

Abstract
A defining characteristic of solid tumors is the capacity to divide aggressively and disseminate under conditions of nutrient deprivation, limited oxygen availability, and exposure to cytotoxic drugs or radiation. Survival pathways are activated within tumor cells to cope with these ambient stresses. We here describe a survival pathway activated by the anti-cancer drug docetaxel in prostate cancer cells. Docetaxel activates STAT3 phosphorylation and transcriptional activity, which in turns induces expression of the PIM1 gene, encoding a serine-threonine kinase activated by many cellular stresses. Expression of PIM1 improves survival of docetaxel-treated prostate cancer cells, and PIM1 knockdown or expression of a dominant-negative PIM1 protein sensitize cells to the cytotoxic effects of docetaxel. PIM1 in turn mediates docetaxel-induced activation of NFκB transcriptional activity, and PIM1 depends in part on RELA/p65 proteins for its prosurvival effects. The PIM1 kinase plays a critical role in this STAT3 → PIM1 → NFκB stress response pathway and serves as a target for intervention to enhance the therapeutic effects of cytotoxic drugs such as docetaxel.
A defining characteristic of solid tumors is the capacity to divide aggressively and metastasize under conditions of nutrient deprivation and limited oxygen availability. These microenvironmental stresses arise from inadequate perfusion as the primary tumor rapidly outgrows its initial blood supply and from dramatic structural abnormalities of tumor vessels that lead to aberrant microcirculation. Survival pathways are activated within tumor cells to cope with these ambient stresses. Examples include stress pathways that respond to hypoxia (1), oxidative stress (2), and unfolded protein/endoplasmic reticulum stresses (3). In addition to these microenvironmental stresses, anti-cancer treatment can cause additional stresses to cancer cells. These added insults call forth additional responses that can augment the survival mechanisms of the malignant cells and impair overall cell kill. Key participants in stress response pathways induced by cytotoxic drugs include AKT- and other kinase-dependent pathways (4–8), NFκB2 pathways (9), and mediators of DNA repair (10).
Among the potential survival proteins in cancer cells are the PIM family of kinases, including the PIM1, PIM2, and PIM3 genes. These small, cytoplasmic serine-threonine kinases function as true oncogenes, promoting the development of cancer in animal models, either alone (11) or synergistically with other oncogenes, such as MYC (12). In normal and malignant cells, PIM kinases are highly regulated at the transcriptional level. Expression is induced by many cellular stresses, including cytokines (13), oncogenes (14), hypoxia (15), heat shock (16), and toxin exposure (17). In addition, PIM kinases are constitutively expressed in a variety of leukemias and lymphomas (18), in head and neck squamous cell carcinomas (19), and in prostate cancer (20–22). Therefore, PIM kinases may mediate in part the process of carcinogenesis. PIM kinases have been shown to promote cell survival in the face of cytokine withdrawal as well as exposure to ionizing radiation and doxorubicin (13, 23, 24). This is accomplished in part through phosphorylation of the proapoptotic protein BAD on serine 112, leading to its sequestration by 14-3-3 (25, 26). It is unknown whether PIM kinases participate in induced cytoprotective responses following treatment of cancer cells with chemotherapeutic agents. Since the PIM1 kinase has been implicated in the development or progression of prostate cancer, we have examined its role in cell responses to docetaxel, the primary cytotoxic agent used to treat prostate cancer (27, 28). We here present data showing that PIM1 expression is induced by docetaxel treatment. Furthermore, PIM1 is a key component of a survival pathway that includes STAT3 and NFκB transcriptional complexes.
EXPERIMENTAL PROCEDURES
Reagents—Docetaxel pharmaceutical grade solution (Sanofi) was diluted in unsupplemented keratinocyte medium (Invitrogen) immediately before each experiment. 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) was prepared as stock solutions in PBS. The following monoclonal antibodies were used: anti-β-ACTIN (clone AC-15; Sigma), anti-PIM1 (clone 12H8; Santa Cruz Biotechnology, Inc., Santa Cruz, CA), anti-BCLxL (clone H-5; Santa Cruz Biotechnology), anti-phospho-STAT3 (Tyr705) (clone 3E2; Cell Signaling), anti-total STAT3 (clone 84; BD Biosciences), anti-GAPDH (clone FL-335; Santa Cruz Biotechnology), anti-PRDX5 (Transduction Laboratories), and anti-human cyclin B1 (clone GNS-1; BD Biosciences).
Cell Culture and Generation of Stable Clones—RWPE-2 prostate epithelial cell lines (ATCC) were maintained in keratinocyte medium (Invitrogen) supplemented with 5 ng/ml human recombinant EGF, 0.05 mg/ml bovine pituitary extract, 100 units/ml penicillin, and 100 μg/ml streptomycin (Media-Tech). DU145 prostate cancer cells were obtained from the ATCC and grown in RPMI1640 medium with 10% fetal bovine serum.
For some experiments, we produced additional pools of prostate cells that overexpressed wild-type or dominant negative PIM1 cDNAs (23) through retroviral transduction. The coding regions for the human PIM1 gene or a dominant-negative variant (NT81) were cloned into the pLNCX retroviral vector (Clontech). To produce infectious viruses, the GP-293 packaging cell line was co-transfected with retroviral backbone plasmids (pLNCX, pLNCX/PIM1, or pLNCX/NT81) and with pVSV-G, a plasmid that expresses the envelope glycoprotein from vesicular stomatitis virus, using the calcium phosphate method. After 48 h of incubation, the medium was collected, and the virus particles were concentrated by centrifugation. Prostate cells were plated at 1 × 105 cells/60-mm plate 16–18 h before infection. Cells were infected with 5 × 104 viral particles/plate in the presence of 8 μg/ml Polybrene. After 6 h of incubation, the virus-containing medium was replaced with fresh medium, and on the next day, 400 μg/ml G418 was added to select stably infected cell populations. After 10 days of selection, stable cell pools were established, and expression of the PIM1 transgenes was verified by Western blot analysis.
For reporter gene assays, RWPE-2 cells stably expressing a NFκB-luciferase reporter plasmid were prepared. The parental cell line was co-transfected with the reporter gene plasmid (Stratagene) and a puromycin resistance plasmid. Puromycin-resistant clones were screened for expression of firefly luciferase in response to stimulation with tumor necrosis factor α (Peprotech). Two highly responsive clones were combined to create a pool. In some experiments, this pool of reporter cells was further infected with PIM1-encoding retroviruses, as described above, and further pools were selected by treatment of the cultures with G418.
Determination of Cell Viability and Apoptosis—To determine cell survival following docetaxel treatment, both short term (MTT) and long term (regrowth) assays were used. For the former, cells were seeded into 96-well plates (1–2 × 104 cells/well) and allowed to adhere overnight. Docetaxel was added, and the cells were incubated for various periods of time. Metabolically active cells were measured by the MTT assay. For regrowth assays, cells were plated at 5 × 104/well of 12-well plates and allowed to adhere overnight. Docetaxel was then added for 24 h. Cells were subsequently trypsinized, and dilutions were plated in fresh medium in 24-well plates and allowed to grow for 6–7 days. Cell numbers were then enumerated by crystal violet staining (29).
To measure caspase activation following docetaxel treatment, the carboxyfluorescein FLICA apoptosis detection kit was used (Immunochemistry Technologies). The stained cells were analyzed with a FACScalibur flow cytometer.
DNA Histogram Analysis—After docetaxel treatment for 24 h, the floating and adherent cells were harvested and combined, washed with PBS, and then fixed with cold 70% ethanol and stored at 4 °C. The cells were then washed with PBS and were resuspended in 1 ml of PBS containing 25 μg/ml propidium iodide, 0.1% Triton X-100, and 40 μg/ml RNase A. After incubation for at least 30 min at 4 °C, the cells were then analyzed by FACScalibur flow cytometer using channel FL3.
