

Abstract
Terpene synthases are responsible for the large diversity of terpene carbon skeletons found in plants. The unique, carbocationic reaction mechanism of these enzymes can form multiple products from a single prenyl diphosphate substrate. Two maize genes were isolated that encode very similar sesquiterpene synthases, TPS6 and TPS11, which both produce β-bisabolene, a common monocyclic sesquiterpene, and β-macrocarpene, an uncommon bicyclic olefin. Investigation of the reaction mechanism showed that the formation of β-macrocarpene proceeds via a neutral β-bisabolene intermediate and requires reprotonation by a proton that may ultimately be abstracted from water. This reprotonation is dependent on the pH and the presence of a Mg2+ cofactor. Mutational analysis of the enzyme demonstrated that a highly conserved tyrosine residue in the active center of the enzymes is important for the protonation process. TPS6 and TPS11 are transcribed both in leaves and roots of maize, but the respective terpene products were only detected in roots. The expression in roots was up-regulated by herbivore damage to the leaves, suggesting a long distance signal transduction cascade between leaves and roots.
Terpenes form the largest group of plant secondary metabolites and are known to have a plethora of different functions both within the plant and in communication with other organisms. Most of the structural diversity among terpenes can be attributed to the terpene synthases, a large enzyme class that catalyzes the conversion of the ubiquitous prenyldiphosphates geranyldiphosphate (GPP),2 farnesyldiphosphate (FPP), or geranylgeranyldiphosphate (GGPP) into a large number of basic terpene skeletons (1). A unique feature of terpene synthases is their capability to produce multiple products with different carbon skeletons from a single prenyldiphosphate substrate (2, 3). For example, δ-selinene synthase and γ-humulene synthase from Abies grandis synthesize 34 and 52 different sesquiterpenes from their farnesyl diphosphate substrate, respectively (4). This unusual behavior is due to an electrophilic reaction mechanism common to all terpene synthases, which is initiated by elimination of the allylic diphosphate from the prenyl diphosphate substrate. The resulting highly reactive carbocationic intermediate undergoes a series of cyclizations, hydride shifts, and other rearrangements until the reaction is terminated by proton loss or the addition of a nucleophile (5).
In addition to highly active, carbocationic intermediates, sesquiterpene synthases like 5-epi-aristolochene synthase (TEAS) from tobacco were also shown to produce stable, enzyme-bound intermediates (6, 7). The structural elucidation of this sesquiterpene synthase provided insight into the reaction mechanism and structure-function relationships in the active center (8). The reaction starts with the dephosphorylation and ionization of the (E,E)-FPP substrate to form a farnesyl cation, which undergoes a 1,10-cyclization to form a stable, enzyme-bound germacrene A intermediate (3). Based on the crystal structure of TEAS, a catalytic triad consisting of a central tyrosine residue (Tyr520) and two aspartate residues (Asp444 and Asp525) was proposed to be involved in the protonation of the germacrene A intermediate (8). In this model, a proton, which is abstracted from carbon atom 13 of the germacrenyl cation to form the neutral intermediate germacrene A, is transferred via the carboxyl group of aspartate 525 and hydroxyl group of tyrosine 520 back to a different position (C6) on the same carbon skeleton to form a second germacrenyl cation (8). An electrophilic attack on C2 results in a second cyclization and a Wagner-Meerwein rearrangement of a methyl group to form the eudesmane-type skeleton. Mutagenesis of tyrosine 520 to phenylalanine resulted in an enzyme that released only germacrene A (9). A similar protonation of an intermediate was postulated for the 5-epi-aristolochene synthase from Capsicum annuum (10), a valencene synthase from Citrus sinensis (11), and a β-selinene synthase from Ocimum basilicum (12), but this has not been experimentally demonstrated.
Terpene production is not only dependent on the path of the terpene synthase reaction but also on the expression and evolutionary history of these enzymes within the plant. To investigate all of these aspects of terpene production, we have been isolating and characterizing a family of terpene synthases in maize, a genetically tractable organism. Maize produces complex, tissue-specific blends of terpenes that have multiple roles in defense against herbivore enemies both above and below ground (13–15). Here, we describe the very similar sesquiterpene synthases TPS6 and TPS11, which both produce β-bisabolene and β-macrocarpene. The formation of the unusual bicyclic sesquiterpene β-macrocarpene proceeds via a neutral β-bisabolene intermediate. The reprotonation requires a proton that may be ultimately abstracted from water and is transferred directly or indirectly via another proton donor to the substrate.
EXPERIMENTAL PROCEDURES
Plant and Insect Material—Plants of the maize (Zea mays L.) variety B73 (KWS seeds, Einbeck, Germany) were grown in commercially available potting soil in a climate-controlled chamber with a 16-h photoperiod, 1 mmol (m2)–1 s–1 of photosynthetically active radiation, a temperature cycle of 22 °C/18 °C (day/night), and 65% relative humidity. 12–15-day-old plants (20–30 cm high, 4–5 expanded leaves) were used in all experiments. Eggs of Spodoptera littoralis Boisd. (Lepidoptera: Noctuidae) were obtained from Aventis (Frankfurt, Germany) and were reared on an artificial wheat germ diet (Heliothis mix; Stonefly Industries, Bryan, TX) for about 10–15 days at 22 °C under an illumination of 750 μmol (m2)–1 s–1. For the S. littoralis treatments, three third instar larvae were enclosed on the middle portion of each plant in a cage made out of two halves of a Petri dish (9-cm diameter) with a circle cut out of each side and covered with gauze to allow for ventilation (16).
Isolation of the Maize Terpene Synthase cDNAs—Sequences with high similarity to plant terpene synthases were identified in BLAST searches of the Plant Genome Database PlantGDB (available on the World Wide Web). Two of these expressed sequence tags showing a high sequence similarity to each other were cloned, sequenced, and extended toward the 5′-end by the Marathon RACE procedure (Clontech) with a cDNA library from herbivore-induced leaves of the maize cultivar B73. The resulting sequences were amplified with the primers H8fwd (TATGGCTGCCCCAACACTAACTACAGA) and H10rev (GATCCTTCACATGAGTAC CGGCTTCACATAAAG) from a cDNA and introduced into the sequencing vector pCR®4-TOPO® (Invitrogen). Sequence alignments were performed with the DNASTAR suite of programs (DNASTAR, Madison, WI). The cDNA sequences contained open reading frames of 1647 bp and were deposited in GenBank™ with the accession numbers AY518315 (tps6-B73) and EU716166 (tps11-B73).
cDNA RACE Library Construction—10-day-old maize plants of the cultivar Delprim were subjected to herbivory by S. littoralis for 4 h. 1 g of leaf material was ground in a mortar to a fine powder in liquid nitrogen and added to 10 ml of Trizol reagent (Invitrogen). The mixture was treated with a Polytron homogenizer (Kinematika AG, Switzerland) for 1 min and incubated for 3 min on ice. Total RNA was isolated according to the manufacturer's instructions. From about 80 μg of total RNA, the mRNA was isolated utilizing poly(T)-coated ferromagnetic beads (Dynal, Sweden). The mRNA was transcribed into cDNA while constructing a Marathon RACE library according to the manufacturer's instructions (Clontech).
