

Abstract
The recognition of terminally misfolded proteins in the endoplasmic reticulum (ER) and the extraction of these proteins to the cytoplasm for proteasomal degradation are determined by a quality control mechanism in the ER. In yeast, Yos9p, an ER lectin containing a mannose 6-phosphate receptor homology (MRH) domain, enhances ER-associated degradation (ERAD) of glycoproteins. We show here that human XTP3-B (hXTP3-B), an ER lectin containing two MRH domains, has two transcriptional variants, and both isoforms retard ERAD of the human α1-antitrypsin variant null Hong Kong (NHK), a terminally misfolded glycoprotein. The hXTP3-B long isoform strongly inhibited ERAD of NHK-QQQ, which lacks all of the N-glycosylation sites of NHK, but the short transcriptional variant of hXTP3-B had almost no effect. Examination of complex formation by immunoprecipitation and by fractionation using sucrose density gradient centrifugation revealed that the hXTP3-B long isoform associates with the HRD1-SEL1L membrane-anchored ubiquitin ligase complex and BiP, forming a 27 S ER quality control scaffold complex. The hXTP3-B short isoform, however, is excluded from scaffold formation. Another MRH domain-containing ER lectin, hOS-9, is incorporated into this large complex, but gp78, another mammalian homolog of the yeast ubiquitin ligase Hrd1p, is not. Based on these results, we propose that this large ER quality control scaffold complex, containing ER lectins, a chaperone, and a ubiquitin ligase, provides a platform for the recognition and sorting of misfolded glycoproteins as well as nonglycosylated proteins prior to retrotranslocation into the cytoplasm for degradation.
In the endoplasmic reticulum (ER)2 quality control system, an elaborate machinery regulates the recognition of terminally misfolded proteins and the extraction of these proteins from the ER into the cytoplasm for degradation by proteasomes. Since most of the proteins synthesized in the ER are modified with N-linked oligosaccharides upon entry into the ER, quality control closely correlates with the processing of sugars on the attached N-glycans, assisted by ER chaperones and lectins (1–4). In yeast, Yos9p, an ER lectin, accelerates ER-associated degradation (ERAD) of glycoproteins (5–8). Yos9p has a mannose 6-phosphate receptor homology (MRH) domain, characterized by its homology to the sugar recognition domain of the mannose 6-phosphate receptor (9). Recent studies investigating the role of Yos9p in glycoprotein ERAD have found that Yos9p forms a complex that includes the ubiquitin ligase Hrd1p/Der3p, Hrd3p, and Kar2p (10–12). Hrd1p is a multimembrane-spanning E3 ubiquitin ligase with a RING domain in the C-terminal cytoplasmic tail that forms a stable, stoichiometric association with Hrd3p (13, 14). HRD1/synoviolin (15, 16) and gp78/AMFR (17) are the mammalian orthologs of Hrd1p. The mammalian homolog of yeast Hrd3p, SEL1L (18), possesses a large luminal domain and a type I transmembrane segment. In addition, several MRH domain-containing proteins of unknown function have been reported in higher eukaryotes and may act as lectins that recognize N-glycans (9).
To understand how ER lectins with MRH domains contribute to ER quality control of glycoproteins in mammals, we have focused on human XTP3-B (hXTP3-B), which has two MRH domains of unknown function (19). We cloned two transcriptional variants of hXTP3-B and examined their effects on ERAD as well as complex formation with the HRD1-SEL1L ubiquitin ligase complex. We propose that the long transcriptional variant of hXTP3-B forms a 27 S ER quality control scaffold complex, providing a platform for the recognition and sorting of misfolded glycoproteins and nonglycosylated proteins, prior to retrotranslocation for degradation.
EXPERIMENTAL PROCEDURES
Antibodies—Antibodies against human SEL1L were generated by immunizing rabbits with a synthetic peptide (APPQQEGPPEQQPPQ) conjugated to keyhole limpet hemocyanin and were used for immunoprecipitation experiments. Rabbit polyclonal antibodies against α1-antitrypsin were purchased from DAKO, and rabbit polyclonal anti-DsRed antibodies were purchased from Clontech. Mouse monoclonal (9E10) and rabbit polyclonal anti-c-Myc antibodies, as well as goat and rabbit polyclonal anti-HA-tag antibodies, were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Mouse monoclonal (M2) and rabbit polyclonal anti-FLAG antibodies were purchased from Sigma. A monoclonal antibody against human SEL1L for immunoblot analysis was obtained from Lifespan Biosciences. Rabbit polyclonal anti-OS-9 was purchased from the Protein Tech Group, mouse monoclonal anti-BiP was obtained from BD Biosciences, and mouse monoclonal anti-p97/VCP was obtained from Affinity Bioreagents. Rabbit polyclonal anti-calnexin antibodies were purchased from StressGen, and a mouse monoclonal anti-actin antibody was obtained from Chemicon.
Plasmid Construction— hXTP3-B and hOS-9 cDNAs were cloned by standard reverse transcription-PCR methods from total RNA prepared from human 293 cells and HepG2 cells. The coding regions of hXTP3-B and hOS-9 were amplified by PCR and tagged with monomeric red fluorescent protein (mRFP), HA, or FLAG at the C terminus by subcloning into the mRFP (20), pMH (Roche Applied Science), or pCMV-Tag4 (Invitrogen) plasmids, respectively. A plasmid expressing human SEL1L was kindly provided by Otsuka Pharmaceutical Co. Ltd. (Tokushima, Japan). NHK-GFP was constructed by amplification of the coding region of NHK from NHK/pREP9 (21) by PCR, followed by subcloning into pEGFP-N1 (Clontech). ER-mCherry was constructed by replacing the enhanced yellow fluorescent protein domain of enhanced yellow fluorescent protein-ER (Clontech) with mCherry (Clontech). NHK-QQQ (22) and HRD1-Myc/gp78-Myc (23) were constructed as described previously.
Small Interfering RNA (siRNA) Sequences—siRNAs targeting hXTP3-B and SEL1L were purchased from Invitrogen (Stealth™ RNA interference (RNAi)). The targeted sequences were as follows: XTP3B-1 (5′–3′), UAUGUGGGUUGUCCCAACAGUGAGC; XTP3B-2 (5′–3′), UUUCCCACUAUCCUUGUCCUCAUGG; XTP3B-3 (5′–3′), UUUCCAUGACAUACUUCGUAAGUCC; SEL1L-1 (5′–3′), AUUCCAAGCCCACUCUGUCCAACUG; SEL1L-2 (5′–3′), UUAACUUGAACUCCUCUCCCAUAGA; SEL1L-3 (5′–3′), AUCUGAUGUACAUUCAUCAUACUCC. Low GC and medium GC Stealth™ RNAi duplexes were used as negative control siRNAs.
Cell Culture and Transfection—Human 293 cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and antibiotics (100 units/ml penicillin G and 0.2 mg/ml streptomycin). Plasmids were transfected using the FuGENE6 transfection reagent (Roche Applied Science) or Lipofectamine 2000 (Invitrogen) according to the methods recommended by the manufacturers. Cells were metabolically labeled and/or harvested 24–30 h after transfection. For siRNA knockdown experiments, RNA was transfected using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's protocol ∼24 h after plating the cells. Following incubation for an additional 24 h, plasmids were transfected using the FuGENE6 transfection reagent, and the cells were metabolically labeled after 24 h.
