

Abstract
Maltose metabolism during the conversion of transitory (leaf) starch to sucrose requires a 4-α-glucanotransferase (EC 2.4.1.25) in the cytosol of leaf cells. This enzyme is called DPE2 because of its similarity to the disproportionating enzyme in plastids (DPE1). DPE1 does not use maltose; it primarily transfers a maltosyl unit from one maltotriose to a second maltotriose to make glucose and maltopentaose. DPE2 is a modular protein consisting of a family 77 glycosyl hydrolase domain, similar to DPE1, but unlike DPE1 the domain is interrupted by an insertion of ∼150 amino acids as well as an N-terminal extension that consists of two carbohydrate binding modules. Phylogenetic analysis shows that the DPE2-type enzyme is present in a limited but highly diverse group of organisms. Here we show that DPE2 transfers the non-reducing glucosyl unit from maltose to glycogen by a ping-pong mechanism. The forward reaction (consumption of maltose) is specific for the β-anomer of maltose, while the reverse reaction (production of maltose) is not stereospecific for the acceptor glucose. Additionally, through deletion mutants we show that the glycosyl hydrolase domain alone provides disproportionating activity with a much higher affinity for short maltodextrins than the complete wild-type enzyme, while absence of the carbohydrate binding modules completely abolishes activity with large complex carbohydrates, reflecting the presumed function of DPE2 in vivo.
During photosynthesis, as much as one-half of the carbon fixed during the day is stored as transitory starch inside chloroplasts for remobilization at night. The pathway by which starch in leaves is converted to sucrose has only recently been elucidated (1) and some questions remain. The breakdown of storage starch upon seed germination takes place essentially outside of cells (cellular integrity is lost) while transitory starch must be broken down in intact chloroplasts and cells (2). Transitory starch must have some phosphate attached in order to be remobilized at night (3–6). During the day, carbon is exported from the chloroplast almost exclusively as triose phosphate, but at night maltose is exported (7, 8). Starch is acted on by β-amylase (9), and the resulting β-maltose is exported from chloroplasts through a novel maltose transporter (10). Plants lacking this transporter accumulate maltose in their plastids (11). In the cytosol, maltose metabolism requires a 4-α-glucanotransferase (EC 2.4.1.25). This enzyme was called the cytosolic D enzyme and, by analogy with the plastidial D enzyme, was believed to have no activity with maltose (12). Arabidopsis thaliana plants lacking this enzyme accumulate up to 100-fold more maltose than wild type and grow significantly more slowly (13, 14). Potato plants in which this enzyme was reduced in activity accumulated high levels of maltose in leaves but leaf starch synthesis and starch metabolism in tubers was not affected (15).
This enzyme is now called disproportionating enzyme 2 (DPE2)3 because of its similarity to the disproportionating enzyme (D enzyme) of plastids (DPE1); it is also known as transglucosidase, amylomaltase, glucosyltransferase, and 4-α-glucanotransferase. The activity of DPE1 transfers a maltosyl unit from maltotriose to a second maltotriose to make a dextrin long enough to be acted on by starch phosphorylase or β-amylase (12). DPE2 can transfer glucosyl units from maltose to glycogen (13, 15, 16) and to a soluble heteroglycan (SHG) found in the cytosol of plant leaves (16, 17). The capacity of Escherichia coli lacking amylomaltase (malQ–) to grow on maltose can be restored by DPE2 of Arabidopsis (16). The role of DPE2 in vivo is presumably to transfer glucosyl units from maltose to a cytosolic SHG, composed of arabinose, galactose, and glucose to a lesser extent (17, 18), during transitory starch degradation (7, 14, 17). Although it has yet to be proven that SHG is the primary substrate of DPE2 in vivo, it is capable of using SHG as an acceptor in vitro. In vitro experiments have often substituted glycogen for SHG, because this too is an effective substrate.
In this study, we examined the mechanism and specificity of DPE2 and several engineered versions of the enzyme to determine which structural aspects give rise to the properties of DPE2 needed for its role in starch conversion to sucrose.
EXPERIMENTAL PROCEDURES
Reagents—Glycogen (type II oyster glycogen (Sigma)) was spun on a Centricon centrifugal filter device with a nominal molecular weight limit (NMWL) of 50 kDa (Millipore) to remove maltodextrins. Uniformly labeled [14C]maltose (610 mCi/mmol) and α-glucose (α-94%, β-6%) were obtained from Sigma. Glucose in anomeric equilibrium was obtained by aging glucose solutions for >1 day. The 94% α-glucose powder was used to make a 10% solution which, when quickly measured, was 93% α (105°) as measured in a Perkin Elmer Polarimeter 341. Spontaneous mutarotation could be observed with rotation falling to 100° within 5 min.
Phylogenetic Analysis—Gene sequences were aligned with ClustalW, and the PAUP program was used to generate the phylogenetic tree using the neighbor joining method. Bootstrap values shown on the branches were generated with 1,000 replicates, and DPE1 was used to root the tree.
