as described above, and the sample was incubated with streptavidin-coupled agarose beads (Sigma-Aldrich). The percentage of E2 remaining soluble following centrifugation of the sample, reflecting nonreduced E2 species that did not react with MPB, was assessed by dot-blot (using a pool of human sera and a standard curve using known amounts of recombinant E2) and quantified by densitometry. The nonreduced species remaining in the reduced preparations was found to be very low, representing <5% of the total E2 in the 1 ΔSS samples and below the threshold of quantitation assay for the three ΔSS samples (Fig. 2) (RSD ≤ 4%). As a control, inactivation of MPB using GSH prior to reaction with reduced E2 essentially abrogated antigen precipitation.
CD81 Binding—Baculovirus-expressed E2 has been shown to be functionally analogous to E2 derived from mammalian cells (26), and E2 binding to CD81, which is central to the complex HCV entry process (27), represents the prevalent measure of a functional E2 conformation (19, 28, 29). Accordingly, the overall conformation of various E2 redox intermediates was monitored using CD81 binding in vitro. Using β-mercaptoethanol, we produced E2 preparations exhibiting known ΔSS, as described above. After reduction, the SH content was verified on an aliquot. The remainder was incubated with iodoacetamide to prevent reoxidation. We verified that the various redox intermediates similarly reacted with the peroxidase-coupled anti-Fc antibodies and that E2 preincubation with soluble CD81 absorbed all binding to the CD81-coated well (not shown). Native E2 and its mock treated counterpart readily bound CD81, indicating authentic folding (Fig. 3). Up to half of the disulfides could be cleaved without preventing binding. Similar results were obtained using E2 HVR1–. Here, and throughout the study, the significance of the results was validated by calculating the RSD value for each sample condition and presenting the maximum RSD value obtained for each set of data, indicated by the symbol ≤ followed by the maximum value obtained for a defined experiment. RSD was found ≤ 2% for each coating condition using redox intermediates derived from either E2 or E2 HVR1–. This shows that the CD81 binding by mature E2 only marginally depends on the integrity of its disulfides and confirms that HVR1 is not essential for CD81 binding (30).
FIGURE 3.

CD81 binding. The envelope proteins were either mock treated (0Δ) or mercaptoethanol-treated to cleave 1–9 disulfides/molecule (1–9Δ) prior to iodoacetamide treatment to prevent reoxidation. CD81 binding was assessed by ELISA using CD81-coated wells. Envelope binding was detected using an anti-Fc peroxidase-coupled system (n = 2; duplicates were performed; means of one experiment are shown).
Reactivity with Monoclonal Antibodies (mAbs)—The conformation of the redox intermediates was also monitored via reactivity with mAbs (31). Saturating amounts of antigens were trapped via the Fc domain using protein G-coated wells. This format, which presents the antigen away from the plastic substratum, was considered particularly suitable for assessing protein conformation. Two conformation-independent (H47 and H52) mAbs (1:300) reacted with E2 containing up to five ΔSS (Fig. 4), whereas full reduction inhibited binding, plausibly as a result of two Cys residues present in each epitope (Fig. 1). H50, a conformation-dependent mAb, exhibited a similar reactivity pattern. Similar data were found for E2 HVR1–. For a given antibody, RSD was ≤2 and 5% for each coating condition using E2 and E2 HVR1–, respectively. We conclude that reduction of up to half of the disulfides of E2 only marginally affects the accessibility to these epitopes, whereas total reduction destroys epitope integrity or accessibility.
FIGURE 4.
