

Abstract
Mitochondrial oxidative phosphorylation provides most cellular energy. As part of this process, cytochrome c oxidase (CcO) pumps protons across the inner mitochondrial membrane, contributing to the generation of the mitochondrial membrane potential, which is used by ATP synthase to produce ATP. During acute inflammation, as in sepsis, aerobic metabolism appears to malfunction and switches to glycolytic energy production. The pro-inflammatory cytokine tumor necrosis factor α (TNFα) has been shown to play a central role in inflammation. We hypothesized that TNFα-triggered cell signaling targets CcO, which is a central enzyme of the aerobic energy metabolism and can be regulated through phosphorylation. Using total bovine and murine hepatocyte homogenates TNFα treatment led to an ∼60% reduction in CcO activity. In contrast, there was no direct effect of TNFα on CcO activity using isolated mitochondria and purified CcO, indicating that a TNFα-triggered intracellular signaling cascade mediates CcO inhibition. CcO isolated after TNFα treatment showed tyrosine phosphorylation on CcO catalytic subunit I and was ∼50 and 70% inhibited at high cytochrome c concentrations in the presence of allosteric activator ADP and inhibitor ATP, respectively. CcO phosphorylation occurs on tyrosine 304 as demonstrated with a phosphoepitope-specific antibody. Furthermore, the mitochondrial membrane potential was decreased in H2.35 cells in response to TNFα. Concomitantly, cellular ATP was more than 35 and 64% reduced in murine hepatocytes and H2.35 cells. We postulate that an important contributor in TNFα-mediated pathologies, such as sepsis, is energy paucity, which parallels the poor tissue oxygen extraction and utilization found in such patients.
Pro-inflammatory cytokine TNFα3 exerts a wide range of inflammatory, immune-modulatory, and metabolic effects. TNFα is associated with various diseases such as sepsis, atherosclerosis, and hepatic failure and initiates its biological effects by binding to high affinity cell surface receptors (TNF receptor types 1 and 2) (1). Receptor ligation in different types of cells is associated with an increased production of reactive oxygen species (ROS). It is well documented that TNFα increases ROS in the mitochondria and that it does so in the cytoplasm in a NADPH-dependent fashion. Mitochondria are considered an early target in TNFα-induced cytotoxicity, because they appear swollen with a reduction in cristae membrane structure in the early course of endotoxinemia and sepsis (2, 3). It was suggested that early mitochondrial dysfunction and inhibition of the oxidative phosphorylation (OxPhos) system play a pivotal role in impaired O2 utilization during inflammatory processes (2, 4). During endotoxinemia and sepsis, glycolytic ATP production was increased (5). Apparently, OxPhos was inhibited, although increased tissue oxygen levels suggest cellular availability of O2 (6), indicating a decrease of oxygen utilization (7, 8).
The formation of the cellular energy carrier ATP is the result of both
anaerobic and aerobic processes. Anaerobic ATP generation is catalyzed by
phosphoglycerate kinase and pyruvate kinase, and GTP is produced by succinyl
coenzyme A synthetase. However, ∼95% of cellular energy is generated
through the aerobic pathway, the mitochondrial OxPhos process that includes an
elaborate electron transport chain (ETC), in which O2 functions as
the terminal electron acceptor and is reduced to water. The flow of electrons
from NADH and FADH2 through the ETC is coupled to the pumping of
protons across the mitochondrial inner membrane, which creates a transmembrane
electrochemical potential (ΔΨm). Through this process, a
proton-motive force is generated that is utilized by ATP synthase to produce
ATP from ADP and phosphate. As a byproduct of the OxPhos process, an estimated
1-2% of cellular O2 is converted into superoxide anions
(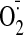 )
(9). OxPhos is regulated at
various sites by numerous factors, including the ATP/ADP ratio
(10), PO2, and the
amount of ROS and nitric oxide
(11,
12), as well as hormonal
influences (13).
)
(9). OxPhos is regulated at
various sites by numerous factors, including the ATP/ADP ratio
(10), PO2, and the
amount of ROS and nitric oxide
(11,
12), as well as hormonal
influences (13).
Cytochrome c oxidase (CcO, complex IV) is the terminal enzyme of the electron transport chain. CcO accepts electrons from cytochrome c and transfers them to molecular oxygen, which is reduced to water. At the same time protons are pumped across the inner mitochondrial membrane leading to the generation of the mitochondrial membrane potential ΔΨm. CcO exhibits the three characteristic regulatory means known to act on key metabolic enzymes: isoform expression, allosteric control, and reversible phosphorylation. There are three liver and heart type isoform pairs of subunits VIa, VIIa, and VIII (reviewed in Ref. 14) in addition to a lung-specific isoform of CcO subunit IV, a testes-specific isoform of subunit VIb, and a third isoform of subunit VIII (15-17); CcO is allosterically regulated through adenine nucleotides and thyroid hormone T2 (reviewed in Ref. 18); and there is clear evidence that CcO can be phosphorylated (19-22). We have recently shown that CcO is targeted for cAMP-dependent phosphorylation on tyrosine 304 of catalytic subunit I in vivo, which leads to strong enzyme inhibition (23). In addition the substrate of CcO, cytochrome c, can be phosphorylated in vivo on tyrosine 97, which also leads to an inhibition of the reaction with CcO (24). The complex regulation of CcO suggests an important role for CcO in the overall regulation of aerobic energy production. This hypothesis is supported by several recent studies suggesting that metabolic flux in the ETC is tightly coupled to CcO under physiological conditions, which assigns the role of the rate-limiting step to CcO (25-28).
The effect of TNFα on the ETC and in particular on CcO has not been studied. We hypothesized that TNFα mediates an early inhibitory effect on the OxPhos system and specifically inhibits CcO, because oxygen utilization is impaired in conditions involving TNFα. We predicted that CcO inhibition in turn leads to a decrease in ΔΨm and, consequently, reduced ATP levels. We here investigated the effect of TNFα on mammalian liver CcO in cell homogenates, isolated mitochondria, and purified CcO. We found that CcO activity was consistently reduced in bovine and murine liver cell homogenates in response to TNFα, whereas CcO activity of isolated mitochondria and isolated CcO in response to TNFα treatment did not change. This indicates that an intact cellular infrastructure is required for signal transduction from the cell surface, triggered by TNFα receptors, to the mitochondria and subsequently to CcO as a terminal target of this pathway. In addition, we show that TNFα treatment leads to reduced ΔΨm and ATP levels. We propose a model in which TNFα-mediated CcO inhibition leads to tissue dysoxia, which suppresses aerobic ATP production and causes a shift to the glycolytic pathway.
EXPERIMENTAL PROCEDURES
Isolation of Bovine and Murine Mitochondria—The chemicals were purchased from Sigma unless otherwise stated. Fresh C57BL/6 mouse liver tissue (2 g) was chopped into small pieces using scissors, whereas frozen cow liver tissue (200 g) was passed through a meat grinder, leaving most cells intact. The samples were supplemented with five volumes of buffer A (250 mm sucrose, 20 mm Tris-Cl (pH 7.4), 2 mm EDTA, 10 mm KF, 2 mm EGTA, 1 mm PMSF) and incubated for 5 min in the absence or presence of 20 ng/ml TNFα at room temperature. Mitochondria were isolated under conditions that preserve the physiological phosphorylation status as previously described (23, 24). Briefly, using 200 g of tissue, mitochondria were isolated at 4 °C, and buffer A was supplemented with unspecific tyrosine phosphatase inhibitor sodium vanadate (1 mm) for all subsequent steps. The samples were further homogenized with a commercial blender using a 5-fold volume of buffer A. After centrifugation (650 × g, 10 min), the supernatant was collected through cheesecloth. The pellet was homogenized and centrifuged to increase mitochondrial yield. Combined supernatants were centrifuged (16,300 × g, 20 min), and mitochondria were resuspended in 200 ml of buffer A using a Teflon homogenizer (150 rpm, four strokes), and buffer A was added to a volume of 2.5 L and centrifuged at low speed (370 × g for 5 min) to remove contaminants. The supernatant was centrifuged (16,300 × g, 20 min) to collect mitochondria, which were washed one more time by resuspension and centrifugation as described above. The mitochondrial pellet was resuspended in buffer A and adjusted to a final protein concentration of 20 mg/ml.