Construction of His-tagged Enzymes—All of the wild-type DPE2 used in this study was His-tagged recombinant protein from the same clone as that used in (16). Protein was expressed in E. coli BL21 (DE3) by growing bacteria in 1 liter of LB broth to an A600 of 0.6 and inducing with 0.5 mm isopropyl-1-thio-β-d-galactopyranoside. Cells were pelleted at 4,000 × g for 30 min and resuspended in lysis buffer, (50 mm NaH2PO4, 300 mm NaCl, 10 mm imidazole, pH 8.0). Cells were lysed by sonication, centrifuged for 25 min at 10,000 × g, and the supernatant was applied to a nickel nitrilotriacetic acid-agarose column (Qiagen), and the protein was washed and eluted with an imidazole gradient ranging from 20–500 mm. When high concentrations of protein were needed, DPE2 was concentrated on a Centricon centrifugal filter device with a NMWL of 30 kDa.
The deletion mutants were constructed the same way as wild type except with forward primers: 5′-ACACAGGATCCAGTGCATTTAAAGATGTC-3′ and 5′-ACACAGGATCCATCGGTAGTAAACAGGAA-3′ (BamHI sites underlined) for the ΔN130 and ΔN248 deletion mutants, respectively. The reverse primer was 5′-ACACACTCGAGTTATGGGTTTGGCTTAGTCG-3′ (XhoI site underlined).
To construct the E758Q site-directed mutant, the wild-type clone was amplified with the forward mutagenic primer 5′-ACACAGAATTCATCTAATATGCTGGCATGTGGGCAGGATCTG-3′ (EcoRI site underlined and mutated codon in bold) and reverse primer 5′-ACACACTCGAGTTATGGGTTTGGCTTAGTCG-3′ (XhoI site underlined). The PCR product was cloned into the vector pGEM-T easy (Promega), and the EcoRI/XhoI-digested product was subcloned into the EcoRI/XhoI-digested wild-type construct DPE2/pET28a, described above.
Anomeric Specificity—The anomeric specificity of DPE2 was measured using an NADP(H)-linked assay previously described (16). The substrates were glycogen (200 μg/ml) and 2.5 μm maltose at anomeric equilibrium. The reaction was initiated with either 25 μg of DPE2 or 5 units of maltose phosphorylase.
Trapping the Covalent Intermediate—The method of Lee and Robyt (19) was used to trap the covalent intermediate except 50 μm DPE2 was incubated with 33 μm [UL-14C]maltose, and the reaction was stopped with 10% (v/v) trifluoroacetic acid. The precipitate was washed five times with 0.1% (v/v) trifluoroacetic acid, resolubilized in 8 m urea, 0.1 m sodium phosphate, 0.01 m Tris-Cl, pH 8.0, and added directly to 3.5 ml of scintillation mixture.
Maltose Production—5 μg of DPE2, 1 mg/ml glycogen, and 50 mm of either α-glucose or glucose at anomeric equilibrium were incubated in 20 mm sodium phosphate buffer, pH 6.8, for 20 min at 4 °C. The reactions were stopped by adding 1 volume of 50 mm sodium phosphate, pH 1.25, and immediately the molar ratios of α- and β-maltose were measured as previously described (20).
Thin Layer Chromatography—TLC analysis was carried out by the method described by Takaha et al. (21). Following 18 h of incubation of 0.37 μm protein with 20 mm maltodextrins, the resulting products were spotted on silica gel TLC plates (Merck) and resolved three times with 1-butanol/ethanol/water (5:5:3). Carbohydrates were visualized with an anisaldehyde/acid alcohol spray reagent (Sigma).
Starch Binding Assay—Corn starch (Sigma) was washed with 100 mm PIPES buffer (pH 6.8), and then 25 μg of protein was added to 20 mg of starch in 500 μl of PIPES buffer; for competition experiments, 10 mg of glycogen was added to the mixture. The mixture was shaken for 30 min on ice and then spun at 16,000 × g for 5 min, and the supernatant was removed. To extract protein from the starch, the pellet was incubated at 75 °C for 10 min in 500 μl of 1% SDS, 100 mm PIPES buffer. The fractions were analyzed by 4–12% SDS-PAGE and stained with 0.1% Coomassie Brilliant Blue.
Initial Rate Kinetics—To compare substrate preferences, the initial rates of glucose production were measured using an NADP(H)-linked assay as described above with substrates (G2-G7) at 2 mm or glycogen at 2 mg/ml. The reactions were initiated with 0.6 μg of protein. To account for differences in mass between the enzymes, the initial rate was converted to a pseudo-first order rate constant (k0), which has units s–1 and is defined in Equation 1,
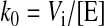 |
(Eq. 1) |
where Vi is the initial rate of glucose production and [E] is the molar concentration of enzyme added to the reaction buffer. The initial rates were fit to a Hanes-Wolff plot in order to obtain the Km and kcat. The lines represent fits to the Michaelis-Menton equation using the experimentally derived Km and kcat.