Metabolic Labeling, Immunoprecipitation, and Western Blotting—After incubation in medium lacking methionine/cysteine for 20 min, cells were pulse-labeled with 8.2 MBq/ml [35S]-Express protein labeling mixture (PerkinElmer Life Sciences) for 15 min, and chased in normal growth medium containing 30 mg/liter methionine and 63 mg/liter cystine-2HCl for the indicated times. For overnight labeling, 6 mg/liter methionine, 13 mg/liter cystine-2HCl, and 4.1 MBq/ml [35S] labeling mixture were added to the medium.
Cells were solubilized in buffer (150 mm NaCl, 50 mm Tris-HCl, pH 7.5) containing 1% Nonidet P-40 or 3% digitonin, supplemented with protease inhibitors. After centrifugation at 12,000 rpm for 20 min at 4 °C, the supernatant was used for immunoprecipitation or Western blotting, as described (24). Radioactivity was quantified by exposing gels to a PhosphorImager (STORM; GE Healthcare) (21).
For Western blot analysis, supernatants were adjusted to 1× in Laemmli buffer and separated by 10% SDS-PAGE. After blotting onto nitrocellulose, the membranes were blocked in Blocking-One solution (Nacalai Tesque, Japan). Antibodies were diluted in phosphate-buffered saline containing 0.1% Tween 20 supplemented with 5% Blocking-One or in the Can Get Signal buffer (TOYOBO) and detected by ECL (GE Healthcare). To detect the multimembrane-spanning ubiquitin ligases HRD1 and gp78 after immunoprecipitation or by Western blotting, samples were incubated in Laemmli buffer at 37 °C for 30 min or 65 °C for 15 min, separated by SDS-PAGE, and processed for quantification or for blotting as described above.
Sucrose Density Gradient Centrifugation—Cells were solubilized in 3% digitonin and centrifuged at 12,000 rpm for 20 min. Supernatants (0.25 ml) were applied to a 10–40% linear sucrose gradient on a 60% sucrose cushion. Centrifugation was performed at 36,000 rpm in a swinging bucket rotor (RPS55; Hitachi, Japan) at 4 °C for 16 h, and 0.25-ml fractions were collected from the top. Aliquots of each fraction (10% of total volume) were separated by SDS-PAGE, followed by Western blotting. A high molecular weight calibration kit for native electrophoresis (GE Healthcare) was used to estimate the molecular weights. The proteins and sedimentation coefficients were as follows: bovine serum albumin (66 kDa), 4.6 S; bovine heart lactate dehydrogenase (140 kDa), 7.7 S; bovine liver catalase (232 kDa), 11.4 S; porcine thyroid thyroglobulin (669 kDa), 19 S. To detect protein interactions in fractions separated by sucrose density gradient centrifugation, the fractions were diluted 2.5-fold with 3% digitonin lysis buffer and immunoprecipitated with various antibodies. Immunoprecipitates were separated by SDS-PAGE, and co-immunoprecipitated proteins were detected by Western blot analysis.
Mass Spectrometric Analysis—Mass spectrometric identification of proteins was performed as previously described (25). Briefly, after SDS-PAGE, proteins were visualized by silver staining, excised from the gel, and subjected to in-gel digestion overnight at 37 °C with trypsin (Promega) in buffer containing 50 mm ammonium bicarbonate (pH 8.0) and 2% acetonitrile. Molecular mass analysis of tryptic peptides was performed by matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry using an Ultraflex TOF/TOF instrument (Bruker Daltonics). Proteins were identified by comparison of the molecular weights determined by MALDI-TOF mass spectrometry and theoretical peptide masses of the proteins registered in NCBInr.
FLAG-tagged hXTP3-B-long transfected into 293 cells was purified by using FLAG-agarose beads (Sigma) after extracting the cells with buffer containing 1% digitonin. Proteins were eluted from the beads with 0.1 m glycine-HCl (pH 3.5).
RESULTS
ER Localization of hXTP3-B—To examine the function of hXTP3-B, we cloned two transcriptional variants of hXTP3-B from cultured human cells by reverse transcription-PCR (Fig. 1A). These two transcripts are reported in the expressed sequence tag data base and differ in the distance between the two MRH domains. The hXTP3-B-short isoform also lacks part of the N terminus of MRH domain 2, including one of the six conserved cysteines within the MRH domain (9). A hydrophobic predicted signal sequence is present at the N terminus of both isoforms. To determine the subcellular localization, hXTP3-B-mRFP was co-transfected with NHK-GFP, a luminal ERAD substrate. Microscopic observations revealed that hXTP3-B was distributed in a fine reticular network in the cytoplasm around the nucleus that co-localized with the distribution of NHK-GFP, indicating its localization in the ER (Fig. 1B). The intracellular distribution was further examined by testing the sensitivity of the hXTP3-B N-linked glycans to Endo H digestion. hXTP3-B has one consensus sequence for N-glycosylation, and the transfected long and short isoforms were both partially N-glycosylated, as indicated by the appearance of doublet bands (Fig. 1C, lanes 1, 3, 7, and 9). The hXTP3-B N-glycan was sensitive to Endo H digestion (Fig. 1C, compare lane 1 with lane 2 and lane 3 with lane 4), confirming its distribution in the ER.
FIGURE 1.
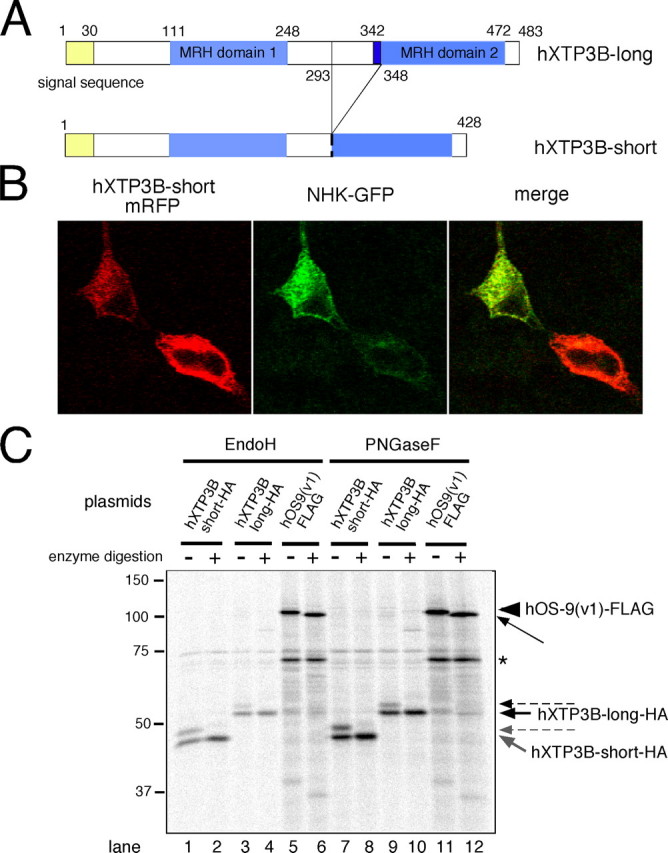
Intracellular localization of transfected hXTP3-B. A, domain organization of hXTP3-B-long and -short isoforms. B, fluorescence microscopy of 293 cells co-transfected with hXTP3-B-short-mRFP and NHK-GFP. After fixing cells with 4% paraformaldehyde at room temperature, mRFP and GFP signals were detected by confocal microscopy (Carl Zeiss). C, Endo H and PNGase F digestion of hXTP3-B and hOS-9. Cultures of 293 cells were transfected with HA-tagged hXTP3-B-long or -short or a FLAG-tagged hOS-9 v1, immunoprecipitated with the appropriate epitope tag antibody, and incubated at 37 °C with Endo H for 3 h or with PNGase F for 1.5 h. The thick arrows indicate the nonglycosylated forms of hXTP3-B, and the dotted arrows show the N-glycosylated forms. The arrowhead corresponds to N-glycosylated hOS-9, and the thin arrow shows nonglycosylated hOS-9. The asterisk indicates a band that binds to Protein G-Sepharose beads nonspecifically.