Luciferase Reporter Assays—Cells (4 × 104/well) were plated in 24-well plates and allowed to adhere overnight. Cells then were untreated or not with docetaxel and incubated for 6 h. The level of luciferase expression was determined in triplicate using a luciferase assay system (Promega) according to the manufacturer's protocol. The luminescent signal was recorded using a plate luminometer (Berthold Technologies). Luciferase activity was normalized to total protein concentrations, as measured by the Bradford method.
Western Blotting—Cells (5–7 × 105) were washed with cold PBS and lysed in 100 μl of lysis buffer (20 mm Tris-HCl, pH 7.5, 1% SDS, 50 mm NaCl, 1 mm EDTA supplied with 1 mm phenylmethylsulfonyl fluoride and protease inhibitor mixture Set V (Calbiochem)). The lysates were sonicated and the protein concentration was measured using the BCA™ Protein assay kit (Pierce). Up to 70 μg of total protein/lane were subjected to 12% SDS-PAGE and transferred to polyvinylidene membranes. The membranes were blocked with 5% skimmed milk in TBST (20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 0.1% Tween 20) and then incubated overnight in 5% skimmed milk or 5% bovine serum albumin in TBST with primary antibodies (dilution 1:1000) at 4 °C with constant shaking. After washing with TBST, the membranes were exposed to peroxidase-coupled secondary antibodies for 1 h at room temperature. Membranes then washed again with TBST. Detection of the protein was performed by using the chemiluminescent SuperSignal West Femto or Pico Maximum Sensitivity substrate (Pierce).
Real Time PCR—Total RNA was extracted with TRIzol reagent (Invitrogen) and single-stranded cDNA was constructed by Superscript III polymerase (Invitrogen) and oligo(dT) primers. Real time PCR was performed using iCycler (Bio-Rad) and SYBR Green PCR master mix reagents (Qiagen). The following primers were used: PIM1 forward, 5′-AACTGGTCTTCCTTTTTGGTT-3′; PIM1 reverse, 5′-TACCATGCCAACTGTACACAC-3′; CFL (cofilin) forward, 5′-GAGCAAGAAGGAGGATCTGGT-3′; CFL reverse, 5′-CAATTCATGCTTGATCCCTGT-3′. The PIM1 primer concentration was 2 μm, and the CFL (cofilin) primer concentration was 0.3 μm per reaction.
STAT3 Decoy and Mutant Control Decoy Oligonucleotide Treatment—The STAT3 decoy and mutant decoy oligonucleotides utilized previously described sequences (30). RWPE-2 cells were seeded into 6-well plates (5–7 × 105 cells/well) and allowed to grow. Twenty-four hours later, the cells were treated with STAT3 decoy oligonucleotide (50 nm) or mutant control oligonucleotide (50 nm) using TransIT®-OligoTransfection Reagent (Mirus). Incubation times of cells with decoy oligonucleotides varied between experiments (see figure legends).
siRNA Studies—In some cases (NFκB siRNA studies), cells were transfected with NFKB1 (p50) siRNA, RELA (p65) siRNA, or control siRNA (Santa Cruz Biotechnology). One day prior to transfection, 5 × 105 cells/well were seeded in 6-well plates. Twenty-four hours later, the cells were transfected with siRNAs using the TransIT-TKO® Transfection Reagent (Mirus) and incubated overnight. The cells were then trypsinized, counted, and plated into 24-well plates (5–7 × 104/well) for luciferase assay, performed 24 h later (48 h after transfection). Alternately, for immunoblot analysis, the cells were plated in 6-well plates, transfected with siRNAs, and lysed after 48 h after transfection. For docetaxel treatment, the cells were seeded into a 96-well plate (1–2 × 104 cell/well, 100-μl total volume) and allowed to adhere for 12 h. They were then transfected with siRNAs using TransIT-TKO® transfection reagent (Mirus). Twenty-four hours later, docetaxel (100 nm) was added to the cells, and incubation continued for 48 h. The MTT assay was then performed.
Alternately (PIM1 siRNA studies), specific and control siRNA sequences were cloned into pSILENCER (Ambion) plasmid and used for transfection. The PIM1-targeting sequence was 5′-AACATCCTTATCGACCTCAATCGCG-3′, and the control sequence was 5′-GCCTACCGTCAGGCTATCGCGTATC-3′. Plasmids were transiently transfected into RWPE-2 cells with a Nucleofector device (Amaxa) and incubated for 24 h. Then cells were trypsinized and replated with a density of 5 × 105 cells/well in 6-well plates for immunoblot assay and 2 × 104 cells/well into a 96-well plate for cell viability analysis. The next day, 48 h after transfection, 100 nm docetaxel was applied, and then the cells were incubated for an additional 6 h, lysed, and used for an imunoblotting assay to detect PIM1 knockdown. Alternatively, for the cell survival assay, 100 nm docetaxel was added for 24, 48, or 72 h. The cell viability was measured with an MTT assay.
Prostate Cancer Xenografts—Studies were carried out under an Institutional Animal Care and Use Committee-approved protocol. Male NCR nu/nu mice were implanted subcutaneously with 106 DU145 cells, and tumors were allowed to form. Tumor-bearing mice (n = 4) were treated with docetaxel (15 mg/kg) or an equal volume of DMSO. Twenty hours later, the mice were sacrificed, and the tumors were excised and processed for histology and for RNA and protein extraction. Part of the tumor was placed immediately into RNALater solution (Ambion) and stored at –20 °C until RNA extraction with Trizol reagent. Another tumor fragment was minced and ground in cold 1% SDS/Tris, pH 7.5, with protease inhibitors. The proteins were then precipitated with 4 volumes of cold acetone. The pellet was then redissolved in the 1% SDS buffer, and protein concentration was measured. Thirty micrograms was used per gel lane for immunoblot analysis.
RESULTS
Docetaxel Increases Expression of PIM1 mRNA and Protein in Prostate Epithelial Cell Lines—To investigate the effect of docetaxel on the expression of the PIM1 kinase, we treated RWPE-2 prostate epithelial cells with pharmacological concentrations of docetaxel that approximate those observed in plasma within 24 h after drug administration. Docetaxel induced expression of the kinase protein by 3 h, with maximum expression between 6 and 12 h, and then a decline to nearly base-line levels thereafter (Fig. 1A). Quantitative analysis of the densitometry data showed that PIM1 expression increased up to 6.25-fold during this interval. The increase was statistically significant at 3, 6, and 9 h and less significant at later time points. Similar results were seen with either 10 nm (data not shown) or 100 nm docetaxel concentrations.
FIGURE 1.

PIM1 expression is induced by docetaxel in RWPE-2 cells. A, cells were treated with 100 nm docetaxel for the indicated times. PIM1 and β-ACTIN proteins were analyzed by immunoblot analysis. One of three similar blots is shown. Ratio, ratio of PIM1/β-ACTIN from pooled densitometry data from three separate experiments, each normalized to that of untreated cells. p value (**), probability of no difference in ratios (treated versus untreated cells) by paired t test (n = 3). B, cells were treated with 10 or 100 nm docetaxel for the indicated time. Real time PCR was used to measure PIM1 mRNA. Each value represents the mean ± S.D. of nine pooled measurements produced by three independent experiments. Bars, relative -fold increase of PIM1 RNA level (normalized to the RNA level of the housekeeping gene cofilin), compared with untreated control (0 h). **, p < 0.01. p values were calculated by t tests and represent the probability of no difference between the treated and untreated values.