Mapping of Terpene Synthase Genes—The method of Burr and Burr (17) was utilized to map the genes tps6 and tps11 in a population of 48 CM37xT232 and 41 Tx303xCO159 recombinant inbred lines kindly provided by R. Burr. Genomic Southern blots from all individuals of the population were probed with a 1200-bp fragment of tps6-B73 that cross-reacted with tps11 (for a detailed description of the probe, see the Northern blotting procedure below).
Protein Overexpression and Enzyme Assay—For expression in Eschericha coli, the open reading frames (ORFs) of tps6 and tps11 were amplified with the primers H24fwd (ATGGTAACCTGCA TTAGCGCATGGCTGCCCCAACACTAACTA) and H25rev (ATGGTAACCTGCATTATATCACATGAGTACCGGCTTCACATAAAG) and cloned as BspMI fragments into the expression vector pASK-IBA7 (IBA GmbH, Göttingen, Germany). The constructs were introduced into the E. coli strain TOP10 (Invitrogen) and fully sequenced to avoid errors introduced by DNA amplification. Liquid cultures of the bacteria harboring the expression constructs were grown at 37 °C to an A600 of 0.6. Expression was induced by the addition of anhydrotetracycline (IBA GmbH) to a final concentration of 200 μg/liter. After a 20-h incubation at 18 °C, the cells were collected by centrifugation and disrupted by a 4 × 30-s treatment with a sonicator (Bandelin UW2070, Berlin, Germany) in chilled extraction buffer (50 mm Mops, pH 7.0, with 5 mm MgCl2, 5 mm sodium ascorbate, 0.5 mm phenylmethylsulfonyl fluoride, 5 mm dithiothreitol, and 10% (v/v) glycerol). The cell fragments were removed by centrifugation at 14,000 × g, and the supernatant was desalted into assay buffer (10 mm Mops, pH 7.0, 1 mm dithiothreitol, 10% (v/v) glycerol) by passage through a Econopac 10DG column (Bio-Rad).
To determine the catalytic activity of the recombinant proteins, enzyme assays containing 20 μl of the bacterial extract and 80 μl of assay buffer with 10 μm substrate (either GPP, (E,E)-FPP, (Z,E)-FPP, or GGPP), 10 mm MgCl2, 0.2 mm NaWO4, and 0.1 mm NaF in a Teflon-sealed, screw-capped 1-ml GC glass vial were performed. A SPME (solid phase microextraction) fiber consisting of 100 μm polydimethylsiloxane (SUPELCO, Belafonte, PA) was placed into the headspace of the vial for 30 min of incubation at 30 °C. For analysis of the adsorbed reaction products, the SPME fiber was directly inserted into the injector of the gas chromatograph.
For product identification by NMR, four assays each containing 300 μl of bacterial extract, 218 μg of (E,E)-FPP, 10 mm MgCl2, 0.2 mm NaWO4, and 0.1 mm NaF in a total volume of 600 μl of assay buffer were overlaid with 500 μl of n-pentane and incubated for 5 h at 25°C. The reaction products were extracted three times by mixing with each 500 μl of n-pentane. The n-pentane phases were merged and passed through a silica column to remove traces of farnesol and other oxygenated extraction products.
For the determination of substrate Km values, an assay containing 10 μm [1-3H]geranyl or [1-3H](E,E)-farnesyl diphosphate (37 GBq mol–1; American Radiolabeled Chemicals, St. Louis, MO), 10 mm MgCl2, and 0.05 mm MnCl2 in 100 μl of assay buffer was used. The assay was overlaid with 1 ml of n-pentane to trap volatile products and incubated for 15 min at 30 °C. The reaction was stopped by mixing, and 0.5 ml of the n-pentane layer was taken for measurement of radioactivity by liquid scintillation counting in 2 ml of Lipoluma mixture (Packard Bioscience, Groningen, The Netherlands) using a Packard Tricarb 2300TR liquid scintillation counter (3H efficiency = 61%). The Km values for the cofactors Mg2+ and Mn2+ and the influence of different metal ion cofactors on enzyme activity were measured with 10 μm (E,E)-FPP. The Km values were determined using seven substrate concentrations with four repetitions each. Assay results are reported as the mean of three independent replicate assays, and each experiment was repeated 2–3 times with similar results. The enzyme activity was linear for at least 30 min and stable for at least 1 year when stored at –80 °C.
To analyze the protonation reaction, 100-μl aliquots of E. coli extract containing recombinant TPS6 were desalted in assay buffer, lyophilized, and dissolved in 100 μl of D2O (Merck) or 100 μl of H2O, respectively. The freshly dissolved protein samples were immediately analyzed in enzyme assays containing 100 μl of extract, 10 μm (E,E)-FPP, and 10 mm MgCl2. The assays were incubated for 15 min at 30 °C. The reaction products were collected by SPME and analyzed by GC-MS.
Gas Chromatography—A Hewlett-Packard model 6890 gas chromatograph was employed with the carrier gas helium at 1 ml min–1, splitless injection (injector temperature 220 °C, injection volume 1 μl), a DB-WAX column (polyethylene glycol, 30 m × 0.25 mm inner diameter × 0.25 μm film thickness; J & W, Folsom, CA) or a Chrompack CP-SIL-5 CB-MS column ((5% phenyl)-methylpolysiloxane, 25 m × 0.25 mm inner diameter × 0.25 μm film thickness; Varian), and a temperature program from 40 °C (3-min hold) at 5 °C min–1 to 240 °C (3-min hold). The coupled mass spectrometer was a Hewlett-Packard model 5973 with a quadrupole mass selective detector, transfer line temperature 230 °C, source temperature 230 °C, quadrupole temperature 150 °C, ionization potential 70 eV, and a scan range of 40–350 atomic mass units. Products were identified by comparison of retention times and mass spectra with authentic reference compounds as described by Köllner et al. (18). Quantification was performed with the trace of a flame ionization detector operated at 250 °C. A nonyl acetate internal standard was utilized to determine the average and S.E. of 3–6 independent samples.
