

Abstract
Activated coagulation factor V functions as a cofactor to factor Xa in the conversion of prothrombin to thrombin. Based on the introduction of extra carbohydrate side chains in recombinant factor V, we recently proposed several regions in factor Va to be important for factor Xa binding. To further define which residues are important for factor Xa binding, we prepared fifteen recombinant factor V variants in which clusters of charged amino acid residues were mutated, mainly to alanines. The factor V variants were expressed in COS-1 cells, and their functional properties evaluated in a prothrombinase-based assay, as well as in a direct binding test. Four of the factor V variants, 501A/510A/511D, 501A/510A/511D/513A, 513A/577A/578A, and 501A/510A/511D/513A/577A/578A exhibited markedly reduced factor Xa-cofactor activity tested in the prothrombinase assay, and reduced binding affinity as judged by the direct binding assay. These factor Va variants were normally cleaved at Arg-506 by activated protein C, and the interaction between the factor Xa-factor Va complex and prothrombin was unaffected by the introduced mutations. Based on the integration of all available data, we propose a key factor Xa binding surface to be centered on Arg-501, Arg-510, Ala-511, Asp-513, Asp-577, and Asp-578 in the factor Va A2 domain. These residues form an elongated charged factor Xa binding cluster on the factor Va surface.
The prothrombinase complex is one of several multicomponent enzyme complexes that are essential for blood clotting. It is composed of the enzyme factor Xa (FXa)2 and the protein cofactor factor Va (FVa), assembled on the surface of negatively charged phospholipid membranes, in the presence of divalent metal ions (1, 2). The prothrombinase complex catalyzes two cleavages in prothrombin, at Arg-271 and Arg-320, which result in the efficient formation of thrombin. FXa alone can slowly activate prothrombin, but the rate of thrombin formation is enhanced several orders of magnitude by the presence of FVa in the prothrombinase complex (3, 4).
FV circulates in plasma as a 330-kDa single chain procofactor. FV is homologous to factor VIII (FVIII), the two proteins sharing the domain structure A1-A2-B-A3-C1-C2 (5). FV is cleaved and activated by limited proteolysis, at Arg-709, Arg-1018, and Arg-1545, mediated by either thrombin or FXa. Upon activation, the B domain is released, and FVa is formed from the non-covalent calcium-dependent complex between the heavy (A1-A2) and the light (A3-C1-C2) chains (6, 7).
Factor X (FX) circulates in plasma as a 58-kDa two-chain zymogen protein. It is composed of the light chain containing a Gla domain and two EGF-like domains and the heavy chain comprised of an activation peptide and a serine-protease domain. FX is converted to its active form by either the extrinsic pathway factor VIIa/tissue factor complex or the intrinsic pathway tenase complex composed of the phospholipid-bound factor IXa (FIXa) and activated FVIII (FVIIIa) (1, 2).
Both FVa and FXa bind to negatively charged phospholipid membranes via their light chains; the FXa binding mediated by the Gla domain, while the FVa binding depends on the C2 domain with additional contribution from the C1 domain (8, 9). The two membrane-bound proteins form a high-affinity complex, but the details of the interactions taking place at the interface have not been fully characterized yet. Both the heavy and light chains of FVa have been shown to be involved in the binding of FXa, and there are several studies giving some insights into the localization of the FXa binding site on FVa (10–16). In addition, some guidance in the efforts to understand the prothrombinase complex is provided by investigations of the intrinsic pathway tenase complex, i.e. the membrane-bound complex between FVIIIa and FIXa (17–23). Based on peptide inhibition studies, several regions of FVa have been suggested to be involved in FXa binding, i.e. residues 311–325, 323–331, and 493–506 (11, 12, 14). We have used site-directed glycosylation of recombinant FV to demonstrate that regions adjacent to FVa residues 467, 511, 652 in the heavy chain and 1683 in the light chain are involved in FXa binding (16). Recently, Gale et al. demonstrated that FXa bound poorly to a loop swap mutant in which FVa residues 499–505 were replaced by residues 555–561 of FVIIIa, which are known to be part of the FIXa binding site on FVIIIa (10). Taken together with previous peptide studies, the results suggested that residues 499–505 of FVa formed part of an extended FXa binding surface. This is very close to one of the cleavage sites for activated protein C (APC), which inhibits FVa by cleaving at Arg-306, Arg-506, and Arg-679 (2, 7, 24). Cleavage at Arg-506 results in partial loss of FVa activity due to decreased affinity for FXa, whereas cleavages at all sites result in the complete loss of FVa activity.
The aim of this study was to further define which residues in FVa are important for FXa binding. A panel of recombinant FV constructs was created using site-directed mutagenesis of clusters of two to three residues. The residues subjected to mutagenesis were chosen based on our previous results obtained in the site-directed glycosylation study (16) after structural analysis of a theoretical model for the three A domains of FVa (25, 26) and of a new model of the entire human FVa based upon the recently reported x-ray structure of activated protein C-inactivated bovine FVa (27). We also investigated the possible importance of FVa residues 323–331, 323, 324, 330, and 331, which have recently been proposed to be involved in FXa binding (12, 28). A key FXa binding surface is identified, which provides experimental support for a recently proposed model of the prothrombinase complex (29).
EXPERIMENTAL PROCEDURES
Materials—Bsu36I and BspEI were purchased from New England Biolabs (Beverly, MA). T4 DNA ligase was from Roche Applied Science (Mannheim, Germany). Double-stranded DNA sequencing kit was from PerkinElmer (Shelton, CT). Cell culture media (Optimem Glutamax) were from Invitrogen (Carlsbad, CA). Monoclonal antibodies AHV-5146 and AHV 5112 against the heavy and light chains of FVa, respectively, were from Hematologic Technologies Inc (Essex Junction, VT). Egg extracts of phosphatidylcholine (PC) and phosphatidylethanolamine (PE) and brain extracts of phosphatidylserine (PS) were purchased from Avanti Polar Lipids (Alabaster, AL). The chromogenic substrate d-Phe-(pipelocyl)-Arg-pNA (S-2238) was a kind gift of Chromogenix (Milano, Italy).