Isolation of CcO—CcO isolation starting from purified cow liver mitochondria was performed as previously described (23). Per 1 ml of isolated mitochondria, 250 μl of buffer B (1 m KH2PO4, pH 7.4, 10 mm KF, 2 mm EGTA, 1 mm sodium vanadate) were added. Mitochondrial proteins were solubilized by dropwise addition of 1 ml of 20% Triton X-114/1 g of mitochondrial protein under stirring. The membrane fraction was collected by centrifugation for 30 min at 195,000 × g and resuspended with a Teflon glass homogenizer after addition of 100 ml of buffer C (200 mm KH2PO4, pH 7.4, 10 mm KF, 2 mm EGTA, 1 mm sodium vanadate). The sample was centrifuged for 25 min at 195,000 × g, and the pellet was resuspended in 100 ml of buffer D (200 mm KH2PO4, pH 7.4, 5% Triton X-100, 10 mm KF, 2 mm EGTA, and 1 mm sodium vanadate) as above, followed by centrifugation (20 min at 195,000 × g). The CcO-containing supernatant was collected, and CcO was further extracted from the pellet by repeating the previous step two times. The supernatants were combined and diluted with 3 volumes of dH2O. The sample was applied to 300 ml of DEAE Sephacel (fast flow; GE Healthcare) equilibrated with buffer E (50 mm KH2PO4, pH 7.4, 0.1% Triton X-100). The column was washed with 800 ml of buffer E, and the proteins were eluted using a linear gradient from 50 mm to 1 m KH2PO4 (pH 7.4, 0.1% Triton X-100). Eluted fractions were analyzed by spectrophotometer, and CcO-containing fractions were combined. Sodium cholate was added (1% w/v) with stirring and pH was adjusted to pH 7.4. Fractionations were performed with 28% ammonium sulfate for 14 h, 37% for 1 h, and 45% for 5 min; the proteins were collected after each step by centrifugation for 15 min at 27,000 × g. Precipitated proteins were dissolved in 250 mm sucrose, 20 mm Tris-Cl (pH 7.4), 2 mm EDTA and stored at -80 °C after spectrophotometric determination of CcO concentration (29) and purity (30).
Cytochrome c Oxidase Activity Measurements with Isolated CcO, Mitochondria, and Homogenates from Tissue and Cultured Cells—To obtain regulatory-competent CcO, cholate has to be removed because it tightly binds to purified CcO (31) at nucleotide-binding sites (10), and cardiolipin that was damaged or removed during enzyme isolation has to be replaced (32). CcO was dialyzed in the presence of 0.1 mm ATP and a 40-fold molar excess of cardiolipin in 50 mm KH2PO4 (pH 7.4), 1% Tween 20, 2 mm EGTA, 10 mm KF. CcO activity was analyzed in a closed 200-μl chamber equipped with a micro Clark-type oxygen electrode (Oxygraph system; Hansatech). Measurements were performed in the presence of 5 mm ADP, an allosteric activator of CcO, or 5 mm ATP, an allosteric inhibitor of CcO, after incubation with an ATP regenerating system as described (23). The measurements were carried out with 2 μm CcO at 25 °C in the presence of 20 mm ascorbate and increasing amounts of cow heart cytochrome c from 0-40 μm. Oxygen consumption was recorded on a computer and analyzed with the Oxygraph plus software. Turnover number is defined as oxygen consumed (μmol)/(s·CcO (μmol)). Mitochondria, tissue, and cell homogenates were incubated in the presence or absence of TNFα in incubation buffer (250 mm sucrose, 20 mm K-HEPES, pH 7.4, 10 mm MgSO4, 2 mm KH2PO4, 1 mm PMSF, 10 mm KF, 2 mm EGTA, 2 μm oligomycin). CcO activity measurements were performed in 10 mm K-HEPES (pH 7.4), 40 mm KCl, 1% Tween 20, 2 μm oligomycin, 1 mm PMSF, 10 mm KF, 2 mm EGTA using the same experimental setup as above after washing, sonication, and centrifugation of the samples as described (23). Murine hepatocytes were freshly obtained from mice. Protein concentration was determined with the DC protein assay kit (Bio-Rad).
Cell Culture—H2.35 murine hepatocytes were grown in modified Dulbecco's medium (Invitrogen) supplemented with 10% fetal bovine serum and 1000 units penicillin/streptomycin at 37 °C and a 5% CO2 atmosphere. For CcO activity measurements, the cells were washed with phosphate-buffered saline (PBS), harvested by scraping in the presence of 10 ml of PBS, collected by centrifugation (50 × g, 5 min), and washed once more with PBS.
Antibody Production—Customized rabbit polyclonal antibodies were generated against the phosphotyrosine 304 epitope of CcO subunit I by Abgent (San Diego, CA). Two rabbits were immunized with the cysteine-conjugated synthetic peptide GMDVDTRApYFTSAC according to the vendor's protocol. Antibodies recognizing the unphosphorylated peptide were removed by affinity absorption against column-bound cysteine-conjugated synthetic GMDVDTRAYFTSAC peptide. Antibodies were tested on the synthetic phosphorylated and unphosphorylated peptides, on Tyr304-phosphorylated and unphosphorylated CcO (23), and phosphotyrosine-conjugated BSA by Western analysis, revealing a specific signal for the phosphorylated peptide and Tyr304-phosphorylated CcO subunit I in the expected size.
Western Analysis—SDS-PAGE with purified liver CcO or isolated mitochondria was carried out as described previously (33). Protein transfer time on a nitrocellulose membrane was 60 min to facilitate efficient transfer of larger CcO subunits. Antiphosphoserine and -threonine antibodies were sets of four (1C8, 4A3, 4A9, and 16B4) and three (1E11, 4D11, and 14B3) individual monoclonal antibodies (Calbiochem), respectively, whereas a single antiphosphotyrosine antibody was used (4G10; Upstate Biotechnology). CcO subunit IV isoform 1 was detected with a monoclonal antibody (10G8; Molecular Probes). In addition, a customized CcO subunit I phosphotyrosine 304 specific antibody was used. Western analysis was performed with a 1:5000 dilution of primary antibodies and horseradish peroxidase-conjugated secondary antibodies (1:10000 dilution; GE Healthcare), and signals were detected using the ECL plus Western blotting detection kit (GE Healthcare).
