

Abstract
Airway smooth muscle is richly endowed with muscarinic receptors of the M2 and M3 subtype. Stimulation of these receptors inhibits large conductance calcium-activated K+ (BK) channels, a negative feed back regulator, in a pertussis toxinsensitive manner and thus facilitates contraction. The underlying mechanism, however, is unknown. We therefore studied the activity of bovine trachea BK channels in HEK293 cells expressing the M2 or M3 receptor (M2RorM3R). In M2R- but not M3R-expressing cells, maximal effective concentrations of carbamoylcholine (CCh) inhibited whole cell BK currents by 53%. This M2R-induced inhibition was abolished by pertussis toxin treatment or overexpression of the Gβγ scavenger transducin-α. In inside-out patches, direct application of 300 nm purified Gβγ decreased channel open probability by 55%. The physical interaction of Gβγ with BK channels was confirmed by co-immunoprecipitation. Interestingly, inhibition of phospholipase C as well as protein kinase C activities also reversed the CCh effect but to a smaller (∼20%) extent. Mouse tracheal cells responded similarly to CCh, purified Gβγ and phospholipase C/protein kinase C inhibition as M2R-expressing HEK293 cells. Our results demonstrate that airway M2Rs inhibit BK channels by a dual, Gβγ-mediated mechanism, a direct membrane-delimited interaction, and the activation of the phospholipase C/protein kinase C pathway.
The parasympathetic nerves provide the dominant autonomic control of airway smooth muscle (ASM).3 The neurotransmitter acetylcholine exerts its effects by binding to muscarinic receptors of which five subtypes (M1-M5) have been identified. All are members of the heptahelical family of G protein-coupled receptors (1–3). On mammalian ASM, the M2 (M2R) and M3 (M3R) receptor subtypes are expressed with M2R representing the larger population (4, 5). Additionally, M4 receptors (M4R) are present in rabbit small airways and parenchyma (4, 6), but their function remains unknown. M3Rs in ASM are coupled to phospholipase C (PLC)/protein kinase C (PKC) pathway via pertussis toxin (PTX)-insensitive G proteins of the Gq/11 family. The contractile response evoked by M3R stimulation is attributed to the formation of inositol trisphosphate (IP3), the subsequent release of Ca2+ from intracellular stores, the additional influx of extracellular calcium, and the Ca2+-sensitizing effect of PKC (7–11). Stimulation of M2 muscarinic receptors (M2Rs) in ASM inhibits adenylyl cyclase via activation of PTX-sensitive G proteins of the Gi/o family (12, 13), and therefore M2Rs are thought to counteract relaxation that requires activation of adenylyl cyclase, as for example the β2-adrenoreceptor agonist-induced ASM relaxation (14). Recently, however, evidence has been provided that M2Rs participate directly in ASM contraction. In mice with a targeted deletion of the M2R, the muscarinic receptor agonist carbamoylcholine (CCh) induced less ASM contraction than in the wild-type littermates (15). In addition, in mice deleted of the M3R contractile responses in ASM to CCh still occur (16). The mechanism by which the M2Rs in ASM induce contraction is, however, still unknown. In isolated guinea pig tracheal strips, methacholine-induced contractions involved activation of PTX-sensitive Gi/o proteins and required the activity of large conductance, Ca2+-activated potassium channels (BK channels) (17). Because BK channels function as a rheostat, limiting Ca2+ influx through voltage-dependent Ca2+ channels by shifting the membrane potential to more inside-negative values (18, 19), it is conceivable that inhibition of BK channel activity favors contraction. By using the heterologous expression system of HEK293 cells and native tracheal cells, we show herein that Gβγ subunits released from Gi/o proteins upon stimulation of M2 muscarinic receptors inhibit BK channels by a dual mechanism. First, they directly bind to the BK channel pore-forming α subunit or an associated protein and inhibit the open probability. Second, they activate the PLC/PKC pathway, thereby contributing to PKC-dependent BK channel inhibition.
EXPERIMENTAL PROCEDURES
Materials—Carbamoylcholine chloride and dl-noradrenaline hydrochloride were obtained from Sigma, and Ro 31-8220, Gö 6976, and staurosporine were from Merck. U-73122, U-73343, and PMA were purchased from Biomol (Hamburg, Germany); iberiotoxin was from Alomone Laboratories (Jerusalem, Israel); and PTX was from List Biological Laboratories (Campbell, CA). The drugs were either dissolved in physiological saline solution (PSS; see solutions) or in Me2SO. The maximum 0.1% final concentration of Me2SO in the bath solution did not affect BK currents. Solutions with iberiotoxin contained 0.1% bovine albumin fraction V (Sigma). Collagenase type H (lot 014K8605), hyaluronidase type I-S, papain, and 1,4-dithio-d,l-threitol were purchased from Sigma.
Purification of Transducin α Subunits and βγ Dimers— Bleached bovine rod outer segment membranes were prepared from bovine retinae as described previously (20). The bovine retinae were obtained from a local abattoir. Transducin (TD) was eluted from the membranes by hypotonic elution in the presence of 100 μm GTP, and the subunits were separated by affinity chromatography on Blue Sepharose (Bio-Rad). The buffers in which the proteins were eluted from the columns were changed to the intracellular pipette solution (see below) by repeated extensive dialysis, and the proteins were stored in aliquots at -80 °C. The protein concentration was determined according to Bradford with IgG as standard.
Cell Lines and Transfection Procedure—HEK293 cells stably transfected with the human M2R, M3R, and M4R (Ref. 21; M2R, 4.6; M3R, 7.8; and M4R, 2.1 pmol receptor/mg of membrane protein; kindly provided by Dr. D. Meyer zu Heringdorf, Essen, Germany) or the murine α2A-adrenoreceptor (α2A-AR) (Ref. 22; 23.1 pmol of receptor/mg of membrane protein; kindly provided by Dr. L. Hein, Freiburg, Germany) were transiently transfected with the BK channel α subunit (GenBank™ accession number AY033472) in pcDNA3 (23). Some α2A-AR-expressing cells, and for comparison cells expressing the M2R and M4R, were transiently transfected with a bicistronic expression vector encoding for the G protein regulated inward rectifying potassium channel (GIRK) subunits GIRK1 and GIRK4 (Ref. 22; kindly provided by Dr. M. Bünemann, Würzburg, Germany). To evaluate Gβγ-mediated effects by CCh, M2R-expressing cells were transiently co-transfected with the BK channel α subunit and with bovine transducin α (TDα) subcloned into the pAdTrack-CMV vector. In some experiments, HEK293 cells were transiently co-transfected with the BK channel α subunit and either with the human M3R in pcDNA3.1 (UMR cDNA Resource Center) or with the β1 subunit of the BK channel cloned from bovine trachea in pcDNA3 (kindly provided by Dr. Maria Garcia, Department of Ion Channels, Merck Research Laboratories, Rahway, NJ). Stably transfected HEK293 cells were cultured in Dulbecco's modified Eagle's medium/Ham's medium (Biochrom, Berlin, Germany) containing 10% fetal calf serum, 100 units ml-1 penicillin, 100 μg ml-1 streptomycin, and 500 μg ml-1 G418. All other HEK293 cells were cultured in minimum essential medium supplemented with Earle's salts medium (Biochrom, Berlin, Germany) containing 10% fetal calf serum, 100 units ml-1 penicillin, and 100 μg ml-1 streptomycin. HEK293 cells were cultured for 24 h and then transiently transfected with the DNA plasmids by calcium phosphate precipitation for 18 h at 35 °C and 3% CO2. After washing, the cells were cultivated for another 48–72 h at 37 °C and 6% CO2. After the medium was exchanged several times with PSS (see below), the cells were transferred in a 35-mm dish to the stage of an inverted microscope (Zeiss Axiovert 200) for electrophysiological measurements. The transfection efficiency varied between 40 and 70%.