To calculate the second order specificity constant (kcat/Km) for the mutant ΔN130 on various maltodextrins, the following low substrate approximation was used (Equation 2),
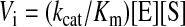 |
(Eq. 2) |
where the substrate concentration [S] was ∼0.03 Km.
The rate of the E758Q mutant was measured with 100 μg/ml glycogen, 2 mm maltose, and 1.5 μg of protein. All rates were measured at room temperature.
RESULTS
Structure and Phylogenetics of the DPE2-type Enzymes—The amino acid sequence of DPE2 indicates that it is a modular protein containing one domain that is similar to glycoside hydrolases of family 77 (GH77) as well as two putative N-terminal carbohydrate binding modules (CBM20) (Fig. 1A). The GH77 domain of DPE2 is split by an insert of 173 amino acids (Fig. 1A). The plastid-localized DPE1 of Arabidopsis lacks both carbohydrate binding domains and the insert in the GH77 domain. In addition to GH77, the only other recognizable motif is a plastid targeting sequence. The E. coli malQ had only the GH77 domain (Fig. 1).
FIGURE 1.
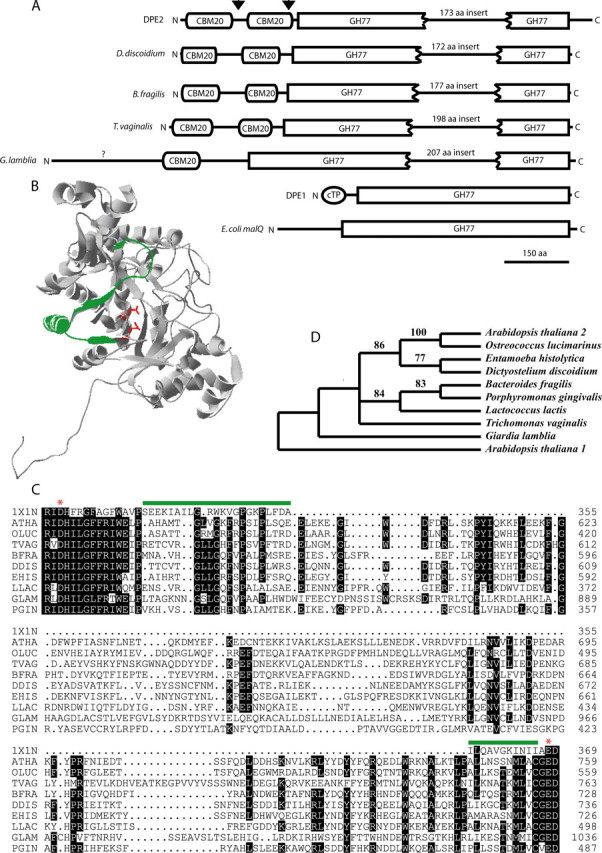
Structure and phylogenetic relationships of the DPE2-type 4-α-glucanotransferases. A, DPE2 is organized into 3 modules with an insert of 173 amino acids splitting the GH77 domain (arrowheads indicate the deletion mutants ΔN130 and ΔN248). B, the structure of DPE1 (PDB: 1X1N) shows the location of the α-helix that has been replaced by 173 amino acids in the DPE2-type enzymes. C, the sequence alignment of the insert from a diverse group of organisms shows its location relative to the two presumed active site residues (red asterisks). The abbreviations and gene accession numbers, respectively are as follows: ATHA (A. thaliana DPE2), NP_181616; OLUC (Ostreococcus lucimarinus) XP_001420502; TVAG (T. vaginalis) XP_001310739; BFRA (Bacteroides fragilis) YP_213214; DDIS (Dictyostelium discoidium) XP_638682; EHIS (Entamoeba histolytica) XP_657483; LLAC (Lactococcus lactis) YP_808707; GLAM (G. lamblia) XP_771503; PGIN (Porphyromonas gingivalis) NP_905029; PDB code 1X1N refers to Solanum tuberosum DPE1. D, phylogenetic relationship of DPE2-type proteins from a wide range of organisms. The sequences were aligned with ClustalW, and the PAUP program was used to generate the phylogenetic tree using the neighbor joining method. Bootstrap values shown on the branches were generated with 1,000 replicates and DPE1 (A. thaliana 1) was used to root the tree.
This arrangement of two CBM20 domains on the N terminus plus a GH77 domain interrupted by a >150 amino acid insert was found in all plants that have been fully sequenced. In addition, a small number of other organisms also had a gene that was predicted to make a protein with this unusual structure. Selected organisms are shown on the tree in Fig. 1. Bacteria cluster together as do the amoeboid Dictyosteleum and Entamoeba. The parasitic flagellated protists Trichomonas vaginalis and Giardia lamblia are not well resolved in the tree. The closest relationship between the plant gene sequence and other occurrences of the gene was with an enzyme from the small green alga Ostreococcus (Fig. 1D). The sequence of the insert was conserved among all members of the DPE2-type enzymes (Fig. 1C). Highly conserved residues occur regularly every three to four residues at both the start and end of the insert consistent with an α-helix, but we have no other information about the structure of this insert.