Buffers—HN buffer was 25 mm Hepes, 150 mm NaCl, pH 7.5. The buffer used during the activation of FV (HNBSACa) was 25 mm Hepes, 150 mm NaCl, 5 mm CaCl2, pH 7.5 containing 0.5% bovine serum albumin. The prothrombinase assay buffer (HNO) was 25 mm Hepes, 150 mm NaCl, 2 mm CaCl2, pH 7.5 containing 0.05% ovalbumin. The EDTA buffer was 50 mm Hepes, 100 mm NaCl, 1% polyethylene glycol, and 20 mm EDTA, pH 7.5.
Proteins—Human α-thrombin was prepared as described (30). Human prothrombin and human FXa were obtained from Kordia (Leiden, Netherlands). BSA and ovalbumin were purchased from Sigma-Aldrich. FXa was radiolabeled with 125I using a lactoperoxidase method as described (31).
Phospholipid Vesicle Preparation—The phospholipid vesicles containing PS/PC (10/90) were prepared as described (16).
Site-directed Mutagenesis—Mutations were introduced into the expression vector pMT2 containing the full-length cDNA of human FV using the QuikChange site-directed mutagenesis kit (Stratagene). For each mutant, two complementary oligonucleotides containing the appropriate mutations were used. The mutated fragments were then isolated by restriction enzymes and used to replace corresponding fragments in the template, essentially as described (16). For each mutant, the sense primers used for mutagenesis and the restriction enzymes used for digestion are shown in supplemental Table S1. The sequences of the fragments were confirmed by DNA sequencing.
Expression and Quantification of Recombinant FV (rFV)—Expression plasmids containing the various FV cDNA constructs were transfected into COS-1 cells using the diethylaminoethyl (DEAE)-dextran method, as previously described (32, 33). After 72 h, the proteins were harvested in serum-free medium (Optimem Glutamax) and concentrated in Vivaspin with a molecular weight cutoff of 100,000 (Vivascience, Carlsbad, CA). Aliquots were frozen at –80 °C. The concentrations of recombinant proteins were determined with both ELISA and PTase assay, as previously described (34). To rule out that the cell culture medium interfered with the ELISA or the prothrombinase assay, plasma-purified FV was diluted in mock medium or in buffer and tested in both assays. No differences related to dilution medium were detectable, indicating that the cell culture medium did not interfere with the assays.
Affinity Purification of Recombinant FVa Variants—The recombinant FVa variants were purified as previously described (33). In brief, a biotinylated monoclonal antibody against the B domain of FV (MK30) was bound to streptavidin-coated magnetic beads. The recombinant variants were incubated with the beads and subjected to a series of washing steps. To release FVa from the beads, the beads were incubated with thrombin and as a result the activated form of FV was released from the beads while the B domain remained associated with the MK-30-coated beads.
Western Blot Analysis of Recombinant Protein—The recombinant FV variants (0.75 nm) in HN buffer with 5 mm CaCl2 were activated by thrombin and subjected to 10% SDS-PAGE under reducing conditions and transferred to polyvinylidene difluoride membranes. Two different monoclonal antibodies were used to detect the proteins, one against the heavy chain (AHV5146) and the other against the light chain (AHV5112). To develop the Western blots, Supersignal West Dura Extended Substrate (Pierce) was used for enhancement according to the manufacturer's instructions. The gel developed with the chemiluminescent technique was exposed and visualized using a LAS 3000 CCD camera (Fuji Film, Tokyo, Japan).
Determination of Apparent Kd of FXa for FVa using the Prothrombinase Assay—The prothrombinase assay was used to determine the apparent Kd for the binding of FXa to the thrombin-activated FV variants, as previously described (16). The formation of membrane-bound FXa·FVag complexes was measured by determining the rates of prothrombin activation, at increasing concentrations of FXa and a fixed concentration of FVa. In this analysis, FVa (20 pm) was preincubated for 4 min with FXa (0.1–50 000 pm) and phospholipid vesicles (50 μm 10/90, PS/PC). The thrombin generation was started by addition of 0.5 μm preheated prothrombin and allowed to continue for 1 min before being stopped by dilution with ice-cold EDTA buffer. Thrombin was quantified using chromogenic substrate S-2238. The apparent Kd for the binding of FXa to FVa was obtained from plots of the rate of thrombin generation as a function of the FXa concentration. The Kd was obtained by fitting the data to Equation 1 for a single site binding isotherm using non-linear least squares regression analysis,
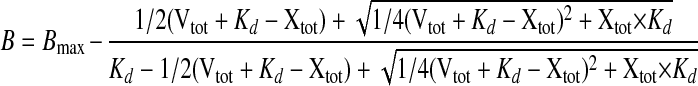 |
(Eq. 1) |
where Xtot is the total concentration of FXa, and Vtot is the total concentration of FVa. The binding maximum, Bmax, set to be the value where the binding of FXa to FVa was saturated. The derivation of the equation is previously described (15).
In control experiments, plasma-purified FVa, diluted in buffer or mock medium, affinity-purified recombinant WT-FVa, and recombinant WT-FVa in conditioned medium behaved very similar with respect to FXa cofactor activity and FXa affinity. Moreover, in the absence of added FV, there was no appreciable thrombin generation.
Determination of Km for Prothrombin Activation by the Prothrombinase Complex—The Km for prothrombin activation by the prothrombinase complexes containing the different FVa variants was determined by varying the prothrombin concentrations, as described (16). FVa (20 pm) was preincubated for 4 min with increasing concentrations of prothrombin (5–2500 nm) and 25 μm phospholipid vesicles (5/95, PS/PC). Thrombin generation was started by the addition of preheated FXa (5 nm final concentration) and allowed to continue for 1 min before being stopped by dilution with ice-cold EDTA buffer. Thrombin was quantified using chromogenic substrate S-2238. The Km was obtained by fitting the data to Equation 2 using non-linear least squares regression analysis.