Animals and Cell Preparation—All of the experimental procedures were carried out according to the animal welfare guidelines of the University Medical Center Hamburg-Eppendorf. C57BL/6 mice of either sex were obtained from a colony bred and maintained at the animal house of the University Medical Center Hamburg-Eppendorf. The tracheae were dissected from mice killed by CO2. After the connective tissue was removed, the tissue was cut with a sharp blade into pieces of ∼1 mm side length and incubated under gentle agitation at 37 °C in Ca2+-free PSS including 0.7 mg ml-1 papain, 1 mg ml-1 1,4-dithio-d,l-threitol, and 1 mg ml-1 fat-free bovine serum albumin. The tissue pieces were transferred 30 min later into PSS containing 50 μm Ca2+, 1 mg ml-1 collagenase, 1 mg ml-1 hyaluronidase, and 1 mg ml-1 albumin and digested for another 8–10 min at 37 °C. Single cells were released by gentle trituration of the digested tissue using a narrow-bore, fire-polished Pasteur pipette and stored in PSS at room temperature. After isolation, >50% of the cells were relaxed, and only these cells were used for the electrophysiological studies. The experiments were conducted within 6 h of cell isolation. A small aliquot of the solution containing the isolated cells was placed in an open perfusion chamber (1 ml) mounted on the stage of an inverted microscope (Zeiss Axiovert 200). The myocytes were allowed to adhere to the bottom of the chamber for 5–10 min and were then superfused at 2–3 ml min-1 with PSS at room temperature.
Recording Techniques—Standard patch clamp recording techniques were used to measure currents in the cell-attached, inside-out, or whole cell patch clamp configuration (24). Patch electrodes were fabricated from borosilicate glass capillaries (MTW 150F; World Precision Instruments, Inc., Sarasota, FL) and filled with prefiltered solutions of different composition (see below). The currents were recorded at room temperature with an EPC-7 amplifier (HEKA Elektronik, Lambrecht, Germany), connected via a 16-bit A/D interface to a pentium IBM clone computer. The signals were low pass filtered (1 kHz) before 5 kHz digitization. Data acquisition and analysis were performed with an ISO-3 multitasking patch clamp program (MFK M. Friedrich, Niedernhausen, Germany). Pipette resistance ranged from 2 to 3 MΩ in whole cell experiments and from 8 to 9 MΩ in the excised patch experiments. The amplitude of single-channel currents was derived from an amplitude distribution histogram. Determination of the average channel open probability (NPo) in patches has been described elsewhere (25). After the patch had been equilibrated for at least 5 min, the drugs were superfused for another 3–5 min before the mean NPo was determined. A multi-barreled perfusion pipette placed 200 μm away from the patch was used to switch the superfusion solution. The holding potential in the whole cell patch clamp experiments was -10 mV.
Solutions—The bath was superfused with PSS containing 127 mm NaCl, 5.9 mm KCl, 2.4 mm CaCl2, 1.2 mm MgCl2, 11 mm glucose, and 10 mm HEPES adjusted to pH 7.4 with NaOH. For whole cell experiments the pipette solution contained 126 mm KCl, 6 mm NaCl, 1.2 mm MgCl2, 5 mm EGTA, 11 mm glucose, 3 mm dipotassium ATP, 0.1 mm Na3GTP, and 10 mm HEPES adjusted to pH 7.4 with KOH. The free Ca2+ concentration was adjusted to 0.3 μm by adding the appropriate amount of CaCl2 as described earlier (25). In some experiments the Ca2+ concentration was increased to 3 μm. Free Ca2+ was checked by fura 2-fluorescence. For inside-out experiments, the extracellular (pipette) solution contained 132 mm KCl, 6 mm NaCl, 1.2 mm MgCl2, 5 mm EGTA, 11 mm glucose, 10 mm HEPES, pH 7.4, adjusted with KOH, and the bath solution (cytosolic surface of the patch) contained 126 mm KCl, 6 mm NaCl, 1.2 mm MgCl2, 5 mm EGTA, 11 mm glucose, 3 mm dipotassium ATP, 0.1 mm Na3GTP, and 10 mm HEPES, pH 7.4, adjusted with KOH. The free Ca2+ concentration was 0.3 μm, and in some experiments it was 1 μm. For the measurement of GIRK currents, whole cell recordings were used with a bath solution containing 120 mm NaCl, 20 mm KCl, 2 mm CaCl2, 1 mm MgCl2, 10 mm HEPES, pH 7.3, adjusted with NaOH. The internal (pipette) solution contained 100 mm potassium aspartate, 40 mm KCl, 5 mm NaCl, 2 mm MgCl2, 5 mm MgATP, 0.1 mm Na3GTP, 2 mm EGTA, and 10 mm HEPES adjusted to pH 7.3 with KOH.
Co-immunoprecipitation—HEK293 cells were seeded in 10-cm culture dishes and transfected with 4 μg of pcDNA3.2-BK channel α subunit, 4 μg of pcDNA6/His-Gβ1IRESγ2 (kind gift of Dr. H. J. Hippe, Heidelberg, Germany), or a combination of both and up to 8 μg of an empty pcDNA control vector. Forty-eight hours after transfection, the cells were lysed with 1000 μl of immunoprecipitation buffer (10 mm Tris-HCl, pH 7.4, 150 mm NaCl, 0.1% Triton X-100, 1 mm phenylmethylsulfonyl fluoride) and homogenized for 30 s. After centrifugation (26,000 × g for 10 min at 4 °C), 40 μl of a 1:1 slurry of protein A-Sepharose (GE Healthcare) was added for preclearing the lysates. The lysates were separated from protein A-Sepharose, and then 4 μg of anti-His6-antibody (Qiagen) was added to each sample and incubated for 30 min on ice. After the addition of 40 μl equilibrated protein A-Sepharose, the samples were gently shaken for 4 h at 4 °C. Then the Sepharose beads were washed three times with immunoprecipitation buffer, and the bound proteins were eluted with 25 μl of sample buffer for 5 min at 95 °C. The precipitated proteins were analyzed by immunoblot with an anti-Gβcom (1:1000 dilution; Santa Cruz Biotechnology) or an anti-BK channel α subunit antibody (26) (1:500 dilution).
Data Analysis—SigmaPlot for windows (Jandel Scientific, version 8) was used for statistical analyses. Significance was determined by paired or unpaired t test or by one-way analysis of variance. When a significant effect was detected with analysis of variance, Student's t test was used for pairwise comparisons. The effects were deemed significant when a p < 0.05 was obtained. The results are expressed as the means ± S.E. where applicable.