In hopes of gaining an understanding of what the function of this insert might be, the structure of potato D-enzyme, DPE1 (PDB code 1X1N) was used to examine where the insert is located. The insert replaces a segment of ∼33 residues in potato DPE1, which makes up the α-4 helix and the β-5 strand of the (α/β)8 barrel. Fig. 1B shows the location of the segment that is replaced in the DPE2 sequence on the recently solved crystal structure of potato DPE1. The segment is at the surface of the enzyme (shown in green) directly opposite from the active site (the two active site acidic residues are shown in red). There is no obvious way in which the insertion could directly interact with substrate at the active site.
Mechanism of the 4-α-Glucanotransferase Reaction—To determine whether or not the mechanism of DPE2 was consistent with other family 77 glycosyl hydrolases, an active site-directed mutant was constructed. The presumed acid/base catalyst glutamate 758 (by homology with amylomaltase from Thermus thermophilus (22)), was converted to a glutamine residue, and the mutant enzyme showed an approximate 2,000-fold decrease in activity relative to wild type with maltose and glycogen as substrates (Table 1).
TABLE 1.
Wild type and mutant activity of the 4-α-glucanotransferase DPE2
DPE2 or E758Q (1.5 μg) were incubated with 100 μg/ml glycogen and 2 mm maltose, and the initial rate of glucose production was measured using an NADP(H)-linked assay as described under “Experimental Procedures.” Values are mean ± S.D. (n = 3).
| Enzyme | Activity |
|---|---|
| μmol glucose min-1 mg-1 protein | |
| Wild type | 5.19 ± 0.35 |
| E758Q | 0.0027 ± 0.0005 |
The mechanism of DPE2 was further investigated by testing for the presence of a covalent intermediate. DPE2 was incubated with [UL-14C]maltose for 30 s and then precipitated with 10% trifluoroacetic acid. The precipitate was washed five times with 0.1% trifluoroacetic acid, and the radioactivity was measured. The precipitate contained a substantial amount of radioactivity (Fig. 2A), which corresponds to glucose bound to ∼2.5% of the enzyme sites.
FIGURE 2.
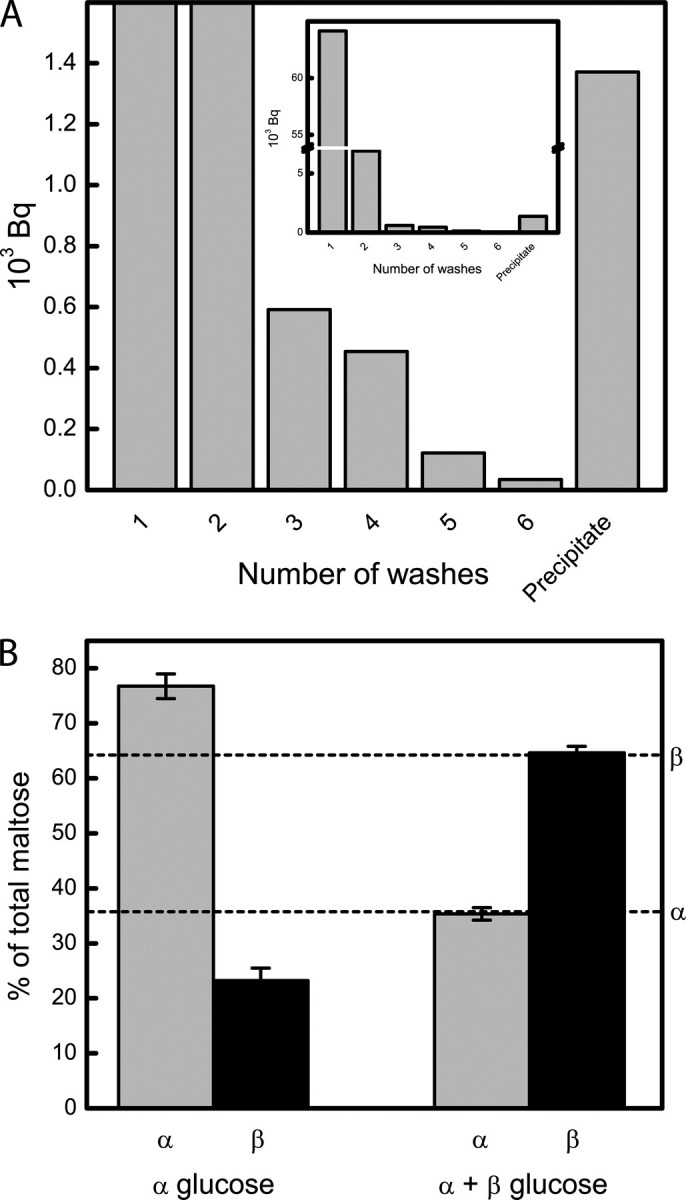
DPE2 forms a substituted enzyme-glucosyl intermediate. A, DPE2 (50 μm) was incubated with [UL-14C]maltose for 30 s and precipitated with 10% trifluoroacetic acid. The precipitate was washed five times with 0.1% trifluoroacetic acid, resolubilized in 8 m urea, and the radioactivity was measured. The covalent intermediate could also be isolated by lowering the pH to 1.25 and filtering the maltose through a Centricon centrifugal filter device with a NMWL of 50 kDa (supplemental Fig. S1). B, DPE2 was incubated with glycogen and either α-glucose or α- and β-glucose at anomeric equilibrium (indicated by dashed lines as reported by Keilin and Hartree (23)) for 20 min, and the ratios of α- and β-maltose were measured.
