

Abstract
RhoA is known to be involved in myogenic differentiation, but whether it acts as a positive or negative regulator is controversial. To resolve this issue, we investigated the differentiation stage-specific roles of RhoA and its effector, Rho-associated kinase, using C2C12 myoblasts. We found that proliferating myoblasts show high levels of RhoA and serum-response factor activities and strong expression of the downstream target of RhoA, myocardin-related transcription factor-A (MRTF-A or MAL); these activities and expression are markedly lower in differentiating myocytes. We further demonstrated that, in proliferating myoblasts, an increase in MRTF-A, which forms a complex with Smad1/4, strikingly activates the expression level of the Id3 gene; the Id3 gene product is a potent inhibitor of myogenic differentiation. Finally, we found that during differentiation, one of the forkhead transcription factors translocates into the nucleus and suppresses Id3 expression by preventing the association of the MRTF-A-Smad complex with the Id3 promoter, which leads to the enhancement of myogenic differentiation. We conclude that RhoA/Rho-associated kinase signaling plays positive and negative roles in myogenic differentiation, mediated by MRTF-A/Smad-dependent transcription of the Id3 gene in a differentiation stage-specific manner.
Myogenic differentiation involves a sequence of processes as follows: withdrawal of myoblasts from the cell cycle, expression of myogenic differentiation markers in myocytes, and formation of multinucleated myotubes by myocyte fusion. These processes are controlled by the myogenic regulatory factor (MRF)2 family and belong to basic helix-loop-helix proteins, consisting of MyoD, Myf5, myogenin, and MRF4 (1-3). The MRFs interact with ubiquitous basic helix-loop-helix proteins such as E12/E47 (E proteins) via their helix-loop-helix motifs, and the basic domains of the dimerized proteins activate transcription by binding to a conserved DNA sequence known as the E-box in the promoter regions of target genes. A family of inhibitors of DNA binding (Id) (Id1, Id2, Id3, and Id4), which has a helix-loop-helix domain but lacks the basic DNA-binding domain, counteracts the active MRF/E protein complexes by forming transcriptionally inactive complexes, consisting of either MRF/Id or E protein/Id (4-6).
The Rho family GTPases are involved in a variety of cytoskeleton-associated cellular events, such as reorganization of the actin cytoskeleton, microtubule dynamics, transcriptional regulation, and cell differentiation, including myogenic differentiation (7). The RhoA-dependent activation of serum response factor (SRF) is required for the expression of MyoD and skeletal (SK) α-actin. Thus, RhoA acts through SRF to positively regulate myogenic differentiation (8-11) and has therefore been referred to as a positive regulator of myogenic differentiation. However, recent studies have revealed that the expression of constitutively active RhoA results in failed myotube formation, and the inactivation of one of the downstream effectors of RhoA, Rho-associated kinase (ROCK), enhances myotube formation (12-15). Thus, apparently conflicting roles of RhoA signaling in myogenic differentiation have been reported.
It is well documented that actin polymerization in response to RhoA signaling enhances SRF activity, as a consequence of the nuclear translocation of myocardin-related transcription factors (MRTF-A/B, also referred to as MAL1/2) (16). MRTF-A is reportedly involved in heart and smooth muscle gene expression (17, 18). Consistent with these findings, the expression of dominant-negative forms of MRTF-A and MRTF-B (dn-MRTF-A and dn-MRTF-B), which inhibit SRF activation, generates abnormally thin muscle fibers in vivo (19) and inhibits myogenic differentiation in vitro (20). The molecular mechanism underlying the contribution of the MRTFs to myogenic differentiation, however, remains unclear.
One of the forkhead family transcription factors, FKHR (also named Foxo1), is required for myocyte fusion (15, 21, 22). Because ROCK directly phosphorylates FKHR in myocytes, resulting in the export of FKHR from the nucleus, the inactivation of RhoA/ROCK signaling is prerequisite for the nuclear localization of FKHR and myocyte fusion (15). In various other cell types, FKHR that is phosphorylated by Akt, but not by ROCK, is also exported from the nucleus, leading to the inhibition of FKHR-mediated transcription (23). However, the target partners of FKHR and its function in myogenic differentiation remain unknown, except for a Foxo1a-cyclic GMP-dependent kinase I interaction (22).
Here, we investigated the role of RhoA/ROCK signaling during myogenic differentiation using C2C12 cells and demonstrated that transcription of the Id3 gene is controlled by MRTF-A/Smad in an FKHR-dependent manner. In proliferating myoblasts, where RhoA activity and MRTF-A expression are relatively high, RhoA-triggered MRTF-A/Smad1/4 pre-dominantly activates transcription of the Id3 gene. Simultaneously, FKHR, possibly phosphorylated by ROCK, is retained in the cytoplasm. In contrast, in differentiating myocytes, where RhoA activity and MRTF-A expression are decreased, FKHR translocates into the nucleus, where it interacts with MRTF-A/Smad1/4, causing the MRTF-A-Smad1/4 complex to dissociate from the Id3 promoter, and thereby suppressing the MRTF-A/Smad-dependent Id3 expression. Taken together, the results of this study uncover why there are conflicting views regarding the role of RhoA/ROCK signaling in myogenic differentiation; differentiation stage-specific RhoA/ROCK signaling and activities of its downstream transcription factors, MRTF-A and FKHR, regulate the Smad-dependent transcription of the Id3 gene.
EXPERIMENTAL PROCEDURES
Reagents and Antibodies—Y-27632 was purchased from Cal-biochem. Tat-C3 was prepared according to a method described elsewhere (24). Commercially available antibodies were as follows: anti-FLAG (M2), anti-α-sarcomeric actin (5C5), and anti-α-tubulin (DM1A) antibodies (Sigma); anti-hemagglutinin (HA) (3F10) antibody (Roche Applied Science); anti-MyoD, anti-myogenin, anti-SRF, anti-Smad1/5/8, and anti-Id3 antibodies (Santa Cruz Biotechnology); anti-myosin heavy chain (MHC) antibody (MF20) (Developmental Studies Hybridoma Bank); anti-FKHR antibody (Cell Signaling); horseradish peroxidase-linked anti-rabbit or -mouse IgG (GE Healthcare); horseradish peroxidase-linked anti-rat IgG (Rockland). Anti-MRTF-A and -MRTF-B antibodies were generated in rabbits using recombinant proteins consisting of amino acids 776-901 of human MRTF-A and amino acids 931-1061 of human MRTF-B as the epitopes. IgGs were affinity-purified from the antisera raised against MRTF-A and MRTF-B on their respective affinity columns and used in this study (25).
Plasmid Constructs—The cDNAs of mouse RhoA, Smad1, Smad4, MRTF-A, and FKHR were amplified by reverse transcriptase (RT)-PCR and inserted into a mammalian expression plasmid, pCS2+ or pcDNA3.1, with the indicated tags. Expression plasmids for the constitutively active form of RhoA (RhoA-V14) and pseudo-phosphorylated Smad1 (26) were constructed by site-directed mutagenesis. The expression plasmid for N-terminally deleted MRTF-A, which acts as a constitutively active form, was constructed as described previously (16). The promoter region of the mouse Id3 gene, spanning from -2000 to +55, was isolated from the genome of C2C12 myoblasts using PCR and inserted into the pGL3-Basic plasmid (Promega) (Id3 (-2000/+55)-Luc). Deletions and mutant derivatives from Id3 (-2000/+55)-Luc, as indicated, were constructed using the PCR-mediated method. The mutations in the Smad-binding element (SBE) were introduced into pGL3-Id3-Luc as follows: the mutant SBE was changed from GTCTG to ATGTA and from CAGAC to CGTGC. The tetracycline-regulated inducible expression plasmids for HA-tagged RhoA-V14 and FLAG-tagged constitutively active MRTF-A (ca-MRTF-A) were constructed in the pTRE-tight expression plasmid (Clontech). The 3×CArG-Luc, referred to as 3D.ALuc (27), which consists of three copies of c-fos serum-responsive element without the ternary complex factor binding element or the basal promoter region of the Xenopus actin gene (28), was constructed to assay the transcriptional activity of SRF. All these constructs were confirmed by sequencing.