RESULTS
Gβγ Subunits Mediate BK Channel Inhibition by M2 and M4 Receptor Activation—A PTX-sensitive inhibitory effect of muscarinic receptor stimulation on BK channel activity has been demonstrated in ASM cells at the single channel level (27, 28). When we elicited whole cell outward currents (Iout) in smooth muscle cells freshly isolated from the mouse trachea (TSMCs) from a holding potential of -10 mV by depolarizing the cells every 5 s for 300 ms from -60 to +80 mV, addition of the specific BK channel inhibiting peptide iberiotoxin (300 nm) revealed that more than 90% of Iout was generated by BK channels (data not shown). The addition of a maximal effective concentration of CCh (10 μm) decreased current densities at all voltages by ∼50% (current densities at +80 mV: control, 135.5 ± 9.2; CCh, 66.7 ± 10.7 pA pF-1, p < 0.01, n = 8). In accordance with the data reported by Kume et al. (27, 28), the inhibitory effect of CCh was completely abolished when TSMCs were exposed to PTX 5 h prior to CCh application. To dissect the effects induced via M2Rs and M3Rs, we transiently expressed the pore-forming BK channel α subunits (KCNMA1) cloned from bovine trachea in HEK293 cells stably expressing the human M2R (21). Whole cell outward currents were then elicited as described above (Fig. 1A). In HEK293 cells not expressing BK channels, a negligible endogenous outward current was obtained which amounted to less than 2% (at + 80 mV) of the current elicited in transfected cells (data not shown). We therefore consider outward currents in transfected HEK293 cells exclusively as BK currents (IBK). As observed in TSMCs, 10 μm CCh suppressed IBK at all voltages by ∼50% (Fig. 1, A and D), and this inhibition was completely abolished when HEK293 cells were exposed to 500 ng ml-1 PTX 5 h prior to CCh application (Fig. 1, B and D). Because this finding indicates that Gi/o proteins also mediate the CCh-induced inhibition of BK channel currents in HEK293 cells, we set up experiments to characterize which subunit of Gi/o is responsible for channel inhibition. We therefore co-expressed the BK channel α subunit and TDα, the Gα subunit of the heterotrimeric G protein that mediates signal transduction in vertebrate photoreceptor cells. TDα has been shown to act as a scavenger of Gβγ dimers dissociated from heterotrimeric Gi/o proteins (29) and thus specifically antagonizes Gβγ-mediated effects, whereas Gαi/o actions are preserved (30, 31). As shown in Fig. 1 (C and D), CCh completely lost its inhibitory effect on BK channel current in TDα-overexpressing cells. When HEK293 cells expressing the M3R and the BK channel α subunit were exposed to 10 μm CCh by utilizing the same experimental protocol, no steady-state inhibition of IBK was obtained (Fig. 2A). Together the findings indicate that the Gi/o-coupled M2R, but not the preferentially Gq/11-coupled M3R (32), is linked to BK channel inhibition via Gβγ dimers. We therefore tested whether the second Gi/o-coupled muscarinic receptor, i.e. the M4R, or stimulation of Gi/o-coupled receptors of different subfamilies can also inhibit steady-state IBK. Whole cell patch clamp experiments in M4R- or α2A-AR-expressing cells revealed that application of 10 μm CCh to the M4R-expressing cells resulted in a significant, approximately 35% decrease of the BK channel current, whereas noradrenaline (10 μm) was ineffective in α2A-AR-expressing cells (Fig. 2, B–D). To confirm that α2A-ARs expressed in HEK293 cells effectively couple to Gi/o proteins, we transiently transfected these cells and, for comparison, also the cells expressing M2R and M4R, with the G protein-gated inwardly rectifying K+ channel subunits GIRK1 and GIRK4. Because functional GIRK channels are activated by Gβγ dimers (33), their activation indicates the agonist-induced release of Gβγ from Gi/o. Agonist-activated currents were measured as inwardly directed K+ currents generated in the whole cell configuration by 300-ms hyperpolarizing pulses from a holding potential of -10 to -90 mV (20 mm external K+). Application of 10 μm noradrenaline and 10 μm CCh induced large currents in the cells expressing the α2A-AR or one of the two muscarinic receptors, respectively (Fig. 3). Interestingly, stimulation of α2A-ARs with noradrenaline generated a significantly larger GIRK current than CCh in M4R- or M2R-expressing cells. Taken together, the results show that all three Gi/o-coupled receptors effectively release Gβγ subunits, but only Gβγ released by activation of Gi/o-coupled muscarinic receptors induces BK channel inhibition. Gβγ dimers regulate the activity of some ion channels such as the GIRK and neuronal voltage-dependent Ca2+ channels by direct interaction (34), whereas other effects require stimulation of Gβγ-dependent effector enzymes like PLCβ isoforms (35, 36) and the subsequent activation of PKC. We therefore set up experiments to analyze the possible contribution of direct and indirect Gβγ-mediated effects to BK channel inhibition.
FIGURE 1.

Inhibition of BK channel currents by CCh in HEK293 cells stably expressing the M2R and transiently transfected with the pore-forming BK channel α subunit. Whole cell BK outward currents (IBK) were elicited from a holding potential of -10 mV by depolarizing the cells every 5 s for 300 ms from -60 to +80 mV in 10-mV increments. A, original current traces before and after application of 10 μm CCh. B and C, abolition of CCh-induced inhibition of IBK after pretreatment of the cells with PTX (B) or after co-transfection with TDα (C). D, summary of the effects of CCh on current densities of IBK at a membrane potential of +80 mV. The bars represent the means ± S.E. of 22 cells (control, Ctr) and 7 cells (PTX and TDα), respectively. The pipette solution contained 0.3 μm Ca2+. ***, p < 0.001 versus control; ns, not significant.
FIGURE 2.
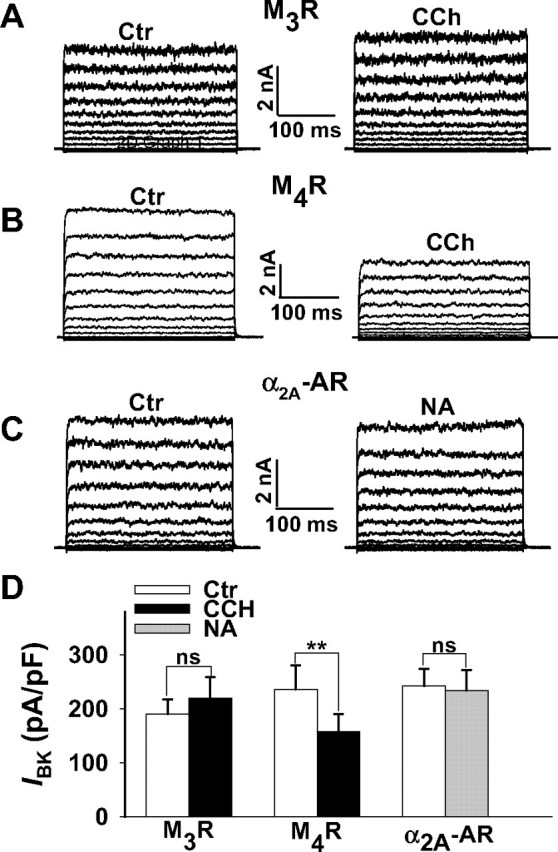
Stimulation of M4Rs but not of M3Rs and α2A-ARs are linked to BK channel inhibition. Whole cell BK outward currents (IBK) were elicited in HEK293 cells transfected with either the M3R(A), the M4R(B), or the α2A-AR (C) and the BK channel α subunit. A–C, original current traces from three representative cells depolarized every 5 s for 300 ms from -60 to +80 mV in 10-mV increments. Holding potential was -10 mV. Currents before (control) and in the presence of 10 μm CCh or 10 μm NA are shown. D, summary of the effects of CCh and NA on current densities of IBK at a membrane potential of +80 mV. The bars represent mean values ± S.E. of 6 (M3R), 8 (M4R), and 10 (α2A-AR) cells. The pipette solution contained 0.3 μm Ca2+. **, p < 0.01 versus control (Ctr); ns, not significant.
FIGURE 3.