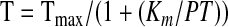 |
(Eq. 2) |
T is the amount of thrombin generated, and Tmax is the maximum thrombin generated at saturating amounts of prothrombin.
Assay for FXa Binding to Membrane-bound FVa using Magnetic Beads—A magnetic bead-based assay for FXa binding to membrane-bound FVa was performed, as described previously (16). In brief, biotinylated phospholipid vesicles were immobilized on the surface of streptavidin-coated magnetic beads. To measure the binding of FXa to membrane-bound FVa, FVa (0.5 nm) was incubated for 10 min with the phospholipid-coated beads (final phospholipids concentration was 625 nm). Increasing concentrations of 125I-FXa (300–5000 pm) were incubated with the beads for 30 min. The binding reaction was stopped by isolation of the magnetic beads using a Dynal MPC-96 plate (Dynal). After ice-cold washing, the amount of FXa that was associated with the beads was measured. Nonspecific binding determined from reactions containing no added FVa was subtracted from the total binding. To estimate the Kd of the FXa binding to FVa, the amount of bound FXa was plotted as a function of added FXa concentration. The Kd was obtained by fitting the data to Equation 3 for a single site binding isotherm using non-linear least squares regression analysis.
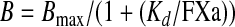 |
(Eq. 3) |
The free concentration of FXa was assumed to be equal to the added concentration of FXa. The binding maximum, Bmax, set to be the value where the binding of FXa to WT FVa was saturated. In control experiments, plasma-purified FVa, diluted in buffer or mock medium, affinity-purified recombinant WT FVa and recombinant WT FVa in conditioned medium behaved very similar with respect to FXa binding. Moreover in the absence of added FV, there was no appreciable binding.
Inactivation of FVa Variants by Activated Protein C—To probe the structure around the APC cleavage site at Arg-506, the following FVa variants (WT, 501A/510A/511D, 501A/510A/511D/513A, 513A/577A/578A, and 501A/510A/511D/513A/577A/578A) were subjected to inactivation by APC, essentially as previously described (35). In brief, the FVa variants were incubated at 0.8 nm with APC (2.5 nm) in the presence of 23 μm phospholipid vesicles (10/20/70 ratio of PS/PE/PC). At intervals, aliquots were drawn, and the remaining FVa activity determined. The kinetic constants for the Arg-506 and Arg-306 cleavage sites were calculated using equations described by Nicolaes et al. (35).
RESULTS
Mutagenesis Strategy—We have previously used N-linked carbohydrate side chain grafting to identify a relatively large region in FVa that is involved in FXa binding (16). To delineate the FXa binding site in further detail, recombinant FV variants were created after site-directed mutagenesis of 25 selected positions (Table 1). Alanine was preferentially chosen as the replacement residue, because alanine is commonly present both in buried and exposed positions in a variety of secondary structures. Moreover, analysis of a three-dimensional model of the three A-domains of FVa made it possible to carefully select the residues to be substituted (26). The probed residues were at solvent-exposed areas where substitutions should be structurally tolerated according to the three-dimensional model. As the FXa binding site appears to be extended and seems to involve many residues, single alanine replacements were not considered to be sufficient for probing the FXa binding. Therefore, the 25 residues chosen to test the binding site of FXa on FVa were combined to two double and eight triple mutants. Two of the triple mutants (501A/510A/511D and 513A/577A/578A) demonstrated impaired FXa binding. To further elucidate the role of these residues, four additional single mutants (501A, 510A, 511D, and 513A) and one quadruple mutant, (501A/510A/511D/513A), as well as one FV variant containing all six mutations (501A/510A/511D/513A/577A/578A) were created.
TABLE 1.
FXa binding to the recombinant FVa variants
Apparent Kd for binding of FXa to the FVa variants estimated the magnetic bead assay and in the prothrombinase assay. The apparent Kd values were calculated by fitting the data shown in Figs. 2 and 3 to an equation describing a single site binding isotherm via non-linear least squares regression analysis. The kcat values were calculated from data of the FXa titration in the prothrombinase assay (Fig. 2). The standard free energies of the mutants were calculated from the equation (ΔG° = -RTlnKd) and the results, relative to WT, were included in the table. The Kd ratio was Kd(mut)/Kd(wt) and the relative binding energies ΔG°(rel) were given by the ratio lnKd(mut)/lnKd(wt). Values are given as mean ± S.E.
|
FVa variant |
Kd magnetic bead |
Kd PTase |
kcat |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| nm | p | Kd ratio | ΔG° (rel) | pm | p | Kd ratio | ΔG° (rel) | s-1 | p | |
| WT | 2.9 ± 0.3 | 1.0 | 1.00 | 208 ± 9.39 | 1.0 | 1.00 | 81.2 ± 1.3 | |||
| 316A/318A/400A | 4.3 ± 0.8 | nsa | 1.5 | 0.98 | 206 ± 33.2 | ns | 1.0 | 1.00 | 42.6 ± 3.1 | ***b |
| 320A/321A/323A | 2.3 ± 0.4 | ns | 0.8 | 1.01 | 245 ± 16.9 | ns | 1.2 | 0.99 | 57.9 ± 8.8 | *** |
| 368A/652A/655A | 7.0 ± 1.6 | **c | 2.4 | 0.95 | 276. ± 13.1 | *d | 1.3 | 0.99 | 59.7 ± 5.4 | *** |
| 461A/467A/499A | 6.0 ± 0.9 | ** | 2.1 | 0.96 | 284 ± 25.9 | ** | 1.4 | 0.99 | 49.5 ± 3.6 | *** |
| 501A/510A/511D | 13.9 ± 5.4 | ** | 4.8 | 0.92 | 1120 ± 21.6 | *** | 5.4 | 0.92 | 26.7 ± 1.2 | *** |
| 501A | 6.0 ± 1.0 | ** | 2.1 | 0.96 | 219 ± 17.2 | ns | 1.1 | 1.00 | 91.5 ± 1.8 | ** |
| 510A | 4.7 ± 1.1 | ns | 1.6 | 0.98 | 297 ± 16.9 | ** | 1.4 | 0.98 | 75.8 ± 0.7 | ns |
| 511D | 7.6 ± 1.1 | *** | 2.6 | 0.95 | 637 ± 62.8 | *** | 3.1 | 0.95 | 80.2 ± 2.1 | ns |
| 513A/577A/578A | 45.2 ± 11.2 | *** | 15.6 | 0.86 | 1303 ± 88.8 | *** | 6.3 | 0.92 | 9.4 ± 0.4 | *** |
| 513A | 3.0 ± 0.2 | ns | 1.1 | 1.00 | 703 ± 53.0 | *** | 3.4 | 0.95 | 32.8 ± 0.9 | *** |
| 577A/578A | 2.2 ± 0.4 | ns | 0.8 | 1.01 | 127 ± 7.8 | ** | 0.6 | 1.02 | 43.2 ± 2.5 | *** |
| 501A/510A/511D/513A | 96.8 ± 39 | ** | 33.4 | 0.82 | 2770 ± 92.9 | *** | 13.4 | 0.88 | 13.1 ± 0.7 | *** |
| 501A/510A/511D/513A/577A/578A | NDe | *** | ND | ND | 6370 ± 312 | *** | 31.0 | 0.85 | 8.5 ± 0.4 | *** |
| 1551A/1650A/1683E | 6.0 ± 1.0 | ** | 2.1 | 0.96 | 221 ± 19.1 | ns | 1.1 | 1.00 | 72.1 ± 3.9 | * |
| 1650A/1665A | 9.2 ± 2.0 | ** | 3.2 | 0.94 | 203 ± 19.3 | ns | 1.0 | 1.00 | 68.9 ± 2.1 | ** |
ns, not significant.