Cell Cultures and Transfection—C2C12 mouse myoblasts were maintained in Dulbecco's modified Eagle's medium supplemented with 20% fetal calf serum (growth medium (GM)). To induce myogenic differentiation, the culture medium was shifted to Dulbecco's modified Eagle's medium supplemented with 2% horse serum (differentiation medium (DM)) at the indicated times. HEK293T cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum. Transfection of the indicated expression plasmids into these cells was performed using Lipofectamine 2000 (Invitrogen). Stable transfectants of C2C12 cells carrying the tetracycline-regulated inducible expression system of HA-RhoA-V14 and FLAG-ca-MRTF-A were isolated as follows. First, we selected C2C12 stable transfectants carrying the pTet-Off plasmid by geneticin. Second, we introduced the expression plasmid, pTER-HA-RhoA-V14 or pTRE-FLAG-ca-MRTF-A into stable C2C12-pTet-Off transfectants and isolated cells carrying both the pTet-Off plasmid and the respective expression plasmid (C2C12-HA-RhoA-V14 and C2C12-FLAG-ca-MRTF-A), by selection with hygromycin B.
Promoter Assays—Proliferating C2C12 myoblasts cultured in GM were co-transfected with the indicated plasmid and pSV-β-gal (Promega). Luciferase and β-galactosidase activities were assayed 2 days after changing the medium from GM to DM as follows. The cell extracts were prepared by passive lysis buffer (Promega) according to the manufacturer's instructions and then assayed for luciferase activity using the luciferase kit (Promega). The promoter activity was expressed in relative units normalized to the β-galactosidase activity in the cell extracts. These assays were done in triplicate and were performed independently at least three times.
Immunocytochemistry—The cells were fixed with 4% formal-dehyde for 30 min, permeabilized, and blocked with 0.1% Triton X-100 and 0.2% bovine serum albumin in phosphate-buffered saline, for 1 h at room temperature. The cells were then incubated with the anti-MHC antibody for 2 h followed by Alexa Fluor 568 anti-mouse IgG (Molecular Probes), with or without Hoechst 33258, for 2 h at room temperature. Fluorescence images were collected using a cooled charge-coupled device camera (Roper Scientific, Tucson, AZ), mounted on an Olympus IX-70 microscope with the appropriate filters, and MetaMorph software.
GTP-bound RhoA Pulldown Assay—RhoA activity was determined by a GTP-bound RhoA pulldown assay using the Rho activation assay kit (Upstate Biotech), according to the manufacturer's protocol.
Chromatin Immunoprecipitation (ChIP) Assay—ChIP assays were carried out using the ChIP assay kit (Upstate Biotech), according to the manufacturer's protocol with some modifications. DNAs isolated from the input chromatin fragments and those from chromatin fragments precipitated by the anti-MRTF-A antibody or control IgG were subjected to PCR using primers flanking the SBE motif in the mouse Id3 promoter (-531 to -316). These primer sequences were as follows: Id3 sense primer, CTCTGGTCACAAGATAATTCC; Id3 antisense primer, GCGCCCAAGTTCTCTGAG.
Gel-shift Assay—A probe containing the SBE motif sequence of the Id3 promoter was amplified by PCR and end-labeled with [32P]dCTP by the Klenow fragment. The sequence of the sense strand of this probe was AATTCCTGACGCCAGTGAGTCTGGAGGTCAGACGAGCAGCAAATTGGGGA. The gel-shift assay was carried out using whole-cell extracts from C2C12 myoblasts (29).
Semiquantitative RT-PCR—The total RNAs were extracted from C2C12 cells cultured under the indicated conditions, and the expression level of Id1, Id2, Id3, twist, myostatin, c-fos, and c-myc mRNAs was quantified by RT-PCR normalized to the expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA, as described previously (30). The specific primer sets were as follows: Id1 sense primer, TGGACGAGCAGCAGGTGAACG; Id1 antisense primer, GCACTGATCTCGCCGTTCAGG; Id2 sense primer, AGCCTTCAGTCCGGTGAGGTCC; Id2 antisense primer, TCAGATGCCTGCAAGGACACG; Id3 sense primer, TGCTACGAGGCGGTGTGCTG; Id3 antisense primer, AGTGAGCTCAGCTGTCTGGATCGG; twist sense primer, CCTCGGACAAGCTGAGCAAGAT; twist antisense primer, CTAGTGGGACGCGGACATGGA; myostatin sense primer, ACGCTACCACGGAAACAATC; myostatin antisense primer, TGGTCCTGGGAAGGTTACAG; c-fos sense primer, GGATTTGACTGGAGGTCTGC; c-fos antisense primer, AAGTAGTGCAGCCCGGAGTA; c-myc sense primer, CAGCAGAGCGAGCTGCAGC; c-myc antisense primer, TCTTTGCGCGCAGCCTGGTA; HA-RhoA-V14 sense primer, TACCCATACGATGTTCCAGAT; HA-RhoA-V14 antisense primer, CTAAACTATCAGGGCTGTCG; GAPDH sense primer, TCTTCACCACCATGGAGAAGG; GAPDH antisense primer, GAAGGCCATGCCAGTGAG.
Quantitative Real Time RT-PCR—The expression of Id3 mRNA in C2C12 cells was analyzed by real time RT-PCR using SYBR GreenER qPCR SuperMix (Invitrogen). The levels of Id3 mRNA were normalized to GAPDH mRNA expression. The primers used in these analyses are as follows: Id3 sense primer, TCCTGGCACCTCCCGAAC; Id3 antisense primer, TAAGTGAAGAGGGCTGGGTTAAG; GAPDH sense primer, CGTGCCGCCTGGAGAAAC; GAPDH antisense primer, TGGGAGTTGCTGTTGAAGTCG.
Knockdown Using siRNA—Proliferating C2C12 cells were transfected with siRNA using Lipofectamine RNAiMAX (Invitrogen) and were cultured in GM for 2 days. The siRNAs against Id3 were purchased from Sigma, and their sequences were as follows: Id3 siRNA1, 5′-GCAUGGAUGAGCUUCGAUCTT-3′; Id3 siRNA2, 5′-CUGGUCAGCAGCUGGGCAATT-3′. In control experiments, scrambled siRNA (Santa Cruz Biotechnology) was used.
Immunoprecipitation, Immunoblotting, and Cellular Fractionation—The whole-cell extracts were prepared from C2C12 and HEK293T cells transfected with the indicated expression plasmids by lysis in buffer containing 50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 0.1% Nonidet P-40, 5% glycerol, and protease inhibitor mixture tablets (Roche Applied Science), and sonication with a Digital Sonifier (Branson). The extracts thus obtained were incubated with the indicated antibodies for 3 h at 4 °C, and the immune complexes were collected by incubation with protein A- or protein G-Sepharose beads for 1 h at 4 °C. Proteins in the immunoprecipitates were detected by immunoblotting using the indicated antibodies. The target proteins were detected with a SuperSignal chemiluminescence detection kit (Pierce). In immunoblotting analysis of the Id3 protein, we used Can Get Signal (TOYOBO) to detect chemifluorescent signals. The cytosol and nuclear fractions were prepared as described elsewhere (15).
DNA Affinity Binding Assay—The proteins translated by in vitro transcription/translation systems (Promega) were incubated with a double-stranded biotinylated DNA probe (1 mg) in gel-shift binding buffer (29) containing 5 mg of poly(dI-dC), 20 mg of herring sperm DNA, and 0.5% Nonidet P-40 for 30 min on ice (31). Streptavidin M-280 Dynabeads (Dynal) were then added, and the mixture was further incubated with rotation for 2 h at 4 °C. The DNA-bound proteins were analyzed by immunoblotting with the indicated antibodies. The sequence of the sense strand of this probe was as follows: AATTCCTGACGCCAGTGAGTCTGGAGGTCAGACGAGCAGCAAATTGGGGA.