To explore whether docetaxel-mediated induction of PIM1 expression was transcriptionally regulated, real time reverse transcription-PCR analysis was used (Fig. 1B). Docetaxel induced up-regulation of the PIM1 transcript level by 2–4-fold in RWPE-2 cells treated with either 10 or 100 nm drug.
RWPE cells are immortalized and transformed from normal prostate epithelium. To determine if other human prostate cancer cells showed docetaxel-induced up-regulation of PIM1, we studied DU145 cells in culture and as xenografts in immunodeficient mice (Fig. 2). DU145 cells also showed time-dependent up-regulation of PIM1 protein in response to docetaxel treatment (Fig. 2A). Onset of the response was similar to that seen in RWPE cells. However, elevated levels of PIM1 protein persisted and indeed increased at least to 24 h after drug addition. Mice with DU145 xenografts were also treated with docetaxel or vehicle (DMSO) by intraperitoneal injection (Fig. 2B). Tumors harvested 20 h after drug administration showed a marked increase in PIM1 protein, compared with loading control proteins GAPDH and PRDX5. In addition, real time PCR analysis of tumor RNA showed a significant increase in human PIM1 mRNA in the tissue from drug-treated mice (Fig. 2C).
FIGURE 2.
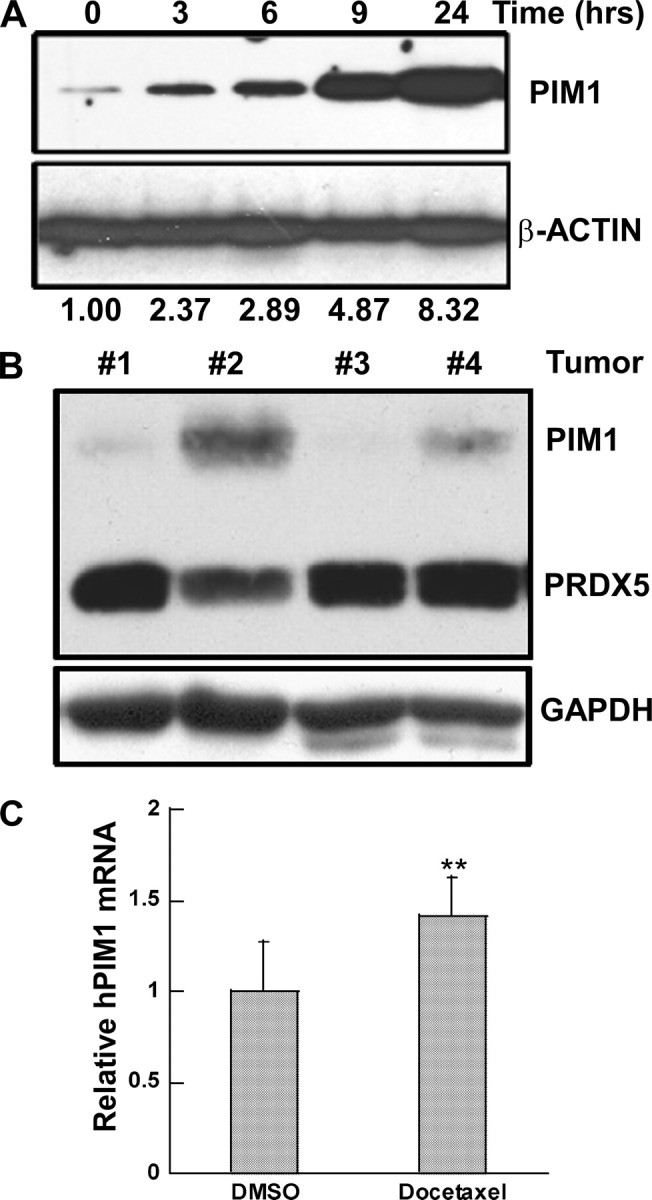
PIM1 expression is induced by docetaxel in DU145 cells. A, cells were treated with docetaxel (100 nm) for the indicated time and then analyzed by immunoblot analysis for PIM1 and β-ACTIN proteins. Ratio, ratio of PIM1/β-ACTIN from densitometry analysis, normalized to that of untreated cells. B, immunoblot analysis of PIM1, PRDX5, and GAPDH proteins in lysates of DU145 tumor tissue. Tumors 1 and 3 were from mice treated with 0.1 ml of DMSO intraperitoneally. Tumors 2 and 4 were from mice treated with docetaxel, 15 mg/kg in 0.1 ml of DMSO intraperitoneally. The upper panel was probed sequentially with antibodies to the 33-kDa PIM1 protein and the 17-kDa PRDX5 protein. The blot was then stripped and probed with antibody to the GAPDH protein. C, real time PCR analysis of human PIM1 mRNA in DU145 tumor tissue. Equal amounts of RNA from tumors 1 and 3 were mixed as a DMSO-treated pool, as were tumors 2 and 4 (docetaxel-treated pool), followed by reverse transcription and amplification. Each bar is the mean ± S.D. of six pooled measurements from two independent experiments. **, p < 0.01 that the increased PIM1 mRNA following docetaxel treatment was the result of chance, calculated by paired t test.
Previous studies suggest that the PIM1 protein increases during the G2/M phase of the cell cycle (31). Since docetaxel treatment has been reported to cause G2/M arrest, it was possible that the increase in PIM1 protein that accompanies drug treatment might merely reflect a change in cell cycle distribution. We used DNA histogram analysis to identify changes in cell cycle distributions in RWPE-2 cells after docetaxel treatment (Fig. 3A). There was no overall increase in the G2/M cell population after 24 h of low dose (10 nm) docetaxel treatment, compared with vehicle-treated cells (p = 0.31 for no difference, based on six independent experiments). A large increase in G2/M cells was observed after treatment of RWPE-2 cells with a higher concentration (100 nm) of docetaxel for 24 h. Variable G2/M arrest was confirmed by immunoblotting to detect expression of cyclin B1 (a G2/M phase marker). There was no change in cyclin B1 expression within 24 h after 10 nm docetaxel treatment, but a time-dependent increase of cyclin B1 protein was apparent after 100 nm docetaxel exposure (Fig. 3B, right). During both treatments, however, PIM1 expression increased between 3 and 12 h of exposure, independent of the extent of G2/M arrest and cyclin B1 expression.
FIGURE 3.

Independence of PIM1 expression and cell cycle arrest. A, DNA histogram analysis of RWPE-2 cells after docetaxel 10 or 100 nm treatments for 24 h. sG1, a sub-G1 cell population with less than 2 n DNA content. G1 and G2, the appearance of cells in G0/G1 or G2/M phases of the cell cycle. B, immunoblot analysis of cyclin B1 and PIM1 expression after docetaxel 10 nm (B, left) or 100 nm (B, right) treatment at various time points.
Endogenous and Enhanced Expression of PIM1 Protects Prostate Epithelial Cells from Docetaxel-induced Cell Death and Apoptosis—To determine whether PIM1 can protect prostate cells from docetaxel-triggered cell death, we infected RWPE-2 and DU145 cells with retroviruses encoding a PIM1 cDNA (pLNCX/PIM1) or an empty retrovirus (pLNCX). Pools of stably transduced cells were selected, treated with docetaxel for up to 72 h, and then analyzed by MTT assay to measure metabolically active cells. Enforced expression of wild-type PIM1 kinase was able to consistently improve survival of RWPE-2 and DU145 cells, as reflected by the MTT assay, at time points up to 72 h after the start of docetaxel exposure (Fig. 4).
FIGURE 4.