Effect of hXTP3-B on ER Quality Control—To investigate the involvement of hXTP3-B in ER quality control, we transfected 293 cells with hXTP3-B and examined the effect on the degradation of NHK, a terminally misfolded variant of the human α1-antitrypsin glycoprotein (21, 26, 27). hXTP3-B retarded NHK degradation, and the effect was more prominent in the presence of the hXTP3-B long form transcriptional variant than the short form variant (Fig. 2A). We also examined the effect of hXTP3-B on the degradation of a nonglycosylated ERAD substrate, NHK-QQQ, which lacks all three NHK N-glycosylation sites (22). Unexpectedly, the hXTP3-B-long isoform greatly delayed NHK-QQQ degradation, whereas the hXTP3-B-short isoform only marginally affected degradation (Fig. 2B). Neither hXTP3-B variant affected the secretion or intracellular transport of wild-type α1-antitrypsin (Fig. 2C). These data suggest that the ER lectin XTP3-B-long is involved not only in glycoprotein ERAD but also in quality control of misfolded non-glycosylated proteins.
FIGURE 2.
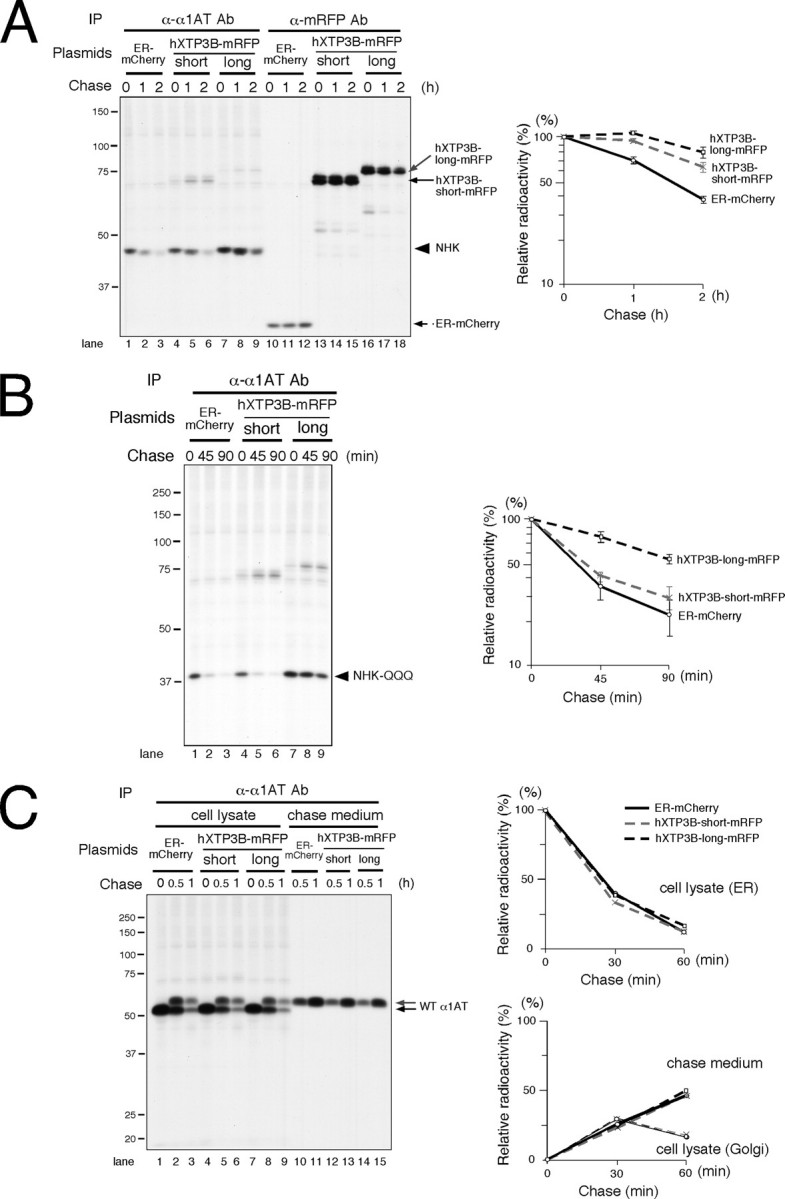
Effect of hXTP3-B on ERAD. A, effect of hXTP3-B on the terminally misfolded glycoprotein NHK. NHK degradation in transfected 293 cells was examined by immunoprecipitation (IP) of cell lysates pulse-labeled with [35S]methionine/cysteine. Immunoprecipitates were separated by 10% SDS-PAGE. The positions of the molecular weight standards are shown on the left. Quantitative analysis by phosphorimaging is shown on the right; error bars indicate S.E. (n = 3). The C terminus of hXTP3-B was tagged with mRFP. ER-mCherry, which expresses mCherry in the ER, was transfected as a control. B, effect of hXTP3-B on nonglycosylated NHK-QQQ degradation. NHK-QQQ degradation was examined as in A. C, secretion and intracellular transport of wild-typeα1-antitrypsin. Quantitative analysis of a typical experiment is shown on the right. Disappearance of the ER form (upper graph), detection of the Golgi form (in cell lysate fractions, gray arrow in the left panel), and secretion into the medium (lower graph) are shown.
Association of SEL1L with HRD1 and BiP—To investigate the role of XTP3-B in ER quality control, we first analyzed formation of complexes between SEL1L and the membrane-embedded ubiquitin ligases HRD1 and gp78, which are both orthologs of yeast Hrd1p. HRD1-SEL1L complex formation was detected in cells solubilized with 3% digitonin buffer (Fig. 3A, lanes 6–9), as previously reported (28). In contrast, gp78 did not associate with SEL1L (Fig. 3A, lanes 10–13). The interaction between SEL1L and HRD1 was disrupted when the cells were extracted in buffer containing 1% Nonidet P-40 (supplemental Fig. 1), indicating an intramembrane or a weak interaction between the two proteins. The association of BiP with the HRD1-SEL1L complex was also observed only when cells were extracted with 3% digitonin and not with 1% Nonidet P-40 (Fig. 3A, supplemental Fig. 1), suggesting a weak interaction. The previously reported stable binding of gp78 and p97/VCP (17), which is resistant to 1% Nonidet P-40, was confirmed (Fig. 3A, lanes 11 and 13, and supplemental Fig. 1; also see Fig. 3C). SEL1L should migrate with a molecular mass of ∼100 kDa on SDS-PAGE, and three bands near the 100-kDa molecular weight standard were detected in immunoprecipitates probed with anti-SEL1L (Fig. 3A, even-numbered lanes). To discriminate between SEL1L and proteins tightly associated with SEL1L, immunoprecipitates were digested with Endo H or PNGase F, since human SEL1L has five putative N-glycosylation sites. The electrophoretic mobility of the middle band (Fig. 3B, lanes 1, 3, 7, and 9, thin black arrows) increased substantially with glycosidase digestion (Fig. 3B, gray arrows; compare lane 1 with lane 2, lane 3 with lane 4, lane 7 with lane 8, lane 9 with lane 10, lane 13 with lane 14, and lane 15 with lane 16). The electrophoretic mobilities of the other two proteins did not change significantly. Hence, the middle band at ∼100 kDa was identified as SEL1L. HRD1-Myc was not glycosylated, as previously reported (Fig. 3B, compare lane 5 with lane 6 and lane 11 with lane 12) (16).