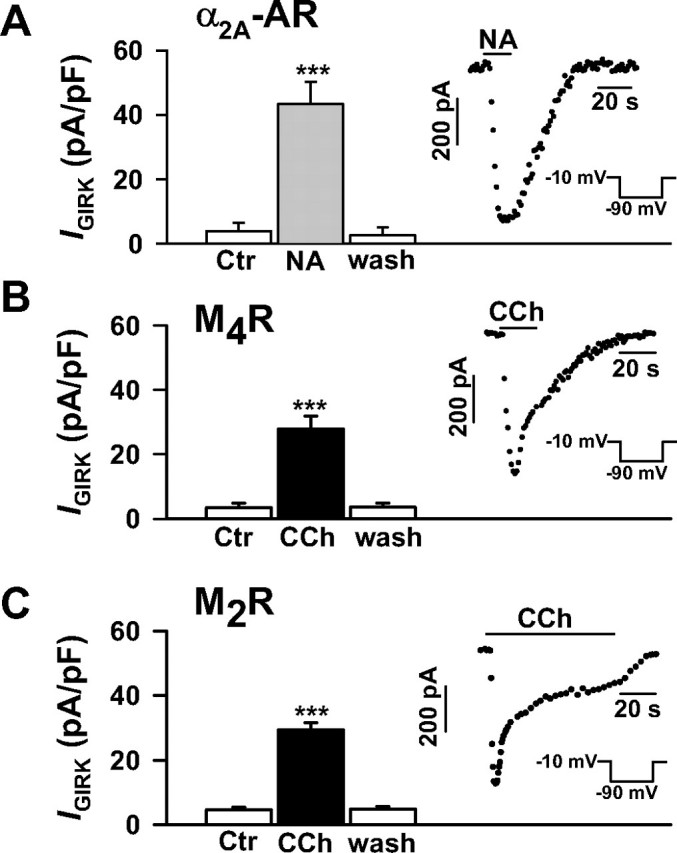
Agonist-induced GIRK currents in HEK293 cells stably expressing the M2R, M4R, and α2A-AR. The cells were transiently transfected with the G protein-gated inwardly rectifying K+ channel subunits GIRK1 and GIRK4. GIRK currents were elicited in the whole cell configuration by 300-ms hyperpolarizing pulses from a holding potential of -10 to -90 mV (20 mm external potassium). Note that the currents (IGIRK) are inward currents. GIRK current densities are strongly enhanced by 10 μm NA in the α2A-AR-expressing cells (A; n = 6), and by 10 μm CCh in the M4R-expressing (B; n = 10) and M2R-expressing cells (C; n = 7). Insets, time course of agonist-induced GIRK currents. ***, p < 0.001 versus control (Ctr).
Gβγ Dimers Directly Inhibit BK Channels—To study the possibility that Gβγ dimers directly inhibit BK channel activity, experiments on inside-out patches from HEK293 cells expressing BK channel α subunits were carried out. In the presence of symmetrically high potassium (140 mm) and at a holding potential of +60 mV, the cytosolic surface of an inside-out patch was sequentially superfused with purified G protein subunits either alone or in combination (Fig. 4, A and B). When 1 μm TDα was applied first as a negative control, channel open probability (NPo) did not change. Instead, NPo rapidly decreased from 0.67 to 0.39 when the superfusion solution contained 300 nm TDβγ. The effect of Gβγ was restricted to BK channel open probability, and the single channel conductance remained unchanged. The inhibitory effect of TDβγ was stable as long as the G protein was present but returned to the control level (0.74) when 1 μm TDα as scavenger was additionally applied. When the effect of 300 nm TDβγ alone was evaluated in 15 patches, a decrease of NPo by 55.3 ± 5.3% was obtained (Fig. 4C). This decrease was not significantly altered by increasing the Ca2+ concentration of the intracellular (bath) solution to 1 μm (inhibition by 51.7 ± 5.7%, n = 6) or by expressing the regulatory β1-subunit of the BK channel together with BKα (inhibition by TDβγ: 53.4 ± 4.4%, n = 9). The experiments presented so far, do not exclude the possibility that PLCβ isozyme(s) sensitive to activation by Gβγ dimers and present in inside-out patches may have mediated TDβγ-induced inhibition of NPo. Inside-out patches were therefore superfused for 10 min with the PLC inhibitor U-73122 (2.5 μm) first and then additionally with 300 nm TDβγ. The resulting inhibition of NPo by 53.7 ± 7.1% (n = 7) was not significantly different from the inhibition in the absence of the PLC inhibitor (Fig. 4C). U-73122 by itself had no significant effect on NPo (data not shown). Together the results indicate that TDβγ inhibits BK channels in inside-out patches independent of the intracellular Ca2+ concentration, the additional expression of the regulatory β1 subunit, or an activation of PLCβ isozymes. We thus determined whether free Gβγ dimers physically associate with BK channel α subunits in an intact cellular context by co-immunoprecipitation. Cell lysates from transiently transfected HEK293 cells expressing either recombinant BK channel α subunits, His-tagged Gβ1γ2 (GHisβ1γ2), or their combination, were used for immunoprecipitation with an anti-His tag antibody. BKα was co-precipitated only from lysates of cells co-expressing GHisβ1γ2 and BKα (Fig. 5). In summary, the results obtained in inside-out patches and by co-immunoprecipitation suggest that the interaction of Gβγ dimers with BKα or an as yet unidentified adapter protein(s) reduces BK channel open probability.
FIGURE 4.

Direct inhibition of BK channel open probability (NPo) in HEK293 cells by TDβγ. Single channel NPo of BK channels was recorded in inside-out patches from HEK293 cells transiently transfected with the BK channelα subunit. Holding potential was +60 mV, and the intracellular (bath) Ca2+ concentration was 0.3 μm. A, NPo as influenced by the sequential superfusion of the cytosolic surface of a single patch with 1 μm TDα and 300 nm TDβγ alone and in combination. B, traces show representative single channel current recordings taken at the times indicated by the corresponding numbers in A. The channel openings are upward deflections. The dashed lines represent numbers of channels in the open state, and c indicates the closed state level. C, the inhibitory effect of TDβγ on NPo is not influenced by raising the intracellular (bath) Ca2+ concentration from 0.3 to 1 μm, by co-transfecting the BK channelα subunit with theβ1 subunit, or by inhibition of PLCβ with 2.5 μm U-73122. The bars represent the mean values ± S.E., n = 15 (BKα 0.3 μm Ca2+), n = 6 (BKα 1 μm Ca2+), n = 9 (BKα+β 0.3 μm Ca2+), and n = 7 (U-73122). Ctr, control.
FIGURE 5.

Binding of Gβγ subunits to the BK channel α subunits in HEK293 cells. HEK293 cells were transfected with plasmids encoding the BK channel α subunit, GHisβ1γ2 (4 μg of DNA each), and up to 8 μg of an empty pcDNA control vector. After cell lysis, the expressed GHisβ1γ2 was precipitated with an anti-His antibody and analyzed by immunoblot with a polyclonal anti-Gβ antibody. Co-precipitated BK channel α subunits were detected with a polyclonal anti-BKα antibody. Equal expression of GHisβ1γ2 and BK channel α subunit constructs in the cell lysates was verified. IP, immunoprecipitation.