The enantiomers of β-bisabolene were separated and identified by GC-MS using a heptakis (2,3-di-O-methyl-6-O-t-butyldimethylsilyl)-β-cyclodextrin (35% in OV1701, w/w) column (30 m × 0.25 mm × 0.125-μm film; BGB Analytik, Adliswil, Switzerland) operated with helium (2 ml min–1) as carrier gas, splitless injection (220 °C, 2-μl volume), and a column temperature of 115 °C. A racemic mixture of (S)- and (R)-β-bisabolene was prepared by dehydration of racemic α-bisabolol at 140 °C with acidic aluminum oxide as catalyst (19). The β-bisabolene enantiomers were identified by comparison with the bergamot (Citrus bergamia) essential oil, which contains only the (S)-enantiomer. The β-macrocarpene was kindly provided as an authentic standard by L. Cool (20).
The enantiomers of β-macrocarpene were separated and identified by GC-MS performed with a Hewlett Packard HP6890 gas chromatograph interfaced to a MasSpec 2 magnetic sector mass spectrometer (Micromass, Manchester, UK) using a heptakis (2,3-di-O-methyl-6-O-t-butyldimethylsilyl)-β-cyclodextrin (35% in OV1701, w/w) column (30 m × 0.25 mm × 0.125-μm film; BGB Analytik). Helium was used as a carrier gas (constant flow 1 ml min–1), and samples were injected split (1 ml) at 200 °C. Compounds were eluted with a temperature program: 40 °C for 2 min, heated at 3 °C/min to 200 °C with 5-min hold. Electron ionization-MS data were recorded in positive ion mode using 70 eV ionization energy.
Nuclear Magnetic Resonance Spectroscopy—1H NMR, 1H,1H COSY, HMBC, and HMQC spectra were measured at 300 K on a Bruker Avance 500 NMR spectrometer (Bruker Biospin, Rheinstetten, Germany) using a cryogenically cooled 5-mm TXI 1H{13C} probe. The operating frequency was 500.13 MHz for acquisition of 1H NMR and 125.75 MHz for 13C NMR spectra. Benzene-d6 was used as a solvent, and tetramethyl siloxane was used as an internal standard.
Analytical Data—(–)-β-Macrocarpene: 1H NMR (500 MHz, benzene-d6): δ 5.46 (1H, br m, H-5), 5.42 (1H, br m, H-12), 2.07 (2H, m, H-6), 2.04 (1H, m, H-1), 2.03 (2H, m, H-11), 1.97 (1H, m, H-3a), 1.88 (1H, m, H-3b), 1.73 (1H, m, H-2a), 1.69 (2H, m, H-8), 1.65 (3H, br s, H-15), 1.48 (1H, m, H-2b), 1.29 (2H, t, J = 6.8 Hz, H-10), 0.919 (3H, s, H-14), 0.915 (3H, s, H-13). 13C NMR (125 MHz, benzene-d6, data obtained from HSQC and HMBC spectra): δ 141.3 (C-7), 133.5 (C-4), 121.6 (C-5), 118.3 (C-12), 41.7 (C-1), 40.9 (C-8), 35.7 (C-10), 31.1 (C-6), 31.0 (C-3), 29.2 (C-9), 28.5 (C-14), 28.3 (C-13), 28.2 (C-2), 23.8 (C-15), 23.6 (C-11). Electron ionization-MS m/z (relative intensity): 204 (47) [M]· +189 (27) [M-CH3]+, 175 (10), 162 (9) [M-C3H6]·+, 148 (16) [M-C4H8]·+, 136 (83) [M-C5H10] +, 121 (67), 107 (53), 105 (20), 95 (25), 94 (34), 93 (100), 92 (37), 81 (16), 80 (53), 79 (49), 77 (16), 69 (14), 67 (18), 65 (9), 55 (18), 53 (13). The numbering of the β-macrocarpene carbon atoms refers to that described previously (20).
Site-directed Mutagenesis—For site-directed mutagenesis, the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) was used according to the manufacturer's instructions. The PCR-based mutagenesis protocol was performed with the tps11-B73 cDNA cloned into the expression vector pASK-IBA7 and primers containing the desired mutations (Y522F-fwd, CTGTAGACTACATGTTCAAGGAAGCTGATAAG; Y522F-rev, CTTATCAGCTTCCTTGAACATGTAGTCTACAG; D526N-fwd, CATGTACAAGGAAGCTAATAAGTACACTGTCTC; D526N-rev, GAGACAGTGTACTTATTAGCTTCCTTGTACATG). The mutagenized constructs were fully sequenced.
RNA Hybridization—Plant RNA was prepared with the RNeasy plant minikit (Qiagen, Hilden, Germany) according the manufacturer's instructions. A 1200-bp fragment was used as a probe, generated by linear PCR with the primer 5′-CACCAGTGTACCTCCAATGCTTATGAGT-3′ and the complete ORF as a template. The probe was labeled with [32P]adenosine triphosphate using the Strip-EZ PCR procedure (Ambion, TX). Blotting on a Nytran-Plus nylon membrane (Schleicher & Schuell), hybridization, and washing were carried out following standard procedures. The blots were exposed to BioMax MS1 film (Eastman Kodak Co.) with an intensifying screen.
Terpene Extraction—The plant material was harvested, frozen in liquid nitrogen, and ground to a fine powder in a mortar. 3 g of tissue were extracted with 10 ml of n-pentane for 1 h at room temperature with constant rotation. After centrifugation at 1800 × g for 5 min to sediment the tissue, 800 ng of nonyl acetate were added to the n-pentane supernatant as an internal standard, and the solution was cleared with 25 mg of activated charcoal. The n-pentane was concentrated under a stream of nitrogen to a volume of 200 μl and stored at –20 °C overnight to remove waxes and other high molecular weight lipids by precipitation. The n-pentane phase containing the terpenes was analyzed by GC-MS and GC-flame ionization detection.
RESULTS
Isolation of the Maize Terpene Synthase Genes tps6 and tps11—The identification and biochemical characterization of terpene synthases in maize is crucial to our understanding of terpene-based plant defense reactions in this species. We have already characterized tps1, tps4, tps5, tps10, and tps23 (14, 15, 18, 21). To identify additional terpene synthases, we screened the public maize sequence data base PlantGDB for sequences with similarity to these genes. Two of the identified sequences were extended by RACE PCR to obtain the complete ORFs. The resulting ORFs (1,647 nucleotides each) were amplified from cDNA of the maize inbred line B73 and designated tps6-B73 and tps11-B73. The genes have 96% nucleotide identity to each other (Fig. 1). The gene tps6-B73 also shares 99% nucleotide identity with umi2, a gene with unknown function previously isolated from the maize variety Early Golden Bantam (22). Most likely, tps6-B73 and umi2 are alleles of each other. The deduced amino acid sequences of TPS6 and TPS11 contain the highly conserved terpene synthase sequence elements DDXXD (residues 302–306) and RXR (residues 265–267) (Fig. 1). No signal peptide was apparent in the N-terminal region of the proteins, indicating that both enzymes are located in the cytoplasm and likely to be sesquiterpene synthases (1). Mapping of tps6 and tps11 within a population of T232xCM37 and CO159xTx303 inbred lines (17) displayed only one segregating band in all individuals, indicating that tps6 and tps11 are in close proximity to each other. The genes were mapped to the short arm of chromosome 10 near the marker npi105A (Fig. 2A). No recombination was observed between both genes and another pair of terpene synthases, tps4 and tps5 (98% nucleotide identity to each other), which map to the same region (18). Since there is about 58% sequence identity among the pairs (tps6 and tps11 versus tps4 and tps5) (Fig. 2B), there may have been at least two duplication events in this region of the chromosome.