***, p <0.001.
**, p = 0.001-0.01.
*, p = 0.01-0.05.
ND, not determined as the binding of FXa to the 501A/510A/511D/513A/577A/578A variant was too low to allow calculation of a Kd.
Expression and Characterization of the Recombinant FV Variants—The expression level, as determined by ELISA, was around 200 ng/ml for all of the FV variants, except for the 329A/330A/379A mutant, which consistently showed 10-fold lower expression. This indicated that, except for this mutant, all the substitutions were tolerated. According to the three-dimensional model, residues Glu-329, Glu-330, and Lys-379 could be part of an electrostatic network, which suggest that one or more of the mutations could destabilize the structure locally and reduce the expression level. Because of the very low expression level, the 329A/330A/379A mutant was not further characterized.
As part of the characterization, the recombinant FV variants were activated by thrombin and analyzed by Western blotting using monoclonal antibodies against heavy and light chains (Fig. 1). All FV mutants were activated by thrombin to similar extents as WT FV with formation of both heavy and light chains.
FIGURE 1.

Characterization of the FVa variants by Western blotting. The recombinant FV variants (0.25 μg/ml, 0.75 nm) in HN buffer with 5 mm CaCl2 were activated with thrombin (0.5 unit/ml) for 10 min in section A and 30 min at 37 °C for section B and analyzed on 10% SDS-PAGE under reducing conditions. After transfer to membranes, the heavy and light chains were detected using monoclonal antibodies AHV5146 (A) and AHV 5112 (B), respectively. Chemiluminescent enhancement was used to develop the Western blots. The asterisk marks the 220-kDa band that represents a small amount of FV that was not cleaved at Arg-1545; this band contained less than 10% of the light chain immune reactivity in most experiments. No higher molecular weight bands were seen on the heavy chain blot (A), demonstrating complete cleavage at Arg-709.
Assessment of FXa Binding to FVa using a Prothrombinase Assay—The ability of the FVa variants to support prothrombin activation was tested at increasing FXa concentrations (Fig. 2). The dose response curves were used to calculate the apparent Kd of FXa binding to the different FVa variants. WT FVa yielded an apparent Kd of ∼200 pm (Table 1), which is similar to that reported for plasma-purified FVa (35, 36). Three of the triple mutants and one double mutant (316A/318A/400A, 320A/321A/323A, 1551A/1650A/1683E, and 1650A/1665A) yielded Kd values that were not significantly different from those obtained with WT FVa, indicating the mutations did not impair the formation of the prothrombinase complex. Two of the triple mutants (386A/652A/655A and 461A/467A/499A) yielded slightly but significantly increased Kd values, whereas 501A/510A/511D and 513A/577A/578A functioned poorly as FVa cofactors (note that higher FVa concentrations were used for these two triple mutants in Fig. 2 than for WT FVa) and the Kd values were significantly higher than for WT FVa. To investigate whether combinations of mutations from the two dysfunctional triple variants would completely block the FXa cofactor activity, two additional FVa variants were prepared, 501A/510A/511D/513A and 501A/510A/511D/513A/577A/578A. Both of them were severely functionally defective and even though five times higher FVa concentrations were used (as compared with WT FVa) little thrombin was generated (Fig. 2). The Kd for the combined variant with six sites being mutated was ∼30 times higher than that of WT FVa (Table 1). To dissect the relative contribution of the different substitutions, four single substitution variants (501A, 510A, 511D, and 513A) and a double mutant (577A/578A) were constructed. The 501A variant was equally efficient at WT FVa, whereas the remaining three variants yielded significantly higher Kd values than WT FVa, in particular the 511D and 513A variants. It was noteworthy that the 577A/578A variant yielded significantly decreased Kd suggesting an increased affinity for FXa.
FIGURE 2.

FXa titration in the prothrombinase assay. The recombinant FV variants (1.25 nm) were incubated with thrombin (0.5 unit/ml) for 10 min at 37 °C and then diluted (20 pm final concentration for all mutants except 501A/510A/511D, 501A/510A/511D/513A, 513A/577A/578A, and 501A/510A/511D/577A/578A that were used at concentrations indicated in the figure) and incubated for 4 min with FXa (1–50,000 pm) and 10/90 PS:PC phospholipid vesicles (50 μm) at 37 °C. Thrombin generation was started by the addition of prothrombin (0.5 μm). After 1 min., the reactions were stopped by dilution with ice-cold EDTA buffer. The generated thrombin was determined with the chromogenic substrate S-2238. The activity was expressed as percentage of maximum activity generated by WT-FV. The control was mock medium. Each data point represents the mean of three independent experiments performed in duplicate. Error bars represent ± S.D.