The M2R-induced Activation of the PLC/PKC Contributes to the Inhibition of BK Channels—It is well known that M2Rs couple to the PLC/PKC pathway by Gi/o-mediated stimulation of Gβγ-sensitive PLCβ isoforms (35–37) of which the β3 isoform is present in HEK293 cells.4 Although the inhibition by directly applied Gβγ dimers on NPo was quantitatively similar to the Gβγ-mediated inhibition of steady-state BK channel currents (compare Figs. 1D and 4C) and was unaffected by the PLC inhibitor U-73122 (Fig. 4C), this does not exclude the possibility that a PTX-sensitive and Gβγ-mediated coupling of the M2Rto the PLC/PKC pathway contributes to the effect of CCh on the steady-state currents in the whole cell configuration. We therefore exposed M2R-expressing cells to U-73122 or its inactive analog U-73343 and measured whole cell current densities. As shown in Fig. 6, the CCh-induced inhibition at +80 mV in U-73343-treated cells (51.2 ± 3.3%, n = 6) was not significantly different from inhibition in control cells (Fig. 1). In the presence of U-73122, however, the CCh-induced inhibition was significantly attenuated to 19.4 ± 4.1% (n = 9). U-73122 alone induced no direct inhibitory effect on the BK current (data not shown). To examine a possible contribution of the PLC-induced PKC activation to CCh-induced BK channel inhibition, we measured IBK in transfected HEK293 cells exposed to the PKC inhibitors Ro 31-8220 and Gö 6976 and the broad spectrum protein kinase inhibitor staurosporine (Fig. 7A). At +80 mV, 10 μm CCh inhibited BK channel currents in the presence of 1 μm Ro 31-8220, 1 μm Gö 6976 and 0.1 μm staurosporine by 26.3 ± 3.6% (n = 8) 22.7 ± 3.1% (n = 7), and 22.6 ± 3.4% (n = 10), respectively. Because Gö-6976 selectively inhibits Ca2+-dependent PKC isozymes, and the inhibitors with the broader spectrum did not significantly differ in their efficacy to diminish CCh-induced inhibition, a contribution of protein kinases other than Ca2+-dependent PKCs seemed unlikely. To further confirm the role of Ca2+-activated PKCs, experiments were performed in HEK293 cells exposed to 10-fold higher Ca2+ concentrations in the pipette filling solution or to the PKC activator PMA. Whereas the CCh-induced inhibition of IBK at +80 mV was 52.3% in control cells (0.3 μm Ca2+), the inhibition significantly increased to 77.8 ± 2.8% (n = 8) when the pipette solution contained 3 μm Ca2+ (Fig. 7B). PMA (100 nm) decreased BK channel currents at all potentials (Fig. 7C, 44.7 ± 6.3%, n = 6, at +80 mV). This inhibitory effect of PMA was completely abolished by 1 μm Gö 6976 (Fig. 7D) or 1 μm Ro 31-8220 (data not shown). Taken together, the findings indicate that stimulation of M2Rs inhibits BK channel currents in HEK293 cells by a dual mechanism that in part requires activation of the Gβγ-PLCβ-PKC signaling pathway as well as an interaction of Gβγ with the BK channel complex.
FIGURE 6.
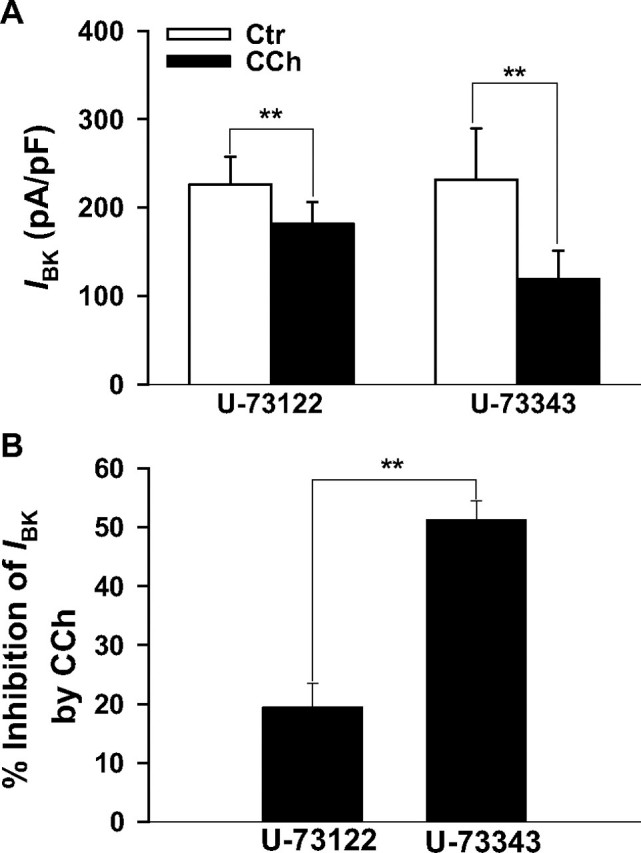
Activation of PLC partially contributes to the CCh-induced inhibition of BK currents in transfected HEK293. Whole cell BK currents (IBK) were elicited as described. Inhibition of IBK by 10 μm CCh is attenuated by the PLC inhibitor U-73122 (2.5 μm) but remains unopposed in the presence of the inactive analog U-73343 (2.5 μm). A, bars represent the mean current densities of cells exposed to CCh in the presence of U-73122 (n = 9) or U-73343(n = 6). B, same data as in A but presented as the percentage of inhibition of current densities. The pipette solution contained 0.3 μm Ca2+. **, p < 0.01. Ctr, control.
FIGURE 7.

Activation of PKC contributes to the CCh-induced inhibition of BK currents in HEK293 cells stably expressing the M2R. Whole cell BK currents (IBK) were elicited as described. A, attenuation of the CCh-induced inhibition (as a percentage) of IBK by the specific PKC inhibitors Ro 31-8220 (1 μm) and Gö 6976 (1 μm) and by the broad spectrum protein kinase inhibitor staurosporine (0.1 μm). The bars represent the mean values ± S.E. of 22 cells (control (Ctr), same cells as in Fig. 1D), 8 (Ro 31-8220), 7 (Gö 6976), and 10 (staurosporine) cells. B, the CCh-induced IBK inhibition is enhanced by increasing the Ca2+ concentration in the pipette filling solution from 0.3 to 3 μm. The bars represent the mean values ± S.E. of 22 cells (0.3 μm Ca2+; same cells as control in A) and 8 cells (3 μm Ca2+). The cells in A and B were clamped from -10 to +80 mV. *, p < 0.05; **, p < 0.01. C and D, effects of 100 nm phorbol 12-myristate, 13-acetate (PMA) on current-voltage relationships of IBK in the absence (C) and in the presence of the PKC inhibitor Gö 6976 (1 μm; D) are shown. The mean current densities of six cells, respectively, are plotted in C and D against the respective test potential. Note that Gö 6976 has no effect on IBK of its own. Whole cell currents were evoked by applying a 300-ms depolarizing pulse every 5 s in 10-mV increments from a holding potential of -10 mV. The insets in C and D show representative IBK recordings elicited by 300-ms pulses from a holding potential of -10 to +80 mV. The pipette solution contained 0.3 μm Ca2+ if not indicated otherwise.
CCh-induced Inhibition of BK Channels in Tracheal Smooth Muscle Cells—To verify that the signal transduction pathways disclosed by the experiments reported above do not only occur in a heterologous expression system but also under physiological conditions in ASM, we tested the influence of PTX, U-73122, Gö 6976, and Ro 31-8220 on CCh-induced inhibition of Iout in TSMCs (Fig. 8). As already mentioned, PTX treatment completely abolished the CCh-induced inhibition (51.0 ± 1.6%, n = 8, at +80 mV) at all potentials. Application of the PKC inhibitors Ro 31-8220 and Gö 6976 (1 μm each) or the PLC inhibitor U-73122 (2.5 μm) for 10 min did not significantly affect Iout. When CCh was added thereafter, inhibition of current densities was significantly smaller in the presence of Gö 6976 (20.4 ± 4.8%; n = 6), Ro 31-8220 (25.0 ± 4.0%; n = 7), and U-73122 (27.1 ± 4.2%; n = 6) than in control cells, resembling partial inhibition in M2R-expressing HEK293 cells. Importantly, direct application of TDβγ to TSMC in the inside-out configuration inhibited single channel NPo to a similar (∼50%) extent as in transfected HEK293 cells (Fig. 8C). This inhibition was not significantly affected by prior pretreatment with the PLC inhibitor U-73122 (data not shown). These results clearly demonstrate that the inhibitory coupling between muscarinic receptors and BK channels is highly similar in specifically M2R-expressing HEK293 cells and freshly isolated TSMCs.
FIGURE 8.