In-gel Tryptic Digest and Protein Identification by Mass Spectrometry—The tryptic digest was performed as described (34). Briefly, the bands were excised from Coomassie-stained gels, cut into 1-mm cubes, and incubated in 50 μl of acetonitrile (Merck) for 10 min. Acetonitrile was removed, and gel particles were swollen in 150 μl of 10 mm dithiothreitol (Merck), 10 mm NH4HCO3 (Merck) and incubated for 1 h at 60 °C. After cooling to room temperature, the solution was replaced by 150 μl of 50 mm iodoacetamide, 10 mm NH4HCO3 and incubated for 45 min at room temperature in the dark with occasional vortexing. The solution was removed, and gel pieces were washed with 150 μl of 10 mm NH4HCO3, which was replaced by acetonitrile to dehydrate gel pieces, which were dried under vacuum. Trypsin (bovine, sequencing grade; Roche Applied Science) was diluted in digestion buffer (10 mm NH4HCO3) to 0.1 μg/μl. Gel pieces were covered with digestion buffer and incubated overnight at 37 °C. The supernatant was transferred to a tube, and gel pieces were incubated for 10 min with occasional mixing in 50 μl of elution buffer (50% CH3CN/50% HCOOH; Merck). Elution was repeated two more times, and supernatants were combined and dried under vacuum. For further cleavage of peptides, tryptic fragments were incubated with 10% cyanogen bromide (w/v) in 100 μl of 70% formic acid (Merck) overnight at room temperature in the dark. 300 μl of dH2O were added, and the samples were dried under vacuum. The peptide mixture was dissolved in 0.1% trifluoroacetic acid/dH2O (Applied Biosystems) and separated by reversed phase HPLC on a Phenomenex 150 × 1-mm column with a flow rate of 0.16 ml/min. The gradient used was 0-60% buffer B in 60 min, 80% buffer B in 70 min (buffer A, 0.1% trifluoroacetic acid/dH2O; buffer B, 0.08% trifluoroacetic acid/CH3CN). Matrix-assisted laser desorption ionization time-of-flight analysis of the tryptic/CNBr HPLC peaks was performed on a Bruker mass spectrometer in positive ion mode. For mass fingerprint analysis, each raw spectrum was opened in Bruker Xtof (version 5.1.5) applying advanced baseline correction and noise removal. The mass spectrometer was calibrated with calibration II standard (Bruker Daltonics), nine peptides (m/z 757.3992, 1046.5418, 1296.6848, 1347.7354, 1619.8223, 1758.9326, 2093.0862, 2465.1983, and 3174.4710; quadratic mode). Filter peak list for monoisotopic masses only was enabled; the peak detection threshold was manually adjusted above background. Peak lists were analyzed with the Mascot public interface and searched against NCBInr data base (parameters: trypsin and CNBr were selected, with one potential missed cleavage; Mascot mass fingerprint matrix-assisted laser desorption ionization was selected as the instrument type, oxidized methionine was selected as variable modification, and carbamidomethylated cysteine was selected as fixed modification; MH+ monoisotopic masses and peptide tolerance were 100 ppm; taxonomy field was set as “other mammalia,” because of the bovine origin of the samples). The results were scored using probability-based Mowse score (protein score) is defined as 10 × log(p), where p is the probability that the observed match is a random event. Only significant protein scores were chosen.
ATP Assay—Fresh mouse liver tissue (50 mg) was partially disrupted in 800 μl of incubation buffer with a Teflon microtube pestle by applying 10 strokes. The suspension was incubated for 5 min in the presence or absence of 20 ng/ml TNFα. H2.35 cells were collected by scraping after applying similar incubation conditions and immediately stored in aliquots at -80 °C until measurement. ATP was released using the boiling method, by adding 300 μl of boiling buffer (100 mm Tris-Cl, pH 7.75, 4 mm EDTA) and immediate transfer to a boiling water bath for 2 min. The samples were put on ice and sonicated. The samples were diluted 300-fold, and 50 μl were utilized to determine the ATP concentration using the ATP biolumescence assay kit HS II (Roche Applied Science) according to the manufacturer's protocol. The experiments were performed in triplicates, and the data were standardized to the protein concentration using the DC protein assay kit (Bio-Rad).
Measurement of Mitochondrial Membrane Potential ΔΨm—Cultured H2.35 cells were grown to 75% confluence. To assess relative changes in mitochondrial membrane potential (ΔΨm), the cells were collected after trypsinization and washed once with PBS. The cells were incubated for 5 min with 20 ng/ml TNFα or 10 μm valinomycin in PBS at 37 °C. After incubation the cells were washed with PBS and incubated for 10 min in PBS containing 1 μm JC-1 (Molecular Probes). JC-1 is able to selectively enter mitochondria, occurs as a monomer at low concentration or at low membrane potential, and emits green fluorescence (35). At higher membrane potential JC-1 forms aggregates that show red fluorescence. Mitochondrial membrane potential ΔΨm was measured with a FACScan (Becton-Dickinson) flow cytometer equipped with a 488-nm argon ion laser, and the data obtained were analyzed with the Cell Quest software. Fluorescence was measured using a 585 ± 42-nm band-pass filter, and 10,000 events were analyzed in each run.
RESULTS
TNFα Leads to Strong Cytochrome c Oxidase Inhibition in Bovine Hepatocytes—TNFα activates several pathways such as inflammatory, apoptotic, and survival pathways. Several recent studies suggested that TNFα may target mitochondria. However, most of these studies addressed late effects of TNFα, such as increased apoptosis and decreased ETC activity that was observed after 24-48 h (36, 37). TNFα may also exert early effects in the cell at the level of the mitochondrial OxPhos system. Because CcO is the terminal and possibly rate-liming enzyme in the ETC, we investigated the early effect of TNFα on CcO activity. As a model system, we first tested the effect of TNFα on chunky cow liver homogenates. Under those conditions most of the cells present are intact, allowing signal transduction in a physiological cellular context. Both optimal incubation time and the concentration of TNFα were determined in a series of preliminary tests (not shown). Five min of incubation at a concentration of 20 ng/ml TNFα was found to produce a strong and consistent response and was applied during all experiments. CcO activity was measured by adding increasing amounts of substrate cytochrome c in the presence of allosteric activator ADP or inhibitor ATP. After TNFα treatment CcO activity was 57 and 54% inhibited at high cytochrome c substrate concentrations in the presence of ADP and ATP, respectively (Fig. 1A). At a more physiologically meaningful cytochrome c concentration of 6 μm, a number that has been reported for mitochondrial heart muscle particles (38), the inhibitory effect is even more pronounced, exceeding 75% inhibition for both ADP-stimulated and ATP-inhibited respiration. This inhibitory effect was not caused by changes in CcO protein amount as shown by Western analysis (Fig. 1B).
FIGURE 1.
TNFα leads to CcO inhibition in cow liver tissue. A, cow liver tissue was partially homogenized leaving most cells intact and incubated in the presence (squares) or absence (triangles) of 20 ng/ml TNFα for 5 min at 20 °C. CcO activity was measured in the presence of 5 mm ATP (closed symbols, inhibited) or ADP (open symbols, stimulated) by the addition of increasing amounts of cytochrome c. Specific activity is defined as consumed O2 (μmol)/(min·total protein (mg)). Shown are representative results of three independent experiments. TN, turnover number. B, Western analysis using an antibody against CcO subunit IV (isoform 1) indicates no changes of CcO amount after short term TNFα treatment. The mitochondria were isolated with or without TNFα treatment (see above). Thirty μg of total mitochondrial protein was loaded, and CcO subunit IV was detected with a monoclonal antibody.
To confirm that the response to TNFα is not species specific, we tested its effect on total cell homogenates of freshly isolated murine hepatocytes and the murine hepatocyte cell line H2.35. Hepatocytes were incubated in the presence or absence of TNFα (20 ng/ml). After solubilization, the activity of CcO was measured in the presence of ADP or ATP. As expected, TNFα exposure also led to strong inhibition of CcO in murine hepatocytes (Fig. 2). Similar results were obtained with H2.35 cells (data not shown).
FIGURE 2.

Effect of TNFα on CcO activity is not species-dependent. Fresh mouse liver tissue was partially homogenized and incubated in the presence or absence of 20 ng/ml TNFα for 5 min at 20 °C. CcO activity was measured in the presence of allosteric effectors ATP (closed symbols) or ADP (open symbols) as described in Fig. 1. TN, turnover number.
Purified Bovine Liver Cytochrome c Oxidase Is Not Inhibited by TNFα—Hormones are capable of directly binding to CcO and changing enzyme activity and mitochondrial energy production as has been shown for thyroid hormone T2, which binds to subunit Va and abolishes the allosteric ATP-inhibition in bovine heart CcO (13). To identify whether the inhibitory effect of TNFα on CcO is due to direct interaction with this enzyme or is mediated through cell signaling, we used purified CcO from bovine liver. Incubation of isolated CcO with TNFα did not change the activity of CcO in comparison with the control in the presence of ATP or ADP (Fig. 3).
FIGURE 3.

TNFα does not inhibit isolated CcO. CcO isolated under standard conditions (see “Experimental Procedures”) was incubated in the presence or absence of TNFα. Turnover number (TN) is defined as consumed O2 (μmol)/(s·CcO (μmol)).