The low thrombin generation was not only an effect of poor FXa binding but also attributed to decreased catalytic activity of the generated PTase complexes (Table 1). In particular the FVa variants with multiple mutations around the APC cleavage site at Arg-506 demonstrated severely decreased kcat values. To investigate whether the mutations around the Arg-506 site disturbed the three-dimensional structure of this region and in particular the APC cleavage site, the FVa variants with combined mutations around the Arg-506 site were subjected to degradation by APC, and the kinetic parameters determined. The rate constants (m–1 s–1) for cleavage at Arg-506 by APC of these FVa variants were similar to that determined for WT FVa (WT FVa, 2.2 × 107; 501A/510A/511D, 1.4 × 107; 501A/510A/511D/513A, 1.9 × 107; 513A/577A/578A, 0.9 × 107; 501A/510A/511D/513A/577A/578A, 2.6 × 107).
Prothrombin Interaction with FVa Variants—To investigate whether the FVa variants demonstrated an attenuated interaction with prothrombin, the Km for the prothrombin activation reaction was determined (Table 2). To assure that the Km reflected the interaction between prothrombin and the FVa·FXa complex rather than with only the phospholipids membrane, high concentrations of FVa (200 pm) and suboptimal phospholipid vesicles were employed. Control experiments performed in the absence of FVa, i.e. with only phospholipids and FXa, yielded a Km for prothrombin activation that was severalfold higher (data not shown). This implies that the calculated Km reflects prothrombin interaction with the FVa·FXa complex. Prothrombinase complexes generated with the different FVa variants yielded Km values for prothrombin similar to that calculated for WT FVa. The results suggest that the substituted residues are not involved in the interaction with prothrombin.
TABLE 2.
Prothrombin titration in prothrombinase assay yielding Km values
The FVa variants were incubated for 4 min with increasing concentrations of prothrombin (2.5-5000 nm) and 5/95 PS:PC phospholipid vesicles (25 μm) at 37 °C. The FVa variants were used at 200 pm final concentrations. Thrombin generation was started by addition of FXa (5 nm final concentration). After 1 min, the reactions were stopped with EDTA, and the generated thrombin determined as described under “Experimental Procedures.” None of the Km values determined for the various FVa variants were significantly different from the WT FVa.
| Factor V variant | Mean ± S.D. | na |
|---|---|---|
| WT | 206 ± 23 | 14 |
| 316A/318A/400A | 229 ± 34 | 3 |
| 320A/321A/323A | 210 ± 29 | 3 |
| 386A/652A/655A | 205 ± 33 | 3 |
| 461A/467A/499A | 200 ± 48 | 3 |
| 501A | 193 ± 18 | 3 |
| 510A | 187 ± 15 | 3 |
| 511D | 190 ± 30 | 3 |
| 501A/510A/511D | 244 ± 5 | 3 |
| 513A | 224 ± 20 | 3 |
| 513A/577A/578A | 199 ± 29 | 3 |
| 577A/578A | 181 ± 40 | 3 |
| 501A/510A/511D/513A | 196 ± 8 | 3 |
| 501A/510A/511D/513A/577A/578A | 181 ± 12 | 4 |
| 1551A/1650A/1683E | 202 ± 15 | 4 |
| 1650A/1665A | 223 ± 36 | 3 |
n, number of determinations.
Direct Analysis of FXa Binding to FVa Variants—The FXa titration in the functional prothrombinase reflected the ability of FXa to interact with FVa/prothrombin on the phospholipids surface rather than with membrane-bound FVa alone. The assay also depended on the catalytic activity of the FXa·FVa complex. To directly evaluate the ability of the various FVa constructs to bind FXa, a binding assay using radiolabeled FXa was used (Fig. 3 and Table 1). In the assay, phospholipid-coated beads were incubated with the different FVa variants and with increasing concentrations of radiolabeled FXa. The WT FVa bound FXa with an apparent Kd of around 3 nm, which is in reasonable agreement with results on record (37, 38). The FV variants that had decreased function in the prothrombinase assay also demonstrated reduced direct FXa binding. Two triple mutants (316A/318A/400A and 320A/321/323A) were not significantly different from WT FVa, three triple mutants (386A/652A/655A, 461A/467A/499A, and 1551A/1650A/1683E) and one double mutant (1650A/1665A) were mildly affected. The variants with severely affected functions also demonstrated very weak binding affinity with more than 10-fold increase in Kd. In particular, the variants mutated at four or more sites at positions 501, 510, 511, 513, 577, and 578 bound FXa very poorly. The four single substitution variants (501A, 510A, 511D, and 513A) and the double mutant (577A/578A) further dissected the relative contribution of the different substitutions. The Kd values for 501A and 511D variants were approximately doubled as compared with WT FVa, whereas the 510A, 513A, and 577A/578A variants essentially behaved like WT FVa.
FIGURE 3.

Direct binding assay of radiolabeled FXa to membrane-bound FVa. Phospholipid-coated beads were incubated for 10 min in the presence of the FVa variants (0.5 nm). Increasing concentrations of 125I-FXa (0.3–5 nm) were added. After 30 min of incubation, the beads were isolated and washed before being counted. The amount of bound 125I-FXa was determined and plotted versus the amount of added 125I-FXa. Nonspecific binding determined from parallel incubation mixtures containing no added FVa was subtracted from all reactions. The control was an incubation mixture with mock medium. Each data point represents the mean of three independent experiments performed in duplicate. Error bars represent ± S.D.