RESULTS
RhoA/ROCK Signaling Orchestrates Myogenic Differentiation in a Stage-specific Manner—As described in the Introduction, conflicting roles of RhoA signaling in myogenic differentiation have been reported. We hypothesized that RhoA signaling may be differentially involved in myogenic differentiation in a stage-specific manner. To test our hypothesis, we studied the effects of RhoA on myogenic phenotypes at both the proliferating and differentiating stages (Fig. 1A). First, we analyzed the roles of RhoA and ROCK in myogenic differentiation using the cell-permeable Rho-specific inhibitor, tat-C3, and the ROCK inhibitor, Y-27632. As shown in Fig. 1B, treatment of the proliferating myoblasts with tat-C3 or Y-27632 suppressed the formation of myotubes after myogenic differentiation was induced, as determined by monitoring the expression of MHC and myotube formation. In contrast, when these treatments were given after the cells had started differentiating (treatment during the last 2 days in DM), MHC-positive myotube formation was markedly enhanced. In accordance with previous studies (12, 15), treatment with Y-27632 during the entire differentiation period (all 4 days in DM) also enhanced myogenic differentiation (data not shown). We performed the additional experiments using Y-27632, because its effect on myotube formation was greater than that of tat-C3 (Fig. 1B). Consistent with the morphological changes it caused, treatment with Y-27632 during differentiation enhanced the expression levels of myogenic markers, such as myogenin, SK α-actin, and MHC proteins, whereas these enhancements were not observed when the Y-27632 treatment was given during proliferation (Fig. 1C). MyoD expression in the Y-27632-treated proliferating myoblasts was down-regulated compared with that in control cells.
FIGURE 1.
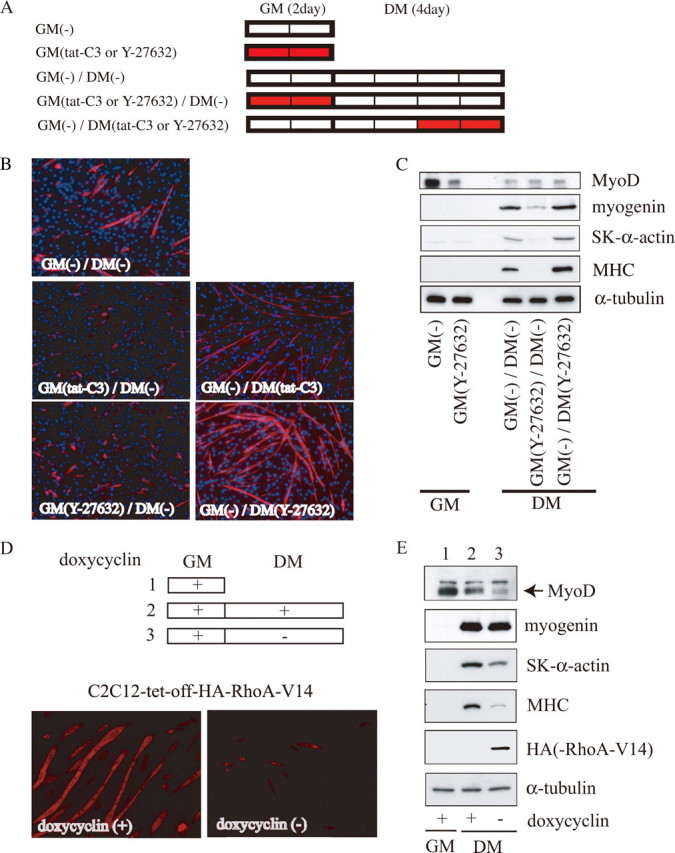
Biphasic roles of RhoA/ROCK signaling in myogenic differentiation. A, experimental procedures are presented schematically. C2C12 myoblasts were cultured in GM for 2 days and then in DM for 4 days, to induce myogenic differentiation. The red bars show the duration of treatment with the indicated inhibitors (5 μg/ml tat-C3 or 25 μm Y-27632). B, cells (4-day cultures in DM) were stained using an anti-MHC antibody (red) and Hoechst 33258 (blue). Magnification is ×100. C, expression of myogenic differentiation markers is shown. The whole-cell extract from C2C12 cells cultured under the indicated conditions was subjected to immunoblotting using the indicated antibodies. The protein content in each sample was normalized to the amount of α-tubulin protein. Representative results from at least three experiments are shown. D, effect of the forced activation of RhoA on differentiating myocytes. The experimental procedures are presented schematically at the top. Stable C2C12 transfectants (C2C12-tet-off-HA-RhoA-V14) were cultured in GM containing 1 μg/ml doxycycline for 3 days (lane 1) and then in DM for 5 days with (lane 2) or without (lane 3) 1 μg/ml doxycycline. The cells (5-day cultures in DM) were stained using the anti-MHC antibody (bottom). Magnification is ×100. E, expression of myogenic differentiation markers is shown. The whole-cell extract from C2C12 cells cultured under the indicated conditions was subjected to immunoblotting using the indicated antibodies, as described in the legend for Fig. 1C. Lanes 1-3 correspond to those in D.
To verify whether RhoA signaling regulates myogenic differentiation, we isolated stable C2C12 cell lines carrying a tetracycline-inducible expression system for constitutively active RhoA, RhoA-V14. The induction of RhoA-V14 expression during differentiation strikingly inhibited myotube formation (Fig. 1D) and the expression of SK α-actin and MHC, but it did not affect myogenin expression (Fig. 1E). These results indicated that RhoA/ROCK signaling in proliferating myoblasts is prerequisite for maintaining the capacity of myogenic differentiation, which features the up-regulation of MyoD expression, but RhoA/ROCK signaling suppresses differentiation in differentiating myocytes.
RhoA and SRF Activities and MRTF-A Expression Are Down-regulated during Myogenic Differentiation—We next examined the endogenous activities of RhoA and its downstream molecules during myogenic differentiation. Consistent with a previous study (15), we found the RhoA activity was decreased during differentiation (Fig. 2A). The activity of SRF, which is a well characterized downstream target of RhoA, rapidly decreased in association with the RhoA activity (Fig. 2B). The expression level of an SRF cofactor, MRTF-A, also decreased after the induction of myogenic differentiation, although the levels of SRF and another cofactor, MRTF-B, remained unchanged (Fig. 2C). Thus, the strong activation of RhoA and SRF and of MRTF-A expression in proliferating myoblasts and their down-regulation in differentiating myocytes is adapted to effect the appropriate myogenic processes.
FIGURE 2.

Changes in RhoA activity and its downstream events during myogenic differentiation. A, time-dependent changes in RhoA activity measured by a rhotekin pulldown assay are shown. Immunoblotting using anti-RhoA antibodies shows the amount of GTP-bound active RhoA protein associated with rhotekin (upper panel) and the total amount of RhoA protein (lower panel) in each whole-cell extract from C2C12 cells cultured under the indicated conditions (left). The relative RhoA activity, i.e. the image intensity of GTP-bound active RhoA protein normalized to that of total RhoA protein, is presented graphically (right). The intensity was measured using NIH Image software. Each value is expressed relative to the value for myoblasts cultured in GM, which was set as 1.0, and represents the mean ± S.D. of the results from three independent experiments. B, change in transcriptional activity of SRF is shown. C2C12 myoblasts, cultured in GM, were transfected with 3×CArG-Luc and pSV-β-gal, and cell extracts prepared at the indicated periods were assayed for luciferase activity as described under “Experimental Procedures.” The relative luciferase activities normalized to the β-galactosidase activity are shown. Each value represents the mean ± S.D. of the results from three independent experiments. C, expression levels of SRF and its co-factors, MRTF-A/B, are shown. The whole-cell extract from C2C12 cells cultured under the indicated conditions was analyzed by immunoblotting using the indicated antibodies (left). The signal intensity is presented graphically (right). The gray, white, and black bars represent the signal intensity of MRTF-A, MRTF-B, and SRF, respectively. Each value is expressed relative to the value for myoblasts cultured in GM, which was set as 1.0, and represents the mean ± S.D. of the results from three independent experiments. D means culture day.