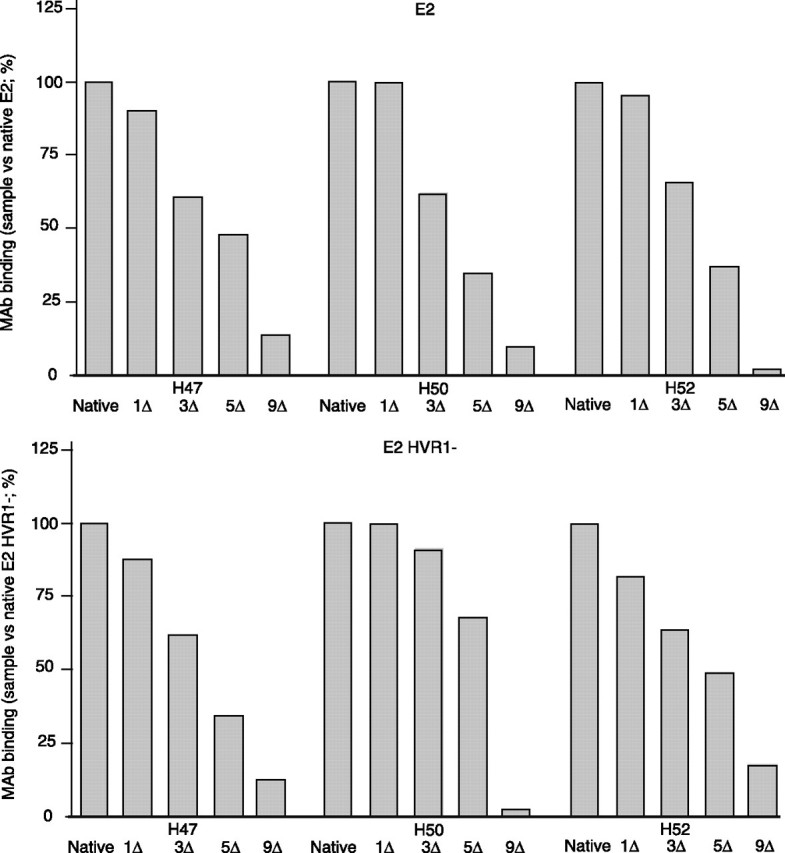
Reactivity with mAbs. The envelope proteins were either mock treated (0Δ) or mercaptoethanol-treated to cleave 1–9 disulfides/molecule (1–9Δ), treated using iodoacetamide, and then trapped via protein G on plastic wells. mAb binding (1:300) was detected using an anti-Fab′ peroxidase-coupled system (n = 2; duplicates were performed; means of one experiment are shown).
Virus Entry—To assess the biological significance of these results, we addressed the influence of the redox state on virus entry by monitoring the infectivity of HCVpps encoding a GFP marker whose expression following entry is quantified by flow cytometry (24). During virus cell incubation, the presence of 0.04 and 0.2 mm DTT, producing two and four to five ΔSS in E2, respectively, did not inhibit infection, whereas >1 mm DTT, fully reducing E2, did (Fig. 5). These data correlated with those of the CD81 binding assay except that reduction of half of E2 disulfides diminished by 50% CD81 binding in vitro but did not affect infectivity. It may be that the more rigorous ELISA procedure removed interactions between CD81 and E2 made unstable following reduction yet were tolerable for infection. We concluded that E2 function in vivo is able to tolerate the cleavage of several disulfides. Moreover, partial reduction of E2 failed to improve entry, in contrast to other viral envelopes for which fusogenicity is triggered by limited reduction (32). When virus was preincubated with >1 mm DTT before its addition to cell culture, and DTT then diluted, entry occurred. This suggested that some of the reduced disulfides reoxidized and restored fusion competence. In support of this hypothesis, incubation with iodoacetamide following DTT treatment to prevent reoxidation prevented the recovery of infection following dilution. We also examined whether disulfide reshuffling occurs during entry; 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) concentrations (1–2 mm) used to block free thiols on protein (22, 23, 33), hence the activity of oxidoreductases, had no effect (Fig. 5), suggesting that free sulfhydryls and redox isomerases are not involved during cell entry. We then studied whether controlled reduction of the HCVpp surface in conjunction with structure destabilization by endosomal pH promotes fusion, as for bovine viral diarrhea virus (8). We confirmed inhibition of infection by Bafilomycin A1, which could not be rescued by pH 3–7 (34), and also found that Bafilomycin A1 treatment and virus binding at 4 °C followed by mild reduction (0.04–0.2 mm) at 37 °C and acidic pH levels did not improve infection (not shown). Controls were performed; preincubation of MLVpps (33) with DTNB or DTT prevented infection in a parallel assay, as expected of the inhibition of the redox activity of the envelope endogenous CXXC motif and the reported dependence of MLV entry on the integrity of the envelope redox state (32, 33). RSD within each condition was ≤8 and 6% for HCVpps and MLVpps, respectively. We conclude that HCVpp infectivity is only weakly dependent on the redox state of E2. Because structural differences may exist between E2 presented at the surface of HCVpp produced in human embryonic kidney 293 cells and E2 on the authentic HCV particle produced in Huh7.5 cells, and there may be different surface environments (e.g. lipoprotein