DISCUSSION
Multiple protein-protein and protein-phospholipid interactions between FXa, FVa, prothrombin, and phospholipid are involved in the formation of the prothrombinase complex and the activation of prothrombin. Relatively little is known about the structure-function relationships involved in the prothrombinase complex. In an effort to identify the regions of FVa that contain the binding site for FXa, we have previously investigated the influence of FXa binding to FVa by introducing N-linked glycosylation sites at selected residues in FVa. Regions around residues 467, 511, and 652 in the heavy chain of FVa, as well as 1683 in the light chain were found to be important for the interaction with FXa (16). A relatively large area is presumably affected by each carbohydrate side chain and in an effort to further identify the FXa binding site, we have now utilized site-directed mutagenesis to modify protein side chains that possibly contribute to the FXa binding site. With this approach we have been able to pinpoint areas on the FVa surface that are part of the FXa binding site. Based on this information as well as other available knowledge and three-dimensional structures of FVa and FXa, we recently performed theoretical docking experiments and created models of the FXa·FVa complex (29).
A total of 25 surface-exposed residues in FVa were probed in this investigation by site-directed mutagenesis. In a majority of the recombinant constructs, alanine was used to replace the wild-type amino acid residue. Except for the 329A/330A/379A FV variant, all the substitutions appeared to be tolerated, as the FV expression levels were similar to that of WT FV. Furthermore, the FV variants were equally activated by thrombin yielding the same pattern as WT FV on Western blotting using monoclonal antibodies for heavy and light chains. The influence of the introduced mutations on the FXa-FVa interaction was tested both with direct binding assay, and in a functional prothrombinase-based assay that involved analysis of the rate of prothrombin activation at increasing FXa concentrations. The PTase assay yielded Kd values that were at least 10-fold lower than the direct binding assay, which presumably was due to the contribution to the binding affinity of prothrombin. However, two types of assays yielded similar relative changes in binding in response to the introduced mutations (Table 1). FV variants having combinations of mutations at positions 501, 510, 511, 513, 577, and 578 exhibited markedly reduced FXa cofactor activity due to both decreased binding affinity and severely impaired catalytic activity (low kcat).
The present results suggest that a key binding surface for FXa includes FVa Arg-501, Arg-510, Ala-511, Asp-513, Asp-577, and Asp-578. These residues are relatively close in space in the three-dimensional model of FVa and form an elongated charged cluster (Fig. 4). The 510, 511, and 513 residues form the central core of this cluster contributing to the free energy of the binding and the 501A/510A/511D/513A variant was severely affected with regard to its FXa cofactor activity (low kcat) and binding properties (Table 1). Each individual residue was tested with the separate mutations 510A, 511D, and 513A. These three FV variants were mildly affected with at most 2–3-fold increased Kd. This observation supports the rational of our approach to combine the mutations, as single alanine replacements might not be sufficient to probe the apparently rather extended FXa binding site. Asp-577 and Asp-578 are close to the key binding area, but these residues do not contribute free energy to the binding, because the double variant 577A/578A demonstrated slightly lower Kd. However, when combined with the 513A mutation, the resulting FVa variant bound FXa weakly, and the kcat of the bound FXa was severely affected.
FIGURE 4.
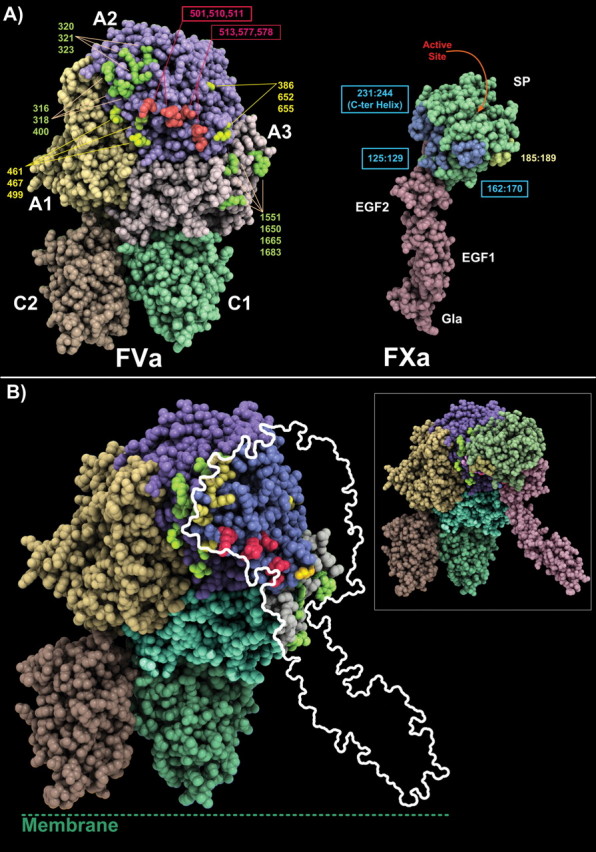
Three-dimensional structures of FVa model and FXa experimental structure and proposed model of the FVa·FXa complex. A, the FVa model is shown with its A1 domain, yellow, the A2 domain, magenta, and the A3, gray, while the C1-C2 domains are in green and light orange, respectively. Some key residues probed in this study or by other research groups are shown. FVa variants found to be of major importance in the present work for FXa binding are red. The FXa model is shown (serine protease domain, SP, and EGF1 and EGF2) with the light chain in light pink and the SP domain in green. Several key regions important for the interaction as described in our previous docking study (29) are colored to facilitate the reading of the figure. B, proposed model of the FVa·FXa complex. The data obtained in the present investigation support our previously generated model of the complex (29). To the left and with about the same orientation as in A, the model of the complex is shown. The FVa structure is shown in CPK representation while FXa is shown as a ribbon. To the right, the contours of FXa are projected onto the FVa molecule to facilitate the visualization of the mutations. In this model, the FVa hot-spot (510 area) makes direct contact with the SP domain of FVa, while the other residues probed by mutagenesis or peptide studies surround this region.