In the presence of glycogen and α-glucose (α-94%, β-6%) DPE2 made primarily α-maltose (α-77%, β-23%, we presume that some spontaneous mutarotation occurred during the reaction). In an equilibrium mixture of glucose (α-36%, β-64%) (23), DPE2 made a similar anomeric ratio of maltose (α-35%, β-65%) (Fig. 2B). Thus, the anomeric configuration of maltose formed depends on the anomeric configuration of glucose, and so glucose must be added to form the reducing end of the maltose.
Anomeric Specificity—It has previously been hypothesized that DPE2 may be specific for β-maltose (20) and Dumez et al. (24) showed that there was no DPE2 activity on a zymogram when a crude extract of Arabidopsis protein was incubated with glycogen and α-maltose. The anomeric specificity for maltose was tested by relying on the specificity of maltose phosphorylase for the α-anomer (25). In a mixture of α- and β-maltose at anomeric equilibrium (approximately equimolar, Ref. 20) and glycogen, maltose phosphorylase acted on half of the maltose, and DPE2 was able to act on the other half (Fig. 3, left side). In the reverse order, DPE2 acted on half of the maltose, and the remaining maltose was judged to be α-maltose based on its reaction with maltose phosphorylase (Fig. 3), which is specific for α-maltose (20).
FIGURE 3.

DPE2 is specific for β-maltose. The reaction of DPE2 and maltose phosphorylase (MPL) is shown with glycogen (0.2 mg/ml) and 2,500 pmol of maltose at anomeric equilibrium. The reaction on the left was initiated with 5 units of maltose phosphorylase followed by 25 μg of DPE2 (as indicated by arrows), and the reaction on the right was initiated with 25 μg of DPE2 followed by 5 units of maltose phosphorylase. The reaction on the right was repeated five times.
The Mutant ΔN130 Is Unable to Use Glycogen but Has High Disproportionating Activity with Maltodextrins—The function of the carbohydrate binding modules was tested with two deletion mutants, ΔN130 (deletion of one CBM20 domain) and ΔN248 (deletion of both CBM20 domains) (Fig. 1A). The ΔN248 mutant could only be purified at low concentrations but we did not detect any significant difference in substrate preferences between the two mutants (Fig. 4A and supplemental Fig. S2). With maltose and various maltodextrin acceptors, ΔN130 had a preference for transferring glucosyl units from maltose to short maltodextrins. Wild-type DPE2, as was previously shown (16) does not efficiently transfer glucosyl units to short maltodextrins but prefers highly branched polymers such as glycogen (Fig. 4A). The increased activity that ΔN130 had on short maltodextrins was due to a large increase in binding affinity at the acceptor site (>100-fold decrease in Km; Fig. 4B). The second order rate constant (kcat/Km) for the disproportionation of various maltodextrins was measured based on the production of glucose. The data show that ΔN130 can produce glucose from maltodextrins (G3–G7) at low substrate concentration (0.1 mm) with a slight preference for short maltodextrins (Fig. 4C). Additionally, the mutant ΔN130 showed an approximate 10-fold increase in affinity for maltotriose when maltose was present in the buffer (Fig. 4D). Thin layer chromatography was used to confirm the reaction was a disproportionating reaction and not a hydrolysis reaction (Fig. 4E).
FIGURE 4.

The mutant ΔN130 has high disproportionating activity on short maltodextrins but is unable to use glycogen. A, the rate of glucose production for ΔN130 and wild-type DPE2 were compared on a variety of maltodextrin acceptors (2 mm) or 2 mg/ml glycogen with 2 mm maltose as a glucosyl donor. B, the Michaelis-Menten plot with maltoheptaose as an acceptor and maltose as a donor based on the rate of glucose production was measured to compare acceptor sites of the mutant ΔN130 (•) and wild type (Δ). C, the apparent specificity constants (kcat/Km) were measured for the ΔN130 mutant with various maltodextrins. The rate constants are based on the rate of glucose production. D, the Michaelis-Menten plot for ΔN130 is shown to compare activity with maltotriose in the presence (•) or absence (○) of 5 mm maltose. E, the products of ΔN130 and wild-type DPE2 were analyzed by thin layer chromatography. F, starch binding assay of wild-type and ΔN130 DPE2. Protein (25 μg) was incubated with corn starch (40 mg/ml) and, where indicated, with 20 mg/ml glycogen for 30 min on ice, and the supernatant and pellet fractions were analyzed by SDS-PAGE followed by Coomassie Brilliant Blue staining.