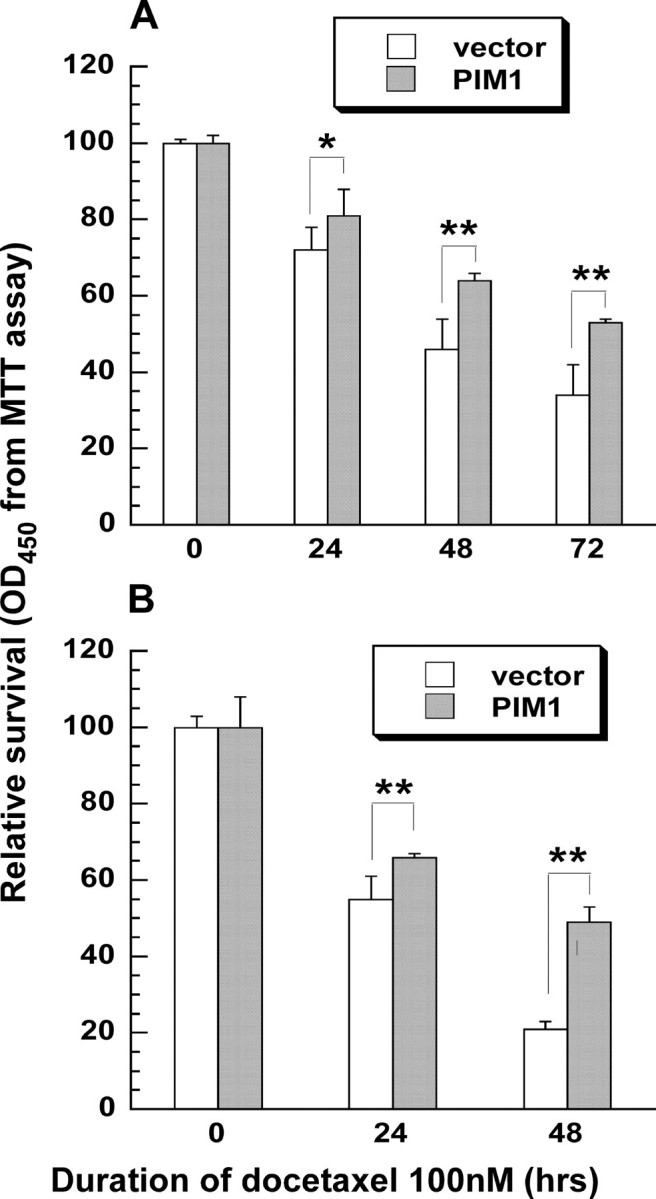
PIM1 expression protects prostate cells from docetaxel-induced cell death. Cells transfected with pLNCX (vector) or pLNCX/PIM1 (PIM1) constructs were treated with docetaxel for up to 72 h, and then cell viability was determined by the MTT assay. Results were normalized to the values for untreated cells. Each value represents the mean ± S.D. of nine measurements pooled from three independent experiments. p values were determined by t tests for comparisons between PIM1- and vector-transfected cells treated similarly. **, p < 0.01, indicating that the chance for no difference between PIM1- and vector-transfected cells was less than 1%. *, p < 0.05. A, RWPE-2 cells. B, DU145 cells.
To determine if ambient levels of PIM1 can protect prostate cells from docetaxel toxicity, we transiently introduced plasmids encoding control and PIM1-specific siRNA sequences into target cells. Control siRNA was unable to block the docetaxel-induced increase in PIM1 expression. In contrast, PIM1 siRNA substantially prevented the increase in kinase expression following drug exposure (Fig. 5, A and C). Down-regulation of endogenous PIM1 kinase expression led to enhanced cell kill up to 72 h after drug application (Fig. 5, B and D). The drug sensitization was statistically significant at every time point. To confirm the protective effect of endogenous PIM1 kinase, we also introduced a dominant negative enzyme (PIM1/NT81) into RWPE-1 and RWPE-2 cells by retroviral transduction. This truncated protein was expressed well (supplemental Fig. 1S). As was seen with the knockdown experiments, the NT81 mutant kinase also sensitized cells to the cytotoxic effect of docetaxel. These experiments clearly demonstrate that ambient levels of PIM1 are protective against docetaxel-induced cell death.
FIGURE 5.

Knockdown of PIM1 expression with siRNA sensitizes prostate cells to docetaxel-induced cell death. A, RWPE-2 cells were transfected with plasmids encoding control siRNA or PIM1 siRNA and treated with 100 nm docetaxel for 6 h. Expression of PIM1 andβ-ACTIN proteins was analyzed by immunoblotting. B, RWPE-2 cell viability measured with an MTT assay after docetaxel treatment for up to 72 h. C, as in A but with DU145 cells. D, as in B but with DU145 cells treated for up to 48 h. Each value is the mean ± S.D. of nine measurements pooled from three independent experiments. p values were determined by t tests for comparisons between PIM1 siRNA- and control siRNA-treated cells.
Docetaxel has previously been shown to induce cell death in part by apoptosis (32–35). Therefore, we measured caspase activation by a fluorescent caspase activity assay in drug-treated cells as an index of docetaxel cytotoxicity. The wild-type PIM1 kinase decreased drug-induced caspase activation, consistent with its previously demonstrated survival activity (supplemental Fig. 2S). The dominant negative PIM1 kinase markedly enhanced drug-induced caspase activation.
The docetaxel effect reflected by the MTT and caspase assays was not great, and its reversal by PIM1 expression, although statistically significant, was still quantitatively modest. These data reflect the fact that docetaxel does not produce massive, immediate apoptotic cell death. To better measure the protective effects of PIM1 kinase on the proliferative potential of docetaxel-treated cancer cells, we used a regrowth assay (Fig. 6). RWPE-2/PIM1 and RWPE-2/NT81 cells were treated with various concentrations of docetaxel for 24 h and then were trypsinized and plated in fresh medium (without drug) and allowed to grow for 6–7 days. Cell growth was then quantified by staining with crystal violet dye. Docetaxel produced dose-dependent inhibition of growth in both cell lines. However, growth inhibition was up to 8-fold greater in the RWPE-2/NT81 cells, particularly at drug concentrations of 5 nm or higher. Thus, the presence of biologically active PIM1 kinase markedly inhibited docetaxel-induced cell death.
FIGURE 6.

PIM1 kinase protects RWPE-2 prostate cells with proliferative potential from docetaxel cytotoxicity. RWPE-2/PIM1 (wild-type PIM1) and RWPE-2/NT81 cells (dominant negative PIM1) were treated with docetaxel for 24 h. Additional cultures of the same cells were treated similarly but without docetaxel. Cells were then trypsinized, diluted, replated in fresh medium, and allowed to grow for 6–7 days. Cultures were then fixed with formalin and stained with crystal violet to measure cell growth. Comparisons of the regrowth of treated cultures were made with similar, untreated cells, but comparisons were always made between wells with A570 values on the linear part of a cell density standard curve. Each point represents the mean ± S.D. of 9–18 measurements pooled from two independent experiments. Untreated cell cultures were assigned a relative regrowth value of 1.0.
The STAT3 Transcription Factor Mediates Induction of PIM1 by Docetaxel—To identify mechanisms by which docetaxel could induce PIM1 expression, we examined the activation status of STAT3 and STAT5 transcriptional factors, known mediators of PIM1 transcription, after docetaxel treatment of RWPE-2 cells. STAT5 was not consistently phosphorylated in RWPE-2 cells (data not shown). The level of phospho-STAT3 (Tyr705) was strongly and rapidly increased after 10 and 100 nm treatment of RWPE-2 cells (Fig. 7A) (data not shown), whereas the total amount of STAT3 protein was not changed. Docetaxel induced phosphorylation of STAT3 simultaneously with up-regulation of PIM1 expression. These results suggested that docetaxel-induced expression of PIM1 may be dependent of activation of the STAT3 transcriptional factor.