TNFα Treatment of Isolated Mitochondria Does Not Lead to Cytochrome c Oxidase Inhibition—There have been a few studies proposing that TNFα may bind to a corresponding receptor on the mitochondrial membrane, which triggers effects within the mitochondria rather than through the known TNFα cell surface receptors (39, 40). To test whether TNFα exerts its inhibitory effect on CcO through a possible mitochondrial receptor or whether this effect is triggered through cell signaling upstream of the mitochondria, we purified mitochondria from freshly isolated murine hepatocytes. Incubation of mitochondria with TNFα (20 ng/ml) did not change the activity of CcO in comparison with the controls in the presence of ADP or ATP (Fig. 4). Thus the inhibitory effect of TNFα on CcO requires intact cells, which contain TNFα receptors. We conclude that CcO inhibition by TNFα is mediated through a cell signaling cascade, presumably involving TNFα cell surface receptors.
FIGURE 4.

TNFα does not lead to CcO inhibition in isolated mitochondria. Mouse liver mitochondria were freshly isolated. TNFα-treated (squares) and untreated (triangles) mitochondria showed no difference for ADP-stimulated (open symbols) and ATP-inhibited (closed symbols) respiration (see Fig. 1 for details). TN, turnover number.
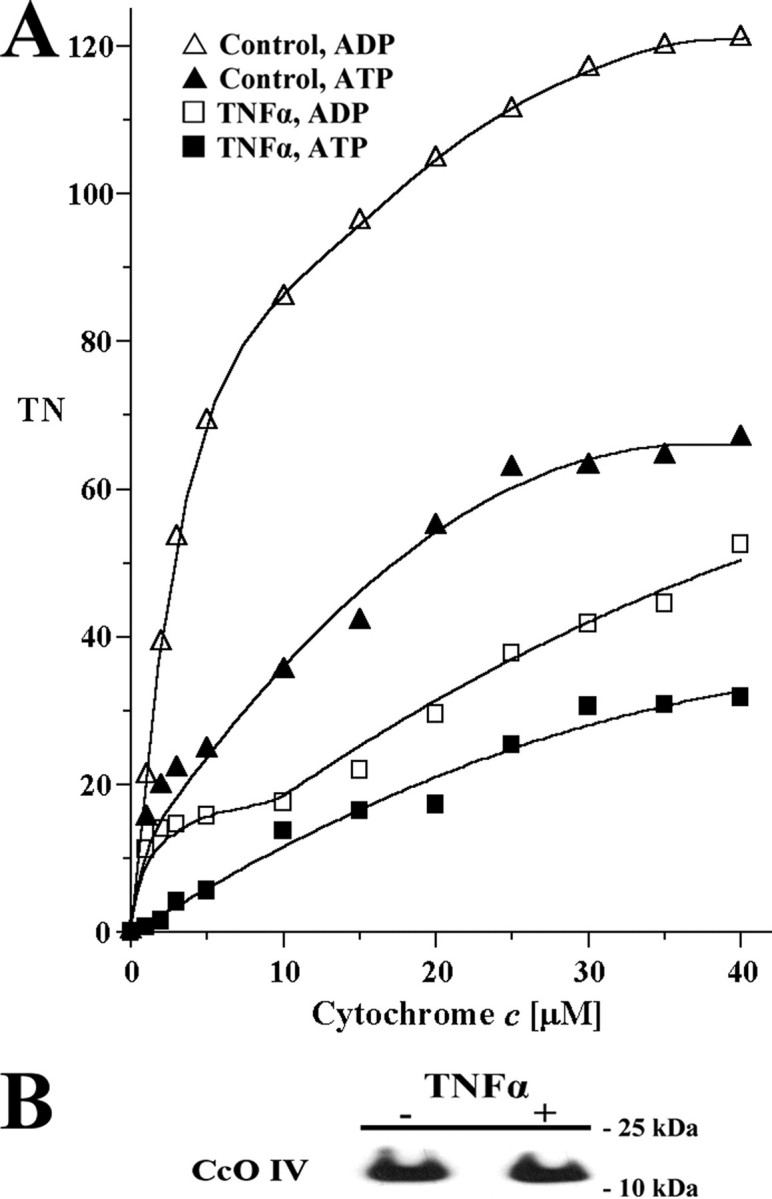
TNFα Treatment of Tissue Leads to Tyrosine Phosphorylation at Cytochrome c Oxidase Subunit I—Because TNFα-induced cell signaling leads to downstream phosphorylation of a variety of proteins and because an inhibitory effect of TNFα on CcO requires an intact cell structure, we hypothesized that TNFα may lead to a downstream activation of a kinase targeting CcO. We isolated cow liver CcO side by side in the presence or absence of 20 ng/ml TNFα under conditions that preserve the physiological phosphorylation status (see “Experimental Procedures”). During the CcO purification we included protein phosphatase inhibitors and calcium chelator EGTA to prevent activation of calcium-dependent protein phosphatases. Isolated CcO was subjected to SDS-PAGE (Fig. 5A) followed by Western analysis with monoclonal antiphosphoserine, -thereonine, and -tyrosine antibodies. Western analysis with antiphosphoserine and antiphosphothreonine antibodies did not indicate differences in serine or threonine phosphorylation (not shown). The only difference observed between control and TNFα CcO was tyrosine phosphorylation of a band in the size of subunit I for the TNFα-treated fraction (Fig. 5B).
FIGURE 5.
Isolation of CcO in the presence of TNFα leads to tyrosine phosphorylation of CcO subunit I at tyrosine 304. Cow liver CcO was isolated side by side after incubation of tissue with (+) or without (-) TNFα. A, CcO subunits were separated by SDS-PAGE, and protein bands were stained with Coomassie Blue. Subunits are as indicated. B, Western analysis with a phosphotyrosine-specific antibody revealed a strong signal only for the TNFα-treated sample at the size of subunit I (lane 4). Lane 1, phosphotyrosine-conjugated BSA (positive control; produces a broad, smeared signal); lane 2, ovalbumin (negative control); lane 3, CcO isolated in the absence of TNFα. C, a customized polyclonal antibody raised against the phosphorylated tyrosine 304 epitope of CcO subunit I produced a strong signal for the TNFα treated sample (lane 4) but not for control CcO (lane 3). Lanes 1-4 as in B. Note that in contrast to B, phosphotyrosine-conjugated BSA serves as a negative control in C, indicating that the epitope surrounding phosphotyrosine 304 is required for binding. Box, the synthetic phosphopeptide GMDVDTRApYFTSAC (+), which was used to generate the phosphoepitope-specific antibody and the unphosphorylated peptide GMDVDTRAYFTSAC (-; 100 ng each) were spotted on a membrane and subjected to Western analysis using the phosphotyrosine 304-specific antibody. A strong and specific signal was observed for the phosphorylated peptide (+). Shown are representative results of five independent experiments.
Gel slices corresponding to the tyrosine-phosphorylated band were excised for analysis by mass spectrometry. A tryptic digest followed by cyanogen bromide cleavage and subsequent mass spectrometry yielded a sequence coverage of 33% (supplemental Table S1) and indicated that CcO subunit I corresponds to the protein band that was tyrosine-phosphorylated based on Western analysis.
Tyrosine 304 of CcO Subunit I Is the Site of TNFα-induced Phosphorylation—Recently, we have shown that CcO subunit I can be phosphorylated on tyrosine 304 in a cAMP-dependent manner, resulting in a strong inhibitory effect on CcO activity (23), and we hypothesized that the same residue can be phosphorylated after TNFα treatment. Given the difficulties of assigning phosphorylation sites on CcO subunit I by mass spectrometry (see “Discussion”), we decided to generate phosphoepitope-specific antibodies against the phosphotyrosine 304 epitope of subunit I (see “Experimental Procedures”). The antibodies were generated against the synthetic peptide GMDVDTRApYFTSA and affinity-purified through absorption of antibodies to the unphosphorylated peptide. The antibody was first tested with the synthetic phosphorylated and unphosphorylated peptides in addition to tyrosine 304 phosphorylated and unphosphorylated CcO purified as previously described (23), and a highly specific signal was observed only with the phosphorylated peptide (Fig. 5C, box) and the phosphorylated enzyme in the expected size of subunit I. No signal was obtained with unphosphorylated CcO and phosphotyrosine-conjugated BSA, indicating that the amino acids adjacent to phosphotyrosine 304 also define specificity. The antibody was then applied to TNFα phosphorylated and control CcO, revealing a strong signal for the TNFα-induced enzyme but no signal for the control CcO and the negative controls (Fig. 5C).