FIGURE 1.

Amino acid sequence comparison of the maize terpene synthases TPS6 and TPS11 with TEAS from N. tabacum. Amino acids identical in all three genes are marked by black boxes. The highly conserved metal cofactor binding region is labeled DDXXD. Amino acids involved in catalysis are marked with arrowheads.
FIGURE 2.

The terpene synthases tps6 and tps11 are located in close proximity to tps4 and tps5 on chromosome 10. A, mapping with recombinant inbred lines shows one locus for tps6 and tps11 as well as tps4 and tps5 on maize chromosome 10. Likelihood of the odds (LOD) scores of the closest marker loci are shown. 10S and 10L indicate the short and long arm of chromosome 10, respectively. B, the dendrogram represents the amino acid sequence relationships of seven terpene synthases from maize. Sequence alignment was performed by the ClustalW method with default settings in the program module MegAlign of the DNASTAR package. Bootstrap values (n = 1000) are indicated as percentages. The accession numbers of the sequences are AAO18435 (TPS1), AAS88571 (TPS4), AAS88572 (TPS5), AAX99146 (TPS10), and EU259634 (TPS23).
TPS6 and TPS11 Both Produce the Bicyclic Sesquiterpene (–)-β-Macrocarpene—To determine the catalytic activity of the putative terpene synthases TPS6 and TPS11, we expressed both ORFs in E. coli. The partial purified recombinant enzymes were incubated with the potential substrates GPP, FPP, and GGPP. Although no enzymatic activity was observed in the presence of GGPP, both proteins were able to convert GPP and FPP into monoterpenes and sesquiterpenes, respectively (Fig. 3). In the presence of FPP, both TPS6 and TPS11 produced an uncommon sesquiterpene hydrocarbon as the major product along with the minor products, β-bisabolene and (E)-β-farnesene (Fig. 3A). Spectra of the major TSP6 product showed a well pronounced molecular [M]· +ion at m/z 204 with two unusual neutral losses of 56 Da (C4H8) and 68 Da (C5H8) formed by retro-Diels-Alder reactions. Catalytic hydrogenation using 10% platinum on carbon for 1 h formed a mixture of two monoenes, and extended hydrogenation for 6 h resulted in one saturated major product ([M]· +at m/z 208). The necessity of extended hydrogenation indicated two triply or quadruply substituted C=C double bonds in the TPS6 product. To fully identify this compound, a large scale enzyme assay was performed, and ∼1 mg of the reaction product was analyzed by NMR. The 1H NMR spectrum displayed characteristic signals of two geminal methyl groups (δ 0.915 and 0.919), a broad singlet (δ 1.65) of a methyl group on a double bond, and broad singlets attributable to the two olefinic protons (δ 5.46 and 5.42). Starting from these signals, 1H,1H-homocorrelation (1H,1H COSY) and 1H,13C-heterocorrelation NMR experiments (HSQC, HMBC) were used to assign the structure of the bicyclic sesquiterpene as 4′,5,5-trimethyl-1,1′-bi(cyclohexane)-1,3′-diene. A compound of this structure, named (S)-β-macrocarpene, was recently reported from Cupressus macrocarpa (20). The 1H NMR data of an authentic sample of this compound matched those of our sample isolated from maize. Enantiomeric analysis of the β-macrocarpene product (Fig. 4) and the β-bisabolene product (Fig. 5) revealed that the (S)-enantiomer was almost exclusively present in both cases. In the presence of GPP, both TPS6 and TPS11 catalyzed the formation of the acyclic monoterpenes, β-myrcene and linalool, along with minor amounts of the cyclic compounds limonene, α-thujene, sabinene, and α-terpinolene (Fig. 3, B and C).
FIGURE 3.

Terpene products of the terpene synthases TPS6 and TPS11 measured in vitro. The enzymes were expressed in E. coli, extracted, and incubated with the substrates GPP and FPP. The resulting terpene products were identified by gas chromatography coupled to mass spectrometry. A, sesquiterpene products of TPS6 and TPS11. B, monoterpene products of TPS6 and TPS11. C, the products were identified as (E)-β-farnesene (1), β-macrocarpene (2); β-bisabolene (3), β-myrcene (4), limonene (5), β-phellandrene (6), α-terpinolene (7), and linalool (8) by comparison of their retention times and mass spectra with those of authentic standards.
FIGURE 4.

Stereochemical analysis of the β-macrocarpene produced by TPS6. The separation and identification of the (S)- and (R)-enantiomers of β-macrocarpene was performed by gas chromatography on a chiral column with authentic standards as described under “Experimental Procedures.” A, the traces of the MS detector are shown for the product of TPS6 (dotted line) and a β-macrocarpene standard (solid line). B, mass spectra of the authentic 1′R and 1′S β-macrocarpene standards and the TPS6 enzyme product.
FIGURE 5.

Stereochemical analysis of the β-bisabolene product of TPS6. The separation and identification of the (S)- and (R)-enantiomers of β-bisabolene were performed by gas chromatography on a chiral column with authentic standards as described under “Experimental Procedures.” The upper trace is that of the TPS6 products, whereas the lower trace depicts the authentic standards.
The Catalytic Activity of TPS6 Is Strongly Affected by Metal Cofactors and pH—To determine the metal cofactor requirement of the terpene synthases, we measured the enzyme activity in the presence of different divalent metal ions at two different concentrations. The activity of TPS6 required a divalent metal ion, with Mg2+ and Mn2+ being the only effective species of those tested. Optimum activity was obtained with 5 mm Mg2+ and 5 mm Mn2+ (Fig. 6A). Although the Km for Mn2+ was significantly lower than for Mg2+ (Table 1), the enzyme is more likely to operate with a Mg2+ cofactor in planta, because the concentration of Mg2+ in plant cells is about 2 orders of magnitudes higher than Mn2+ (23). The nature of the divalent metal cofactor has often been shown to influence the product specificity of terpene synthases (18, 24, 25). In the presence of Mn2+, the product spectrum of TPS6 was shifted toward an increased production of (S)-β bisabolene and a decreased production of (S)-β-macrocarpene (Fig. 6B).
FIGURE 6.