association) and requirements for fusion in either system, we similarly tested the sensitivity of HCVcc entry to a reducing environment. We confirmed the weak effect of reducing medium on infection (Fig. 5; RSD ≤ 4%). We did not test the IAA treatment following full reduction because the corresponding IAA concentration was cytotoxic. Only low titer HCVcc preparations were obtained, and sufficient dilution of the IAA-treated sample was not possible. As a control, the addition of an anti-CD81 antibody (JS-81, BioSciences; 5 mg/liter) specifically inhibited infection.
FIGURE 5.

Virus entry. DTT concentrations cleaving 2–9 disulfides/E2 (Δ) were present in the medium during incubation of target cells with HCVpp/HCVcc (Coinc DTT). Alternatively, HCVpp/HCVcc were pretreated prior to dilution (>1:100) and infection (Preinc DTT). In some experiments, iodoacetamide (IAA; a 3 m excess versus DTT) was added following DTT preincubation. For infection, cells and virions expressing GFP (HCVpp)/YFP (HCVcc) were co-incubated for 4–5 h before washing and culture for 72 h. GFP/YFP expression resulting from virus entry was quantified by flow cytometry (100%, signal using untreated particles) (n = 3, HCVpps; n = 4, HCVccs); duplicates were performed; means of one experiment are shown). In some assays, DTNB (1–2 mm) was also added during the co-incubation step of cells and HCVpp/HCVcc. Amphotropic MLV 4070A pseudoparticles expressing GFP were used in control infections in parallel to the use of HCVpp. The anti-CD81 JS-81 antibody and an irrelevant antibody were used as controls for HCVcc infections. *, not done.
Reactivity with Patient Sera—These results suggested that mild reduction of E2 allows the exposure of otherwise hidden epitopes without gross denaturation as indicated by maintenance of its bioactivity. Accordingly, we studied the reactivity of various redox intermediates for sera from patients infected with HCV-1a isolates. Each serum was assayed with saturating amounts of redox intermediates within the same experiment, and antibody binding was performed under identical conditions using the same batches of reduced antigen for all sera. Sera 1–3 were non-neutralizing, whereas sera 4–6 prevented HCVpp infection at 1:100 final dilution (not shown). The reactivity of the sera (1:100) against native E2 was similar to that against E2 HVR1–(Fig. 6). Two groups of sera were identified; for sera 1, 3, and 5, partial reduction of E2 leading to 3 ΔSS improved binding, whereas complete reduction prevented antibody interaction. The reactivity against E2 HVR1– was somewhat different; reactivity was also improved following partial reduction, although binding to the fully reduced antigen remained substantial. The rest of sera reacted similarly with the various antigens. Similar patterns were obtained for a range of dilutions (1:100–1:3,000). For a given serum, RSD was ≤5 and 4% for each coating condition using E2 and E2 HVR1–, respectively. We conclude that binding of IgG from human sera involves both disulfide-dependent and independent epitopes, the latter being responsible for a large part of the binding. Our results also indicated that epitopes can be unmasked following moderate reduction, whereas HVR1 deletion often improved IgG recognition, especially to non-disulfide-dependent epitopes such as the fully reduced antigen.