These results show for the first time that CcO subunit I is tyrosine-phosphorylated in response to TNFα treatment. This suggests tyrosine kinase and phosphatase involvement in the regulation of oxidative phosphorylation in response to inflammation at the level of CcO.
TNFα-induced Phosphorylation of Cytochrome c Oxidase Leads to Inhibition of the Isolated Enzyme—To test the effect of this phosphorylation on enzyme kinetics, we compared respiration of CcO isolated after TNFα treatment and control CcO. Subunit I-phosphorylated CcO is ∼50 and 70% inhibited at high cytochrome c substrate concentrations in the presence of allosteric activator ADP or inhibitor ATP, respectively (Fig. 6, compare open and closed triangles and squares, respectively). At a more physiological cytochrome c concentration of 6 μm (38), we observed a 68% inhibition for both ADP-stimulated and ATP-inhibited respiration. The curves of subunit I phosphorylated CcO showed pronounced sigmoidal tendencies (Fig. 6).
FIGURE 6.

CcO isolated in the presence of TNFα is inhibited and shows pronounced sigmoidal kinetics. Cow liver tissue was incubated in the presence or absence of TNFα. Mitochondria and subsequently CcO were isolated under conditions that preserve the physiological phosphorylation status (see “Experimental Procedures”). The activity of solubilized CcO pretreated to restore regulatory properties as described (23) was measured in the presence of 5 mm ATP (closed symbols) or ADP (open symbols). Turnover number (TN) is defined as consumed O2 (μmol)/(s·CcO (μmol)).
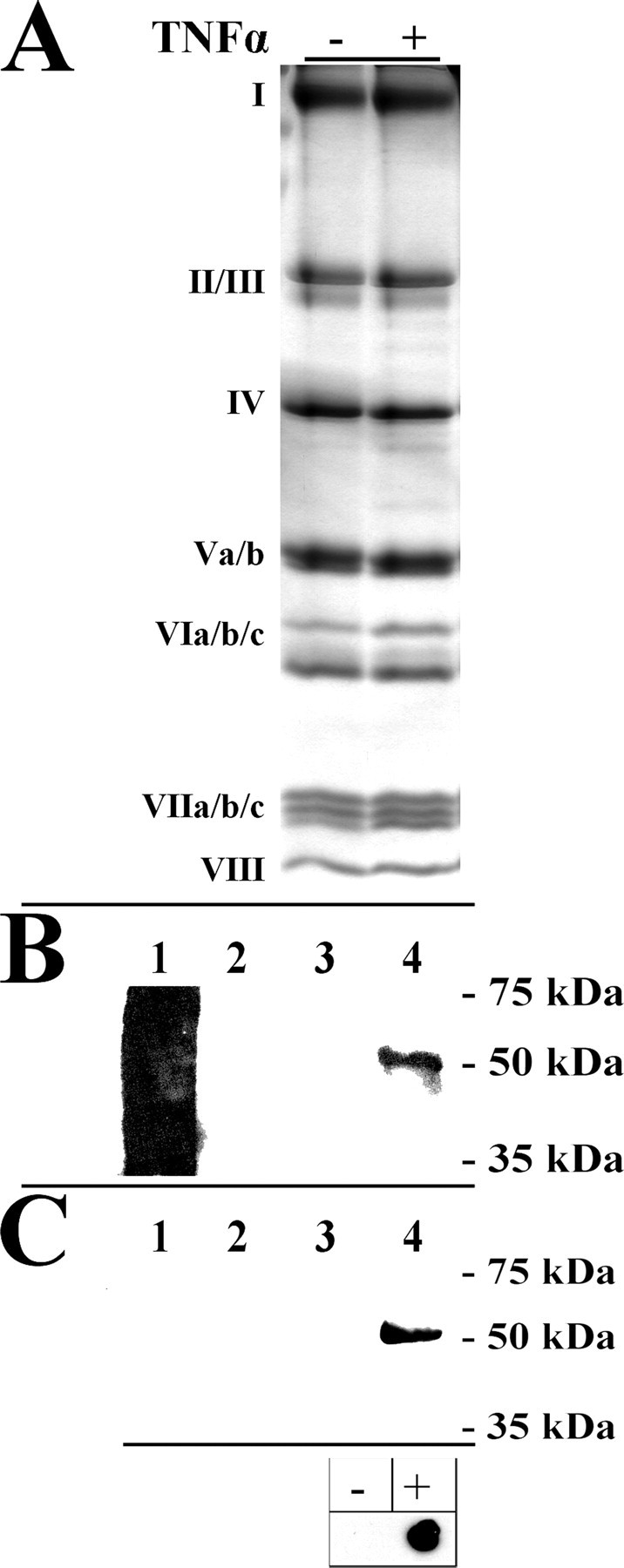
To further support the link between CcO tyrosine phosphorylation and enzyme inhibition, we analyzed CcO activity and phosphorylation in a time-dependent manner. Because signaling operates quickly and because initial experiments showed immediate effects on cell respiration after 5 min using a concentration of 20 ng/ml TNFα, bovine liver was incubated with a lower concentration of 10 ng/ml TNFα for 0, 2, 5, and 12 min followed by mitochondria isolation. Respiration measurements showed a time-dependent decrease in CcO activity, which was 35 and 55% inhibited after 2 and 5 min, respectively (Fig. 7B, compare squares, triangles, and crosses). Under these conditions CcO inhibition was maximal (71%) after 12 min of incubation (Fig. 7B, circles) and could not be further amplified by incubation for 20 min (not shown). Western blot analysis with our phosphoepitope-specific antibody showed a time-dependent increase in tyrosine 304 phosphorylation (Fig. 7A).
FIGURE 7.
Phosphorylation of tyrosine 304 correlates with changes in CcO activity. To assess a time-dependent change in CcO phosphorylation (A) and respiration (B), bovine liver tissue was treated with a lower concentration of 10 ng/ml TNFα for the indicated time periods, followed by immediate mitochondria isolation and measurement of respiration. Specific activity is defined as consumed O2 (μmol)/(min·total mitochondrial protein (mg)) (B). Western blot analysis of subsequently isolated CcO (6 μg/lane) using the phosphotyrosine 304-specific antibody showed a time-dependent increase of CcO subunit I phosphorylation, which was maximal after 12 min (A). C, correlation between tyrosine 304 phosphorylation (A) and CcO specific activity at maximal turnover (B, at 30 μm cytochrome c) are presented time-dependently. Films from Western analysis were scanned and intensities of individual bands analyzed with ImageQuant software (version 5, Molecular Dynamics) correcting for background signal. The signal obtained after 12 min was arbitrarily set to 100%. TN, turnover number.
A representation of CcO activity at maximal turnover together with the degree of tyrosine 304 phosphorylation in a single diagram (Fig. 7C) illustrates the negative correlation. For example, the signal intensity for tyrosine 304 phosphorylation at 5 min is 30% decreased compared with the signal at 12 min, which is accompanied by a 56% increase in CcO activity at 5 min compared with 12 min. A further decrease of 31% of Western signal at 2 min compared with 5 min results in an additional 68% activation. Both curves show saturation behavior at later time points in agreement with our finding that CcO phosphorylation could not be further increased via longer incubation periods.
TNFα Treatment Decreases the Mitochondrial Membrane Potential in H2.35 Cells—CcO couples redox chemistry with proton pumping creating the mitochondrial membrane potential ΔΨm together with ETC complexes I and III. It was previously shown that TNFα induces a drop of ΔΨm (41), and we hypothesized that applying our specific conditions that led to a decrease in CcO activity via subunit I phosphorylation would lead to a reduction of the mitochondrial membrane potential. JC-1 is a fluorescent probe, which is often used to analyze changes of ΔΨm via flow cytometry. JC-1 selectively enters mitochondria, and changes in the ratio of green and red fluorescence indicate relative changes of ΔΨm. H2.35 cells treated with TNFα for 5 min showed a clear decrease of red to green fluorescence, indicating a decrease of the mitochondrial membrane potential (Fig. 8B). The drop in ΔΨm observed with TNFα was qualitatively similar to the drop of ΔΨm in cells that were treated with the potassium ionophore valinomycin, which depolarizes ΔΨm (Fig. 8A).