Inactivation of ROCK Results in the Down-regulation of Id3 Expression—To study the mechanism underlying the enhanced myogenic differentiation caused by Y-27632 (Fig. 1), we analyzed the expressional changes of factors that are known to be inhibitory for myogenic differentiation, such as Ids1-3 (4, 32-34), twist (35), myostatin (36), c-fos (37), and c-myc (38) (Fig. 3A). In agreement with previous reports (4, 32-34), the expression levels of Id1, Id2, and Id3 mRNAs were significantly down-regulated during myogenic differentiation. The Id3 mRNA, but not the mRNA for twist, myostatin, c-fos, or c-myc, was further down-regulated in response to Y-27632 treatment. These results suggest that the Y-27632 treatment-induced enhancement of myogenic differentiation might have been due to the down-regulation of Id3 expression. To confirm the specific inhibition of ROCK by Y-27632, siRNA-mediated ROCK1 and ROCK2 knockdown was carried out. The expression of Id3 was decreased by knockdown of ROCK in proliferating myoblasts (data not shown). Furthermore, we addressed a role of RhoA, upstream effector of ROCK, in the transcription of Id3 gene. The induction of RhoA-V14 increased the expression of Id3 in tetracycline-regulated C2C12 cells (Fig. 3B). Both analyses by semi-quantitative and real time RT-PCR revealed that the up-regulation of Id3 expression is modest in proliferating myoblasts but is marked in differentiating myocytes. These results indicated that the transcription of Id3 gene is up-regulated by RhoA/ROCK1 signaling but is not regulated by the pathway mediated through ROCK2.
FIGURE 3.

The expression of myogenic inhibitors in Y-27632-treated differentiating myocytes. A, semiquantitative RT-PCR was performed using total RNA isolated from C2C12 cells cultured under the indicated conditions (4 days in DM) to evaluate the mRNA levels for the indicated myogenic inhibitor genes. The culture conditions are described in the legend for Fig. 1A. PCR cycle number and the size of the PCR product were as follows: Id1, 28, 276 bp; Id2, 26, 350 bp; Id3, 26, 286 bp; twist, 34, 194 bp; myostatin, 38, 398 bp; c-fos, 32, 347 bp; c-myc, 32, 371 bp; GAPDH, 20, 398 bp. The mRNA level for the myogenic inhibitors was normalized to the level of GAPDH mRNA. B, stable C2C12 transfectants (C2C12-tet-off-HA-RhoA-V14) were cultured in GM followed by 4 days in DM with or without 1 μg/ml doxycycline. Semiquantitative RT-PCR (upper panel) and real RT-PCR (lower panel) were carried out using total RNAs extracted from the indicated culture condition. PCR cycle number of Id3 was 24 and 28.
Forced Activation of MRTF-A in Differentiating Myocytes Inhibits Myogenic Differentiation via the Induction of Id3 Expression—Because the expression of MRTF-A was reduced in differentiating myocytes compared with its high levels in proliferating myoblasts (Fig. 2C), we focused on downstream events in the RhoA/MRTF-A pathway. To examine the effect of MRTF-A on differentiating myocytes, we isolated stable C2C12 cell lines carrying a tetracycline-inducible expression system for constitutively active MRTF-A (ca-MRTF-A). Using these cell lines, we found that the induction of ca-MRTF-A after the start of differentiation markedly suppressed myotube formation (Fig. 4A) as well as the expression of myogenic differentiation markers, including myogenin, SK α-actin, and MHC (Fig. 4B). We also analyzed the expression levels of factors that are inhibitory for myogenic differentiation. Id3 expression was solely up-regulated at both the protein and mRNA levels in myocytes expressing ca-MRTF-A during differentiation, but the other inhibitory factors examined did not show increased expression (Fig. 4, B and C). Taken together, these results suggest that MRTF-A-induced Id3 expression is critical for the RhoA/ROCK1 signaling-dependent inhibition of myogenic differentiation.
FIGURE 4.
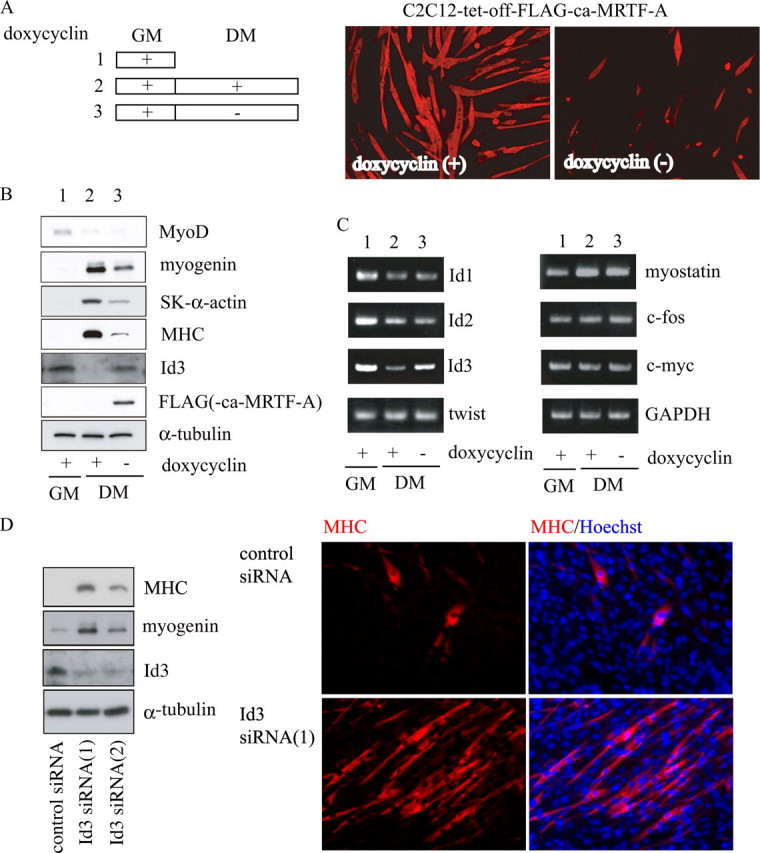
Effect of forced ca-MRTF-A expression on myogenic differentiation. A, experimental procedures are presented schematically at left. Stable C2C12 transfectants (C2C12-tet-off-FALG-ca-MRTF-A) were cultured under the conditions described in the legend for Fig. 1D. Cells (5-day cultures in DM) were stained with an anti-MHC antibody (right). Magnification is ×100. B and C, expression levels of myogenic differentiation markers and myogenic inhibitors are shown. B, whole-cell extracts from C2C12 cells cultured under the indicated conditions (5 days in DM) were subjected to immunoblotting using the indicated antibodies. C, total RNA from C2C12 cells cultured under the indicated conditions (5 days in DM) was analyzed by RT-PCR as described in the legend for Fig. 3. Lanes 1-3 (B and C) correspond to those in A. D, C2C12 cells were transfected with siRNAs against Id3 (Id3 siRNA1 and -2) and were cultured in GM for 2 days. The whole-cell extracts were subjected to immunoblotting using the indicated antibodies. The cell morphology and expression of MHC were also analyzed by staining with an anti-MHC antibody (red) and Hoechst 33258 (blue). Magnification is ×200.
To ask the degree of negative contribution of Id3 during myogenic differentiation, we carried out siRNA-mediated knockdown of Id3. It has been reported that confluent culture of C2C12 myoblasts could differentiate into skeletal muscle marker-positive myotubes even in GM (39). We then examined the effects of siRNA against Id3 on C2C12 cultures in GM. The expression of myogenin and MHC was markedly increased in proliferating myoblasts treated with siRNAs against Id3, Id3 siRNA1, and Id3 siRNA2. Compared with the siRNA2, the siRNA1 showed more potent effect on promotion of myogenic differentiation (Fig. 4D, left panel). Even under growing conditions, MHC-positive myotube was apparent in C2C12 cultures treated with Id3-siRNA1 (Fig. 4D, right panel); the population of nuclei in MHC-positive myocytes was 27.58 ± 4.21%. By contrast, in C2C12 cultures treated with control siRNA, such percentage was low (6.32 ± 2.70%), and fused myocytes were hardly found. These results further support the above findings that Id3 would play a critical role in maintaining a myoblast state.
MRTF-A/Smad-mediated Transcription of the Id3 Gene—To analyze the mechanism underlying the MRTF-A-induced Id3 expression, we constructed several reporter plasmids carrying different portions of the promoter region of the Id3 gene (Fig. 5A) and examined the effects of MRTF-A on their promoter activities in differentiating myocytes. The ca-MRTF-A markedly enhanced the promoter activity of Id3 (-2000/+55)-Luc in a dose-dependent manner (Fig. 5B). We further characterized the MRTF-A-responsive element using a series of deletion derivatives from Id3 (-2000/+55)-Luc. The ca-MRTF-A caused equal transactivation of the promoter activities of Id3 (-2000/+55)-Luc (5.9-fold activation) and Id3 (-524/+55)-Luc (5.8-fold activation), but its effect on the activity of Id3 (-279/+55)-Luc was reduced to a negligible level (Fig. 5C). We next performed ChIP assays to determine whether MRTF-A is functionally involved in the transcription of the Id3 gene. As shown in Fig. 5D, endogenous MRTF-A in proliferating myoblasts physically associated with the promoter region of the Id3 gene, indicating that MRTF-A plays a vital role in the regulation of Id3 transcription in vivo.