FIGURE 6.

Reactivity with patient-sera. The envelope proteins were either mock treated (0Δ) or mercaptoethanol-treated to cleave 3–9 disulfides/molecule (3–9Δ) prior to iodoacetamide treatment. IgG binding (1:100) was detected using envelope-coated wells and an anti-Fab′ peroxidase coupled system (n = 4; duplicates were performed; means of one experiment are shown).
Antigenicity in Mice—Because our data indicated a role for disulfide-masked epitopes in antibody recognition, we asked whether immunization with redox intermediates might beneficially alter the immune response to E2. Accordingly, mice were immunized with each redox intermediate on three occasions over a 6-week period. End point titration of the resulting sera was done using wells coated with E2. The titer was expressed as the reciprocal of the highest dilution that resulted in specific binding. Serum titers (Mean, ≈4,000; range, 1,500–5,200) and slopes of titration were found to be similar for all sera. The polyvalent serum response was then assessed by ELISA and HCVpp neutralization assays. For E2, the unreduced, mock treated antigen generated a serum that bound well to native E2, as well or marginally less to redox intermediates but showed essentially no binding to the fully reduced species (1:200; Fig. 7). In contrast, sera raised against reduced E2 showed an increased reactivity with the moderately reduced species, generally by 2–3-fold, compared with reactivity with native E2. Binding to the fully reduced antigens was also improved, especially for the HVR1– species, because reactivity against the fully reduced HVR1– species could approach levels observed for native E2. Similar patterns of reactivity were observed (i) at higher dilutions (1:1,000–1:5,000), (ii) when E2 HVR1– and its redox intermediates were used for immunization, and (iii) when the antigen was trapped on the plate via protein G (not shown). For a given serum, RSD was ≤5 and 6% for each coating condition using E2 and E2 HVR1–, respectively. We observed that the reactivity pattern was similar within pairs of animals immunized using the same immunogen, RSD within each animal pair being ≤23 and 26% for coating using E2 and E2 HVR1–, respectively. We conclude that epitopes hidden on native E2 were unmasked following reduction, and immunization using reduced preparations improved the ability of the resulting sera to recognize reduced antigens.
FIGURE 7.

Antigenicity in mice. The mice were immunized using either mock treated (0Δ) or mercaptoethanol-treated envelope proteins (3–9Δ) prior to iodoacetamide treatment. IgG binding (1:200) was detected using envelope-coated wells and an anti-Fab′ peroxidase-coupled system (n = 2; duplicates were performed; means of one experiment are shown).
Neutralizing Capacity—We next studied whether changes in neutralization accompanied the changes in immunoreactivity observed for these sera and tested their ability (1:100) to inhibit HCVpp infection, a widely accepted surrogate of HCV neutralization (reviewed in Ref. 10). In contrast to the use of native antigens and irrespective of HVR1 deletion, immunization using reduced immunogens elicited a neutralizing activity against H77 pseudotyped HCVpps similar to a potent neutralizing patient serum (serum 5) (Fig. 8; for a given serum, RSD was ≤16%; within pair of animals immunized using the same antigen, RSD was ≤12%). Similar conclusions were obtained when HCVpps derived from the 1b Con1 envelope (not shown). The nonspecific neutralizing capacity of the sera was determined using RD114 enveloped particles (RSD, ≤15%).
FIGURE 8.

Neutralizing capacity. The neutralizing capacity of sera from mice immunized using either mock treated (0 ΔSS) or mercaptoethanol-treated E2 or E2 HVR1– (3–9 ΔSS) was calculated versus the situation observed using nonimmune sera. HCVpps were incubated with serum dilutions (1:20) prior to infection of Huh7 cells (1:100 final dilution). GFP expression was monitored as described for Fig. 5. The nonspecific neutralizing capacity of sera was determined using RD114 pseudotyped particles. The neutralizing capacity of two human sera is also presented (non-neutralizing serum, patient 2; neutralizing serum, patient 5) (n = 2; duplicates were performed; means of one experiment are shown).