FIGURE 7.

PIM1 expression following docetaxel treatment is dependent on STAT3. A, immunoblots of RWPE-2 cells treated with 100 nm docetaxel for the indicated time. B, RWPE-2 cells were transfected with STAT3 mutant control oligonucleotides or STAT3 decoy oligonucleotides and incubated for 16 h. Cells were then treated with 100 nm docetaxel for 6 h, lysed, and analyzed by immunoblotting for PIM1, phospho-STAT3, and total STAT3.
To determine if docetaxel induces PIM1 expression in a STAT3-dependent manner, we used double-stranded STAT3 decoy oligonucleotides (30) to selectively abrogate STAT3 transcriptional activity. RWPE-2 cells were incubated with wild-type or mutant sequence STAT3 decoys for 48 h. PIM1 expression was then analyzed by immunoblotting (supplemental Fig. 3S). STAT3 decoys, but not mutant decoys, decreased PIM1 expression, as well as expression of the known STAT3 target gene BCLxL. These results demonstrate that STAT3 transcriptional activity controlled basal PIM1 gene expression in RWPE-2 prostate cells. STAT3 decoy treatment was not associated with decreased levels of either STAT3 protein or tyrosine-phosphorylated STAT3.
To further define the role of STAT3 transcriptional activity in docetaxel-dependent PIM1 expression, we treated RWPE-2 cells with STAT3 or mutant decoy oligonucleotides for 18 h. Docetaxel was then added for an additional 6 h. As shown (Fig. 7B), the STAT3 decoy did not prevent docetaxel-induced phosphorylation of STAT3 but did inhibit the effect of the drug on PIM1. In contrast, the mutant oligonucleotides had no effect on PIM1 expression. These results identify STAT3 as an upstream mediator through which docetaxel induces expression of the PIM1 kinase.
Docetaxel Activates NFκB Transcriptional Activity in a PIM1-dependent Manner—Inhibition of the NFκB transcriptional complex sensitizes prostate cancer cells to paclitaxel (another taxane) and enhances drug-induced apoptosis (36). We hypothesized that the protective role of PIM1 in docetaxel-induced apoptosis could be mediated through activation of NFκB transcriptional activity as well. We initially investigated the effect of PIM1 expression on NFκB transcriptional activity. RWPE-2 cells stably expressing an NFκB-dependent luciferase expression plasmid were infected with retroviruses encoding PIM1 or empty retrovirus only. Enhanced expression of PIM1 consistently increased NFκB transcriptional activity about 2-fold (supplemental Fig. 4S).
We then treated the NFκB reporter cell line with docetaxel. Cells were incubated for 6 h with docetaxel and then were assayed for luciferase activity. Docetaxel increased NFκB-directed luciferase expression in a concentration-dependent manner (Fig. 8A). Co-expression of a dominant negative PIM1 protein substantially blocked drug-induced activation of NFκB transcriptional activity at each docetaxel concentration.
FIGURE 8.

Protective effect of PIM1 against docetaxel cytotoxicity depends in part on NFκB transcriptional activity. A, expression of a dominant negative PIM1 (NT81) decreases docetaxel-induced activation of NFκB. RWPE-2 cells stably expressing an NFκB-luciferase reporter gene were infected with retroviruses carrying pLNCX vector or pLNCX/NT81 constructs. Pools were selected with G418 and then treated for 6 h with docetaxel. Luciferase activity was determined. Each bar represents the mean ± S.D. of triplicate determinations of one of three similar experiments. p values were calculated by t tests. **, p < 0.01, showing that the chance of no difference in luciferase activity between vector and NT81-transduced cells is less than 1%. B, p50/NFKB1 and p65/RELA siRNAs inhibit NFκB transcriptional activity in RWPE-2 cells with high or low PIM1 expression. RWPE-2/NFκB-luciferase/PIM1 cells were transfected with control siRNA, siRNAs targeting p50/NFKB1 (si p50), or p65/RELA (si p65). After 48 h, the luciferase activity was measured and compared with that of RWPE-2/NFκB-luciferase/pLNCX cells transfected similarly. Each bar represents the relative luciferase activity of the various cells compared with that of vector-transduced cells treated with control siRNA. The values are the mean ± S.D. of six measurements pooled from two independent experiments. **, p ≤ 0.01 for no difference; *, indicates p < 0.05 for no difference. C, inhibition of NFκB activation by siRNA increases docetaxel-induced cell death. RWPE-2/pLNCX and RWPE-2/PIM1 cells were transfected with the indicated siRNAs and allowed to rest for 24 h. Docetaxel (100 nm) was then added for 48 h. Cell survival was then estimated by MTT assay. Each bar represents the mean ± S.D. of six measurements pooled from two independent experiments. p values were calculated by t test and represent comparisons between PIM1-expressing cells and vector control cells as well as among cells treated with different types of siRNAs. D, MTT survival data from C for docetaxel-treated cells are represented following normalization of the data to the values for control siRNA-treated cells. In this analysis, survival of cells transfected with NFκB-targeting siRNAs is shown as a percentage of the values for the same cells treated with control siRNA. Each bar presents the mean ± S.D. of six measurements pooled from two independent experiments. p values were calculated by t test and represent the likelihood that there is no difference in the sensitizing effect of the siRNA between vector- and PIM1-transduced cells.
The Protective Effect of PIM1 Expression from Docetaxel-induced Death Depends in Part on NFκB Activation—To determine if PIM1 enhances survival of docetaxel-treated cells through NFκB activation, we used siRNA to inhibit expression of the RELA (p65) and NFKB1 (p105, p50) proteins, the two components of the major NFκB complex. Fig. 8B showed that basal and PIM1-dependent activation of NFκB was decreased by p65/RELA and p50/NFKB1 siRNAs. Immunoblotting confirmed the knockdown of the corresponding p65/RELA and p50/NFKB1 proteins (supplemental Fig. 5S).
A survival analysis, based on the MTT assay, was then performed on docetaxel-treated cells (Fig. 8, C and D). With all siRNA treatments, RWPE-2/PIM1 cells showed improved survival compared with that of cells infected with pLNCX virus alone (Fig. 8C). The p65/RELA and p50/NFKB1 siRNAs reduced survival of both cell lines. The p50/NFKB1 siRNA did not significantly impair the survival of docetaxel-treated RWPE-2/pLNCX cells, whereas it did have a significant effect on RWPE-2/PIM1 cells. In contrast, p65/RELA siRNAs significantly enhanced docetaxel cell kill in both cell lines. These data suggested that cells with high expression of PIM1 (RWPE-2/PIM1) might be more sensitive to the effects of NFκB siRNAs than were cells with low levels of PIM1 (RWPE-2/pLNCX). We then reanalyzed the data by normalizing the survival of p65/RELA and p50/NFKB1 siRNA-treated cells to that of cells treated with docetaxel and control siRNA (Fig. 8D). The p65/RELA and p50/NFKB1 siRNAs enhanced docetaxel-induced cell kill of RWPE-2/PIM1 cells to a greater extent than they enhanced kill of RWPE-2/pLNCX (vector only) cells. This enhancement was of borderline significance for p50/NFKB1 siRNA (p = 0.057) but was highly significant for p65/RELA siRNA. These results demonstrate that the p65/RELA and p50/NFKB1 proteins mediate resistance to docetaxel cell kill. Their effects are more pronounced in prostate cells with higher PIM1 levels that in similar cells with lower amounts of PIM1. These data demonstrate that the ability of PIM1 to decrease docetaxel-induced cell kill depends in part on the p65/RELA, and possibly the p50/NFKB1, protein.