FIGURE 3.
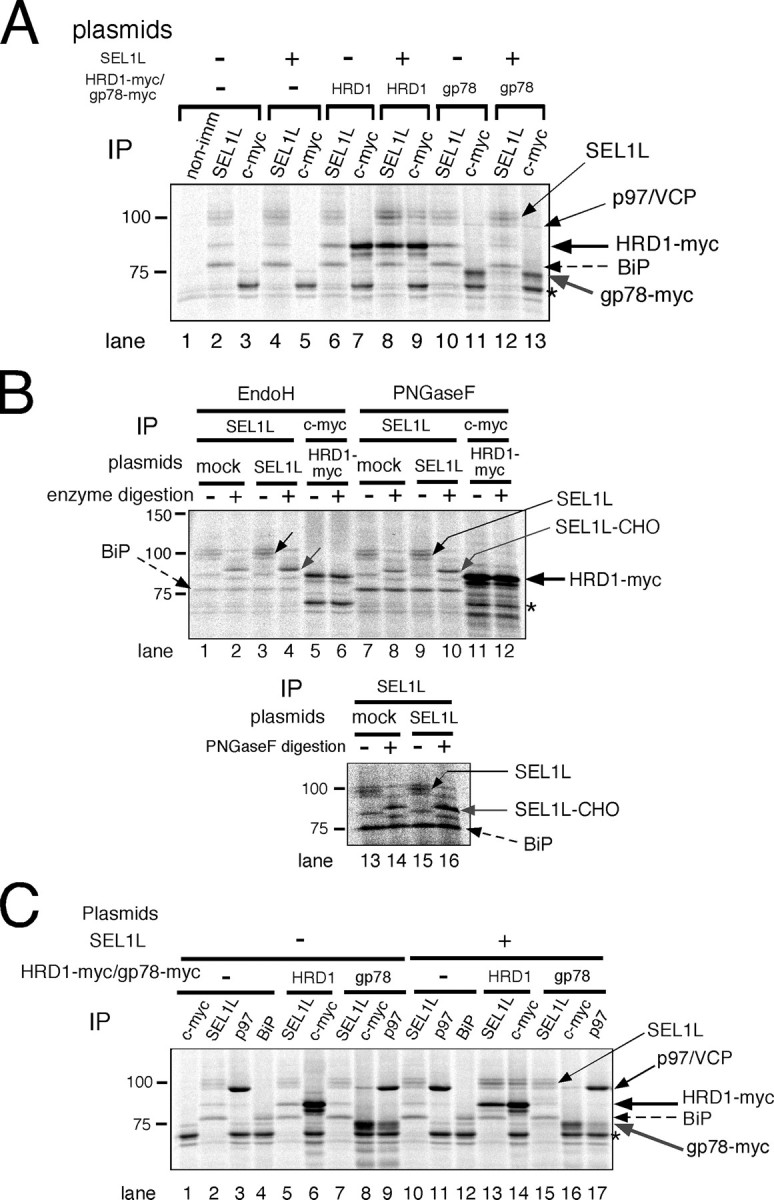
HRD1-SEL1L complex formation. A, HRD1-SEL1L complex formation in 3% digitonin lysis buffer. Cultures of 293 cells were transfected with mock, SEL1L, and/or HRD1-Myc/gp78-Myc. After metabolic labeling for 3 h, cell lysates were immunoprecipitated with the indicated specific antibodies. The asterisk indicates a band nonspecifically detected by Protein G-Sepharose beads. non-imm, nonimmune rabbit serum. B, Endo H and PNGase F digestion of SEL1L and HRD1-Myc. N-Glycosidase digestion was performed as described in the legend to Fig. 1B. The results of two independent experiments are shown in the upper (lanes 1–12) and lower (lanes 13–16) panels.*, as in A. C, identification of endogenous SEL1L, BiP, and p97/VCP by immunoprecipitation. Cells transfected with the indicated plasmids were labeled with [35S]methionine/cysteine for 3 h and extracted with 3% digitonin. The electrophoretic mobilities of proteins immunoprecipitated with the indicated antibodies (IP) are compared.
Identification of proteins that co-immunoprecipitated with SEL1L, HRD1, and gp78 was verified using specific antibodies and sensitivity to Endo H or PNGase F digestion (Fig. 3, B and C). Cells transfected with the indicated plasmids were metabolically labeled and extracted with 3% digitonin. Co-immunoprecipitation of gp78-Myc with p97/VCP was confirmed using antibodies against c-Myc (Fig. 3C, lane 16) and p97/VCP (lane 17). The ∼75-kDa protein that co-immunoprecipitated with SEL1L was identified as BiP, since its electrophoretic mobility was the same as in immunoprecipitates probed with anti-BiP (Fig. 3C, lane 4 versus lane 5 and lane 12 versus lane 13). BiP is not a glycoprotein, and the ∼75-kDa protein was also resistant to glycosidase digestion (Fig. 3B, compare adjacent lanes with or without glycosidase digestion). Thus, SEL1L forms a complex with HRD1 but does not associate with gp78.
Association of hXTP3-B-long with the HRD1-SEL1L Complex and BiP—We next examined the interaction between hXTP3-B and the HRD1-SEL1L ubiquitin ligase complex after extracting the cells with 3% digitonin. Remarkably, the hXTP3-B-long isoform co-immunoprecipitated with SEL1L and BiP, whereas the short form variant did not interact with either protein (Fig. 4A, compare lane 4 with lane 6). Additionally, EDEM1, which accelerates glycoprotein ERAD (21), did not interact with the HRD1-SEL1L complex (lanes 8 and 9). hXTP3-B-long co-immunoprecipitated with SEL1L in both digitonin and Nonidet P-40 lysis buffers (Fig. 4B, lanes 4 and 11), whereas the interaction between hXTP3-B-long and HRD1 was disrupted in the presence of 1% Nonidet P-40 (Fig. 4B, compare lane 6 with lane 13), suggesting that hXTP3-B-long binds directly to SEL1L, whereas its association with HRD1 is indirect. The interaction between hXTP3-B-long and HRD1 was further assessed by immunoprecipitation followed by Western blotting. In immunoprecipitates of hXTP3-B-long-HA, HRD1-Myc was recovered (Fig. 5A, lane 5), but gp78-Myc was not (lane 6). Thus, hXTP3-B-long forms a complex with HRD1 but not with gp78. Although partially degraded HRD1-Myc was detected with anti-c-Myc in the cell extract (Fig. 5A, lane 2), only full-length HRD1-Myc co-immunoprecipitated with hXTP3-B-long (compare lanes 2 and 5), suggesting that the N-terminal transmembrane segment of HRD1 is important for the interaction between these proteins. Although reciprocal co-immunoprecipitation of hXTP3-B-long and HRD1 was detected when cells were lysed in 3% digitonin lysis buffer (Fig. 5B, lanes 6 and 9), these proteins were dissociated by extraction in 1% Nonidet P-40 (Fig. 5B, compare lane 6 with lane 15 and lane 9 with lane 18). Therefore, hXTP3-B-long, but not hXTP3-B-short, interacts with SEL1L to form a complex containing the HRD1-SEL1L ubiquitin ligase.