DISCUSSION
We addressed here the role of disulfide bonding in the function and antigenicity of HCV E2-glycoprotein.
First, we studied the overall structure of the various E2 redox intermediates by monitoring their CD81 binding ability as an index of their functional conformation. The in vitro format used here was preferred because it prevented lectin or charge-based interaction interfering with E2 binding in a cell surface binding assay. Reduction of up to half of the nine disulfides of E2 had no significant effect on receptor binding, a result that contrasts strongly with the lack of receptor binding observed for human immunodeficiency virus envelope with a single reduced disulfide (22, 23). Data obtained from mAb binding further supported the marginal influence of disulfide bonding on the mature E2 structure.
As shown with serum-derived virus, HCV uses various processes to enter target cells, and HCVpp infection of Huh7-cells is a surrogate model for most entry pathways, including the CD81 route used here, its sole lack being entry mediated by virus when associated with lipoproteins (11, 12, 30, 35–39). In this system, CD81 and E2 clearly have a major role because anti-E2 antibodies and soluble CD81 interfere with virus entry (24, 39). We observed here that cell entry occurred in conditions where up to half of the disulfides are cleaved within E2. In addition, the lack of effect of DTNB indicates that disulfide reshuffling mediated by oxidoreductase activities does not take place during entry. A recent report showed that 10 mm DTT reduction at low pH did not trigger HCV infection (40). We also found that a combination of low pH and mild reducing conditions did not enhance HCV entry.
Altogether, CD81 and mAb binding experiments and HCVpp experiments showed the relative independence of E2 function in relation to its redox state. These data were supported by experiments with HCVcc.
These observations contrast with that observed for the envelope glycoproteins of several viruses (reviewed in Ref. 7) and is intriguing because the many viruses displaying a strong dependence on redox state for function of their envelopes and the very few that are poorly sensitive appear to fuse virus cell membranes by similar mechanisms. Severe acute respiratory syndrome coronavirus is an example of a virus whose envelope function is poorly sensitive to reduction (33). However, its native, mature envelope protein S exhibits four unpaired cysteines, auguring the weak dependence. This is strongly contrasted by mature E2, where the Cys residues were shown here to be all involved in disulfides and yet have poor impact on fusion. We cannot rule out, however, the possibility that the disulfides that can be cleaved without effect on HCV entry play a role during biosynthesis or assembly. It is possible this role could be studied using site-directed mutagenesis, but this approach presents many drawbacks to study the function of Cys residues. In addition to structure and folding influence, Cys mutations may also affect major steps of biosynthesis such as routing, oligomerization, and surface presentation, rendering it difficult to distinguish between the loss of entry because of the involvement of Cys in structure maintenance and the loss of entry because of interference with other events.
HVR-1 exhibits positively charged residues and plays a role in glycosaminoglycan or scavenger receptor class B, type 1 binding, high density lipoprotein-enhanced HCVpp infectivity, and attenuation of neutralization (41–45). We found here that HVR1 deletion did not significantly modify the sensitivity of E2 to reduction and had no influence on its ability to interact with CD81 or mAbs, a result that is in agreement with the fact that deletion of the variable regions of E2 is compatible with CD81 binding (30, 46).
We then examined the relationship between redox state and antigenicity. We observed that the majority of the immune response in patient sera was directed against epitopes that were resistant to partial reduction. Moreover, half of the sera tested had significantly better binding to E2 species where up to half of the disulfides were reduced when compared with native E2. Increased reactivity with increasing reduction may result from uncovering more epitopes. However, a degree of overall structure is clearly important, because complete reduction prevented reactivity, and sera preferentially reacted with only the marginally reduced forms of E2.
These data stimulated a preliminary investigation of the influence of redox state on E2 immunogenicity in mice. In general, reduction of the E2 immunogen improved the ability of the resulting sera to recognize the homologous reduced antigen. More importantly, and in contrast to the use of native antigens, reduced immunogens were found to elicit neutralizing antibody response. This suggests that some epitopes capable of eliciting a neutralizing response are masked on the disulfide-bonded structure of E2 but are revealed following mild reduction to elicit neutralizing antibodies. Understanding the partially reduced structure may therefore inform vaccine design.