Wild-type DPE2 could bind to starch and glycogen had very little, if any, ability to dissociate DPE2 from the starch pellet (Fig. 4F; replicate experiments showed small amounts of DPE2 in the supernatant, data not shown). Removal of the carbohydrate binding module nearest to the N terminus completely abolished starch binding (Fig. 4F).
DISCUSSION
Gene and Protein Domain Structure—All fully sequenced plant genomes have a gene with the predicted domain structure of two N-terminal CBM20 domains and a GH77 domain interrupted by a large insert. A small number of similar genes were found in a few widely diverse organisms including bacteria and primitive eukaryotes. The insert in the GH77 domain showed significant conservation at the amino acid level. Many of the organisms form symbiotic relationships, often parasitic. The presence in basal eukaryotes and very sparse occurrence in bacteria could indicate that lateral gene transfer accounts for the bacteria in which this gene is found and that it is primarily a eukaryotic gene.
The metabolic function of DPE2 bridges the endosymbiont (chloroplast) and host (the rest of the plant cell). Perhaps the proteins in other organisms are also involved in metabolism that bridges the symbiont and host metabolism but the role of these proteins in organisms other than plants is not currently known.
Glycoside hydrolases GH77 appear to be closely related to the α-amylase family GH13 and both have been shown to use a ping-pong mechanism that involves a covalent glucosyl-enzyme intermediate (22, 26). DPE2 formed a covalent intermediate as expected. However, the DPE2 GH77 domain was interrupted by a large insert that replaced roughly all of the amino acid residues between the aspartate and glutamate active site residues that participate in catalysis. The function of this insert is not clear. It is not the reason that DPE2 can use maltose because MalQ can use maltose, and it does not have the insert. The use of glycogen was a function of the carbohydrate binding domain. But despite the lack of obvious function, the insert in the GH77 domain was found in several other organisms including some bacteria. The insert appears to have α-helix character at the beginning and end. A large number of amino acids are strictly conserved across bacteria, basal eukaryotes, and plants.
The Mechanism of DPE2—In the reverse reaction (Fig. 5), glucose makes up the reducing end of maltose, and it retains its anomeric configuration. This indicates that DPE2 will add or remove glucosyl units to or from the non-reducing end of glycogen (or SHG). Fettke et al. (17) has shown that DPE2 and a cytosolic glucan phosphorylase are capable of acting on the same sites of the SHG. Glucan phosphorylases are thought to interact with the non-reducing end of polyglucans (27), which is consistent with DPE2 adding glucosyl units to and from the non-reducing end of glycogen or SHG. The isolation of the covalent intermediate as well as the loss of activity in the E758Q mutant is consistent with a common mechanism for the DPE2-type enzymes and other GH77 enzymes. Thus, in the conversion of maltose to glucose, Glu-758 would protonate the α-1,4-glycosidic oxygen in β-maltose while Asp-563 simultaneously attacks C1 of the non-reducing end glucosyl unit, forming a covalent bond. β-Glucose then leaves the active site which allows Glu-758 to deprotonate the C4-hydroxyl group at the non-reducing end of a glycogen branch, activating it for nucleophilic attack on C1 of the enzyme bound glucosyl unit to reform an α-1,4-glycosidic bond.
FIGURE 5.

The proposed reaction mechanism of DPE2. The schematic shows the transfer of one glucosyl unit from maltose to an α-glucan polymer yielding one free glucose. The proposed role of Glu-758 is to protonate the α-1,4-glycosidic oxygen in maltose while Asp-563 simultaneously attacks C1 of the non-reducing end glucosyl unit, forming a covalent bond. Glucose then leaves the active site which allows Glu-758 to deprotonate the C4-hydroxyl group at the non-reducing end of an α-glucan, activating it for nucleophilic attack on C1 of the enzyme-bound glucosyl unit to reform an α-1,4-glycosidic bond.