DISCUSSION
The present study assessed the up-regulation of PIM1 expression following docetaxel treatment of prostate epithelial cells. The drug effect was seen in both engineered and spontaneously transformed prostate cancer cells. Furthermore, the effect was documented in both cultured cells and tumor xenografts, suggesting that it is a physiologically significant response. Apoptosis is involved in the antitumor effects of docetaxel, both in cultured cells and in clinical settings (34, 35, 37). Our results demonstrated that PIM1 inhibited docetaxel-induced apoptosis. Recent work has indicated that other modes of cell death may also contribute significantly to the overall therapeutic response to docetaxel (33). Whether PIM1 modulates these other forms of docetaxel-induced cell death requires further investigation.
Cellular stressors are known to activate survival pathways. Among these stressors are a wide variety of antineoplastic agents, such as cytotoxic drugs (including taxanes (6, 7, 10, 39)), tyrosine- and serine-threonine kinase inhibitors (4, 5), and triterpenes, such as betulinic acid (38). These agents are capable of transiently activating kinases and other survival mediators, such as AKT, ERK1, and NFκB transcriptional activity. It appears that drug-induced activation of survival signaling pathways can impair the cytotoxic effects of chemotherapy drugs both in vivo and in vitro (9, 40), and inhibition of activated kinases can potentiate cytotoxic drug cell kill (40–44).
Our data document the existence of a STAT3 → PIM1 → NFκB survival pathway that is activated by docetaxel and mediates a form of docetaxel resistance. The linear relationships among the pathway components were established by temporal correlations as well as by blocking experiments using siRNAs, dominant negative proteins, and oligonucleotide decoys. Resistance to docetaxel has previously been ascribed to tubulin mutations (45) as well as to MDR-dependent effects (46, 47) and to limited tissue penetration (48). Fewer data exist to implicate transient or acquired resistance mediated through survival pathways. A previous report has shown that stable overexpression of STAT1 is associated with docetaxel resistance in prostate cancer cell lines (49). In addition, genetic inhibition of EGFR expression has been shown to sensitize head and neck cancer cells to docetaxel (50). The involvement of PIM kinases in induced resistance to cytotoxic drugs has not been documented thus far. However, activation of AKT has often been described (6, 51). The PIM kinases and AKT kinases have been described as mediating separate but parallel survival pathways (52). At times they also phosphorylate the same substrates. Thus, the involvement of PIM kinases in induced resistance to cytotoxic drugs may be anticipated in cells where the kinase is expressed.
DU145 cells showed a more prolonged PIM1 response following docetaxel treatment than did RWPE-2 cells. This may reflect the greater degree of transformation in the DU145 cells, which are hyper-diploid and form tumors readily. Such cells might have constitutive activation of multiple signaling pathways. For this reason, we performed mechanistic studies in the weakly transformed, nearly diploid RWPE-2 cells, which may offer a simpler cancer model.
The mechanism through which docetaxel activates the STAT3 → PIM1 → NFκB pathway is unknown at present. Docetaxel induces an increase in reactive oxygen species (ROS), as do many cytotoxic drugs (53). This form of oxidizing stress inhibits phosphatase activity, leading to an increase in tyrosine phosphorylation of multiple proteins (54–56). Transactivation of receptor-type tyrosine kinases (such as the EGFR) has been shown in cells stressed by ROS and by cytotoxic agents, including paclitaxel (57–59). Docetaxel can transactivate the EGFR, and EGFR inhibitors can act synergistically with taxanes to enhance cancer cell kill (43, 51). However, we continue to see expression of pSTAT3 or PIM1 proteins following docetaxel treatment of RWPE-2 cells pretreated with an EGFR inhibitor (data not shown). ROS have previously been shown to activate JAK kinase signaling in some cell lines, possibly providing a mechanism for STAT activation as well (60, 61). ROS can also activate STAT proteins without JAK kinase activation (62). Regardless of the most proximal mediators, activated STAT3 is a known mediator of ROS-induced survival signals. Furthermore, STAT transcription factors are known upstream mediators of PIM1 transcription, at least in hematopoietic cells (63–65). Our data demonstrate that STAT3 regulates PIM1 expression in prostate cells as well. The decoy studies establish a linear relationship between STAT3 and PIM1 as downstream mediators of docetaxel survival signals. Since prostate cancer cells frequently express activated STAT3 and PIM1, this relationship may occur constitutively as well (66–68).
Our identification of a drug-induced signaling pathway leading to NFκB activation is consistent with the known effects of docetaxel (69–71). Although many prostate cancer cell lines show constitutive activation of NFκB transcriptional complexes, docetaxel can further increase NFκB transcriptional activity (70). Our studies indicate that, in RWPE-2 cells, docetaxel activates NFκB in a PIM1-dependent manner. Previous reports have shown that the related PIM2 kinase can activate NFκB activity (72), although alternative opinions about the PIM1 kinase have been presented (73). PIM2 activates NFκB activity through phosphorylation and activation of the COT/TPL2 kinase, a kinase with known IκB kinase-like activity (72). Clarification as to whether the PIM1 kinase acts through this mechanism or through another pathway will require further studies.
A decrease in NFκB expression or activity would be predicted to increase docetaxel-induced cell death in both RWPE-2/pLNCX and RWPE-2/PIM1 cell lines (36), and this was in fact seen (Fig. 8, B and C). However, cells with higher expression of the PIM1 kinase were more sensitive to the blockage of NFκB function (Fig. 8D). Compared with their effects in RWPE-2/pLNCX cells, p65/RELA siRNAs were significantly more effective at potentiating docetaxel-induced death in RWPE-2/PIM1 cells. P50/NFKB1 siRNAs were also more active against cells with high levels of PIM1, but the effect was of borderline significance. These data suggest that the prosurvival effect of PIM1 kinase in docetaxel-treated cells probably involves members of the NFκB transcriptional complex, particularly p65/RELA. The observation that inhibition of NFκB only partially enhances docetaxel-induced cell death in PIM1-expressing cells is consistent with the ability of the kinase to protect cells through other mechanisms as well as the incomplete knockdown of the target protein in RWPE-2 cells. Nevertheless, the result demonstrates that PIM1, like PIM2 (72), can mediate NFκB activation and that PIM1 also requires NFκB transcriptional activity for the development of the full drug resistance phenotype.
The survival response induced by low concentrations of docetaxel is reminiscent of the concept of hormesis. A controversial body of literature documents that stressors (including radiation, gases, toxins, exercise, and others) can produce biphasic dose-response curves in various assay systems (74, 75). At low doses, a protective (hormetic) response is generated, whereas at high doses, toxicity is the result. Hormesis has been invoked to explain the beneficial effects of calorie restriction, exercise, and various phytochemicals in disease prevention. In many cancer cells lines, cytotoxic agents also generate a classic biphasic hormetic dose-response curve (76–78). Fig. 6 demonstrates the same phenomenon in our experimental system. There is a 24% increase in regrowth/survival of RWPE-2/PIM1 cells treated with low concentrations (0.5–1.0 nm) of docetaxel, compared with the survival of untreated RWPE-2/PIM1 cells (p < 0.001). In contrast, survival of RWPE-2/NT81 cells treated similarly is worse than that of RWPE-2/PIM1 cells, and there is no enhancement of survival at low drug concentrations. Our data strongly suggest that the PIM1 kinase participates in cytotoxic drug-induced hormesis. PIM1 is also increased in response to a wide variety of cellular stressors: growth factors, oncogenes, heat, radiation, toxins, oxidative stress, and hypoxia. Thus, one may postulate that PIM1 is a general mediator of hormesis, protective stress responses induced by low level environmental stresses. Recently, small molecule inhibitors of the PIM kinases have been described in vitro and in cell-based systems (79–81). Targeting the PIM1 kinase may be a beneficial addition to a traditional docetaxel-based chemotherapy regimen. However, it will be important to determine if the same maneuver will increase normal tissue toxicity as well.