It is noteworthy that even though the FXa binding site is severely affected by the FVa variants containing multiple mutations around the Arg-506 site, the kinetics of the APC-mediated cleavage at this site is not influenced by the mutations. This suggest that the FXa binding site does not involve the same interaction sites on FVa as APC, even though FXa binding protects the Arg-506 site from APC-mediated cleavage (32). The results also suggest that the three-dimensional structure of the region around the Arg-506 sites is not compromised by the introduced mutations. This is in line with the findings of Gale et al. (10) who reported that their 499–505 FV loop swap mutant impaired FXa binding but not degradation by APC. Apparently, the loop surrounding the APC cleavage site at Arg-506 is flexible, and the three-dimensional structure tolerates multiple mutations.
In the site-directed glycosylation study, we observed that introduction of a carbohydrate side chain at residue 467 yielded a FVa that almost completely abolished FXa binding (16). To further define which residues contribute to the FXa binding, we constructed the 461A/467A/499A variant. In both the direct binding assay and the functional FXa titration assay, this molecule only demonstrated a small reduction of the FXa cofactor binding activity. These results suggest that this region is close to the FXa binding site but that the involved residues are not crucial for the binding. Similar observations were made for another region surrounding residue 652 from the carbohydrate side chain grafting study, and the 386A/652A/655A FV variant was therefore prepared. This FV variant exhibited slightly impaired FXa cofactor activity and FXa binding affinity (about 2-fold). Several reports suggest an important role for the light chain of FVa in binding of FXa (13, 16). However, there is little information on which part of the light chain could be involved in this interaction. In the site-directed glycosylation study, the area surrounding residue 1683 was proposed to be part of the FXa binding site (16). Based on this data, two FV variants 1551/1650A/1683E and 1650A/1665A were created. In the functional FXa titration assay, both mutants were essentially normal, whereas in the direct binding assay the Kd values were moderately higher than that of WT FVa (2-fold for the 1551/1650A/1683E FV variant, and 3-fold for the 1650A/1665A FV variant). These data suggest that these residues should be located at the FVa-FXa interface but that their contribution to the free energy of binding is moderate.
Studies using synthetic peptides have indicated regions 311–325 and 323–331 to potentially be important for FXa binding (12, 14). To investigate the potential contribution of this area for FXa binding, three FV variants were constructed: 316A/318A/R400A, 320A/321A/323A, and 329A/330A/379A. As discussed, 329A/330A/379A was excluded because of very low expression levels. The other two variants 316A/318A/400A and 320A/321A/323A were not significantly different from the WT FVa. As seen in Fig. 4, these residues are close to the main binding region (area of residue 510) of FXa but they most likely do not directly contact the enzyme.
The above-mentioned experimental results were projected onto the previously reported three-dimensional model of the FVa·FXa complex (Fig. 4) (29). The model selected in our previous docking study remains fully consistent with the present data. The serine protease domain of FXa covers and interacts directly with the FVa 510 area. Thus, the FVa binding interface is formed by the 510 area, which constitutes a central core critical to the interaction while the other residues are located at the periphery, forming a ring-like structure surrounding the main contact point.
In conclusion, the present study highlights several important residues involved in FXa binding. These residues are all charged, suggesting that electrostatic interactions play a major role in the formation of the FXa·FVa complex. An extended and key FXa binding surface is centered on the A2 domain around residues Arg-501, Arg-510, Ala-511, Asp-513, Asp-577, and Asp-578. This is in agreement with previous reports suggesting that a region surrounding the APC cleavage site Arg-506 is involved in FXa binding (10, 11, 16, 39) and with the observation that FVa in the assembled prothrombinase complex is protected from APC-mediated cleavage at Arg-506, whereas the Arg-306 site is fully exposed to APC (32). The FVa 510 region represents a key functional epitope with a few amino acids contributing the majority of the interaction energy, thereby forming a clear hot-spot on the surface of FVa. Hot-spot areas are usually surrounded by a ring of residues that contribute mildly to the interaction energy. This situation appears to be the case for the FVa·FXa complex. Taken together, the available data improve the understanding of the relationship between the structure and the function of FVa with regard to the prothrombinase complex, a major procoagulant complex that could be the target of future anticoagulant molecules.
Supplementary Material
This work was supported by grants from the Swedish Medical Research Council (Grant 07143), a Senior Investigator's Award from the Foundation for Strategical Research, by research funds from the University Hospital Malmö, by research grants from the INSERM Institute (to B. V.) (Avenir), and the INSERM/region Ile de France (to B. V. and L. A.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Table S1.
Footnotes
The abbreviations used are: FXa, activated factor X; FV, factor V; FVa, activated factor V; FVIII, factor VIII; FVIIIa, activated factor VIII; FIXa, activated factor IX; FX, factor X; FVIIa, activated factor VII; PPACK, Phe-Pro-Arg-chloromethylketone; APC, activated protein C; PE, phosphatidylethanolamine; PS, phosphatidylserine; PC, phosphatidylcholine; WT, wild type; ASA, accessible surface area; ELISA, enzyme-linked immunosorbent assay; EGF, epidermal growth factor.