FIGURE 5.

MRTF-A-dependent activation of the Id3 promoter. Transcriptional activity of the Id3 promoter in differentiating myocytes was measured as described under “Experimental Procedures.” A, Id3 promoter constructs used in this analysis are illustrated. The effects of ca-MRTF-A co-expression on the promoter activity of Id3 (2000/+55)-Luc (B), its derivatives (C), and Id3 (-524/+55)-Luc bearing a mutated SBE (G) are shown. The luciferase activities were expressed relative to that in empty plasmid-transfected C2C12 myocytes, which was set as 1.0 (B and G). C, luciferase activity of Id3 (-2000/+55) in cells co-transfected with empty plasmid was set as 1.0. D, ChIP assay shows the interaction of endogenous MRTF-A with the promoter region of the Id3 gene in proliferating C2C12 myoblasts. The extracted chromatin fragments were immunoprecipitated (IP) with the indicated antibodies, and the precipitated genomic DNA was analyzed by PCR using primers for the Id3 promoter region containing the SBE. The size of the PCR product was 216 bp. PCR amplification was also performed prior to immunoprecipitation for the input control. E, 32P-labeled SBE of the Id3 promoter was incubated with the whole-cell extracts of proliferating C2C12 myoblasts (lanes 1 and 3-6) or Smads-depleted whole-cell extracts prepared by pretreatment with anti-Smad1/5/8 antibody (lane 2) in the presence of following additives: 30-fold excess amounts of indicated cold competitors (lanes 3 and 4), an anti-Smad1/5/8 antibody (Ab) (lane 5) or control IgG (lane 6). The reactants were subjected to 5% PAGE. Arrow indicates the SBE-Smad complex. mut, mutant; wt, wild type. F, depletion of Smad1/5/8 in whole-cell extracts of proliferating C2C12 myoblasts was characterized by immunoblotting using anti-Smad1/5/8 antibody. The Smads in C2C12 whole-cell extracts were depleted by immunoprecipitation using anti-Smad1/5/8 antibody. Samples applied to immunoblotting were as follows: Smads-depleted C2C12 whole-cell extracts (lane 1), nondepleted one (lane 2), and immunoprecipitates by anti-Smad1/5/8 antibody (lane 3) or control IgG (lane 4). H, interaction between Smad1 and MRTF-A was analyzed using in vitro-translated proteins. HA-Smad1 and FLAG-MRTF-A proteins were co-incubated, and their interaction was analyzed by immunoprecipitation followed by immunoblotting using the indicated antibodies. I, indicated tagged proteins were translated in vitro and then incubated with a biotinylated-DNA probe containing the SBE of the Id3 promoter. The proteins bound to the probe were collected with streptavidin-conjugated magnetic beads, and the SBE-interacting proteins were analyzed by immunoblotting using the indicated antibodies.
The region of the Id3 promoter spanning from -524 to +55 contains the SBE and its neighboring sequences, which are conserved among humans, mice, and Xenopus (40). A previous study using one of smooth muscle marker genes showed that transcription of the SM22α gene was enhanced by its association with myocardin/Smad3 in an SRF/CArG-box motif-independent manner (41). These findings led us to speculate that activation of the Id3 promoter for MRTF-A might be mediated through Smads. We therefore performed gel-shift assays to determine whether Smads interact with the SBE in the Id3 promoter region. The upper shifted band (Fig. 5E, lane 1) was detected only when the SBE probe was incubated with whole-cell extracts from C2C12 myoblast; incubation with the whole-cell extracts depleted of Smads did not form this band (Fig. 5E, lane 2). The intensity of this band was diminished by an excess amount of cold mutant SBE probe (Fig. 5E, lane 3) but by cold SBE mutant one (lane 4). Furthermore, treatment with anti-Smad 1/5/8 antibodies (Fig. 5E, lane 5) but not with control IgG (lane 6) attenuated the formation of this band. These results strongly suggest that Smad1, -5, or -8, which are known regulators for Id3 expression under BMP signaling (42), interacts with the SBE in the Id3 promoter region. We then introduced mutations in the SBE within Id3 (-524/+55)-Luc to evaluate the contribution of the SBE to the MRTF-A-dependent activation of the Id3 promoter. Compared with the wild-type construct, the mutations within the SBE markedly reduced the promoter activity (Fig. 5G). These results indicate that the SBE acts as a critical cis-element in the Id3 promoter in cooperation with MRTF-A.
We next examined the interaction of MRTF-A with Smad, because MRTF-A requires a DNA-binding transcription factor such as Smads to exert its activity. As shown in Fig. 5H, in vitro-translated MRTF-A protein interacted directly with Smad1, which was used as representative of Smad1, -5, and -8, suggesting that Smad1 and MRTF-A might form a complex on the Id3 promoter. To test this possibility, we performed DNA affinity binding assays (31), in which the sequence of the Id3 promoter containing the SBE was biotinylated and then incubated with in vitro-translated MRTF-A, pseudo-phosphorylated-Smad1 (constitutively active Smad1: ca-Smad1) and Smad4. In contrast to the signal caused by the incubation of MRTF-A and the biotinylated-SBE (which was nonspecific, because MRTF-A cannot bind to DNA), the signal intensity of the interaction of MRTF-A with the biotinylated SBE was significantly increased by the addition of ca-Smad1 and Smad4, indicating that MRTF-A interacts with the SBE via Smad1 and Smad4 (Fig. 5I).
FKHR Relieves the MRTF-A/Smad-dependent Transcription of the Id3 Gene in Differentiating Myocytes—Because Id3 expression was down-regulated after the induction of myogenic differentiation (Figs. 3 and 4), the MRTF-A-dependent transactivation of the Id3 gene must be repressed in differentiating myocytes. To elucidate this mechanism, we analyzed the localization of MRTF-A at different differentiation stages and found that it was mainly in the cytoplasm and partly in the nucleus at both the proliferation (cytosol 72%, nucleus 28%) and differentiation stages (cytosol 75%, nucleus 25%) (Fig. 6A). No changes in its localization during the differentiation process suggest that some other mechanism regulates the Id3 transcription.
FIGURE 6.

Time-dependent effect of FKHR on MRTF-A-induced activation of the Id3 promoter during myogenic differentiation. A, myoblasts (GM) and differentiated myocytes (DM) cultured for 4 days in DM were separated into the cytoplasmic (C) and nuclear (N) fractions, followed by immunoblotting for MRTF-A. Immunoblotting for HSP-90 and histone H2B indicates control for a cytoplasmic protein and a nuclear protein, respectively. B, cells (cultured in GM and GM containing 25 μm of Y-27632 and 4day cultures in DM) were stained using the anti-FKHR antibody. Magnification is ×600. C, proliferating C2C12 myoblasts were transfected with Id3 (-2000/+55)-Luc, pSV-β-gal, ca-MRTF-A expression plasmid (80 (+) or 160 ng (++)) and increasing amounts of FKHR expression plasmid (0, 80, 160, or 320 ng). The transfected plasmids were adjusted to a constant amount by the addition of empty plasmid. Luciferase activity was measured at the proliferation (24 h after transfection in GM) and differentiation (2 days in DM) phases. The luciferase activities were expressed relative to that in empty plasmid-transfected C2C12 cells, which was set as 1.0.