In conclusion, it is interesting to observe that partial reduction of E2 has significant consequences for the immune response, whereas essentially no effect was observed in a variety of tests to detect structural changes. A parallel can be made with recent data showing that the removal of glycans increases the ability of patient sera to inhibit cell entry without inhibiting CD81 binding (47). The summation of observations on antibody reactivity and neutralization experiments highlights a major but previously unrecognized role for the disulfides of mature E2, epitope masking and leads to the suggestion that careful manipulation of E2 redox status may improve the immunogenicity of vaccine candidates.
Acknowledgments
We thank J. Dubuisson for providing mAbs, R. Bartenschlager for the pFK-venus-Jc1 plasmid, C. Rice for the Huh7.5 cell line, and B. Boson for help in infection experiments.
This work was supported by Agence Nationale de Recherche sur le SIDA (ANRS) Grant HCV 2006–2007 (to E. F. and R. B. (sole support), and I. M. J., and F. L. C.), a grant from the Ligue Nationale Contre le Cancer (to F. L. C.), European Union Grant LSHB-CT-2004-005246 (to F. L. C.), and funds from the Wellcome Trust (to I. M. J.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: HCV, hepatitis C virus; HVR, hypervariable region; MPB, 3-(N-maleimidylpropionyl)biocytin; RSD, relative standard-deviation; pp, pseudotyped particles; HCVcc, cell culture-derived HCV; MLV, murine leukemia virus; GFP, green fluorescent-protein; YFP, yellow fluorescent protein; DTT, dithiothreitol; ΔSS, number of reduced disulfide bonds/antigen; mAb, monoclonal antibody; DTNB, 5,5′-dithiobis-2-nitrobenzoic acid; ELISA, enzyme-linked immunosorbent assay.
References
- 1.Benham, A. M. (2005) Antioxid. Redox Signal. 7 835–838 [DOI] [PubMed] [Google Scholar]
- 2.Jessop, C. E., Chakravarthi, S., Watkins, R. H., and Bulleid, N. J. (2004) Biochem. Soc. Trans. 32 655–658 [DOI] [PubMed] [Google Scholar]
- 3.Ferrari, D. M., and Soling, H. D. (1999) Biochem. J. 339 1–10 [PMC free article] [PubMed] [Google Scholar]
- 4.Betz, S. F. (1993) Protein Sci. 2 1551–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jensen, P. E. (1995) Semin. Immunol. 7 345–353 [DOI] [PubMed] [Google Scholar]
- 6.Hogg, P. J. (2003) Trends Biochem. Sci. 28 210–214 [DOI] [PubMed] [Google Scholar]
- 7.Fenouillet, E., Barbouche, R., and Jones, I. M. (2007) Antioxid. Redox Signal. 9 1009–1034 [DOI] [PubMed] [Google Scholar]
- 8.Krey, T., Thiel, H. J., and Rümenapf, T. (2005) J. Virol. 79 4191–4200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garry, R. F., and Dash, S. (2003) Virology 307 255–265 [DOI] [PubMed] [Google Scholar]
- 10.Bartosch, B., and Cosset, F. L. (2006) Virology 348 1–12 [DOI] [PubMed] [Google Scholar]
- 11.von Hahn, T., and Rice, C. M. (2008) J. Biol. Chem. 283 3689–3693 [DOI] [PubMed] [Google Scholar]
- 12.Poumbourios, P., and Drummer, H. E. (2007) Antivir. Chem. Chemother. 18 169–189 [DOI] [PubMed] [Google Scholar]
- 13.Youn, J. W., Park, S. H., Lavillette, D., Cosset, F. L., Yang, S. H., Lee, C. G., Jin, H. T., Kim, C. M., Shata, M. T., Lee, D. H., Pfahler, W., Prince, A. M., and Sung, Y. C. (2005) Hepatology 42 1429–1436 [DOI] [PubMed] [Google Scholar]