The Glycosyl Hydrolase Domain of DPE2—The mechanism of DPE2 indicates that there are two conformational states of the active site, the unbound active site, and the covalently bound intermediate state. We define the donor site as the free enzyme active site, whereas the acceptor site represents subsites +1 and higher in the glucosyl-enzyme intermediate state. Two unique features of the donor site in wild-type DPE2 compared with other GH77 enzymes are that it is specific for the β-anomer of maltose (Fig. 3), and carbohydrate binding is prevented beyond subsite –1 (Ref. 17 and Fig. 4E). Unlike the wild-type enzyme, ΔN130 can produce glucose from maltodextrins in the absence of maltose (Fig. 4C). We cannot rule out the possibility that, due to an increased affinity for maltodextrins, the mutant is transferring single glucosyl moieties stepwise until the formation and subsequent disproportionation of maltose yields one free glucose. However, the reactions were carried out at a relatively low substrate concentration (∼0.03 Km) and given that the production of glucose from maltoheptaose would require six disproportionation reactions with each subsequent donor substrate being at a lower concentration than the previous, the final donor substrate (maltose) would be at a vanishingly low concentration. Thus, we would predict the initial rates of glucose production to differ by several orders of magnitude when comparing maltotriose and maltoheptaose as substrates. The other possibility is that carbohydrate binding is no longer inhibited beyond subsite –1, and ΔN130 is transferring larger maltooligosyl moieties to yield glucose in a single or few steps, i.e. when the size of the maltooligosyl moiety transferred is one less than the degree of polymerization of the maltodextrin. The latter explanation is consistent with the data in Fig. 4C, showing that the initial rates of glucose production from maltodextrins (G3–G7) are of approximately the same order of magnitude. We propose that the CBM20 domain restricts carbohydrate binding beyond subsite –1 in some way.
The acceptor site has been dramatically altered in the ΔN130 mutant relative to wild type DPE2. DPE2 shows optimum activity when highly branched water soluble polyglucans are used as an acceptor (16). By contrast, ΔN130 was unable to use glycogen as an acceptor; however, it binds to short maltodextrins with a much higher affinity than wild-type DPE2, as judged by >100-fold decrease in Km, with only a small decrease in kcat (Fig. 4B). Therefore, the CBM20 domain makes a large contribution to the specificity of the donor site by decreasing the affinity for maltodextrins, while increasing the affinity for complex carbohydrates.
The glycosyl hydrolase domain of ΔN130 appears to be similar to amylomaltases from other species in its affinity for carbohydrates. Kaper et al. (28, 29) determined the Km for maltotriose with amylomaltases from T. thermophilus and P. aerophilum to be 2.2 and 3.7 mm, respectively. We obtained a Km of 3.16 mm for maltotriose with ΔN130 (Fig. 4D). The presence of maltose causes an increased affinity for maltotriose (Fig. 4D). Given the proposed role of DPE2 in maltose metabolism, it is likely that maltose makes an extremely effective donor substrate in the mutant ΔN130, and the increased affinity for maltotriose is a measure of affinity for the acceptor site. Another possibility is that maltose can allosterically regulate maltodextrin binding.
The Carbohydrate Binding Module of DPE2 Is Functionally Related to the CBM20 Domain—CBM20 domains are typically on the C terminus of amylases and bind starch, while non-amylase enzymes sometimes have N-terminal CBM20 domains (30), notably the laforin dual specificity phosphorylase that can dephosphorylate complex carbohydrates (31) and is important in the human disease lafora. DPE2 represents an unusual case in which there are two adjacent CBM20 domains positioned N-terminally to the glycoside hydrolase domain. Because DPE2 is located in the cytosol of plants (13), which lacks starch, the CBM20 domains must bind something else. The in vivo function could be to bind the SHG, proposed to be part of the pathway from starch to sucrose in photosynthetic cells (7, 17).
It was confirmed that the CBM20 domain nearest to the N-terminal has retained its capacity to bind starch, despite the fact that starch is localized to the plastid (Fig. 4F). It could not be confirmed that the adjacent CBM20 has carbohydrate binding properties, as the removal of a single CBM20 completely eliminated starch binding. It is possible that the two domains work together as a single functional unit.
In summary, DPE2 of plants and enzymes from a small number of other unrelated organisms can act on maltose and have an unusual but conserved domain structure. DPE2 may play an important role in regulating metabolism in the cytosol of cells from plant leaves by preventing maltose from accumulating in maltodextrins and instead, directing it to a highly branched SHG where downstream enzymes can phosphorylytically cleave off glucosyl units for conversion to sucrose. The role of the homologues in non-photosynthetic organisms is not yet known.
Supplementary Material
Acknowledgments
We thank Xiaoyong Li (MSU Chemistry) and Sean Weise for measuring the rotation of the glucose used in this work.
This work was supported by United States Dept. of Energy Grant DE-FG02-04ER15565. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
Footnotes
The abbreviations used are: DPE2, disproportionating enzyme 2; SHG, soluble heteroglycan; PIPES, 1,4-piperazinediethanesulfonic acid; NMWL, nominal molecular weight limit.