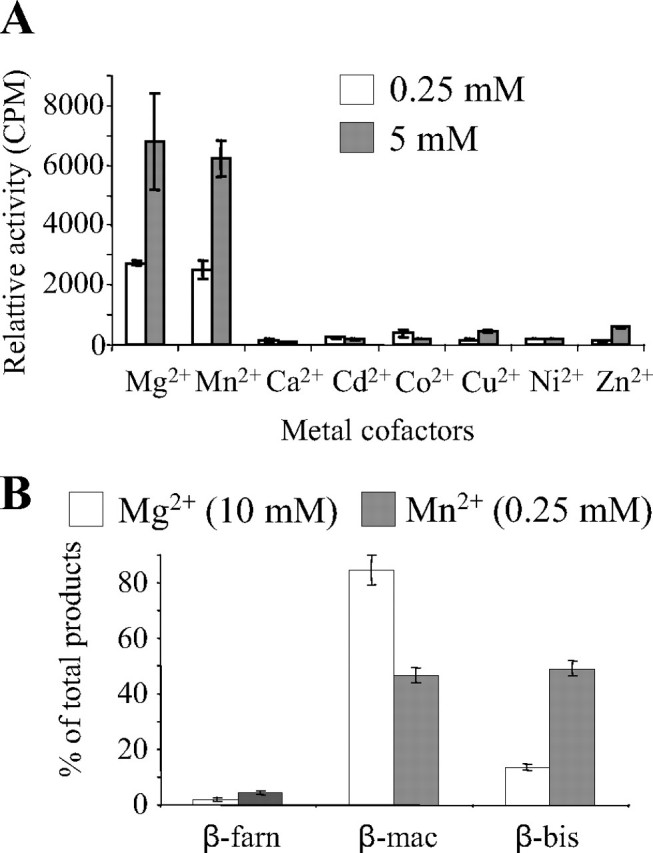
Divalent metal ion cofactors affect the velocity and product specificity of TPS6. A, the catalytic activity of TPS6 after heterologous expression in E. coli was measured in the presence of various divalent metal ions at 0.25 or 5.0 mm. Means and S.E. of triplicate assays are shown. B, the relative proportions of the major reaction products were determined in the presence of 10 mm Mg2+ or 0.25 mm Mn2+. The compounds are designated as follows. β-farn, (E)-β-farnesene; β-mac, β-macrocarpene; β-bis, β-bisabolene. Means and S.E. of triplicate assays are shown.
TABLE 1.
Kinetic constants for TPS6-B73 heterologously expressed in E. coli
|
Km |
Vmax relative |
||||
|---|---|---|---|---|---|
| FPPa | GPPa | Mg2+b | Mn2+b | FPP/GPP | Mg2+/Mn2+ |
| 2.1 μm | 1.1 μm | 130.7 μm | 23.4 μm | 100:14 | 100:100 |
Values for FPP and GPP were measured in the presence of 10 mm Mg2+ and 0.05 mm Mn2+.
Values for Mg2+ and Mn2+ were measured with 10 μm FPP.
TPS6 was active in a broad pH range with an optimum at pH 7.0 and half-maxima at pH 6.2 and pH 8.6 in the presence of 5 mm Mg2+ (Fig. 7). Within a pH range from 5.0 to 8.0, the major product was (S)-β-macrocarpene, but higher pH values favored the formation of (S)-β-bisabolene. Maximum production of (S)-β-macrocarpene and (S)-β-bisabolene was observed at pH 7.0 and pH 8.5, respectively (Fig. 7). TPS6 accepts the monoterpene precursor GPP with a Km similar to that of FPP, but the monoterpene products were produced at a lower velocity (Table 1).
FIGURE 7.

Enzymatic activity and product specificity of TPS6 is affected by pH. The catalytic activity of TPS6 after heterologous expression in E. coli was determined at different pH values of the assay buffer. Means and S.E. of triplicate assays are shown.
TPS6 and TPS11 Catalyze a Unique Protonation of a Stable β-Bisabolene Intermediate—Based on the mechanism of TEAS (7, 8), a reaction mechanism for (S)-β-macrocarpene formation has been proposed in which a neutral (S)-β-bisabolene intermediate is reprotonated to initiate the second cyclization (20). Reprotonation of the neutral germacrene A intermediate in TEAS is dependent on a catalytic triad of three amino acids, tyrosine 520, aspartate 444, and aspartate 525 (8, 9). Two of these amino acids are conserved in TPS6 and TPS11, corresponding to tyrosine 522 and aspartate 526, respectively.
To test whether these residues are involved in the reprotonation of the (S)-β-bisabolene intermediate, we selectively mutated these amino acids in TPS11 to phenylalanine and asparagine, respectively, and tested the expressed mutant enzymes for activity. The alteration of tyrosine to phenylalanine at position 522 reduced the production of (S)-β-macrocarpene to trace amounts (Fig. 8). The enzyme formed (S)-β-bisabolene almost exclusively, indicating a crucial role for tyrosine 522 in the protonation reaction. The overall activity of the mutated enzyme is dramatically reduced, which suggests an additional function of tyrosine 522 in the early reaction steps leading to the formation of (S)-β-bisabolene. This reduction of enzyme activity was alleviated in the presence of the (Z,E)-isomer of farnesyl diphosphate, indicating that tyrosine 522 is involved in the initial isomerization of the (E,E)-nerolidyl cation to its (Z,E)-isomer. A similar role for the corresponding tyrosine residue in the isomerization step was also observed for the maize terpene synthase TPS4 (26). The alteration of aspartate 526 to asparagine inactivated the enzyme completely, suggesting that this amino acid is necessary for the first steps of the reaction mechanism or affects the overall structure of the protein.
FIGURE 8.

Single amino acid changes can alter TPS11 product formation. Two amino acid alterations, Y522F and D526N, were introduced into the active site of TPS11. The mutated enzymes were expressed in E. coli, purified, and incubated with FPP. Total ion chromatograms of GC-MS analyses of the terpene products are shown. The sesquiterpene olefins were identified by comparison of their mass spectra and retention times with those of authentic standards. 1, β-macrocarpene; 2, β-bisabolene.
To identify the origin of the proton involved in the protonation of (S)-β-bisabolene, we analyzed the enzymatic activity of TPS6 in the presence of deuterium oxide. The water was removed by lyophilization, and the protein was redissolved in D2O. The terpene spectrum in the presence of D2O was identical to that produced in the presence of water (Fig. 9A). Analysis of the reaction products by mass spectrometry revealed an incorporation of one deuterium atom into the (S)-β-macrocarpene structure, which increased the molecular mass by one unit to m/z 205 (Fig. 9B). The position of the deuterium atom was elucidated by comparing the mass spectra of the deuterated and undeuterated (S)-β-macrocarpene products (Fig. 9B). Both spectra possessed a substantial m/z 148 that results from a retro-Diels-Alder reaction eliminating 2-methylpropene, which includes C10–C13 numbered according to the starting FPP (Fig. 10). Since the mass of this charged fragment in the deuterated product remains the same, the deuterium atom must reside in the neutral 2-methylpropene cleavage product, consistent with the proposed mechanism, which suggests it to be at C10 (Fig. 10). On the other hand, the mass spectrum of the second terpene product, (S)-β-bisabolene, was identical in the presence of D2O and water. These results support a reaction mechanism in which a water-derived proton is incorporated during reprotonation of the (S)-β-bisabolene intermediate (Fig. 10).