FIGURE 4.

Association of hXTP3-B with the membrane-anchored ubiquitin ligase complex. A, co-immunoprecipitation of hXTP3-B-long and -short with SEL1L. Cells mock-transfected or transfected with HA-tagged hXTP3-B or mouse EDEM1 (mEDEM1) were metabolically labeled for 16 h prior to extraction with 3% digitonin. non-imm, nonimmune rabbit serum. B, comparison of complex formation by hXTP3-B-long and HRD1 in lysis buffer containing 3% digitonin or 1% Nonidet P-40. Cells were labeled for 16 h.
FIGURE 5.

Complex formation by hXTP3-B and HRD1. A, immunoblot of 293 cells co-transfected with hXTP3-B-long-HA and HRD1-Myc/gp78-Myc. Cells were solubilized with 3% digitonin, and immunoprecipitates of hXTP3-B (HA-tagged) were separated and blotted with anti-c-Myc or anti-HA antibodies (lanes 4–6). Aliquots of the lysates (10% of the total volume) used for immunoprecipitation (IP) were loaded as input controls (lanes 1–3). Partially degraded fragments of HRD1-Myc were detected (lane 2), but only full-length HRD1-Myc co-immunoprecipitated with hXTP3-B (lane 5). B, indirect association of hXTP3-B and HRD1. Cells transfected with FLAG-tagged hXTP3-B and HRD1-Myc were extracted in either 3% digitonin (left) or 1% Nonidet P-40 (right) and subjected to immunoprecipitation followed by Western blotting.
Incorporation of hXTP3-B-long in a 27 S Complex—To further characterize the complex formed by hXTP3-B-long, HRD1-SEl1L, and BiP, cell extracts were subjected to sucrose density gradient centrifugation. The hXTP3-B-long isoform fractionated into two distinct peaks (Fig. 6, third panel, brackets), whereas the hXTP3-B-short isoform sedimented only in the lighter fractions (Fig. 6, second panel). Thus, hXTP3-B-long is incorporated into a high molecular weight complex, and the sedimentation coefficient of this complex was estimated to be ∼27 S by comparison with the sedimentation of protein standards (see “Experimental Procedures”). We also compared the distribution of transfected EDEM1, which migrated more broadly with a peak at 11 S, suggesting that EDEM1 primarily segregates into complexes that do not contain hXTP3-B (Fig. 6, bottom panel).
FIGURE 6.

Fractionation of hXTP3-B and mouse EDEM1 (mEDEM1) by sucrose density gradient centrifugation. Cells transfected with hXTP3-B or mouse EDEM1 were extracted with 3% digitonin and fractionated using a 10–40% sucrose density gradient. Fractions were Western blotted with anti-FLAG (for hXTP3-B-short and -long) or anti-HA (for mouse EDEM1) antibodies to determine the sedimentation profiles of hXTP3-B-short and -long and mouse EDEM1. The brackets indicate the two separate regions of the gradient where hXTP3-B-long was detected. Fractions containing high molecular weight calibration proteins are indicated at the bottom by their estimated sedimentation coefficients (arrows). The positions of molecular weight standards for SDS-PAGE are shown on the left.
Mass spectrometric analysis of FLAG-tagged hXTP3-B-long revealed that hXTP3-B-long associates with SEL1L, HRD1, and another MRH domain-containing protein, hOS-9 (29–32), which is a human homolog of yeast Yos9p and has one MRH domain. Four transcriptional variants (v1–v4) have been reported in the expressed sequence tag data base (see Fig. 8A), but the function of hOS-9 in ER quality control has not been reported. We probed sucrose density gradient fractions with antibodies against hOS-9 and SEL1L. Endogenous hOS-9 and SEL1L were distributed in two separate peaks and co-sedimented with the 27 S high molecular weight complex containing FLAG-tagged XTP3-B-long (Fig. 7A, rightmost panels, brackets). Examination of membrane-embedded ubiquitin ligases revealed that gp78 sedimented separately from SEL1L and hXTP3-B-long (Fig. 7B, rightmost panels), whereas HRD1 was more broadly distributed than fractions enriched for SEL1L, suggesting that HRD1 forms complexes in addition to the HRD1-SEL1L complex (Fig. 7B, third row of panels). Thus, we conclude that hXTP3-B-long co-sediments with HRD1-SEL1L and hOS-9 but not with gp78 or EDEM1.
FIGURE 8.

Co-immunoprecipitation of hOS-9 with SEL1L and BiP. A, domain organization of hOS-9 variants 1 and 2. Variants lacking the 15-amino acid region indicated by the arrow in variants 1 and 2 correspond to variants 3 and 4, respectively. B, complex formation by hOS-9 and SEL1L. Cells were transfected with FLAG-tagged hOS-9 v1 or v2, labeled for 3 h, and extracted with 1% Nonidet P-40 (left) or 3% digitonin (right). The extracts were analyzed by immunoprecipitation (IP) with specific antibodies. non-imm, nonimmune rabbit IgG. The asterisk indicates a nonspecific signal detected by Protein G-Sepharose beads.
FIGURE 7.

High molecular weight complex formation by hXTP3-B-long. A, co-fractionation of endogenous hOS-9 and SEL1L with hXTP3-B-long. Lysates of cells transfected with hXTP3-B with or without HRD1-Myc/gp78-Myc were fractionated by sucrose density gradient centrifugation as in Fig. 6 and were subjected to immunoblotting with anti-FLAG to detect hXTP3-B-long or with antibodies against endogenous proteins. The position of SEL1L is indicated by the arrowheads, and the two hOS-9 variants are indicated by arrows. The high molecular weight fractions containing hXTP3-B-long are indicated by the brackets. The asterisks denote nonspecific signals detected by the anti-OS-9 antibody. B, distribution of HRD1 and gp78. Fractions were probed with antibodies against the epitope tags (FLAG for hXTP3-B and c-Myc for HRD1 and gp78) or endogenous proteins (OS-9, SEL1L, and p97/VCP). The arrows and arrowheads are as in A. gp78-Myc is indicated by the gray arrowhead.