FIGURE 8.

TNFα treatment leads to a decrease in the mitochondrial membrane potential. Murine H2.35 cells were incubated for 5 min with TNFα or potassium ionophore valinomycin in PBS. Relative membrane potentials were determined using the fluorescent probe JC-1. Measurements were performed with a FACScan (Becton-Dickinson) flow cytometer equipped with a 488-nm argon ion laser, and data obtained were analyzed with the Cell Quest software. Fluorescence was measured using a 585 ± 42-nm band-pass filter, and 10,000 cells were analyzed in each run. In comparison with the untreated cells (Control), TNFα treatment leads to membrane depolarization (B) similar to that observed with valinomycin (A).
ATP Levels in Murine Hepatocytes and H2.35 Cells Are Strongly Decreased after TNFα Treatment—Using freshly isolated murine hepatcytes and H2.35 cells, we assessed whether inhibition of CcO and the reduction of ΔΨm are translated into lower ATP level in the cells. The cells were incubated for 5 min in the presence or absence of 20 ng/ml TNFα. ATP levels were significantly decreased in both murine hepatocytes and H2.35, namely 35 and 64% (Fig. 9).
FIGURE 9.

Effect of TNFα on cellular ATP levels. Mouse liver tissue and H2.35 cells were incubated with 20 ng/ml TNFα for 5 min at 20 °C. ATP concentrations were measured using the bioluminescence method. ATP concentrations of the control samples were set to 100%. ATP levels were reduced by 35 and 64% in liver tissue and H2.35 cells, respectively.
DISCUSSION
TNFα is a cytokine involved in various cell functions, such as differentiation, inflammation, and apoptosis. TNFα plays a critical role in different disease processes such as liver failure, ischemic reperfusion injury (42), and sepsis (43). Several studies demonstrated that many of these signaling cascades, such as the TNF-related apoptosis-inducing ligand and caspase-8 pathways are directly linked to the mitochondrial electron transport machinery (44, 45). However, the underlying mechanism of this process has not yet been described. There is ample evidence that mitochondria are involved in endotoxemia and sepsis. Interestingly, the only known genetic predictor for survival during sepsis is mitochondrial DNA composition. It was recently shown that haplogroup H, which is common in Europeans, confers a more than 2-fold increased chance of survival compared with patients with a different haplogroup (46), suggesting that mitochondrial function plays a key role in inflammation. There are other suggestions that the cytotoxic activity of TNFα is mediated by damage to mitochondria and even that the outcome of sepsis may depend on the extent of mitochondrial damage (2). At the functional level it has long been known that TNFα induces the production of lactate in vitro (47) and in vivo (48), indicating a switching from aerobic to glycolytic energy metabolism. Callahan and Supinski (49) determined that in endotoxin-treated rats, ETC components complexes I, II, and IV were down-regulated both at the transcript and protein level within 24 h after treatment. Even as early as 1 h after TNFα treatment, morphological changes were observed in mitochondria, whereas other organelles did not show any damage (3). Another study using a mouse model for sepsis indicated a long term inhibitory effect on CcO after 6-24 h and concomitantly reduced levels of CcO (50). However, all of these studies did not consider changes in the phosphorylation state of OxPhos complexes in the context of inflammation.
Signaling molecules exert long and short term effects. Short term effects are mediated through phosphorylation events that occur on the seconds to minutes time scale. Such immediate effects do not involve changes in protein mass that, because of a relatively slow protein turnover, usually require hours or days. Our study was designed to address the short term effect of TNFα on cellular respiration and to identify a regulatory mechanism, which is not based on changes in CcO protein levels (Fig. 1B). We show that TNFα has an early and profound inhibitory effect on CcO activity in different cell systems (Figs. 1, 2, and 6). This inhibition occurs in the presence of both allosteric effectors ADP and ATP. The TNFα-mediated effect requires intact cells, because the CcO activity from purified enzyme or isolated mitochondria treated with TNFα was not inhibited (Figs. 3 and 4).
Inhibition of CcO is due to tyrosine phosphorylation of subunit I (Figs. 5B and 7A), which was the only difference found by comparison with control CcO. A newly generated antibody against tyrosine 304 of CcO subunit I indicated that TNFα-mediated phosphorylation occurs on the same residue we previously identified in a cAMP-dependent context (Fig. 5C). Although it cannot be ruled out that an additional tyrosine residue of subunit I can be targeted for phosphorylation, we have not identified peptide fragments via mass spectrometry, suggesting the presence of further phosphorylation sites. It is noteworthy in this context that the analysis of CcO subunit I by mass spectrometry is a difficult problem, perhaps because of its hydrophobicity, which might explain the inefficient digestion and low peptide fragment yield as indicated by the low sequence coverage of only 6% in our previous study (23). Other proteomic studies failed to detect subunit I via mass spectrometry in mouse heart, liver, brain, and kidney, whereas most other CcO subunits were detected in multiple tissues (51). Although we here improved our protocol to increase sequence coverage of subunit I more than 5-fold to 33% using trypsin digestion in combination with cyanogen bromide cleavage (see “Experimental Procedures”), we were not able to detect the tyrosine 304-containing peptide for analysis by mass spectrometry in this study. Therefore, this approach needs further optimization to include all peptides of interest.
Numerous studies have focused on the role of cytosol- and mitochondria-derived ROS in TNFα signaling. These findings are clearly important for our understanding of TNFα-mediated cell toxicity (for current reviews see Refs. 52 and 53). However, one other important aspect of mitochondrial dysregulation induced by TNFα has not been addressed at the molecular level, and we here propose energy paucity as an essential hallmark of TNFα signaling in pathological conditions (Fig. 10); CcO inhibition leads to an overall backup of electron flow through the ETC, which results in reduced ΔΨm levels. Because ΔΨm is required for ATP synthesis, cellular energy drops. Eventually, cellular functions cannot be supported, which can lead to organ failure as observed in sepsis.
FIGURE 10.
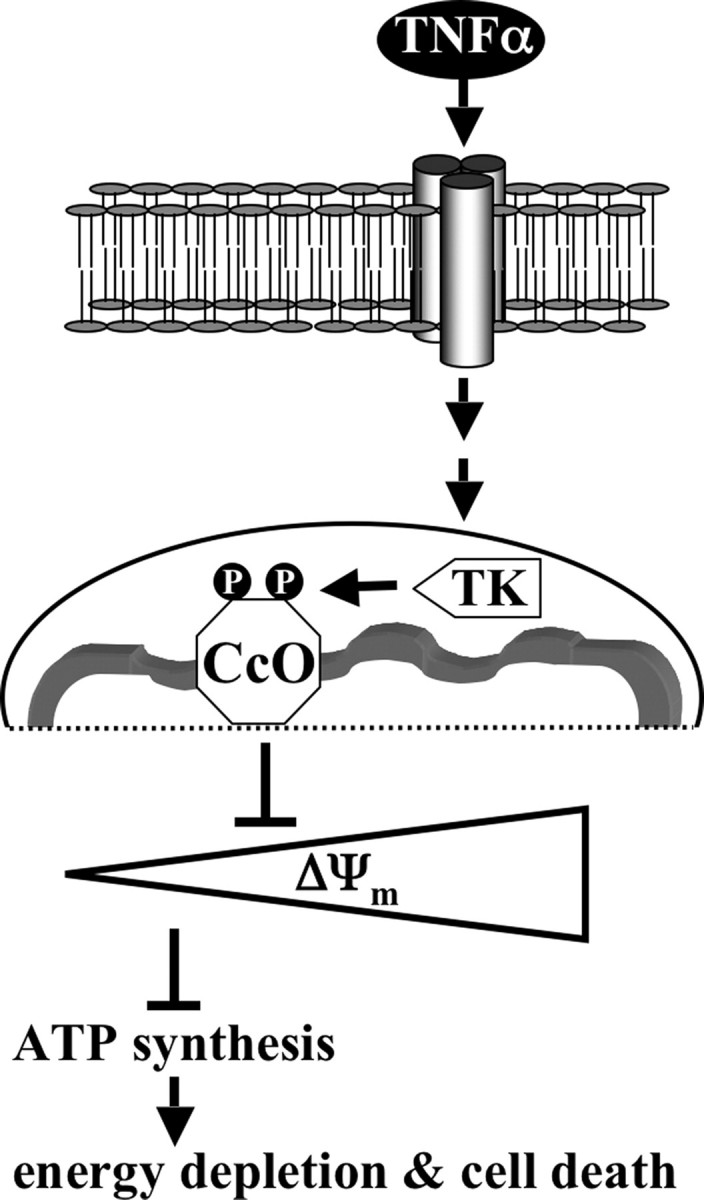
Model of TNFα-mediated CcO inhibition. TNFα binds to its cognate cell surface receptors, which initiates receptor ligation triggering the intracellular TNFα signaling cascade. As part of this process a mitochondrial tyrosine kinase (TK) is activated, which phosphorylates CcO subunit I on tyrosine 304. CcO inhibition leads to a decrease of the mitochondrial membrane potential ΔΨm. This in turn provides fewer substrate protons for ATP synthase, resulting in lowered ATP levels, which over extended periods of time leads to cell death caused by energy depletion.