Bois and Grosveld (21) reported that the forkhead transcription factor FKHR is required for myotube formation by myoblasts in primary culture, in which FKHR translocates from the cytoplasm to the nucleus in response to the induction of myogenic differentiation. They also demonstrated that the phosphorylation-dependent nuclear export of FKHR is not mediated by phosphatidylinositol 3-kinase/Akt signaling. Nishiyama et al. (15) showed that the inactivation of RhoA/ROCK signaling is prerequisite for the nuclear localization of FKHR in C2C12 cells, because ROCK directly phosphorylates FKHR in vitro. Another report showed that a different forkhead transcription factor, Foxo4, interacts with myocardin and represses myocardin-dependent transcription in smooth muscle cells (43). We therefore examined the effect of FKHR on the MRTF-A-induced Id3 expression. First, we confirmed the localization of FKHR. Consistent with previous reports (15, 21), FKHR translocated from the cytoplasm to the nucleus in response to induction of myogenic differentiation and also accumulated into the nucleus in proliferating myoblasts treated with Y-27632 (Fig. 6B). We next examined the effect of FKHR on the MRTF-A-induced activation of the Id3 promoter. FKHR showed no apparent effect on MRTF-A-induced activation of the Id3 promoter in proliferating myoblasts but markedly inhibited this activation in differentiating myocytes in a dose-dependent manner (Fig. 6C), indicating that FKHR serves as a differentiating myocyte-specific repressor of the MRTF-A-dependent transcription of the Id3 gene.
Immunoprecipitation followed by immunoblotting analysis revealed that the exogenous MRTF-A and FKHR proteins associated with each other in HEK293T cells (Fig. 7A), and endogenous MRTF-A potently interacted with FKHR in differentiating myocytes and slightly in proliferating myoblasts (Fig. 7B). We further identified the mutual interacting domains of MRTF-A and FKHR using in vitro-translated proteins (Fig. 7C). We found that the N-terminal domain (residues 1-330) of FKHR and the central domain (residues 201-626) of MRTF-A, including the basic, SAP, and leucine zipper domains, physically interacted with each other.
FIGURE 7.

A, interaction between exogenously expressed FKHR and MRTF-A is shown. HEK293T cells were transfected with expression plasmids for HA-FKHR and FLAG-MRTF-A and cultured for 24 h. The whole-cell extracts were immunoprecipitated (IP) using control IgG or the indicated anti-tag protein antibodies. The separated proteins in the input and the immunoprecipitated (IP) samples were probed with the indicated anti-tag protein antibodies. B, interaction between endogenous FKHR and MRTF-A in C2C12 cells is shown. The whole-cell extracts from proliferating C2C12 myoblasts (GM) and differentiating myocytes (2 days after the induction of myogenic differentiation by DM) were immunoprecipitated using control IgG or an anti-FKHR antibody. Input and immunoprecipitation samples were subjected to immunoblotting using anti-MRTF-A and anti-FKHR antibodies. C, mapping of the interactive domains of FKHR (left) and MRTF-A (right) with each other. In vitro-translated HA-FKHR derivatives, the N-terminal domain (residues 1-330) and C-terminal domain (residues 331 to 652) of FKHR, were incubated with in vitro-translated FLAG-MRTF-A, and their interactions were analyzed by immunoprecipitation (IP) using an anti-HA antibody followed by immunoblotting (IB) with the indicated antibodies (left). The inclined arrows indicate the HA-FKHR derivatives. Reciprocally, in vitro-translated FLAG-MRTF-A derivatives, the N-terminal domain (residues 1-221), central domain (residues 201-626), and C-terminal domain (residues 627-929), were incubated with in vitro-translated HA-FKHR, and their interactions were analyzed as described above (right). D, in vitro-translated FLAG-MRTF-A, HA-Smad1, and FKHR were incubated under the indicated combinations, and the effect of FKHR on the interaction between MRTF-A and Smad1 was analyzed. Separated proteins in the input and immunoprecipitation using anti-HA antibody samples were subjected to immunoblotting with the indicated antibodies. E, in vitro-translated HA-ca-Smad1, HA-Smad4, FLAG-MRTF-A, and HA-FKHR were incubated with a biotinylated-DNA probe containing the SBE of the Id3 promoter under the indicated combinations, and the effect of FKHR on the association of MRTF-A and Smads with the probe was analyzed as described in the legend for Fig. 5I.
To gain further insight into the inhibitory effect of FKHR on the MRTF-A-dependent transactivation of the Id3 gene, we analyzed whether FKHR influences the interactions among MRTF-A, Smad1, and the SBE within the Id3 promoter. FKHR did not affect the degree of MRTF-A binding to Smad1 in vitro (Fig. 7D). We next examined the effect of FKHR on the binding of the complex consisting of MRTF-A and Smad1/4 to the SBE. The binding of this complex to the Id3 promoter probe, which included the SBE, was strongly inhibited by FKHR (Fig. 7E), indicating that FKHR represses the MRTF-A/Smad-induced transactivation of the Id3 gene by abrogating the binding of the MRTF-A and Smad1/4 complex to the SBE within the Id3 promoter.
DISCUSSION
Here we demonstrated that RhoA/ROCK signaling plays stage-specific roles in myogenic differentiation. In the proliferation stage, high levels of RhoA activity and MRTF-A expression bring about enhanced SRF activity and MyoD expression, which are required for the cells to retain their myogenic capacity. Even under these conditions, MRTF-A also markedly transactivates the Id3 gene, leading to the inhibition of the MRF action, including that of MyoD, by Id3. Thus, although proliferating myoblasts retain their myogenic differentiation capacity, the Rho/ROCK-mediated up-regulation of Id3 simultaneously suppresses premature differentiation. Because Id3-siRNA-treated C2C12 cells showed multinucleated terminal differentiation even under proliferating conditions (Fig. 4D), the negative contribution of Id3 to myogenic differentiation is significant.
After the induction of differentiation, when RhoA activity and MRTF-A expression are reduced, FKHR translocates into the nucleus and forms a complex with MRTF-A-Smad, which inactivates transcription of the Id3 gene so that myogenic differentiation can proceed. The low levels of MRTF-A expression and SRF activity may also be sufficient to induce myogenic differentiation and maintain the differentiated state, as revealed by the up-regulation of myogenic differentiation marker expressions and myotube formation. We summarize these findings schematically in Fig. 8.
FIGURE 8.

Model for the phase-dependent regulation of Id3 expression by MRTF-A in association with FKHR and RhoA/ROCK-signaling during myogenic differentiation. The detailed description is provided in the text.
Multiple Functions of RhoA/ROCK Signaling in Myogenic Cells—The Rho family of small GTPases is involved in a variety of cellular events, including skeletal muscle differentiation (44). RhoA signaling is reported to be a positive regulator of myogenic differentiation, because the inactivation of RhoA decreases the expression of MyoD and inhibits myogenic differentiation (8). In contrast, activated RhoA blocks myotube formation, and the inactivation of ROCK promotes myogenic differentiation (12, 15). In this study, we discovered why conflicting views of the role played by RhoA/ROCK signaling in myogenic differentiation have arisen. Stage-specific analysis was critical for evaluating the roles of RhoA/ROCK signaling in myogenic differentiation. The inhibition of this signaling in proliferating myoblasts suppressed myogenic capacity because of the down-regulation of SRF activity and MyoD expression. In contrast, the inhibition of RhoA/ROCK signaling in differentiating myocytes promoted myogenic differentiation, which is agreement with previous studies reported by Castellani et al. (12) and Nishiyama et al. (15) (Fig. 1). Consistent with this, the endogenous RhoA and SRF activities and MRTF-A expression were decreased in differentiating myocytes (Fig. 2).
Relationships between SRF and Myogenic Differentiation—Actin polymerization in response to RhoA signaling positively affects the nuclear translocation of MRTF-A/B, leading to an increase in SRF activity (16, 45). We demonstrated that despite stage-specific changes in the RhoA activity and the expression level of MRTF-A between myoblast proliferation and myogenic differentiation, the localization of MRTF-A was, however, unchanged at both stages. Its localization, mainly to the cytoplasm and partly to the nucleus, suggested that its nuclear export might be predominant over its import (46). During proliferation, in which the levels of RhoA activity and MRTF-A expression were increased, myoblasts showed high levels of SRF activity and MyoD expression, but they did not express myogenic differentiation markers, such as α-sarcomeric actin and MHC, and they did not form multinucleated myotubes. In contrast, in differentiating myocytes, in which the expression levels of myogenic differentiation markers were markedly up-regulated, MRTF-A expression and endogenous SRF activity were severely decreased (Figs. 1 and 2). Thus, a RhoA-dependent high level of SRF activity in proliferating myoblasts is required for the expression of MyoD, which plays a vital role in retaining the myogenic capacity of the cells, whereas the low activity of SRF could be sufficient for the strong expression of myogenic differentiation markers in differentiating myocytes. However, it remains unclear whether MRTFs are involved in MyoD expression. Because the distal regulatory region of the MyoD gene contains a conserved CArG-box motif (47), MRTF-A/B and SRF are likely to be involved in MyoD expression. As shown in Fig. 4, the forced activation of MRTF-A during myogenic differentiation suppressed the further differentiation into myotubes. We demonstrated that this suppression was because of ca-MRTF-A-dependent transactivation of the Id3 gene (Fig. 5). In support of this idea, Selvaraj and Prywes (48) identified Id3 as a possible target gene for MRTF-A by microarray analysis.