- 14.Grollo, L., Torresi, J., Drummer, H., Zeng, W., Williamson, N., and Jackson, D. C. (2006) Antivir. Ther. 11 1005–1014 [PubMed] [Google Scholar]
- 15.Zhang, P., Wu, C. G., Mihalik, K., Virata-Theimer, M. L., Yu, M. Y., Alter, H. J., and Feinstone, S. M. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 8449–8454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Law, M., Maruyama, T., Lewis, J., Giang, E., Tarr, A. W., Stamataki, Z., Gastaminza, P., Chisari, F. V., Jones, I. M., Fox, R. I., Ball, J. K., McKeating, J. A., Kneteman, N. M., and Burton, D. R. (2007) Nat. Med. 14 25–27 [DOI] [PubMed] [Google Scholar]
- 17.Mondelli, M. U., Cerino, A., Segagni, L., Meola, A., Cividini, A., Silini, E., and Nicosia, A. (2001) Antiviral Res. 52 153–159 [DOI] [PubMed] [Google Scholar]
- 18.Hofmann, W. P., Sarrazin, C., Kronenberger, B., Schonberger, B., Bruch, K., and Zeuzem, S. (2003) J. Infect. Dis. 187 982–987 [DOI] [PubMed] [Google Scholar]
- 19.Chan-Fook, C., Jiang, W. R., Clarke, B. E., Zitzmann, N., Maidens, C., McKeating, J. A., and Jones, I. M. (2000) Virology 273 60–66 [DOI] [PubMed] [Google Scholar]
- 20.Yoo, B. J., Selby, M. J., Choe, J., Suh, B. S., Choi, S. H., Joh, J. S., Nuovo, G. J., Lee, H. S., Houghton, M., and Han, J. H. (1995) J. Virol. 69 32–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen, H., Xu, X., and Jones, I. M. (2007) Retrovirology 4 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barbouche, R., Miquelis, R., Jones, I. M., and Fenouillet, E. (2003) J. Biol. Chem. 78 3131–3136 [DOI] [PubMed] [Google Scholar]
- 23.Barbouche, R., Lortat-Jacob, H., Jones, I. M., and Fenouillet, E. (2005) Mol. Pharm. 67 1111–1118 [DOI] [PubMed] [Google Scholar]
- 24.Lavillette, D., Tarr, A. W., Voisset, C., Donot, P., Bartosch, B., Bain, C., Patel, A. H., Dubuisson, J., Ball, J. K., and Cosset, F. L. (2005) Hepatology 41 265–274 [DOI] [PubMed] [Google Scholar]
- 25.Koutsoudakis, G., Herrmann, E., Kallis, S., Bartenschlager, R., and Pietschmann, T. (2007) J. Virol. 81 588–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Owsianka, A., Clayton, R. F., Loomis-Price, L. D., McKeating, J. A., and Patel, A. H. (2001) J. Gen. Virol. 82 1877–1883 [DOI] [PubMed] [Google Scholar]
- 27.Zeisel, M. B., Koutsoudakis, G., Schnober, E. K., Haberstroh, A., Blum, H. E., Cosset, F. L., Wakita, T., Jaeck, D., Doffoel, M., Royer, C., Soulier, E., Schvoerer, E., Schuster, C., Stoll-Keller, F., Bartenschlager, R., Pietschmann, T., Barth, H., and Baumert, T. F. (2007) Hepatology 46 1722–1731 [DOI] [PubMed] [Google Scholar]
- 28.Owsianka, A. M., Tarr, A. W., Keck, Z. Y., Li, T. K., Witteveldt, J., Adair, R., Foung, S. K., Ball, J. K., and Patel, A. H. (2008) J. Gen. Virol. 89 653–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tani, H., Komoda, Y., Matsuo, E., Suzuki, K., Hamamoto, I., Yamashita, T., Moriishi, K., Fujiyama, K., Kanto, T., Hayashi, N., Owsianka, A., Patel, A. H., Whitt, M. A., and Matsuura, Y. (2007) J. Virol. 81 8601–8612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roccasecca, R., Ansuini, H., Vitelli, A., Meola, A., Scarselli, E., Acali, S., Pezzanera, M., Ercole, B. B., McKeating, J., Yagnik, A., Lahm, A., Tramontano, A., Cortese, R., and Nicosia, A. (2003) J. Virol. 