References
- 1.Mann, K. G., Butenas, S., and Brummel, K. (2003) Arterioscler. Thromb. Vasc. Biol. 23 17–25 [DOI] [PubMed] [Google Scholar]
- 2.Dahlback, B. (2005) J. Intern. Med. 257 209–223 [DOI] [PubMed] [Google Scholar]
- 3.Nesheim, M. E., Taswell, J. B., and Mann, K. G. (1979) J. Biol. Chem. 254 10952–10962 [PubMed] [Google Scholar]
- 4.Rosing, J., Tans, G., Govers-Riemslag, J. W., Zwaal, R. F., and Hemker, H. C. (1980) J. Biol. Chem. 255 274–283 [PubMed] [Google Scholar]
- 5.Kane, W. H., and Davie, E. W. (1988) Blood 71 539–555 [PubMed] [Google Scholar]
- 6.Mann, K. G., and Kalafatis, M. (2003) Blood 101 20–30 [DOI] [PubMed] [Google Scholar]
- 7.Nicolaes, G. A., and Dahlback, B. (2002) Arterioscler. Thromb. Vasc. Biol. 22 530–538 [DOI] [PubMed] [Google Scholar]
- 8.Nicolaes, G. A., Villoutreix, B. O., and Dahlback, B. (2000) Blood Coagul. Fibrinolysis 11 89–100 [PubMed] [Google Scholar]
- 9.Saleh, M., Peng, W., Quinn-Allen, M. A., Macedo-Ribeiro, S., Fuentes-Prior, P., Bode, W., and Kane, W. H. (2004) Thromb. Haemost. 91 16–27 [DOI] [PubMed] [Google Scholar]
- 10.Gale, A. J., Yegneswaran, S., Xu, X., Pellequer, J. L., and Griffin, J. H. (2007) J. Biol. Chem. 282 21848–21855 [DOI] [PubMed] [Google Scholar]
- 11.Heeb, M. J., Kojima, Y., Hackeng, T. M., and Griffin, J. H. (1996) Protein Sci. 5 1883–1889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kalafatis, M., and Beck, D. O. (2002) Biochemistry 41 12715–12728 [DOI] [PubMed] [Google Scholar]
- 13.Kalafatis, M., Xue, J., Lawler, C. M., and Mann, K. G. (1994) Biochemistry 33 6538–6545 [DOI] [PubMed] [Google Scholar]
- 14.Kojima, Y., Heeb, M. J., Gale, A. J., Hackeng, T. M., and Griffin, J. H. (1998) J. Biol. Chem. 273 14900–14905 [DOI] [PubMed] [Google Scholar]
- 15.Steen, M., and Dahlback, B. (2002) J. Biol. Chem. 277 38424–38430 [DOI] [PubMed] [Google Scholar]
- 16.Steen, M., Villoutreix, B. O., Norstrom, E. A., Yamazaki, T., and Dahlback, B. (2002) J. Biol. Chem. 277 50022–50029 [DOI] [PubMed] [Google Scholar]
- 17.Fay, P. J., Beattie, T., Huggins, C. F., and Regan, L. M. (1994) J. Biol. Chem. 269 20522–20527 [PubMed] [Google Scholar]
- 18.Jenkins, P. V., Dill, J. L., Zhou, Q., and Fay, P. J. (2004) Biochemistry 43 5094–5101 [DOI] [PubMed] [Google Scholar]
- 19.Jenkins, P. V., Dill, J. L., Zhou, Q., and Fay, P. J. (2004) J. Thromb. Haemost. 2 452–458 [DOI] [PubMed] [Google Scholar]
- 20.Jenkins, P. V., Freas, J., Schmidt, K. M., Zhou, Q., and Fay, P. J. (2002) Blood 100 501–508 [DOI] [PubMed] [Google Scholar]
- 21.Lenting, P. J., van de Loo, J. W., Donath, M. J., van Mourik, J. A., and Mertens, K. (1996) J. Biol. Chem. 271 1935–1940 [DOI] [PubMed] [Google Scholar]
- 22.Mertens, K., Celie, P. H., Kolkman, J. A., and Lenting, P. J. (1999) Thromb. Haemost. 82 209–217 [PubMed] [Google Scholar]
- 23.Celie, P. H., Van Stempvoort, G., Jorieux, S., Mazurier, C., Van Mourik, J. A., and Mertens, K. (1999) Br. J. Haematol. 106 792–800 [DOI] [PubMed] [Google Scholar]
- 24.Dahlback, B., and Villoutreix, B. O. (2005) Arterioscler. Thromb. Vasc. Biol. 25 1311–1320 [DOI] [PubMed] [Google Scholar]
- 25.Pellequer, J. L., Gale, A. J., Getzoff, E. D., and Griffin, J. H. (2000) Thromb. Haemost. 84 849–857 [PubMed] [Google Scholar]
- 26.Villoutreix, B. O., and Dahlback, B. (1998) Protein Sci. 7 1317–1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adams, T. E., Hockin, M. F., Mann, K. G., and Everse, S. J. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 8918–8923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Singh, L. S., Bukys, M. A., Beck, D. O., and Kalafatis, M. (2003) J. Biol. Chem. 278 28335–28345 [DOI] [PubMed] [Google Scholar]
- 29.Autin, L., Steen, M., Dahlback, B., and Villoutreix, B. O. (2006) Proteins 63 440–450 [DOI] [PubMed] [Google Scholar]
- 30.Lundblad, R. L., Uhteg, L. C., Vogel, C. N., Kingdon, H. S., and Mann, K. G. (1975) Biochem. Biophys. Res. Commun. 66 482–489 [DOI] [PubMed] [Google Scholar]
- 31.Dahlback, B., and Stenflo, J. (1978) Biochemistry 17 4938–4945 [DOI] [PubMed] [Google Scholar]
- 32.Norstrom, E. A., Tran, S., Steen, M., and Dahlback, B. (2006) J. Biol. Chem. 281 31486–31494 [DOI] [PubMed] [Google Scholar]
- 33.Sun, Y. H., Tran, S., Norstrom, E. A., and Dahlback, B. (2004) J. Biol. Chem. 279 47528–47535 [DOI] [PubMed] [Google Scholar]
- 34.Norstrom, E., Thorelli, E., and Dahlback, B. (2002) Blood 100 524–530 [DOI] [PubMed] [Google Scholar]
- 35.Nicolaes, G. A., Tans, G., Thomassen, M. C., Hemker, H. C., Pabinger, I., Varadi, K., Schwarz, H. P., and Rosing, J. (1995) J. Biol. Chem. 270 21158–21166 [DOI] [PubMed] [Google Scholar]
- 36.Camire, R. M., Kalafatis, M., and Tracy, P. B. (1998) Biochemistry 37 11896–11906 [DOI] [PubMed] [Google Scholar]
- 37.Krishnaswamy, S. (1990) J. Biol. Chem. 265 3708–3718 [PubMed] [Google Scholar]
- 38.Thiec, F., Cherel, G., and Christophe, O. D. (2003) J. Biol. Chem. 278 10393–10399 [DOI] [PubMed] [Google Scholar]
- 39.Gale, A. J., Xu, X., Pellequer, J. L., Getzoff, E. D., and Griffin, J. H. (2002) Protein Sci. 11 2091–2101 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.