Supplementary Material
The work was supported by Department of Defense/CDMRP Prostate Cancer Program Award W81XWH-04-1-0087. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1S–5S.
Footnotes
The abbreviations used are: NFκB, nuclear factor κB; PBS, phosphate-buffered saline; MTT, 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; siRNA, short interfering RNA.
References
- 1.Welsh, S. J., Koh, M. Y., and Powis, G. (2006) Semin. Oncol. 33 486–497 [DOI] [PubMed] [Google Scholar]
- 2.Dairkee, S. H., Nicolau, M., Sayeed, A., Champion, S., Ji, Y., Moore, D. H., Yong, B., Meng, Z., and Jeffrey, S. S. (2007) Oncogene 26 6269–6279 [DOI] [PubMed] [Google Scholar]
- 3.Feldman, D. E., Chauhan, V., and Koong, A. C. (2005) Mol. Cancer Res. 3 597–605 [DOI] [PubMed] [Google Scholar]
- 4.Burchert, A., Wang, Y., Cai, D., von Bubnoff, N., Paschka, P., Muller-Brusselbach, S., Ottmann, O. G., Duyster, J., Hochhaus, A., and Neubauer, A. (2005) Leukemia 19 1774–1782 [DOI] [PubMed] [Google Scholar]
- 5.Shi, Y., Yan, H., Frost, P., Gera, J., and Lichtenstein, A. (2005) Mol. Cancer Ther. 4 1533–1540 [DOI] [PubMed] [Google Scholar]
- 6.di Palma, A., Matarese, G., Leone, V., Di Matola, T., Acquaviva, F., Acquaviva, A. M., and Ricchi, P. (2006) Mol. Cancer Ther. 5 1318–1324 [DOI] [PubMed] [Google Scholar]
- 7.Ling, X., Bernacki, R. J., Brattain, M. G., and Li, F. (2004) J. Biol. Chem. 279 15196–15203 [DOI] [PubMed] [Google Scholar]
- 8.Okano, J., and Rustgi, A. K. (2001) J. Biol. Chem. 276 19555–19564 [DOI] [PubMed] [Google Scholar]
- 9.Cusack, J. C., Jr., Liu, R., and Baldwin, A. S., Jr. (2000) Cancer Res. 60 2323–2330 [PubMed] [Google Scholar]
- 10.Anderson, K. M., Alrefai, W. A., Anderson, C. A., Ho, Y., Jadko, S., Ou, D., Wu, Y. B., and Harris, J. E. (2002) Anticancer Res. 22 75–81 [PubMed] [Google Scholar]
- 11.van Lohuizen, M., Verbeek, S., Krimpenfort, P., Domen, J., Saris, C., Radaszkiewicz, T., and Berns, A. (1989) Cell 56 673–682 [DOI] [PubMed] [Google Scholar]
- 12.Verbeek, S., van Lohuizen, M., van der Valk, M., Domen, J., Kraal, G., and Berns, A. (1991) Mol. Cell Biol. 11 1176–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lilly, M., Le, T., Holland, P., and Hendrickson, S. L. (1992) Oncogene 7 727–732 [PubMed] [Google Scholar]
- 14.Nieborowska-Skorska, M., Hoser, G., Kossev, P., Wasik, M. A., and Skorski, T. (2002) Blood 99 4531–4539 [DOI] [PubMed] [Google Scholar]
- 15.Teh, B. G. (2004) Hokkaido J. Med. Sci. 79 19–26 [PubMed] [Google Scholar]
- 16.Shay, K. P., Wang, Z., Xing, P. X., McKenzie, I. F., and Magnuson, N. S. (2005) Mol. Cancer Res. 3 170–181 [DOI] [PubMed] [Google Scholar]
- 17.DaSilva, L., Cote, D., Roy, C., Martinez, M., Duniho, S., Pitt, M. L., Downey, T., and Dertzbaugh, M. (2003) Toxicon 41 813–822 [DOI] [PubMed] [Google Scholar]
- 18.Amson, R., Sigaux, F., Przedborski, S., Flandrin, G., Givol, D., and Telerman, A. (1989) Proc. Natl. Acad. Sci. U. S. A. 86 8857–8861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beier, U. H., Weise, J. B., Laudien, M., Sauerwein, H., and Gorogh, T. (2007) Int. J. Oncol. 30 1381–1387 [PubMed] [Google Scholar]
- 20.Dhanasekaran, S. M., Barrette, T. R., Ghosh, D., Shah, R., Varambally, S., Kurachi, K., Pienta, K. J., Rubin, M. A., and Chinnaiyan, A. M. (2001) Nature 412 822–826 [DOI] [PubMed] [Google Scholar]
- 21.Valdman, A., Fang, X., Pang, S. T., Ekman, P., and Egevad, L. (2004) Prostate 60 367–371 [DOI] [PubMed] [Google Scholar]
- 22.Xu, Y., Zhang, T., Tang, H., Zhang, S., Liu, M., Ren, D., and Niu, Y. (2005) J. Surg. Oncol. 92 326–330 [DOI] [PubMed] [Google Scholar]
- 23.Lilly, M., Sandholm, J., Cooper, J. J., Koskinen, P. J., and Kraft, A. (1999) Oncogene 18 4022–4031 [DOI] [PubMed] [Google Scholar]
- 24.Pircher, T. J., Zhao, S., Geiger, J. N., Joneja, B., and Wojchowski, D. M. (2000) Oncogene 19 3684–3692 [DOI] [PubMed] [Google Scholar]
- 25.Yan, B., Zemskova, M., Holder, S., Chin, V., Kraft, A., Koskinen, P. J., and Lilly, M. (2003) J. Biol. Chem. 278 45358–45367 [DOI] [PubMed] [Google Scholar]
- 26.Aho, T. L., Sandholm, J., Peltola, K. J., Mankonen, H. P., Lilly, M., and Koskinen, P. J. (2004) FEBS Lett. 571 43–49 [DOI] [PubMed] [Google Scholar]
- 27.Tannock, I. F., de Wit, R., Berry, W. R., Horti, J., Pluzanska, A., Chi, K. N., Oudard, S., Theodore, C., James, N. D., Turesson, I., Rosenthal, M. A., and Eisenberger, M. A. (2004) N. Engl. J. Med. 351 1502–1512 [DOI] [PubMed] [Google Scholar]
- 28.Petrylak, D. P., Tangen, C. M., Hussain, M. H., Lara, P. N., Jr., Jones, J. A., Taplin, M. E., Burch, P. A., Berry, D., Moinpour, C., Kohli, M., Benson, M. C., Small, E. J., Raghavan, D., and Crawford, E. D. (2004) N. Engl. J. Med. 351 1513–1520 [DOI] [PubMed] [Google Scholar]
- 29.Kueng, W., Silber, E., and Eppenberger, U. (1989) Anal. Biochem. 182 16–19 [DOI] [PubMed] [Google Scholar]
- 30.Leong, P. L., Andrews, G. A., Johnson, D. E., Dyer, K. F., Xi, S., Mai, J. C., Robbins, P. D., Gadiparthi, S., Burke, N. A., Watkins, S. F., and Grandis, J. R. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 4138–4143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liang, H., Hittelman, W., and Nagarajan, L. (1996) Arch. Biochem. Biophys. 330 259–265 [DOI] [PubMed] [Google Scholar]