Co-immunoprecipitation of hOS-9 with SEL1L and BiP—We next examined the incorporation of hOS-9 into the 27 S high molecular weight complex by binding to SEL1L. We cloned the four transcriptional variants of hOS-9 (v1–v4) by reverse transcription-PCR from HepG2 and 293 cells (Fig. 8A). These transcriptional variants have alternatively spliced regions near the C terminus. HepG2 cells expressed v1 and v2 mRNA, whereas 293 cells expressed all four transcripts. In cells extracted with 1% Nonidet P-40, hOS-9 v1 and v2 co-immunoprecipitated with endogenous SEL1L (Fig. 8B, compare lane 4 with lane 5 and lane 6 with lane 7). Co-immunoprecipitation of BiP was also detected in cells solubilized with 3% digitonin. These data suggest that the two human MRH domain-containing proteins, hOS-9 and hXTP3-B-long, are incorporated into the 27 S complex through their interactions with SEL1L.
Physical Association of SEL1L, HRD1, and hXTP3-B-long—Although the association of SEL1L, HRD1, and hXTP3B-long was consistently detected by immunoprecipitation followed by Western blotting (Fig. 5) and by co-immunoprecipitation of radiolabeled proteins (Figs. 3 and 4), additional unidentified radiolabeled proteins were detected in the co-precipitates when cells were labeled for 3–16 h (Figs. 3 and 4). To more clearly assess the physical association of SEL1L, HRD1, and hXTP3-B-long, we performed immunoprecipitations using pooled fractions from sucrose density gradient fractionations. Reciprocal co-immunoprecipitations of SEL1L, HRD1, and hXTP3-B-long were obtained from the pooled fractions 14–17 in which the 27 S high molecular weight complex sedimented (Fig. 9A), suggesting a physical association between the proteins.
FIGURE 9.

Physical interaction between SEL1L, HRD1, and hXTP3-B. A, co-immunoprecipitation of SEL1L, HRD1, and hXTP3-B-long from fractions separated by sucrose density gradient centrifugation. Lysates of cells transfected with mock or hXTP3B-long-FLAG + HRD1-Myc were separated by 10–40% sucrose density gradient centrifugation as in Fig. 6, and fractions containing the 27 S high molecular weight complex (fractions 14–17) were pooled and subjected to immunoprecipitation followed by Western blot analysis. Input control lanes (lanes 1 and 2) contained aliquots (10% of the total volume) of the pooled fractions. Since hXTP3-B-long migrates close to the immunoglobulin heavy chains used for immunoprecipitation, short and long exposures of the FLAG blot are shown to allow visualization of FLAG-tagged proteins. B, effect of XTP3-B RNAi on complex formation. Cell cultures were incubated with the indicated siRNA (30 nm) and after 24 h, HRD1-Myc was transfected. After 48 h of RNAi treatment, cells were metabolically labeled for 3 h and then extracted with 3% digitonin. Complex formation was analyzed by immunoprecipitation (IP) with specific antibodies. Two negative control siRNAs (Control-1, low GC content; Control-2, medium GC content) and two specific siRNAs were used. The asterisk denotes a nonspecific band detected by Protein G-Sepharose beads. Calnexin (CNX) immunoprecipitation served as a loading control. C, effect of SEL1L RNAi on complex formation; the same as B, with the exception that hXTP3-B-long-HA was co-transfected with HRD1-Myc.
Next, we knocked down endogenous proteins using RNAi and analyzed the association of SEL1L, HRD1, and hXTP3-B-long by immunoprecipitation. We confirmed the knockdown of transfected hXTP3-B and endogenous SEL1L by Western blot analysis. The knockdown efficiency was greater than 80%, with the exception of the SEL1L-1 siRNA (supplemental Fig. 2 and Fig. 9C). Because the XTP3B-1 and SEL1L-1 siRNAs inhibited cell proliferation, we used the XTP3B-2 and -3 and SEL1L-2 and -3 siRNAs for further analysis. Although the XTP3B-2 and -3 siRNAs knocked down both the hXTP3-B-long and hXTP3-B-short isoforms efficiently (supplemental Fig. 2A), co-immunoprecipitation of SEL1L and HRD1 was not affected (Fig. 9B). In contrast, co-immunoprecipitation of HRD1 with hXTP3B-long was greatly impaired by knockdown of SEL1L (Fig. 9C, compare lanes 9 and 10 and lanes 11 and 12). Co-immunoprecipitation of HRD1 and BiP with SEL1L was also reduced by SEL1L RNAi (Fig. 9C, lanes 1–4). Collectively, these data suggest that SEL1L directly interacts with HRD1, whereas hXTP3-B-long associates with HRD1 through an interaction with SEL1L in the 27 S complex.
Effects of hXTP3-B and SEL1L Knockdown on ERAD—To elucidate the role of the 27 S complex in ERAD, we knocked down hXTP3-B and SEL1L by siRNA and analyzed the kinetics of ERAD substrate degradation in pulse-chase experiments. Although the intracellular degradation of both NHK and NHK-QQQ was largely unaffected by XTP3-B RNAi (Fig. 10, A and B), knockdown of SEL1L strongly inhibited the degradation of both NHK and NHK-QQQ (Fig. 10, C and D). These data indicate that the 27 S complex is involved in ERAD pathways for both glycosylated and nonglycosylated proteins.
FIGURE 10.

Effects of hXTP3-B and SEL1L knock-down on ERAD. A, NHK degradation following XTP3-B siRNA; the same as Fig. 9B, with the exception that NHK was transfected 24 h after RNAi, and cells were pulse-chased 48 h after RNAi, as described in the legend to Fig. 2. One representative result is shown on the left, and quantitative analysis is shown on the right, with error bars indicating the S.E. (n = 3). B, NHK-QQQ degradation following XTP3-B siRNA; the same as in A, with the exception that NHK-QQQ was transfected instead of NHK. C, NHK degradation following SEL1L siRNA. D, NHK-QQQ degradation following SEL1L siRNA.
Effects of Co-expression of hXTP3-B and EDEM1 on NHK Degradation—Since EDEM proteins (EDEM1, -2, and -3) are involved in glycoprotein ERAD by accelerating NHK degradation (21, 22, 33, 34), we examined the effect of co-expressing XTP3-B and EDEM1. Indeed, hXTP3-B-short partially reversed the effect of EDEM1 (Fig. 11A). Similar results were obtained with hXTP3-B-long and EDEM1 (data not shown). These data suggest that EDEM1 and hXTP3-B act in the same pathway of NHK degradation.
FIGURE 11.

Involvement of hXTP3-B and EDEM1 in the mammalian ER quality-control pathway. A, effect of co-expression of hXTP3-B and EDEM1 on NHK ERAD. NHK degradation was examined by pulse-chase in cells co-transfected with hXTP3-B-short and EDEM1. Quantitative analysis is shown on the right, with error bars indicating the S.E. (n = 3). B, a model for the mammalian ER quality control scaffold. Only the components analyzed in this study are shown. Both misfolded glycoproteins and nonglycosylated proteins are recruited to the scaffold assembled on the HRD1-SEL1L ubiquitin ligase complex, which also includes hXTP3-B-long, hOS-9, and BiP.