The PTX-sensitive, CCh-induced inhibition of BK channels in mouse tracheal smooth muscle cells involves direct interaction of Gβγ with BK channels and activation of the PLC/PKC pathway. A and B, whole cell outward currents (Iout) were elicited from a holding potential of -10 mV by depolarizing the cells every 5 s for 300 ms from -60 to +80 mV in 10-mV increments. A, current-voltage relationships of Iout before (control, Ctr) and after the application of 10 μm CCh are shown (10 cells from five mice). B, abolition of the CCh-induced inhibition of Iout after pretreatment of the cells with PTX (15 cells from eight mice). Insets in A and B, representative original current recordings before and in the presence of CCh. C, direct inhibition of BK channel open probability (NPo) by TDβγ. Single channel NPo of BK channels was recorded in inside-out patches. Holding potential was +80 mV, and the intracellular (bath) Ca2+ concentration was 0.3 μm. TDβγ (300 nm) before and after heat denaturation was sequentially applied to the cytosolic surface of a single patch. Ctr, control trace. The traces show representative single channel current recordings. Channel openings are upward deflections. D, attenuation of the CCh-induced inhibition (in percent) of Iout by the specific inhibitors U-73122 (2.5 μm), Ro 31-8220 (1 μm), and Gö 6976 (1 μm). The bars represent the mean values ± S.E. of cells in the absence (Ctr; 10 cells from four mice) and in the presence of either U-73122 (6 cells from two mice), Ro-31-8220 (6 cells from three mice), or Gö 6976 (7 cells from three mice). The cells were clamped from a holding potential of -10 to +80 mV. The pipette solution contained 0.3 μm Ca2+. ***, p < 0.001 versus control (Ctr).
DISCUSSION
The results presented in this study demonstrate, for the first time, that Gβγ dimers released from Gi/o proteins upon activation of M2Rs are capable of inhibiting BK channel activity by two different mechanisms. First, they directly interact with the BK channel α subunit or an associated protein and reduce the channel open probability NPo. Second, they activate Ca2+-dependent PKCs via the PLCβ pathway that additionally contribute to the inhibition of channel activity. We obtained evidence that this dual mechanism occurs similarly in native smooth muscle cells isolated from mouse trachea and in HEK293 cells expressing recombinant receptors together with the pore-forming BK channel α subunits that, after forming tetrameric channels, retain their characteristic biophysical properties and are functionally responsive to various protein kinases (23, 38, 39). Therefore our data indicate that this regulation is a more general phenomenon, and the sensitivity of the M2R-induced inhibition of BK channel activity to PTX (40) and TDα expression (29) in transfected HEK293 cells demonstrates that the presence of the G protein-coupled receptor and the BK channel α subunit is sufficient to establish this regulation in a background containing Gi/o proteins, Gβγ-activated PLCβ isoforms, and Ca2+-activated PKCs.
The evidence for a direct membrane-delimited inhibition of BK channel activity by Gβγ dimers is based on experiments in cell-free inside-out patches from transfected HEK293 cells as well as mouse TSMCs exposed to TDα, TDβγ, or their combination. The results unambiguously demonstrate that TDβγ but not TDα inhibits BK channel open probability by 55%, and this action can be reversed by formation of the inactive heterotrimer. The TDβγ-induced inhibition was not influenced by varying the intracellular (bath) Ca2+ concentration, by inhibition of PLCβ, or by co-expressing the α subunit of the BK channel with the auxiliary β1 subunit (KCNMB1) whose presence in the channel complex confers an increased voltage- and calcium sensitivity toward the pore-forming α subunit (41). These findings indicate that in cell-free inside-out patches, the activation of Ca2+-activated PKC isoforms does not contribute to channel inhibition. Together with the co-immunoprecipitation experiments, the data clearly argue for a mechanism whereby channel inhibition by Gβγ occurs through direct interaction with the α subunit of the BK channel or a closely associated intermediary. BK channels therefore join the group of directly Gβγ-modulated ion channels that so far consist of G protein-coupled inwardly rectifying K+ channels (GIRK or Kir3) (33), and neuronal voltage-dependent Ca2+ channels that give rise to P/Q-, N-, and R-type currents (Cav2) (34).
Interestingly, in both of these ion channels, PKC-dependent phosphorylation contributes to the complex regulation (42, 43). There are several reports, mainly on arterial smooth muscle cells (44–46), but also on rat pituitary tumor cells (47), indicating that PKC inhibits BK channels (48). The inhibition likely occurs by phosphorylation of Ser/Thr residues. Several putative PKC phosphorylation sites have been identified within the COOH-terminal “tail” of the BK channel α subunit (23, 49). In the present study, we found that the two PKC inhibitors Ro 31-8220 and Gö 6976 (50) significantly attenuated the CCh-induced BK channel inhibition. In addition, the PKC activator PMA inhibited BK channel activity in transfected HEK293 cells by ∼50%, and this effect was completely eliminated by Ro 31-8220 and Gö 6976. Ro 31-8220 is an unspecific blocker of many PKC isozymes, whereas Gö 6976 preferentially inhibits Ca2+-dependent PKCs (50, 51). Because both PKC inhibitors were equally effective, and CCh-induced BK channel inhibition increased (Fig. 7B) when the Ca2+ concentration was raised, Ca2+-dependent PKCs are likely involved. Furthermore, the finding that staurosporine, a broad spectrum inhibitor of several protein kinases including PKC, produced the same inhibitory effect as the specific PKC inhibitors likely excludes the additional contribution of other protein kinases. It is well established that the M2R can stimulate the activity of Gβγ-sensitive PLCβ isoforms by activation of Gi/o proteins (35–37). Because activation of PLCβ and the resulting production of the endogenous PKC activator diacylglycerol and Ca2+-releasing IP3 are thought to be the major source of activation of Ca2+-dependent PKCs, it was not surprising that the PLC inhibitor U-73122 blunted the CCh-induced inhibition of BK channel activity to a similar extent as inhibition of PKCs. In line with this interpretation, the functional coupling of M2Rs to PLC-IP3-induced Ca2+ release via Gi/o proteins has been clearly demonstrated by CCh-induced Ca2+ transients in transfected and fura-2-loaded Chinese hamster ovary cells. In contrast to M3R-expressing Chinese hamster ovary cells, these CCh-evoked Ca2+ transients in M2R-expressing cells were sensitive to PTX (52). Taken together, our results show a contribution of the M2R-Gi/oβγ-PLCβ-PKC pathway to CCh-induced inhibition of BK channel activity of ∼50%.
Experiments designed to determine whether BK channel inhibition is a general principle of Gi/o-coupled receptors showed that stimulation of recombinant M4Rs could also inhibit BK channel activity, whereas stimulation of the α2A-AR by NA was without response. This lack was not due to a missing or insufficient coupling between the α2A-AR and Gi/o proteins. Within the same cells, α2A-AR stimulation largely activated transiently expressed GIRK channels, which could also be activated by M2R and M4R stimulation. The maximal effect was largest in the α2A-AR-expressing cells, followed by the M2R-expressing cells, and the effect was weakest in the M4R-expressing cells, which strictly correlated to the amount of recombinantly expressed receptors (21, 22). The specificity of Gi/o-coupled muscarinic receptors to inhibit BK channels is intriguing. The reason for this may be a highly localized signal transduction complex of receptor, G protein, and ion channel (Fig. 9). Indeed, the ability of BK channels to assemble specifically into macromolecular complexes with other transmembrane proteins such as the β2-adrenoreceptor (53) and voltagegated calcium channels (54) has been shown before.
FIGURE 9.