References
- 1.Smith, A. M., Zeeman, S. C., and Smith, S. M. (2005) Annu. Rev. Plant Biol. 56 73–98 [DOI] [PubMed] [Google Scholar]
- 2.Zeeman, S. C., Delatte, T., Messerli, G., Umhang, M., Stettler, M., Mettler, T., Streb, S., Reinhold, H., and Kotting, O. (2007) Funct. Plant Biol. 34 465–473 [DOI] [PubMed] [Google Scholar]
- 3.Reimann, R., Ritte, G., Steup, M., and Appenroth, K. J. (2002) Physiol. Plant 114 2–12 [DOI] [PubMed] [Google Scholar]
- 4.Ritte, G., Lloyd, J. R., Eckermann, N., Rottmann, A., Kossmann, J., and Steup, M. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 7166–7171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kötting, O., Pusch, K., Tiessen, A., Geigenberger, P., Steup, M., and Ritte, G. (2005) Plant Physiol. 137 242–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ritte, G., Scharf, A., Eckermann, N., Haebel, S., and Steup, M. (2004) Plant Physiol. 135 2068–2077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu, Y., and Sharkey, T. D. (2006) Plant Cell Environ. 29 353–366 [DOI] [PubMed] [Google Scholar]
- 8.Weise, S. E., Weber, A., and Sharkey, T. D. (2004) Planta 218 474–482 [DOI] [PubMed] [Google Scholar]
- 9.Scheidig, A., Fröhlich, A., Schulze, S., Lloyd, J. R., and Kossmann, J. (2002) Plant J. 30 581–591 [DOI] [PubMed] [Google Scholar]
- 10.Nittylä, T., Messerli, G., Trevisan, M., Chen, J., Smith, A. M., and Zeeman, S. C. (2004) Science 303 87–89 [DOI] [PubMed] [Google Scholar]
- 11.Lu, Y., Steichen, J. M., Weise, S. E., and Sharkey, T. D. (2006) Planta 224 935–943 [DOI] [PubMed] [Google Scholar]
- 12.Kakefuda, G., and Duke, S. H. (1989) Plant Physiol. 91 136–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chia, T., Thorneycraft, D., Chapple, A., Messerli, G., Chen, J., Zeeman, S., Smith, S. M., and Smith, A. M. (2004) Plant J. 37 853–863 [DOI] [PubMed] [Google Scholar]
- 14.Lu, Y., and Sharkey, T. D. (2004) Planta 218 466–473 [DOI] [PubMed] [Google Scholar]
- 15.Lloyd, J. R., Blennow, A., Burhenne, K., and Kossmann, J. (2004) Plant Physiol. 134 1347–1354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu, Y., Steichen, J. M., Yao, J., and Sharkey, T. D. (2006) Plant Physiol. 142 878–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fettke, J., Chia, T., Eckermann, N., Smith, A., and Steup, M. (2006) Plant J. 46 668–684 [DOI] [PubMed] [Google Scholar]
- 18.Yang, Y., and Steup, M. (1990) Plant Physiol. 94 960–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee, S. B., and Robyt, J. F. (2001) Carbohydr. Res. 336 47–53 [DOI] [PubMed] [Google Scholar]
- 20.Weise, S. E., Kim, K. S., Stewart, R. P., and Sharkey, T. D. (2005) Plant Physiol. 137 756–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takaha, T., Yanase, M., Okada, S., and Smith, S. M. (1993) J. Biol. Chem. 268 1391–1396 [PubMed] [Google Scholar]
- 22.Barends, T. R. M., Bultema, J. B., Kaper, T., van der Maarel, M. J. E. C., Dijkhuizen, L., and Dijkstra, B. W. (2007) J. Biol. Chem. 282 17242–17249 [DOI] [PubMed] [Google Scholar]
- 23.Keilin, D., and Hartree, E. F. (1952) Biochem. J. 50 341–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dumez, S., Wattebled, F., Dauvillee, D., Delvalle, D., Planchot, V., Ball, S. G., and D'Hulst, C. (2006) Plant Cell 18 2694–2709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shirokane, Y., Ichikawa, K., and Suzuki, M. (2000) Carbohydr. Res. 329 699–702 [DOI] [PubMed] [Google Scholar]
- 26.Uitdehaag, J. C. M., Mosi, R., Kalk, K. H., van der Veen, B. A., Dijkhuizen, L., Withers, S. G., and Dijkstra, B. W. (1999) Nat. Struct. Mol. Biol. 6 432–436 [DOI] [PubMed] [Google Scholar]
- 27.Palm, D., Klein, H. W., Schinzel, R., Buehner, M., and Helmreich, E. J. M. (1990) Biochemistry 29 1099–1107 [DOI] [PubMed] [Google Scholar]
- 28.Kaper, T., Leemhuis, H., Uitdehaag, J. C. M., Van der Veen, B. A., Dijkstra, B. W., Van der Maarel, M. J. E. C., and Dijkhuizen, L. (2007) Biochemistry 46 5261–5269 [DOI] [PubMed] [Google Scholar]
- 29.Kaper, T., Talik, B., Ettema, T. J., Bos, H., Van der Maarel, M. J. E. C., and Dijkhuizen, L. (2005) App. Env. Microbiol. 71 5098–5106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Machovic, M., Svensson, B., MacGregor, E. A., and Janecek, Š. (2005) Eur. J. Biochem. 272 5497–5513 [DOI] [PubMed] [Google Scholar]
- 31.Worby, C. A., Gentry, M. S., and Dixon, J. E. (2006) J. Biol. Chem. 281 30412–30418 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.