FIGURE 9.

A water molecule is involved in the reprotonation of the β-bisabolene intermediate by TPS6. The enzyme was expressed in E. coli, purified, lyophilized, and dissolved in H2O or D2O, respectively. A, GC-MS traces of products formed from FPP in the presence of H2O and D2O are shown. B, mass spectra of β-macrocarpene in the presence of H2O and D2O.
FIGURE 10.

Reaction mechanism for the formation of sesquiterpene products by TPS6 and TPS11 proposed by Cool (20) and demonstrated in this work. The substrate FPP as well as the two major products (S)-β-macrocarpene and (S)-β-bisabolene are highlighted with boxes. The numbering of the carbon atoms is derived from that of the FPP substrate.
TPS6 and TPS11 Are Predominantly Active in the Roots of Maize—To determine the contribution of TPS6 and TPS11 to maize terpene production, we measured transcript accumulation in different organs by RNA hybridization with a probe that detects both genes (Fig. 11A). Transcripts were absent in undamaged leaves but were detected after 16 h of feeding by the generalist lepidopteran, Spodoptera littoralis. In roots, the transcripts were found both in damaged plants fed upon by S. littoralis and in undamaged controls, but the transcript level was greater after feeding.
FIGURE 11.

Expression analysis of TPS6 and TPS11 in planta. A, transcript accumulation of tps6 and tps11 in roots and leaves of 14-day-old seedlings that were undamaged (ctr) or treated with S. littoralis for 12 h (dam). The bottom panel shows an ethidium bromide-stained agarose gel with total RNA as control for equal RNA loading. B, sesquiterpene hydrocarbons were extracted from leaves and roots of 14 day-old seedlings that were undamaged or treated with S. littoralis for 12 h. Compounds were identified and quantified as described under “Experimental Procedures.” Means and S.E. of six independent measurements are shown. Compounds marked with white bars were found in leaves and roots of both control and damaged plants. Compounds marked with black bars are products of the terpene synthases TPS6 and TPS11. Compounds marked with gray bars are products of TPS10. The compounds are as follows: α-copaene (1), (E)-β-caryophyllene (2), (E)-α-bergamotene (3), sesquisabinene A (4), (E)-β-farnesene (5), germacrene D (6), zingiberene (7), β-macrocarpene (8), β-bisabolene (9), δ-cadinene (10), and sesquiphellandrene (11).
An analysis of the terpene products in the roots showed that both (S)-β-macrocarpene and (S)-β-bisabolene are present (Fig. 11B). The terpene concentration increased about 2-fold after herbivory and therefore seems to correlate with changes in transcript levels. However, in leaves, no (S)-β-macrocarpene was detected after herbivore damage despite the presence of tps6/tps11 transcripts (Fig. 11B). The low amount of (S)-β-bisabolene present in damaged leaves was thus likely to have been produced by another terpene synthase, such as TPS10. This enzyme forms (S)-β-bisabolene as a minor product after herbivore damage along with the major products, (E)-α-bergamotene and (E)-β-farnesene (14). None of the monoterpene products of TPS6 and TPS11 measured in vitro from GPP were detected in leaves and roots, consistent with the fact that these enzymes, which lack signal peptides, are localized in a compartment, such as the cytosol, without a supply of GPP.
DISCUSSION
(S)-β-Macrocarpene Formation by Maize TPS6 and TPS11 Requires a Protonation of the (S)-β-Bisabolene Intermediate—The two terpene synthase genes isolated from maize in this study both encode enzymes that form the unusual, bicyclic sesquiterpene (S)-β-macrocarpene. Site-directed mutagenesis and D2O feeding experiments support a mechanism in which a neutral (S)-β-bisabolene intermediate, a product of a C1–C6 cyclization, is reprotonated to form (S)-β-macrocarpene (Fig. 10). (S)-β-Bisabolene, a side product of enzyme catalysis, has not previously been identified as a neutral intermediate in the carbocation cascade of terpene synthases. Similar reaction mechanisms involving stable intermediates that are products of C1–C10 cyclization were demonstrated earlier for the formation of eudesmane- or selinene-type sesquiterpenes, like 5-epi-aristolochene (3, 4). This indicates that a variety of sesquiterpene skeletons are subject to reprotonation by terpene synthases. Protonation of a carbon-carbon double bond is also employed in carbocation formation in the entry step of the reaction mechanism of certain diterpene synthases, like copalyl diphosphate synthase (27).
The Rate of Reprotonation of the (S)-β-Bisabolene Intermediate by TPS6 and TPS11 Is Dependent on the Divalent Metal Ion Present—In sesquiterpene synthase mechanisms, metal ion cofactors are involved in binding and ionization of the substrate FPP by forming ion bridges between the negatively charged diphosphate group of the substrate and the negatively charged aspartate residues of the highly conserved DDXXD motif in the active site (8). TPS6 and TPS11 can use both magnesium and manganese as a cofactor with a similar rate of enzyme activity. However, in the presence of manganese ions, there was increased formation of (S)-β-bisabolene. A similar effect on product distribution has already been shown for other terpene synthases (18, 24, 25). For example, for the very similar maize terpene synthases TPS4 and TPS5, the replacement of Mg2+ ions by Mn2+ resulted in an increased formation of the acyclic sesquiterpene product, (E)-β-farnesene and a decreased formation of the cyclic products, suggesting that the metal ion cofactor affects the deprotonation rate of the farnesyl cation relative to its rate of isomerization to nerolidyl diphosphate (18). In TPS6 and TPS11, the nature of the cofactor appears to affect the protonation reaction of terpene synthases. Further studies are needed to demonstrate whether the metal ions are directly involved in the protonation or whether they exert an indirect effect on reaction by altering the conformation of the substrate and the active site cavity. Comparing the in vitro assay data of TPS6 and TPS11 with the ratio of (S)-β-macrocarpene and (S)-β-bisabolene products found in maize root tissue suggests that these terpene synthases operate in vivo at nearly neutral pH (Fig. 6) and with a magnesium ion cofactor (Fig. 7), which coincides with the conditions generally present in the cytosol of plant cells, where Mg2+ is more abundant than Mn2+ (23).