Scheme illustrating the inhibition of the negative feedback regulation of BK channels in ASM by stimulation of M2Rs. Occupation of M2Rs by an agonist induces the dissociation of heterotrimeric Gi proteins in Gαi and Gβγ subunits. Gβγ inhibits BK channels (BK) by binding directly to the channel protein or to a closely associated protein with modulatory function. In addition, Gβγ stimulates the β-isozymes of PLC, which in turn catalyzes the formation of IP3 and diacylglycerol (DAG) from phosphatidylinositol bisphosphate. IP3 releases Ca2+ from the sarcoplasmic reticulum through the Ca2+-conducting IP3 receptor. Diacylglycerol accumulates in the plasma membrane and causes, in concert with an elevated cytosolic Ca2+ concentration, translocation and activation of calcium-activated PKCs. Activated PKCs inhibit BK channels, probably by phosphorylating specific Ser/Thr residues of the channel protein. Inhibition of BK channels interferes with the function of BK channels as negative feedback regulators, membrane hyperpolarization is prevented, and more Ca2+ flows into the cell through voltage-dependent Ca2+ channels (VDCC). Together with the IP3-mediated Ca2+-release from the SR, BK channel inhibition helps to enhance the cytosolic Ca2+ concentration and hence contraction. Stimulatory and inhibitory pathways are shown in green and red, respectively.
BK channels are expressed in many cell types where they play a powerful integrative role in the regulation of electrical excitability by conducting repolarizing outward currents (49, 55). Therefore, the question arises of what is the physiological significance of BK channel inhibition as part of the cell response to muscarinic receptor activation in the ASM. Like in other tissues, BK channels in ASM buffer cell excitation in response to excitatory cell signals such as depolarization and an increased Ca2+ concentration. Consequently, inhibition of BK channel activity via M2Rs counteracts this negative feedback loop and thus favors cell excitation in synergism with the M2R-Gi/o-PLCβ-IP3 induced Ca2+ release from the sarcoplasmic reticulum (Fig. 9). Functional experiments support this mechanism. First, CCh-induced contractions in tracheae from mice with targeted deletion of the M2R or the M3R demonstrate that both receptor subtypes importantly contribute to contraction (15, 16). Second, inhibition of BK channels in isolated guinea pig tracheae by charybdotoxin reversed the inhibitory effect of PTX on methacholine-induced contraction (17). Furthermore, under physiological conditions, the well known Gαi/o-dependent inhibitory effect of M2R stimulation on adenylyl cyclase activity that counteracts the β2-adrenoceptor-induced, cAMP-dependent ASM relaxation agrees well and likely contributes to the contraction-reinforcing properties of this receptor subtype.
An issue that still has to be addressed is the question of why Gβγ subunits inhibit BK channels by a dual mechanism, a direct membrane-delimited pathway, and a more elaborate signaling cascade resulting in the activation of PKC? Although a definite answer cannot be given, the answer might be as simple as that one alone is not enough to cause a physiologically relevant effect. The results in our study are pointing in that direction. On the one hand the inhibition of the PLC/PKC pathway diminishes the inhibitory effect of CCh by ∼50%. On the other hand the activation of the PLC/PKC pathway by CCh in HEK293 cells only expressing the M3R is not sufficient to cause an inhibition of steady-state IBK. Taking further into account that the direct application of Gβγ to the channel induced an ∼50% inhibition of NPo independent of the intracellular Ca2+ concentration, the membrane-delimited direct interaction might be more essential. In line with that view, in a complex formed by a specific G protein-coupled receptor (M2RorM4R), the Gi/o protein, and the BK channel (Fig. 9), such a locally delimited pathway would allow for an ultra rapid and efficient coupling, whereas the second messenger formation requiring PKC-dependent inhibition needs more time and thus might be more essential for a second, longer lasting phase of inhibition.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: ASM, airway smooth muscle; BK channel, large conductance Ca2+-activated K+ channel; BKα, pore-forming α subunit of the BK channel; TSMC, tracheal smooth muscle cell; HEK, human embryonic kidney; TD, transducin; GIRK, G protein-gated inwardly rectifying K+ channel; CCh, carbamoylcholine; NA, noradrenaline; PTX, pertussis toxin; M2R, M2 receptor; M3R, M3 receptor; M4R, M4 receptor; PLC, phospholipase C; PKC, protein kinase C; IP3, inositol trisphosphate; PMA, phorbol 12-myristate 13-acetate; PSS, physiological saline solution; AR, adrenoceptor.
M. Schmidt, personal communication.
References
- 1.Bonner, T. I., Buckley, N. J., Young, A. C., and Brann, M. R. (1987) Science 237 527-532 [DOI] [PubMed] [Google Scholar]
- 2.Bonner, T. I., Young, A. C., Brann, M. R., and Buckley, N. J. (1988) Neuron 1 403-410 [DOI] [PubMed] [Google Scholar]
- 3.Caulfield, M. P., and Birdsall, N. J. (1998) Pharmacol. Rev. 50 279-290 [PubMed] [Google Scholar]
- 4.Mak, J. C., and Barnes, P. J. (1990) Am. Rev. Respir. Dis. 141 1559-1568 [DOI] [PubMed] [Google Scholar]
- 5.Haddad, E. B., Mak, J. C. W., Hislop, A., Haworth, S. G., and Barnes, P. J. (1994) Am. J. Physiol. 266 L642-L648 [DOI] [PubMed] [Google Scholar]
- 6.Mak, J. C. W., Haddad, E.-B., Buckley, N. J., and Barnes, P. J. (1993) Life Sci. 53 1501-1508 [DOI] [PubMed] [Google Scholar]
- 7.Grandordy, B. M., Cuss, F. M., Sampson, A. S., Palmer, J. B., and Barnes, P. J. (1986) J. Pharmacol. Exp. Ther. 238 273-279 [PubMed] [Google Scholar]
- 8.Meurs, H., Roffel, A. F., Postema, J. B., Timmermans, A., Elzinga, C. R. S., Kauffman, H. F., and Zaagsma, J. (1988) Eur. J. Pharmacol. 156 271-274 [DOI] [PubMed] [Google Scholar]
- 9.Roffel, A. F., Elzinga, C. R. S., and Zaagsma, J. (1989) Pulm. Pharmacol. 3 47-51 [DOI] [PubMed] [Google Scholar]
- 10.Roffel, A. F., Meurs, H., Elzinga, C. R., and Zaagsma, J. (1990) Br. J. Pharmacol. 99 293-296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eglen, R. M., Hedge, S. S., and Watson, N. (1996) Pharmacol. Rev. 48 531-565 [PubMed] [Google Scholar]