DISCUSSION
In the present study, we analyzed the function of the human MRH domain-containing lectin hXTP3-B and demonstrated its involvement in ER quality control. Based on the current data, we propose a model for mammalian ERAD in which a large 27 S ER quality control scaffold complex, containing the HRD1 ubiquitin ligase, SEL1L, BiP, and the ER lectins hXTP3-B-long and hOS-9, is formed (Fig. 11B). Importantly, this complex provides a scaffold for ERAD pathways for terminally misfolded nonglycosylated proteins as well as improperly folded glycoproteins. The components of this complex are similar to the complex reported in yeast (10–12), suggesting a conserved machinery from yeast to mammals. The degradation of terminally misfolded NHK is prevented by overexpression of hXTP3-B-long, probably due to increased docking on the scaffold complex, which traps misfolded proteins prior to sorting to the retrotranslocation machinery. Interestingly, the hXTP3-B-short variant is excluded from the quality control complex, as indicated by both co-immunoprecipitation and co-sedimentation analyses. The absence of the conserved cysteine in the N-terminal MRH domain 2 and/or the distance between the two MRH domains in the hXTP3-B-short isoform may provide the structural basis for these results, but this hypothesis remains to be demonstrated. The binding of hXTP3-B to nonglycosylated NHK-QQQ was detected by immunoprecipitation (Fig. 2B), consistent with the observation in yeast that Yos9 binds to misfolded CPY* lacking all N-glycans (6), although binding of Yos9p to N-linked oligosaccharides has been demonstrated (8). Since BiP binds to NHK-QQQ, but not NHK (supplemental Fig. 3), we hypothesize that BiP recruits nonglycosylated NHK-QQQ to the quality control scaffold. Alternatively, hXTP3-B may provide the primary binding interface for NHK-QQQ.
The MRH domain-containing lectins hXTP3-B and hOS-9 are both incorporated into the quality control complex. Knockdown of XTP3-B by RNAi did not affect the degradation kinetics of NHK (Fig. 10A), suggesting that these lectins are functionally redundant in ERAD. However, overexpression of hOS-9 v1 or v2 had little effect on NHK degradation (data not shown). Thus, the functional differences between hXTP3-B and hOS-9 are not clear at present. The reported intracellular localization of hOS-9, a cytosolic distribution as well as association with the ER, is consistent with the protein functioning in the cytosol or at the cytoplasmic surface of the ER (31, 32, 35). However, hOS-9 has a putative N-glycosylation site in the MRH domain, and the electrophoretic mobility of hOS-9 increases after digestion with Endo H or PNGase F, indicating its localization in the ER (Fig. 1C, compare lane 5 with lane 6 and lane 11 with lane 12). Thus, we suggest that hOS-9 associates with the quality control complex at the ER luminal surface.
We propose that the interactions between SEL1L and HRD1 and between SEL1L and hXTP3B are direct, whereas the association between hXTP3-B-long and HRD1 is indirect, based on the detergent sensitivity of the co-immunoprecipitations (Figs. 4B and 5B) and the disruption of the HRD1-hXTP3-B-long complex by SEL1L knockdown (Fig. 9C). Since antibodies that recognize endogenous hXTP3-B were not available, we transfected cells with tagged forms of hXTP3-B. It is unlikely that transfected hXTP3-B-long is incorporated into the quality control scaffold as a substrate because of its improper folding, since only very small amounts of hXTP3-B-long sedimented to the bottom fractions of sucrose density-gradients, and these amounts were similar to those of other endogenous proteins (Figs. 6 and 7). Assessment of the folded state of transfected hXTP3-B by measuring its resistance to protease digestion further confirmed that most of the hXTP3-B was productively folded (supplemental Fig. 4).
Inhibition of both NHK and NHK-QQQ degradation by SEL1L knockdown (Fig. 10, C and D) strongly suggests the involvement of the 27 S quality control scaffold complex in ERAD. Overexpression of SEL1L did not affect the ERAD of NHK or NHK-QQQ (data not shown), further suggesting the importance of the SEL1L-containing complex in ERAD. Since several other proteins co-immunoprecipitated with SEL1L (Fig. 3), it is possible that these as yet unidentified proteins are also incorporated into the 27 S quality control complex. Additionally, the stoichiometry of each protein in this large complex is not clear at present. It will be important to address these issues in future experiments.
Accelerated degradation of NHK by EDEM1 was partially reversed by co-expression of hXTP3-B, although EDEM1 is not a component of the quality control complex. Htm1p/Mnl1p, the yeast homolog of mammalian EDEM proteins (36, 37), and EDEM1 are predicted to act as lectins, because they lack α-mannosidase activity (21, 36); however, the lectin activity of EDEM has not yet been verified. Since Yos9p recognizes Man8GlcNAc2 and Man5GlcNAc2 glycans on misfolded CPY* (8), identification of the N-glycan structures that the human ER lectins recognize will clarify the roles of the components of the 27 S complex as well as the EDEM proteins in sorting ERAD substrates to the retrotranslocation machinery.
While this manuscript was in revision, Christianson et al. (38) reported that OS9 and GRP94 deliver NHK to the SEL1L-HRD1 complex for ERAD. Consistent with our study here, they also showed that both OS-9 and XTP3-B associate with the HRD1-SEL1L ubiquitin ligase complex and that XTP3-B recognizes both glycosylated and nonglycosylated proteins as ERAD substrates, presumably as long as they are misfolded. However, our conclusions do not agree with theirs in several respects. First, based on our analyses of the two transcriptional variants of hXTP3-B, we hypothesized that the region of the protein lacking in the short variant is responsible for complex formation with SEL1L. In contrast, Christianson et al. (38) suggest that it is the MRH domains that are critical for the interactions with SEL1L. This is an important discrepancy, since the functions of the MRH domains of these lectins appear to be essential for ERAD. Second, our results indicate that both XTP3-B-long and OS-9 form a large complex containing HRD1-SEL1L, whereas the results of Christianson et al. (38) indicate that the binding of these two lectins to SEL1L is mutually exclusive. Third, we analyzed the association of the ER-resident HSP70 homolog BiP (the ortholog of yeast Kar2p) in the HRD1-SEL1L complex, and our results indicate a possible contribution of BiP in the ERAD of nonglycosylated proteins within the context of the large quality control complex. Although Christianson et al. (38) also detected the association of BiP with XTP3-B and OS-9, their study concentrated on the interaction between OS-9 and GRP94. Thus, further analyses will be required to provide a better understanding of the molecular mechanism of mammalian ERAD.
Supplementary Material
Acknowledgments
We thank K. Kanamori for technical assistance.
Parts of this work were supported by grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (to N. H., I. W., and K. N.) and the Uehara Memorial Foundation (to N. H.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–4.
Footnotes
The abbreviations used are: ER, endoplasmic reticulum; ERAD, ER-associated degradation; hXTP3-B, human XTP3-B; NHK, α1-antitrypsin null (Hong Kong); mRFP, monomeric red fluorescent protein; Endo H, endoglycosidase H; PNGase F, peptide-N-glycosidase F; siRNA, small interfering RNA; RNAi, RNA interference; MRH, mannose 6-phosphate receptor homology; E3, ubiquitin-protein isopeptide ligase; HA, hemagglutinin; MALDI, matrix-assisted laser desorption/ionization; TOF, time-of-flight; GFP, green fluorescent protein; v1–v4, version 1–4, respectively.