Our data suggest that the TNFα pathway activates a mitochondrial tyrosine kinase (Fig. 10) that phosphorylates CcO on tyrosine residue 304 of subunit I. This phosphorylation has a strong impact on the catalytic activity of CcO. We have recently shown that the same residue can be phosphorylated by the cAMP-dependent pathway, leading to strong enzyme inhibition (23). cAMP, as increased during starvation, is another example of a stress signal. Raised cAMP levels lead to the activation of serine/threonine-specific protein kinase A (PKA). Protein kinase A-anchoring proteins (AKAPs) have been shown to localize PKA to cellular compartments in close proximity to its targets to streamline signaling (54). Several studies indicated that AKAPs localize PKA to the outer side of the outer mitochondrial membrane (55-58). Recently, Livigni and co-workers (58) have shown that AKAP121 provides a combined platform for PKA, protein-tyrosine phosphatase PTPD1, and c-Src signaling. Interestingly, in addition to cAMP-dependent subunit I phosphorylation, it has been shown that CcO subunit II can be tyrosine-phosphorylated, mediated by c-Src tyrosine kinase (20). In contrast to subunit I phosphorylation, c-Src-mediated phosphorylation of subunit II leads to CcO activation. Thus opposing signaling pathways with respect to CcO activation/inhibition appear to converge on mitochondrial signaling hubs, which then transmit the signal to the inner mitochondrial compartments. It is possible that additional components will be identified as part of those hubs, including components of the TNFα pathway. This hypothesis is supported by our finding that tyrosine 304 is phosphorylated in response to both cAMP and TNFα, suggesting that both pathways converge on the same terminal target.
We found that TNFα-treated murine hepatocytes and H2.35 cells have lower levels of ATP (Fig. 9), which may be a central element in inflammation-related pathology and death. TNFα caused a decline of ATP concentration in a time-dependent fashion, and cells were almost energy-depleted after 30 min (not shown). The kinases and phosphatases involved in reversible TNFα-mediated CcO phosphorylation may be promising targets for drug development because they act on an end point of cell signaling. Inflammatory conditions, found in sepsis, cardiomyopathy, diabetes, and neurodegenerative disorders, could be targeted for therapy if energy paucity is a central aspect of those conditions.
Supplementary Material
Acknowledgments
We gratefully thank Drs. Lawrence I. Grossman for critical discussions and Jeffrey W. Doan for comments on the manuscript.
This work was supported by the Division of Pulmonary Critical Care and Sleep Medicine and the Center for Molecular Medicine and Genetics. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Table S1.
Footnotes
The abbreviations used are: TNF, tumor necrosis factor; CcO, cytochrome c oxidase; ETC, electron transport chain; JC-1, 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolyl-carbocyanine iodide; OxPhos, oxidative phosphorylation; PKA, protein kinase A; PMSF, phenylmethylsulfonyl fluoride; ROS, reactive oxygen species; dH2O, distilled H2O; PBS, phosphate-buffered saline; BSA, bovine serum albumin; HPLC, high pressure liquid chromatography; AKAP, protein kinase A-anchoring protein.
References
- 1.Locksley, R. M., Killeen, N., and Lenardo, M. J. (2001) Cell 104 487-501 [DOI] [PubMed] [Google Scholar]
- 2.Brealey, D., Brand, M., Hargreaves, I., Heales, S., Land, J., Smolenski, R., Davies, N. A., Cooper, C. E., and Singer, M. (2002) Lancet 360 219-223 [DOI] [PubMed] [Google Scholar]
- 3.Schulze-Osthoff, K., Bakker, A. C., Vanhaesebroeck, B., Beyaert, R., Jacob, W. A., and Fiers, W. (1992) J. Biol. Chem. 267 5317-5323 [PubMed] [Google Scholar]
- 4.Singer, M. (2005) Crit. Care Med. 33 (Suppl. 12) S539-S542 [DOI] [PubMed] [Google Scholar]
- 5.Berg, S., Sappington, P. L., Guzik, L. J., Delude, R. L., and Fink, M. P. (2003) Crit. Care Med. 31 1203-1212 [DOI] [PubMed] [Google Scholar]
- 6.Rosser, D. M., Stidwill, R. P., Jacobson, D., and Singer, M. (1995) J. Appl. Physiol. 79 1878-1882 [DOI] [PubMed] [Google Scholar]
- 7.Kreymann, G., Grosser, S., Buggisch, P., Gottschall, C., Matthaei, S., and Greten, H. (1993) Crit. Care Med. 21 1012-1019 [DOI] [PubMed] [Google Scholar]
- 8.Boneh, A. (2006) Cell Mol. Life Sci. 63 1236-1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richter, C., Park, J. W., and Ames, B. N. (1988) Proc. Natl. Acad. Sci. U. S. A. 85 6465-6467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Napiwotzki, J., Shinzawa-Itoh, K., Yoshikawa, S., and Kadenbach, B. (1997) Biol. Chem. 378 1013-1021 [DOI] [PubMed] [Google Scholar]
- 11.Palacios-Callender, M., Quintero, M., Hollis, V. S., Springett, R. J., and Moncada, S. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 7630-7635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brunori, M., Giuffre, A., and Sarti, P. (2005) J. Inorg. Biochem. 99 324-336 [DOI] [PubMed] [Google Scholar]
- 13.Arnold, S., Goglia, F., and Kadenbach, B. (1998) Eur. J. Biochem. 252 325-330 [DOI] [PubMed] [Google Scholar]
- 14.Hüttemann, M., Lee, I., Liu, J., and Grossman, L. I. (2007) FEBS J. 274 5737-5748 [DOI] [PubMed] [Google Scholar]
- 15.Hüttemann, M., Kadenbach, B., and Grossman, L. I. (2001) Gene (Amst.) 267 111-123 [DOI] [PubMed] [Google Scholar]
- 16.Hüttemann, M., Jaradat, S., and Grossman, L. I. (2003) Mol. Reprod. Dev. 66 8-16 [DOI] [PubMed] [Google Scholar]
- 17.Hüttemann, M., Schmidt, T. R., and Grossman, L. I. (2003) Gene (Amst.) 312 95-102 [DOI] [PubMed] [Google Scholar]
- 18.Ludwig, B., Bender, E., Arnold, S., Hüttemann, M., Lee, I., and Kadenbach, B. (2001) Chembiochem. 2 392-403 [DOI] [PubMed] [Google Scholar]
- 19.Bender, E., and Kadenbach, B. (2000) FEBS Lett. 466 130-134 [DOI] [PubMed] [Google Scholar]