- 12.Jones, C. A., Madison, J. M., Tom-Moy, M., and Brown, J. K. (1987) Am. J. Physiol. 253 C97-C104 [DOI] [PubMed] [Google Scholar]
- 13.Sankary, R. M., Jones, C. A., Madison, J. M., and Brown, J. K. (1988) Am. Rev. Respir. Dis. 138 145-150 [DOI] [PubMed] [Google Scholar]
- 14.Fernandes, L. B., Fryer, A. D., and Hirshman, C. A. (1992) J. Pharmacol. Exp. Ther. 262 119-126 [PubMed] [Google Scholar]
- 15.Stengel, P. W., Gomeza, J., Wess, J., and Cohen, M. L. (2000) J. Pharmacol. Exp. Ther. 292 877-885 [PubMed] [Google Scholar]
- 16.Stengel, P. W., Yamada, M., Wess, J., and Cohen, M. L. (2002) Am. J. Physiol. 282 R1443-R1449 [DOI] [PubMed] [Google Scholar]
- 17.Kume, H., Mikawa, K., Takagi, K., and Kotlikoff, M. I. (1995) Am. J. Physiol. 268 L221-L229 [DOI] [PubMed] [Google Scholar]
- 18.Nelson, M. T., Cheng, H., Rubart, M., Santana, L. F., Bonev, A. D., Knot, H. J., and Lederer, W. J. (1995) Science 270 633-637 [DOI] [PubMed] [Google Scholar]
- 19.Jaggar, J. H., Porter, V. A., Lederer, W. J., and Nelson, M. T. (2000) Am. J. Physiol. 278 C235-C256 [DOI] [PubMed] [Google Scholar]
- 20.Wieland, T., Ulibarri, I., Gierschick, P., and Jakobs, K. H. (1991) Eur. J. Biochem. 196 707-716 [DOI] [PubMed] [Google Scholar]
- 21.Sandmann, J., Peralta, E. G., and Wurtman, R. J. (1991) J. Biol. Chem. 266 6031-6034 [PubMed] [Google Scholar]
- 22.Bünemann, M., Bucheler, M. M., Philipp, M., Lohse, M. J., and Hein, L. (2001) J. Biol. Chem. 276 47512-47517 [DOI] [PubMed] [Google Scholar]
- 23.Zhou, X.-B., Arntz, C., Kamm, S., Motejlek, K., Sausbier, U., Wang, G.-X., Ruth, P., and Korth, M. (2001) J. Biol. Chem. 276 43239-43245 [DOI] [PubMed] [Google Scholar]
- 24.Hamill, O. P., Marty, A., Neher, E., Sakmann, B., and Sigworth, F. J. (1981) Pflügers Arch. Eur. J. Physiol. 391 85-100 [DOI] [PubMed] [Google Scholar]
- 25.Zhou, X.-B., Ruth, P., Schlossmann, J., Hofmann, F., and Korth, M. (1996) J. Biol. Chem. 271 19760-19767 [DOI] [PubMed] [Google Scholar]
- 26.Steffens, F., Zhou, X.-B., Sausbier, U., Sailer, C., Motejlek, K., Ruth, P., Olcese, J., Korth, M., and Wieland, T. (2003) Mol. Endocrinol. 17 2103-2115 [DOI] [PubMed] [Google Scholar]
- 27.Kume, H., and Kotlikoff, M. I. (1991) Am. J. Physiol. 261 C1204-C1209 [DOI] [PubMed] [Google Scholar]
- 28.Kume, H., Graziano, M. P., and Kotlikoff, M. I. (1992) Proc. Natl. Acad. Sci. U. S. A. 89 11051-11055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Federmann, A. D., Conklin, B. R., Schrader, K. A., Reed, R. R., and Bourne, H. R. (1992) Nature 356 159-161 [DOI] [PubMed] [Google Scholar]
- 30.Zhou, X.-B., Wang, G.-X., Hüneke, B., Wieland, T., and Korth, M. (2000) J. Physiol. 524 339-352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou, X.-B., Lutz, S., Steffens, F., Korth, M., and Wieland, T. (2007) Mol. Endocrinol. 21 740-752 [DOI] [PubMed] [Google Scholar]
- 32.Offermanns, S., Wieland, T., Homann, D., Sandmann, J., Bombien, E., Spicher, K., Schultz, G., and Jakobs, K. H. (1994) Mol. Pharmacol. 45 890-898 [PubMed] [Google Scholar]
- 33.Logothetis, D. E., Kurachi, Y., Galper, J., Neer, E. J., and Clapham, D. E. (1987) Nature 325 321-326 [DOI] [PubMed] [Google Scholar]
- 34.De Waard, M., Liu, H., Walker, D., Scott, V. E. S., Gurnett, C. A., and Campbell, K. P. (1997) Nature 385 446-450 [DOI] [PubMed] [Google Scholar]
- 35.Camps, M., Carozzi, A., Schnabel, P., Scheer, A., Parker, P. J., and Gierschik, P. (1992) Nature 360 684-686 [DOI] [PubMed] [Google Scholar]
- 36.Katz, A., Wu, D., and Simon, M. I. (1992) Nature 360 686-689 [DOI] [PubMed] [Google Scholar]
- 37.Ashkenazi, A., Winslow, J. W., Peralta, E. G., Peterson, G. L., Schimerlik, M. I., Capon, D. J., and Ramachandran, J. (1987) Science 238 672-675 [DOI] [PubMed] [Google Scholar]
- 38.Fukao, M., Mason, H. S., Britton, F. C., Kenyon, J. L., Horowitz, B., and Keef, K. D. (1999) J. Biol. Chem. 274 10927-10935 [DOI] [PubMed] [Google Scholar]
- 39.Tian, L., Coghill, L. S., McClafferty, H., MacDonald, S. H.-F., Antoni, F. A., Ruth, P., Knaus, H.-G., and Shipston, M. J. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 11897-11902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holbourn, K. P., Shone, C. C., and Acharya, K. R. (2006) FEBS J. 273 4579-4593 [DOI] [PubMed] [Google Scholar]
- 41.McManus, O. B., Helms, L. M. H., Pallanck, L., Ganetzky, B., Swanson, R., and Leonard, R. J. (1995) Neuron 14 645-650 [DOI] [PubMed] [Google Scholar]
- 42.Zamponi, G. W., Bourinet, E., Nelson, D., Nargeot, J., and Snutch, T. P. (1997) Nature 385 442-446 [DOI] [PubMed] [Google Scholar]
- 43.Voigt, N., Friedrich, A., Bock, M., Wettwer, E., Christ, T., Knaut, M., Strasser, R. H., Ravens, U., and Dobrev, D. (2007) Cardiovasc. Res. 74 426-437 [DOI] [PubMed] [Google Scholar]
- 44.Schubert, R., Noack, T., and Serebrayakov, V. N. (1999) Am. J. Physiol. 276 C648-C658 [DOI] [PubMed] [Google Scholar]
- 45.Taguchi, K., Kaneko, K., and Kubo, T. (2000) Biol. Pharm. Bull. 23 1450-1454 [DOI] [PubMed] [Google Scholar]
- 46.Barman, S. A., Zhu, S., and White, R. E. (2004) Am. J. Physiol. 286 L149-L155 [DOI] [PubMed] [Google Scholar]
- 47.Shipston, M. J., and Armstrong, D. L. (1996) J. Physiol. 493 665-672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schubert, R., and Nelson, M. T. (2001) Trends Pharmacol. Sci. 22 505-512 [DOI] [PubMed] [Google Scholar]
- 49.Toro, L., Wallner, M., Meera, P., and Tanaka, Y. (1998) News Physiol. Sci. 13 112-117 [DOI] [PubMed] [Google Scholar]
- 50.Mackay, H. J., and Twelves, C. J. (2007) Nat. Rev. Cancer 7 554-562 [DOI] [PubMed] [Google Scholar]
- 51.Martiny-Baron, G., Kazanietz, M. G., Mischak, H., Blumberg, P. M., Kochs, G., Hug, H., Marmé, D., and Schächtele, C. (1993) J. Biol. Chem. 268 9194-9197 [PubMed] [Google Scholar]
- 52.Dell'Acqua, M. L., Caroll, R. C., and Peralta, E. G. (1993) J. Biol. Chem. 268 5676-5685 [PubMed] [Google Scholar]
- 53.Liu, G., Shi, J., Yang, L., Cao, L., Park, S. M., Cui, J., and Marx, S. O. (2004) EMBO J. 23 2196-2205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Berkefeld, H., Sailer, C. A., Bildl, W., Rohde, V., Thumfart, J.-O., Eble, S., Klugbauer, N., Reisinger, E., Bischofberger, J., Oliver, D., Knaus, H.-G., Schulte, U., and Fakler, B. (2006) Science 314 615-620 [DOI] [PubMed] [Google Scholar]
- 55.Salkoff, L., Butler, A., Ferreira, G., Santi, C., and Wei, A. (2006) Nat. Neurosci. 5 921-931 [DOI] [PubMed] [Google Scholar]