It has been reported that the expression of dn-MRTF-A or dn-MRTF-B generates abnormally thin muscle fibers in vivo and inhibits myogenic differentiation in vitro (25, 45). These results are inconsistent with our present finding that the forced activation of MRTF-A during differentiation impairs myotube formation (Fig. 4). We explain this contradiction as follows. The expression of dn-MRTF-A/B would down-regulate Id3 expression and simultaneously abort SRF activity, resulting in abnormal myogenesis because of the decrease in MyoD expression and inhibition of myogenic differentiation. On the other hand, ca-MRTF-A would promote the induction of Id3 expression concomitant with SRF-dependent transcription. Thus, the up-regulation of Id3 would counteract the SRF-dependent positive regulation of myogenic differentiation (49, 50). A recent study has demonstrated that forced expression of myocardin in myoblasts markedly inhibits the myogenic differentiation (51). This is because of the inhibition of myogenin promoter by myocardin-induced dysfunction of MyoD and recruitment of histone deacetylase. Considering the similarity in the structure and function between myocardin and MRTF-A, myocardin-triggered up-regulation of Id3 might be an additional reason for the inhibition of myogenic differentiation by myocardin. However, because of extremely low expression levels of myocardin in skeletal muscles (52), there is a concern about the function of endogenous myocardin in myogenic differentiation. Taken together, our findings suggest that a balance between the RhoA/ROCK-dependent activation of positive and negative genes for myogenic differentiation is important for proper myogenesis to occur.
FKHR Negatively Regulates MRTF-A/Smad-dependent Transcription of the Id3 Gene in Differentiating Myocytes—The transcriptional regulation of the Id3 gene has been partially characterized. Wu and Lim (53) reported that Sp2 binding to the GC-box-like motif, which is present within the promoter region (-180/+1 bp) of the Id3 gene, positively regulates the Id3 promoter in proliferating C2C12 myoblasts. von Bubnoff et al. (40) found, using the Xenopus animal cap system, that the Id3 promoter contains a BMP-response element, which includes an SBE and a GC-rich element resembling an OAZ-binding site, and they demonstrated that these elements are necessary for responsiveness to BMP. Smad7 is known to be a negative regulator of BMP (54) and transforming growth factor-β (55) signaling and a positive regulator for myogenic differentiation by directly interacting with MyoD and enhancement of MyoD transcriptional activity (56). We examined whether Smad7 is involved in the regulation of Id3 transcription. As described by Kollias et al. (56), Smad7 expression was increased during C2C12 myogenic differentiation both in mRNA and protein levels. However, Id3 promoter activity was not affected by forced expression of Smad7, indicating that Smad7 would not be involved in the Id3 transcriptional regulation (data not shown). In this way, the Id3 gene is transcriptionally regulated by diverse mechanisms, in a tissue- or stimulus-dependent manner. Here we demonstrated that MRTF-A acts as a positive regulator of Id3 gene transcription and, with Smad 1/4, associates with the SBE in the Id3 promoter (Fig. 5).
FKHR has been reported to be a regulator of myogenic differentiation because it activates the transcription of target genes involved in cell fusion or extracellular matrix remodeling and promotes myoblast fusion (21). The interaction of FKHR with cyclic GMP-dependent protein kinase I causes myoblast fusion by influencing the fusion rate (22). Despite these findings, conflicting results have been reported; normal myogenic differentiation is impaired by the forced expression of constitutively active or wild-type FKHR in skeletal muscle cells (57, 58). FKHR also interacts with androgen receptor and suppresses androgen-induced androgen receptor transcription in prostate cancer cells (59). Thus, this forkhead transcription factor family member is involved in various transcriptional regulations. In this study, we identified a novel function of FKHR as a repressor of the Id3 promoter. We demonstrated here that FKHR plays a positive role in myogenic differentiation through the inhibition of Id3 expression. Foxo4 interacts with both myocardin and SRF, and this ternary complex then aborts the transcription of myocardin/SRF target genes in smooth muscle cells (42). Using myocytes, we revealed that FKHR inhibits MRTF-A/Smad-dependent Id3 transcription by interacting with MRTF-A/Smads and thereby interrupting the binding of MRTF-A/Smads to the SBE in the Id3 promoter. Further study regarding the involvement of FKHR in Id3-dependent developmental, physiological, and pathological processes will be required to elucidate the diverse functions of this forkhead transcription factor.
This work was supported by Grant-in-aid for Scientific Research 15GS0312 (to K. S.) from the Ministry of Education, Science, Sports, and Culture of Japan. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: MRF, myogenic regulatory factor; ROCK, Rho-associated kinase; MRTF-A, myocardin-related transcription factor; Id, inhibitor of DNA binding; FKHR, forkhead in human rhabdomyosarcoma; SRF, serum response factor; MHC, myosin heavy chain; SBE, Smad binding element; GM, growth medium; DM, differentiation medium; ChIP, chromatin immunoprecipitation; RT, reverse transcription; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; HA, hemagglutinin; ca, constitutively active; BMP, bone morphogenetic protein.
References
- 1.Molkentin, J. D., and Olson, E. N. (1996) Curr. Opin. Genet. Dev. 6 445-453 [DOI] [PubMed] [Google Scholar]
- 2.Olson, E. N., and Klein, W. H. (1994) Genes Dev. 8 1-8 [DOI] [PubMed] [Google Scholar]
- 3.Sabourin, L. A., and Rudnicki, M. A. (2000) Clin. Genet. 57 16-25 [DOI] [PubMed] [Google Scholar]
- 4.Benezra, R., Davis, R. L., Lockshon, D., Turner, D. L., and Weintraub, H. (1990) Cell 61 49-59 [DOI] [PubMed] [Google Scholar]
- 5.Jen, Y., Weintraub, H., and Benezra, R. (1992) Genes Dev. 6 1466-1479 [DOI] [PubMed] [Google Scholar]
- 6.Ruzinova, M. B., and Benezra, R. (2003) Trends Cell Biol. 13 410-418 [DOI] [PubMed] [Google Scholar]
- 7.Etienne-Manneville, S., and Hall, A. (2002) Nature 420 629-635 [DOI] [PubMed] [Google Scholar]
- 8.Carnac, G., Primig, M., Kitzmann, M., Chafey, P., Tuil, D., Lamb, N., and Fernandez, A. (1998) Mol. Biol. Cell 9 1891-1902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gauthier-Rouviere, C., Vandromme, M., Tuil, D., Lautredou, N., Morris, M., Soulez, M., Kahn, A., Fernandez, A., and Lamb, N. (1996) Mol. Biol. Cell 7 719-729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takano, H., Komuro, I., Oka, T., Shiojima, I., Hiroi, Y., Mizuno, T., and Yazaki, Y. (1998) Mol. Cell. Biol. 18 1580-1589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wei, L., Zhou, W., Croissant, J. D., Johansen, F. E., Prywes, R., Balasubramanyam, A., and Schwartz, R. J. (1998) J. Biol. Chem. 273 30287-30294 [DOI] [PubMed] [Google Scholar]