References
- 1.Ellgaard, L., Molinari, M., and Helenius, A. (1999) Science 2861882 -1888 [DOI] [PubMed] [Google Scholar]
- 2.Trombetta, E. S., and Parodi, A. J. (2003) Annu. Rev. Cell Dev. Biol. 19 649-676 [DOI] [PubMed] [Google Scholar]
- 3.Helenius, A., and Aebi, M. (2004) Annu. Rev. Biochem. 731019 -1049 [DOI] [PubMed] [Google Scholar]
- 4.Meusser, B., Hirsch, C., Jarosch, E., and Sommer, T. (2005) Nat. Cell Biol. 7 766-772 [DOI] [PubMed] [Google Scholar]
- 5.Buschhorn, B. A., Kostova, Z., Medicherla, B., and Wolf, D. H. (2004) FEBS Lett. 577422 -426 [DOI] [PubMed] [Google Scholar]
- 6.Bhamidipati, A., Denic, V., Quan, E. M., and Weissman, J. S. (2005) Mol. Cell 19 741-751 [DOI] [PubMed] [Google Scholar]
- 7.Kim, W., Spear, E. D., and Ng, D. T. (2005) Mol. Cell 19753 -764 [DOI] [PubMed] [Google Scholar]
- 8.Szathmary, R., Bielmann, R., Nita-Lazar, M., Burda, P., and Jakob, C. A. (2005) Mol. Cell 19 765-775 [DOI] [PubMed] [Google Scholar]
- 9.Munro, S. (2001) Curr. Biol. 11R499 -R50111470418 [Google Scholar]
- 10.Denic, V., Quan, E. M., and Weissman, J. S. (2006) Cell 126349 -359 [DOI] [PubMed] [Google Scholar]
- 11.Carvalho, P., Goder, V., and Rapoport, T. A. (2006) Cell 126361 -373 [DOI] [PubMed] [Google Scholar]
- 12.Gauss, R., Jarosch, E., Sommer, T., and Hirsch, C. (2006) Nat. Cell Biol. 8 849-854 [DOI] [PubMed] [Google Scholar]
- 13.Bays, N. W., Gardner, R. G., Seelig, L. P., Joazeiro, C. A., and Hampton, R. Y. (2001) Nat. Cell Biol. 3 24-29 [DOI] [PubMed] [Google Scholar]
- 14.Deak, P. M., and Wolf, D. H. (2001) J. Biol. Chem. 27610663 -10669 [DOI] [PubMed] [Google Scholar]
- 15.Amano, T., Yamasaki, S., Yagishita, N., Tsuchimochi, K., Shin, H., Kawahara, K., Aratani, S., Fujita, H., Zhang, L., Ikeda, R., Fujii, R., Miura, N., Komiya, S., Nishioka, K., Maruyama, I., Fukamizu, A., and Nakajima, T. (2003) Genes Dev. 172436 -2449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kikkert, M., Doolman, R., Dai, M., Avner, R., Hassink, G., van Voorden, S., Thanedar, S., Roitelman, J., Chau, V., and Wiertz, E. (2004) J. Biol. Chem. 2793525 -3534 [DOI] [PubMed] [Google Scholar]
- 17.Fang, S., Ferrone, M., Yang, C., Jensen, J. P., Tiwari, S., and Weissman, A. M. (2001) Proc. Natl. Acad. Sci. U. S. A. 9814422 -14427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gardner, R. G., Swarbrick, G. M., Bays, N. W., Cronin, S. R., Wilhovsky, S., Seelig, L., Kim, C., and Hampton, R. Y. (2000) J. Cell Biol. 15169 -82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cruciat, C. M., Hassler, C., and Niehrs, C. (2006) J. Biol. Chem. 28112986 -12993 [DOI] [PubMed] [Google Scholar]
- 20.Campbell, R. E., Tour, O., Palmer, A. E., Steinbach, P. A., Baird, G. S., Zacharias, D. A., and Tsien, R. Y. (2002) Proc. Natl. Acad. Sci. U. S. A. 997877 -7882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hosokawa, N., Wada, I., Hasegawa, K., Yorihuzi, T., Tremblay, L. O., Herscovics, A., and Nagata, K. (2001) EMBO Rep. 2415 -422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hirao, K., Natsuka, Y., Tamura, T., Wada, I., Morito, D., Natsuka, S., Romero, P., Sleno, B., Tremblay, L. O., Herscovics, A., Nagata, K., and Hosokawa, N. (2006) J. Biol. Chem. 2819650 -9658 [DOI] [PubMed] [Google Scholar]
- 23.Morito, D., Hirao, K., Oda, Y., Hosokawa, N., Tokunaga, F., Cyr, D. M., Tanaka, K., Iwai, K., and Nagata, K. (2008) Mol. Biol. Cell 191328 -1336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hosokawa, N., Wada, I., Natsuka, Y., and Nagata, K. (2006) Genes Cells 11 465-476 [DOI] [PubMed] [Google Scholar]
- 25.Jensen, O. N., Podtelejnikov, A., and Mann, M. (1996) Rapid Commun. Mass Spectrom. 101371 -1378 [DOI] [PubMed] [Google Scholar]
- 26.Sifers, R. N., Brashears-Macatee, S., Kidd, V. J., Muensch, H., and Woo, S. L. (1988) J. Biol. Chem. 2637330 -7335 [PubMed] [Google Scholar]
- 27.Liu, Y., Choudhury, P., Cabral, C. M., and Sifers, R. N. (1999) J. Biol. Chem. 2745861 -5867 [DOI] [PubMed] [Google Scholar]
- 28.Lilley, B. N., and Ploegh, H. L. (2005) Proc. Natl. Acad. Sci. U. S. A. 10214296 -14301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Su, Y. A., Hutter, C. M., Trent, J. M., and Meltzer, P. S. (1996) Mol. Carcinog. 15 270-275 [DOI] [PubMed] [Google Scholar]
- 30.Kimura, Y., Nakazawa, M., and Yamada, M. (1998) J. Biochem. (Tokyo) 123876 -882 [DOI] [PubMed] [Google Scholar]
- 31.Litovchick, L., Friedmann, E., and Shaltiel, S. (2002) J. Biol. Chem. 27734413 -34423 [DOI] [PubMed] [Google Scholar]
- 32.Baek, J. H., Mahon, P. C., Oh, J., Kelly, B., Krishnamachary, B., Pearson, M., Chan, D. A., Giaccia, A. J., and Semenza, G. L. (2005) Mol. Cell 17 503-512 [DOI] [PubMed] [Google Scholar]
- 33.Mast, S. W., Diekman, K., Karaveg, K., Davis, A., Sifers, R. N., and Moremen, K. W. (2005) Glycobiology 15 421-436 [DOI] [PubMed] [Google Scholar]
- 34.Olivari, S., Galli, C., Alanen, H., Ruddock, L., and Molinari, M. (2005) J. Biol. Chem. 2802424 -2428 [DOI] [PubMed] [Google Scholar]
- 35.Nakayama, T., Yaoi, T., Kuwajima, G., Yoshie, O., and Sakata, T. (1999) FEBS Lett. 453 77-80 [DOI] [PubMed] [Google Scholar]
- 36.Jakob, C. A., Bodmer, D., Spirig, U., Battig, P., Marcil, A., Dignard, D., Bergeron, J. J., Thomas, D. Y., and Aebi, M. (2001) EMBO Rep. 2423 -430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakatsukasa, K., Nishikawa, S., Hosokawa, N., Nagata, K., and Endo, T. (2001) J. Biol. Chem. 2768635 -8638 [DOI] [PubMed] [Google Scholar]
- 38.Christianson, J. C., Shaler, T. A., Tyler, R. E., and Kopito, R. R. (2008) Nat. Cell Biol. 10 272-282 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.