- 20.Miyazaki, T., Neff, L., Tanaka, S., Horne, W. C., and Baron, R. (2003) J. Cell Biol. 160 709-71812615910 [Google Scholar]
- 21.Steenaart, N. A., and Shore, G. C. (1997) FEBS Lett. 415 294-298 [DOI] [PubMed] [Google Scholar]
- 22.Prabu, S. K., Anandatheerthavarada, H. K., Raza, H., Srinivasan, S., Spear, J. F., and Avadhani, N. G. (2006) J. Biol. Chem. 281 2061-2070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee, I., Salomon, A. R., Ficarro, S., Mathes, I., Lottspeich, F., Grossman, L. I., and Hüttemann, M. (2005) J. Biol. Chem. 280 6094-6100 [DOI] [PubMed] [Google Scholar]
- 24.Lee, I., Salomon, A. R., Yu, K., Doan, J. W., Grossman, L. I., and Hüttemann, M. (2006) Biochemistry 45 9121-9128 [DOI] [PubMed] [Google Scholar]
- 25.Villani, G., and Attardi, G. (1997) Proc. Natl. Acad. Sci. U. S. A. 94 1166-1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Villani, G., Greco, M., Papa, S., and Attardi, G. (1998) J. Biol. Chem. 273 31829-31836 [DOI] [PubMed] [Google Scholar]
- 27.Kunz, W. S., Kudin, A., Vielhaber, S., Elger, C. E., Attardi, G., and Villani, G. (2000) J. Biol. Chem. 275 27741-27745 [DOI] [PubMed] [Google Scholar]
- 28.Acin-Perez, R., Bayona-Bafaluy, M. P., Bueno, M., Machicado, C., Fernandez-Silva, P., Perez-Martos, A., Montoya, J., Lopez-Perez, M. J., Sancho, J., and Enriquez, J. A. (2003) Hum. Mol. Genet 12 329-339 [DOI] [PubMed] [Google Scholar]
- 29.von Jagow, G., and Klingenberg, M. (1972) FEBS Lett. 24 278-282 [DOI] [PubMed] [Google Scholar]
- 30.Kadenbach, B., Stroh, A., Ungibauer, M., Kuhn-Nentwig, L., Buge, U., and Jarausch, J. (1986) Methods Enzymol. 126 32-45 [DOI] [PubMed] [Google Scholar]
- 31.Tsukihara, T., Aoyama, H., Yamashita, E., Tomizaki, T., Yamaguchi, H., Shinzawa-Itoh, K., Nakashima, R., Yaono, R., and Yoshikawa, S. (1996) Science 272 1136-1144 [DOI] [PubMed] [Google Scholar]
- 32.Lee, I., and Kadenbach, B. (2001) Eur. J. Biochem. 268 6329-6334 [DOI] [PubMed] [Google Scholar]
- 33.Kadenbach, B., Jarausch, J., Hartmann, R., and Merle, P. (1983) Anal. Biochem. 129 517-521 [DOI] [PubMed] [Google Scholar]
- 34.Shevchenko, A., Wilm, M., Vorm, O., and Mann, M. (1996) Anal. Chem. 68 850-858 [DOI] [PubMed] [Google Scholar]
- 35.Reers, M., Smith, T. W., and Chen, L. B. (1991) Biochemistry 30 4480-4486 [DOI] [PubMed] [Google Scholar]
- 36.Zhu, J., Liu, M., Kennedy, R. H., and Liu, S. J. (2006) Cytokine 34 96-105 [DOI] [PubMed] [Google Scholar]
- 37.Lopez-Armada, M. J., Carames, B., Martin, M. A., Cillero-Pastor, B., Lires-Dean, M., Fuentes-Boquete, I., Arenas, J., and Blanco, F. J. (2006) Osteoarthritis Cartilage 14 1011-1022 [DOI] [PubMed] [Google Scholar]
- 38.Zhao, Y., Wang, Z. B., and Xu, J. X. (2003) J. Biol. Chem. 278 2356-2360 [DOI] [PubMed] [Google Scholar]
- 39.Busquets, S., Aranda, X., Ribas-Carbo, M., Azcon-Bieto, J., Lopez-Soriano, F. J., and Argiles, J. M. (2003) Cytokine 22 1-4 [DOI] [PubMed] [Google Scholar]
- 40.Ledgerwood, E. C., Prins, J. B., Bright, N. A., Johnson, D. R., Wolfreys, K., Pober, J. S., O'Rahilly, S., and Bradley, J. R. (1998) Lab. Investig. 78 1583-1589 [PubMed] [Google Scholar]
- 41.Liu, H., Ma, Y., Pagliari, L. J., Perlman, H., Yu, C., Lin, A., and Pope, R. M. (2004) J. Immunol. 172 1907-1915 [DOI] [PubMed] [Google Scholar]
- 42.Kimura, H., Shintani-Ishida, K., Nakajima, M., Liu, S., Matsumoto, K., and Yoshida, K. (2006) Life Sci. 78 1901-1910 [DOI] [PubMed] [Google Scholar]
- 43.Ding, W. X., and Yin, X. M. (2004) J. Cell Mol. Med. 8 445-454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ashkenazi, A., and Dixit, V. M. (1998) Science 281 1305-1308 [DOI] [PubMed] [Google Scholar]
- 45.Chandel, N. S., Schumacker, P. T., and Arch, R. H. (2001) J. Biol. Chem. 276 42728-42736 [DOI] [PubMed] [Google Scholar]
- 46.Baudouin, S. V., Saunders, D., Tiangyou, W., Elson, J. L., Poynter, J., Pyle, A., Keers, S., Turnbull, D. M., Howell, N., and Chinnery, P. F. (2005) Lancet 366 2118-2121 [DOI] [PubMed] [Google Scholar]
- 47.Lee, M. D., Zentella, A., Vine, W., Pekala, P. H., and Cerami, A. (1987) Proc. Natl. Acad. Sci. U. S. A. 84 2590-2594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tracey, K. J., Lowry, S. F., Fahey, T. J., 3rd, Albert, J. D., Fong, Y., Hesse, D., Beutler, B., Manogue, K. R., Calvano, S., Wei, H., et al. (1987) Surg. Gynecol. Obstet. 164 415-422 [PubMed] [Google Scholar]
- 49.Callahan, L. A., and Supinski, G. S. (2005) J. Appl. Physiol. 99 1120-1126 [DOI] [PubMed] [Google Scholar]
- 50.Levy, R. J., Vijayasarathy, C., Raj, N. R., Avadhani, N. G., and Deutschman, C. S. (2004) Shock 21 110-114 [DOI] [PubMed] [Google Scholar]
- 51.Mootha, V. K., Bunkenborg, J., Olsen, J. V., Hjerrild, M., Wisniewski, J. R., Stahl, E., Bolouri, M. S., Ray, H. N., Sihag, S., Kamal, M., Patterson, N., Lander, E. S., and Mann, M. (2003) Cell 115 629-640 [DOI] [PubMed] [Google Scholar]
- 52.Garg, A. K., and Aggarwal, B. B. (2002) Mol. Immunol. 39 509-517 [DOI] [PubMed] [Google Scholar]
- 53.Liu, Z. G. (2005) Cell Res. 15 24-27 [DOI] [PubMed] [Google Scholar]
- 54.Griffioen, G., and Thevelein, J. M. (2002) Curr. Genet. 41 199-207 [DOI] [PubMed] [Google Scholar]
- 55.Lin, R. Y., Moss, S. B., and Rubin, C. S. (1995) J. Biol. Chem. 270 27804-27811 [DOI] [PubMed] [Google Scholar]
- 56.Newhall, K. J., Criniti, A. R., Cheah, C. S., Smith, K. C., Kafer, K. E., Burkart, A. D., and McKnight, G. S. (2006) Curr. Biol. 16 321-327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen, Q., Lin, R. Y., and Rubin, C. S. (1997) J. Biol. Chem. 272 15247-15257 [DOI] [PubMed] [Google Scholar]
- 58.Cardone, L., Carlucci, A., Affaitati, A., Livigni, A., DeCristofaro, T., Garbi, C., Varrone, S., Ullrich, A., Gottesman, M. E., Avvedimento, E. V., and Feliciello, A. (2004) Mol. Cell. Biol. 24 4613-4626 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.