77 1856–1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Op De Beeck, A., Voisset, C., Bartosch, B., Ciczora, Y., Cocquerel, L., Keck, Z., Foung, S., Cosset, F. L., and Dubuisson, J. (2004) J. Virol. 78 2994–3002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wallin, M., Ekstrom, M., and Garoff, H. (2004) EMBO J. 23 54–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lavillette, D., Barbouche, R., Yao, Y., Boson, B., Cosset, F. L., Jones, I. M., and Fenouillet, E. (2006) J. Biol. Chem. 281 9200–9204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meertens, L., Bertaux, C., and Dragic, T. (2006) J. Virol. 80 11571–11578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Diedrich, G. (2006) FEBS J. 273 3871–3885 [DOI] [PubMed] [Google Scholar]
- 36.Evans, M. J., von Hahn, T., Tscherne, D. M., Syder, A. J., Panis, M., Wölk, B., Hatziioannou, T., McKeating, J. A., Bieniasz, P. D., and Rice, C. M. (2007) Nature 446 801–805 [DOI] [PubMed] [Google Scholar]
- 37.Zheng, A., Yuan, F., Li, Y., Zhu, F., Hou, P., Li, J., Song, X., Ding, M., and Deng, H. (2007) J. Virol. 81 12465–12471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Navas, S., Martin, J., Quiroga, J. A., Castillo, I., and Carreno, V. (1998) J. Virol. 72 1640–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Flint, M., von Hahn, T., Zhang, J., Farquhar, M., Jones, C. T., Balfe, P., Rice, C. M., and McKeating, J. A. (2006) J. Virol. 80 11331–11342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tscherne, D. M., Jones, C. T., Evans, M. J., Lindenbach, B. D., McKeating, J. A., and Rice, C. M. (2006) J. Virol. 80 1734–1741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barth, H., Schnober, E. K., Zhang, F., Linhardt, R. J., Depla, E., Boson, Cosset, F. L., Patel, A. H., Blum H. E., and Baumert, T. F. (2006) J. Virol. 80 10579–10590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scarselli, E., Ansuini, H., Cerino, R., Roccasecca, R. M., Acali, S., Filocamo, G., Traboni, C., Nicosia, A., Cortese, R., and Vitelli, A. (2002) EMBO J. 21 5017–5025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bartosch, B., Verney, G., Dreux, M., Donot, P., Morice, Y., Penin, F., Pawlotsky, J. M., Lavillette, D., and Cosset, F. L. (2005) J. Virol. 9 8217–8229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dreux, M., Pietschmann, T., Granier, C., Voisset, C., Ricard-Blum, S., Mangeot, P. E., Keck, Z., Foung, S., Vu-Dac, N., Dubuisson, J., Bartenschlager, R., Lavillette, D., and Cosset, F. L. (2006) J. Biol. Chem. 281 18285–18295 [DOI] [PubMed] [Google Scholar]
- 45.Forns, X., Thimme, R., Govindarajan, S., Emerson, S. U., Purcell, R. H., Chisari, F. V., and Bukh, J. (2000) Proc. Natl. Acad. Sci. U. S. A. 97 13318–13323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McCaffrey, K., Boo, I., Poumbourios, P., and Drummer, H. E. (2007) J. Virol. 81 9584–9590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Falkowska, E., Kajumo, F., Garcia, E., Reinus, J., and Dragic, T. (2007) J. Virol. 81 8072–8079 [DOI] [PMC free article] [PubMed] [Google Scholar]