- 12.Castellani, L., Salvati, E., Alemà, S., and Falcone, G. (2006) J. Biol. Chem. 281 15249-15257 [DOI] [PubMed] [Google Scholar]
- 13.Charrasse, S., Comunale, F., Grumbach, Y., Poulat, F., Blangy, A., and Gauthier-Rouvière, C. (2006) Mol. Biol. Cell 17 749-759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meriane, M., Roux, P., Primig, M., Fort, P., and Gauthier-Rouvière, C. (2000) Mol. Biol. Cell 11 2513-2528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nishiyama, T., Kii, I., and Kudo, A. (2004) J. Biol. Chem. 279 47311-47319 [DOI] [PubMed] [Google Scholar]
- 16.Miralles, F., Posern, G., Zaromytidou, A. I., and Treisman, R. (2003) Cell 113 329-342 [DOI] [PubMed] [Google Scholar]
- 17.Cen, B., Selvaraj, A., and Prywes, R. (2004) J. Cell. Biochem. 93 74-82 [DOI] [PubMed] [Google Scholar]
- 18.Pipes, G. C., Creemers, E. E., and Olson, E. N. (2006) Genes Dev. 20 1545-1556 [DOI] [PubMed] [Google Scholar]
- 19.Li, S., Czubryt, M. P., McAnally, J., Bassel-Duby, R., Richardson, J. A., Wiebel, F. F., Nordheim, A., and Olson, E. N. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 1082-1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Selvaraj, A., and Prywes, R. (2003) J. Biol. Chem. 278 41977-41987 [DOI] [PubMed] [Google Scholar]
- 21.Bois, P. R., and Grosveld, G. C. (2003) EMBO J. 22 1147-1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bois, P. R., Brochard, V. F., Salin-Cantegrel, A. V., Cleveland, J. L., and Grosveld, G. C. (2005) Mol. Cell. Biol. 25 7645-7656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Birkenkamp, K. U., and Coffer, P. J. (2003) Biochem. Soc. Trans. 31 292-297 [DOI] [PubMed] [Google Scholar]
- 24.Park, J., Kim, J. S., Jung, K. C., Lee, H. J., Kim, J. I., Kim, J., Lee, J. Y., Park, J. B., and Choi, S. Y. (2003) Mol. Cells 16 216-223 [PubMed] [Google Scholar]
- 25.Morita, T., and Mayanagi, T., Sobue, K. (2007) J. Cell Biol. 3 1027-1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qin, B. Y., Chacko, B. M., Lam, S. S., de Caestecker, M. P., Correia, J. J., and Lin, K. (2001) Mol. Cell 8 1303-1312 [DOI] [PubMed] [Google Scholar]
- 27.Copeland, J. W., and Treisman, R. (2002) Mol. Biol. Cell 13 4088-4099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mohun, T., Garrett, N., and Treisman, R. (1987) EMBO J. 6 667-673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brennan, T. J., and Olson, E. N. (1990) Genes Dev. 4 582-595 [DOI] [PubMed] [Google Scholar]
- 30.Hayashi, K., Nakamura, S., Nishida, W., and Sobue, K. (2006) Mol. Cell. Biol. 26 9456-9470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suzuki, T., Fujisawa, J. I., Toita, M., and Yoshida, M. (1993) Proc. Natl. Acad. Sci. U. S. A. 15 610-614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Atherton, G. T., Travers, H., Deed, R., and Norton, J. D. (1996) Cell Growth & Differ. 7 1059-1066 [PubMed] [Google Scholar]
- 33.Melnikova, I. N., and Christy, B. A. (1996) Cell Growth & Differ. 7 1067-1079 [PubMed] [Google Scholar]
- 34.Melnikova, I. N., Bounpheng, M., Schatteman, G. C., Gilliam, D., and Christy, B. A. (1999) Exp. Cell Res. 247 94-104 [DOI] [PubMed] [Google Scholar]
- 35.Spicer, D. B., Rhee, J., Cheung, W. L., and Lassar, A. B. (1996) Science 272 1476-1480 [DOI] [PubMed] [Google Scholar]
- 36.Langley, B., Thomas, M., Bishop, A., Sharma, M., Gilmour, S., and Kambadur, R. (2002) J. Biol. Chem. 277 49831-49840 [DOI] [PubMed] [Google Scholar]
- 37.Li, L., Chambard, J. C., Karin, M., and Olson, E. N. (1992) Genes Dev. 6 676-689 [DOI] [PubMed] [Google Scholar]
- 38.Miner, J. H., and Wold, B. J. (1991) Mol. Cell. Biol. 11 2842-2851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yoshiko, Y., Hirao, K., and Maeda, N. (2002) Am. J. Physiol. 283 C1278-C1286 [DOI] [PubMed] [Google Scholar]
- 40.von Bubnoff, A., Peiffer, D. A., Blitz, I. L., Hayata, T., Ogata, S., Zeng, Q., Trunnell, M., and Cho, K. W. (2005) Dev. Biol. 281 210-226 [DOI] [PubMed] [Google Scholar]
- 41.Qiu, P., Ritchie, R. P., Fu, Z., Cao, D., Cumming, J., Miano, J. M., Wang, D. Z., Li, H. J., and Li, L. (2005) Circ. Res. 97 983-991 [DOI] [PubMed] [Google Scholar]
- 42.Xu, L. (2006) Biochim. Biophys. Acta. 1759 503-513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu, Z. P., Wang, Z., Yanagisawa, H., and Olson, E. N. (2005) Dev. Cell 9 261-270 [DOI] [PubMed] [Google Scholar]
- 44.Travaglione, S., Messina, G., Fabbri, A., Falzano, L., Giammarioli, A. M., Grossi, M., Rufini, S., and Fiorentini, C. (2005) Cell Death Differ. 12 78-86 [DOI] [PubMed] [Google Scholar]
- 45.Kuwahara, K., Barrientos, T., Pipes, G. C., Li, S., and Olson, E. N. (2005) Mol. Cell. Biol. 25 3173-3181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vartiainen, M. K., Guettler, S., Larijani, B., and Treisman, R. (2007) Science 22 1749-1752 [DOI] [PubMed] [Google Scholar]
- 47.L'honore, A., Lamb, N. J., Vandromme, M., Turowski, P., Carnac, G., and Fernandez, A. (2003) Mol. Biol. Cell 14 2151-2162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Selvaraj, A., and Prywes, R. (2004) BMC Mol. Biol. 5 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.French, B. A., Chow, K. L., Olson, E. N., and Schwartz, R. J. (1991) Mol. Cell. Biol. 11 2439-2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Muscat, G. E., Emery, J., and Collie, E. S. (1992) Gene Expr. 2 241-257 [PMC free article] [PubMed] [Google Scholar]
- 51.Long, X., Creemers, E. E., Wang, D. Z., Olson, E. N., and Miano, J. M. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 16570-16575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang, D., Chang, P. S., Wang, Z., Sutherland, L., Richardson, J. A., Small, E., Krieg, P. A., and Olson, E. N. (2001) Cell 105 852-862 [DOI] [PubMed] [Google Scholar]
- 53.Wu, J., and Lim, R. W. (2005) Biochim. Biophys. Acta 1731 13-22 [DOI] [PubMed] [Google Scholar]
- 54.Ishisaki, A., Yamato, K., Hashimoto, S., Nakao, A., Tamaki, K., Nonaka, K., ten Dijke, P., Sugino, H., and Nishihara, T. (1999) J. Biol. Chem. 274 13637-13642 [DOI] [PubMed] [Google Scholar]
- 55.Hayashi, H., Abdollah, S., Qiu, Y., Cai, J., Xu, Y. Y., Grinnell, B. W., Richardson, M. A., Topper, J. N., Gimbrone, M. A., Jr., Wrana, J. L., and Falb, D. (1997) Cell 27 1165-1173 [DOI] [PubMed] [Google Scholar]
- 56.Kollias, H. D., Perry, R. L., Miyake, T., Aziz, A., and McDermott, J. C. (2006) Mol. Cell. Biol. 26 6248-6260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hribal, M. L., Nakae, J., Kitamura, T., Shutter, J. R., and Accili, D. (2003) J. Cell Biol. 162 535-541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kamei, Y., Miura, S., Suzuki, M., Kai, Y., Mizukami, J., Taniguchi, T., Mochida, K., Hata, T., Matsuda, J., Aburatani, H., Nishino, I., and Ezaki, O. (2004) J. Biol. Chem. 279 41114-41123 [DOI] [PubMed] [Google Scholar]
- 59.Fan, W., Yanase, T., Morinaga, H., Okabe, T., Nomura, M., Daitoku, H., Fukamizu, A., Kato, S., Takayanagi, R., and Nawata, H. (2007) J. Biol. Chem. 282 7329-7338 [DOI] [PubMed] [Google Scholar]