Tyrosine 522 Is Important for the Protonation of (S)-β-Bisabolene—We conducted a mutational analysis of TPS11 to determine the amino acids involved in the reprotonation of the (S)-β-bisabolene intermediate. This analysis was guided by the structural elucidation of TEAS from tobacco, a plant terpene synthase known to have a similar reprotonation mechanism (3, 7). Of the three amino acids involved in protonation in TEAS, only two, the central tyrosine and one of the aspartate residues, are conserved in TPS6 and TPS11. Removal of the hydroxyl group on the corresponding tyrosine residue in TPS11 (Tyr522) by conversion to phenylalanine strongly reduced the rate of reprotonation of (S)-β-bisabolene. Additional evidence for the role of tyrosine 522 in reprotonation is the lower proportion of (S)-β-macrocarpene, the eventual product of protonation, formed at higher pH. For example, at pH 8.5, the amount of (S)-β-macrocarpene formed is only half that at pH 8.0, whereas the amount of (S)-β-bisabolene formed is greater than that a pH 8.0. Although the actual pKa for tyrosine 522 is not known, it is reasonable to assume that at pH 8.5 a significant amount of the side chain is present as a phenolate anion and not able to participate in proton donation. However, tyrosine is unlikely to be the direct proton donor to (S)-β-bisabolene, since mutation of this amino acid to phenylalanine does not disable reprotonation completely. With both the (E,E)-FPP and (Z,E)-FPP substrates, a small amount of (S)-β-macrocarpene was detected as a product of the reaction catalyzed by this mutant. Tyrosine 522 might support reprotonation by stabilization of the actual proton donor or substrate via hydrogen bonds and its general acid/base properties.
The TPS6 enzyme products formed in the presence of deuterium oxide demonstrated that the proton donor for reprotonation of the (S)-β-bisabolene intermediate is likely to be a water molecule. Therefore, an intramolecular proton transfer from the C7 of the β-bisabolyl cation to C11 of the (S)-β-bisabolene is unlikely, although a similar mechanism has been proposed for aristolochene synthase from Penicillium roqueforti (28). The water molecule providing the proton to the (S)-β-bisabolene intermediate in TPS6 may be situated inside the active center in association with polar amino acid side chains, where it does not interfere with the early steps of the carbocationic reaction mechanism.
It is also conceivable that the pyrophosphate moiety cleaved from the substrate in the first step of the reaction can act as proton donor. Such a role for pyrophosphate was suggested by a study based on the active site structure of the aristolochene synthase of Aspergillus terreus (29). Since the complexation of the pyrophosphate with Mg2+ versus Mn2+ ions affects the reprotonation activity of TPS6 and TPS11 strongly, its role as proton donor is conceivable. However, the exact nature and location of the proton donor can only be specified by more detailed structural information on the active center.
Alteration of aspartate 525 to asparagine blocked catalysis completely. Interestingly, when the corresponding to asparagine, there was a 500-fold decrease in olefin production (30). More recently, we demonstrated that this amino acid is also involved in the catalysis of early reaction steps by maize terpene synthase TPS4 (26). Therefore, the mutation of aspartate 525 in TPS11 may have disturbed the general binding or ionization of the FPP substrate.
The Two Genes tps6 and tps11 May Be the Result of a Tandem Duplication—Not only do the terpene synthase genes, tps6 and tps11, encode proteins with identical catalytic activity, but the genes have a high degree of identity (96% at the nucleotide level) and also map to the same position on chromosome 10. The location of tps6 and tps11 is also close to a second pair of sesquiterpene synthase genes, tps4 and tps5 (18). However, in contrast to tps6 and tps11, the enzymatic functions of tps4 and tps5 are different due to the alteration of four amino acids in the active center of the enzymes (18). It is likely that both gene pairs are the result of a tandem duplication followed by divergence. For tps6 and tps11, the part of the gene encoding the C-terminal domain (which includes the active site) has a higher ratio of synonymous to nonsynonymous substitutions than the N-terminal domain (dS/dN = 13.7 and 3.9, respectively), suggesting that positive selection pressure helped to conserve the function of both enzymes. However, since tps4 and tps5 have functionally diverged, these genes might have been subject to weaker selection pressure than tps6 and tps11 (dS/dN = 6.5 for the C-terminal domain and 3.6 for the N-terminal domain) (31). Similar scenarios of gene duplication followed by functional divergence have been implicated in the enlargement and diversification of the large terpene synthase gene families found in the fully sequenced genomes of Arabidopsis thaliana (32) and rice (33). Sometimes only a small degree of sequence diversification is necessary to give terpene synthases that function in completely different pathways, such as the rice diterpene synthases involved in gibberellin and phytoalexin metabolism (27).
The Terpene Synthases TPS6 and TPS11 May Be Involved in Plant Defense—The genes encoding TPS6 and TPS11 were shown to be expressed in the root tissue of the maize inbred line B73, and the enzyme products, (S)-β-macrocarpene and (S)-β-bisabolene, were detected in this organ, in confirmation of an earlier report (34). After leaf damage by S. littoralis, both the transcript levels and terpene products are elevated, indicating transduction of a signal from herbivore-damaged leaves to roots, a phenomenon not previously observed in terpene metabolism. Such signal transduction pathways between leaves and roots have been observed for secondary plant metabolites other than terpenes (35). Further work is planned to elucidate the nature of this signal and determine the ecological role of elevated terpene production in roots after leaf damage.
TPS6 shares 99% amino acid identity with the recently described gene umi2 isolated from the maize variety Early Golden Bantam (22). umi2 was identified due to a strong up-regulation of transcript levels in leaves after infestation with Ustilago maydis and Colletotrichum graminicola (22). We expressed umi2 from the variety Early Golden Bantam in a bacterial expression system and confirmed that the enzymatic activity of the protein is identical to that of TPS6 (data not shown). Therefore, it is likely that tps6 and umi2 are alleles of the same gene. Since the maize seedlings in our work were grown in unsterile soils, the expression levels of tps6 and tps11 could have been a result of fungal infestation in the roots. The role of tps6 and tps11 expression in defense against both herbivores and microbial pathogens requires further elucidation.
Despite the presence of gene transcript, no sesquiterpene products of TPS6 or TPS11 were detected in herbivore-damaged leaves. Since other terpene synthases with a similar Km value like TPS10 are active in this tissue, a posttranscriptional mechanism suppressing the formation of active enzymes of TPS6 and TPS11 is likely. A similar posttranscriptional regulation was observed for maize TPS1, a nerolidol synthase, where transcription of a gene encoding an active protein (as measured in vitro) was observed, but no enzyme products were detected in vivo (21). Alternatively, it is conceivable that the TPS6 and TPS11 enzymes are active, but their products were converted to nonvolatile compounds with high efficiency and are therefore not detectable in planta.
Acknowledgments
We thank Larry Cool (Richmond, CA) for a sample of (S)-β-macrocarpene. The ORF of umi2 was kindly provided by Christoph Basse (Marburg, Germany). We are indebted to Jens Wurlitzer for excellent technical assistance. The (Z,E)-FPP was kindly provided by N. Gatto and W. Boland (Max Planck Institute for Chemical Ecology).
The work was supported by German Research Foundation Grant DE8372-2 (to J. D. and J. G.) and funds from the Max Planck Society. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement”in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The nucleotide sequence(s) reported in this paper has been submitted to the GenBank™/EBI Data Bank with accession number(s) AY518315 and EU716166.
Footnotes
The abbreviations used are: GPP, geranyldiphosphate; FPP, farnesyldiphosphate; GGPP, geranylgeranyldiphosphate; TEAS, 5-epi-aristolochene synthase; GC, gas chromatography; MS, mass spectrometry; ORF, open reading frame.
References
- 1.Gershenzon, J., and Kreis, W. (1999) Biochemistry of Plant Secondary Metabolism: Annual Plant Reviews, Vol. 2, pp. 222–299, Sheffield Academic Press, Sheffield, UK [Google Scholar]
- 2.Croteau, R. (1987) Chem. Rev. 87 929–954 [Google Scholar]
- 3.Cane, D. E. (1990) Chem. Rev. 90 1089–1103 [Google Scholar]
- 4.Steele, C. L., Crock, J., Bohlmann, J., and Croteau, R. (1998) J. Biol. Chem. 273 2078–2089 [DOI] [PubMed] [Google Scholar]
- 5.Cane, D. E. (1999) Comprehensive Natural Products Chemistry, Vol. 2, pp. 155–200, Elsevier, Amsterdam [Google Scholar]
- 6.Cane, D. E., Prabhakaran, P. C., Oliver, J. S., and McIlwaine, D. B. (1990) J. Am. Chem. Soc. 112 3209–3210 [Google Scholar]
- 7.Facchini, P. J., and Chappell, J. (1992) Proc. Natl. Acad. Sci. U. S. A. 89 11088–11092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Starks, C. M., Back, K. W., Chappell, J., and Noel, J. P. (1997) Science 277 1815–1820 [DOI] [PubMed] [Google Scholar]
- 9.Rising, K. A., Starks, C. M., Noel, J. P., and Chappell, J. (2000) J. Am. Chem. Soc. 122 1861–1866 [Google Scholar]
- 10.Back, K., and Chappell, J. (1995) J. Biol. Chem. 270 7375–7381 [DOI] [PubMed] [Google Scholar]
- 11.Sharon-Asa, L., Shalit, M., Frydman, A., Bar, E., Holland, D., Or, E., Lavi, U., Lewinsohn, E., and Eyal, Y. (2003) Plant J. 36 664–674 [DOI] [PubMed] [Google Scholar]
- 12.Iijima, Y., Davidovich-Rikanati, R., Fridman, E., Gang, D. R., Bar, E., Lewinsohn, E., and Pichersky, E. (2004) Plant Physiol. 136 3724–3736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rasmann, S., Kollner, T. G., Degenhardt, J., Hiltpold, I., Toepfer, S., Kuhlmann, U., Gershenzon, J., and Turlings, T. C. J. (2005) Nature 434 732–737 [DOI] [PubMed] [Google Scholar]
- 14.Schnee, C., Kollner, T. G., Held, M., Turlings, T. C. J., Gershenzon, J., and Degenhardt, J. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 1129–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kollner, T. G., Held, M., Lenk, C., Hiltpold, I., Turlings, T. C. J., Gershenzon, J., and Degenhardt, J. (2008) Plant Cell 20 482–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Degenhardt, J., and Gershenzon, J. (2000) Planta 210 815–822 [DOI] [PubMed] [Google Scholar]
- 17.Burr, B., and Burr, F. A. (1991) Trends Genet. 7 55–60 [DOI] [PubMed] [Google Scholar]
- 18.Kollner, T. G., Schnee, C., Gershenzon, J., and Degenhardt, J. (2004) Plant Cell 16 1115–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Konig, W. A., Rieck, A., Hardt, I., Gehrcke, B., Kubeczka, K. H., and Muhle, H. (1994) J. High Resolut. Chromatogr. 17 315–320 [Google Scholar]
- 20.Cool, L. G. (2005) Phytochemistry 66 249–260 [DOI] [PubMed] [Google Scholar]
- 21.Schnee, C., Kollner, T. G., Gershenzon, J., and Degenhardt, J. (2002) Plant Physiol. 130 2049–2060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Basse, C. W. (2005) Plant Physiol. 138 1774–1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marschener, H. (1998) Field Crops Res. 56 203–207 [Google Scholar]
- 24.Crock, J., Wildung, M., and Croteau, R. (1997) Proc. Natl. Acad. Sci. U. S. A. 94 12833–12838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Picaud, S., Olofsson, L., Brodelius, M., and Brodelius, P. E. (2005) Arch. Biochem. Biophys. 436 215–226 [DOI] [PubMed] [Google Scholar]
- 26.Kollner, T. G., O'Maille, P. E., Gatto, N., Boland, W., Gershenzon, J., and Degenhardt, J. (2006) Arch. Biochem. Biophys. 448 83–92 [DOI] [PubMed] [Google Scholar]
- 27.Xu, M. M., Wilderman, P. R., and Peters, R. J. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 7397–7401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Allemann, R. K., Young, N. J., Ma, S., Truhlar, D. G., and Gao, J. (2007) J. Am. Chem. Soc. 129 13008–13013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shishova, E. Y., Di Costanzo, L., Cane, D. E., and Christianson, D. W. (2007) Biochemistry 46 1941–1951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Little, D. B., and Croteau, R. B. (2002) Arch. Biochem. Biophys. 402 120–135 [DOI] [PubMed] [Google Scholar]
- 31.MacMillan, J. (1997) Nat. Prod. Rep. 14 221–243 [Google Scholar]
- 32.Aubourg, S., Lecharny, A., and Bohlmann, J. (2002) Mol. Genet. Genom. 267 730–745 [DOI] [PubMed] [Google Scholar]
- 33.Yuan, Y. S., Kollner, T. G., Wiggins, G., Grant, J., Degenhardt, J., and Chen, F. (2008) Plant J., in press [DOI] [PubMed]
- 34.Kollner, T. G., Schnee, C., Gershenzon, J., and Degenhardt, J. (2004) Phytochemistry 65 1895–1902 [DOI] [PubMed] [Google Scholar]
- 35.Erb, M., Ton, J., Degenhardt, J., and Turlings, T. C. J. (2008) Plant Physiol. 146 867–874 [DOI] [PMC free article] [PubMed] [Google Scholar]