- 32.Paoletti, A., Giocanti, N., Favaudon, V., and Bornens, M. (1997) J. Cell Sci. 110 2403–2415 [DOI] [PubMed] [Google Scholar]
- 33.Morse, D. L., Gray, H., Payne, C. M., and Gillies, R. J. (2005) Mol. Cancer Ther. 4 1495–1504 [DOI] [PubMed] [Google Scholar]
- 34.Kramer, G., Schwarz, S., Hagg, M., Havelka, A. M., and Linder, S. (2006) Br. J. Cancer 94 1592–1598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hernandez-Vargas, H., Palacios, J., and Moreno-Bueno, G. (2007) Oncogene 26 2902–2913 [DOI] [PubMed] [Google Scholar]
- 36.Flynn, V., Jr., Ramanitharan, A., Moparty, K., Davis, R., Sikka, S., Agrawal, K. C., and Abdel-Mageed, A. B. (2003) Int. J. Oncol. 23 317–323 [PubMed] [Google Scholar]
- 37.Kraus, L. A., Samuel, S. K., Schmid, S. M., Dykes, D. J., Waud, W. R., and Bissery, M. C. (2003) Invest. New Drugs 21 259–268 [DOI] [PubMed] [Google Scholar]
- 38.Qiu, L., Wang, Q., Di, W., Jiang, Q., Schefeller, E., Derby, S., Wanebo, H., Yan, B., and Wan, Y. (2005) Int. J. Oncol. 27 823–830 [PubMed] [Google Scholar]
- 39.Katoh, M., Koninkx, J., and Schumacher, U. (2000) Cancer Lett. 161 113–120 [DOI] [PubMed] [Google Scholar]
- 40.Van Schaeybroeck, S., Kyula, J., Kelly, D. M., Karaiskou-McCaul, A., Stokesberry, S. A., Van Cutsem, E., Longley, D. B., and Johnston, P. G. (2006) Mol. Cancer Ther. 5 1154–1165 [DOI] [PubMed] [Google Scholar]
- 41.Sirotnak, F. M., Zakowski, M. F., Miller, V. A., Scher, H. I., and Kris, M. G. (2000) Clin. Cancer Res. 6 4885–4892 [PubMed] [Google Scholar]
- 42.Ohta, T., Ohmichi, M., Hayasaka, T., Mabuchi, S., Saitoh, M., Kawagoe, J., Takahashi, K., Igarashi, H., Du, B., Doshida, M., Mirei, I. G., Motoyama, T., Tasaka, K., and Kurachi, H. (2006) Endocrinology 147 1761–1769 [DOI] [PubMed] [Google Scholar]
- 43.Pu, Y. S., Hsieh, M. W., Wang, C. W., Liu, G. Y., Huang, C. Y., Lin, C. C., Guan, J. Y., Lin, S. R., and Hour, T. C. (2006) Biochem. Pharmacol. 71 751–760 [DOI] [PubMed] [Google Scholar]
- 44.Herbst, R. S., Oh, Y., Wagle, A., and Lahn, M. (2007) Clin. Cancer Res. 13 (suppl.) s4641–s4646 [DOI] [PubMed] [Google Scholar]
- 45.Hari, M., Loganzo, F., Annable, T., Tan, X., Musto, S., Morilla, D. B., Nettles, J. H., Snyder, J. P., and Greenberger, L. M. (2006) Mol. Cancer Ther. 5 270–278 [DOI] [PubMed] [Google Scholar]
- 46.Burns, B. S., Edin, M. L., Lester, G. E., Tuttle, H. G., Wall, M. E., Wani, M. C., and Bos, G. D. (2001) Clin. Orthop. Relat. Res. 383 259–267 [DOI] [PubMed] [Google Scholar]
- 47.Baker, S. D., Sparreboom, A., and Verweij, J. (2006) Clin. Pharmacokinet. 45 235–252 [DOI] [PubMed] [Google Scholar]
- 48.Kyle, A. H., Huxham, L. A., Yeoman, D. M., and Minchinton, A. I. (2007) Clin. Cancer Res. 13 2804–2810 [DOI] [PubMed] [Google Scholar]
- 49.Patterson, S. G., Wei, S., Chen, X., Sallman, D. A., Gilvary, D. L., Zhong, B., Pow-Sang, J., Yeatman, T., and Djeu, J. Y. (2006) Oncogene 25 6113–6122 [DOI] [PubMed] [Google Scholar]
- 50.Nozawa, H., Tadakuma, T., Ono, T., Sato, M., Hiroi, S., Masumoto, K., and Sato, Y. (2006) Cancer Sci. 97 1115–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Takabatake, D., Fujita, T., Shien, T., Kawasaki, K., Taira, N., Yoshitomi, S., Takahashi, H., Ishibe, Y., Ogasawara, Y., and Doihara, H. (2007) Int. J. Cancer 120 181–188 [DOI] [PubMed] [Google Scholar]
- 52.Hammerman, P. S., Fox, C. J., Birnbaum, M. J., and Thompson, C. B. (2005) Blood 105 4477–4483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lin, H. L., Liu, T. Y., Chau, G. Y., Lui, W. Y., and Chi, C. W. (2000) Cancer 89 983–994 [PubMed] [Google Scholar]
- 54.Natarajan, V., Scribner, W. M., al-Hassani, M., and Vepa, S. (1998) Environ Health Perspect. 106 Suppl. 5, 1205–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kappert, K., Sparwel, J., Sandin, A., Seiler, A., Siebolts, U., Leppanen, O., Rosenkranz, S., and Ostman, A. (2006) Arterioscler. Thromb. Vasc. Biol. 26 2644–2651 [DOI] [PubMed] [Google Scholar]
- 56.Fiaschi, T., Buricchi, F., Cozzi, G., Matthias, S., Parri, M., Raugei, G., Ramponi, G., and Chiarugi, P. (2007) Hepatology 46 130–139 [DOI] [PubMed] [Google Scholar]
- 57.Frank, G. D., and Eguchi, S. (2003) Antioxid. Redox Signal. 5 771–780 [DOI] [PubMed] [Google Scholar]
- 58.Chen, C. H., Cheng, T. H., Lin, H., Shih, N. L., Chen, Y. L., Chen, Y. S., Cheng, C. F., Lian, W. S., Meng, T. C., Chiu, W. T., and Chen, J. J. (2006) Mol. Pharmacol. 69 1347–1355 [DOI] [PubMed] [Google Scholar]
- 59.Qiu, L., Di, W., Jiang, Q., Scheffler, E., Derby, S., Yang, J., Kouttab, N., Wanebo, H., Yan, B., and Wan, Y. (2005) Int. J. Oncol. 27 1441–1448 [PubMed] [Google Scholar]
- 60.Simon, A. R., Rai, U., Fanburg, B. L., and Cochran, B. H. (1998) Am. J. Physiol. 275 C1640–C1652 [DOI] [PubMed] [Google Scholar]
- 61.Park, S. K., Kim, J., Seomun, Y., Choi, J., Kim, D. H., Han, I. O., Lee, E. H., Chung, S. K., and Joo, C. K. (2001) Biochem. Biophys. Res. Commun. 284 966–971 [DOI] [PubMed] [Google Scholar]
- 62.Liu, T., Castro, S., Brasier, A. R., Jamaluddin, M., Garofalo, R. P., and Casola, A. (2004) J. Biol. Chem. 279 2461–2469 [DOI] [PubMed] [Google Scholar]
- 63.Demoulin, J. B., Van Roost, E., Stevens, M., Groner, B., and Renauld, J. C. (1999) J. Biol. Chem. 274 25855–25861 [DOI] [PubMed] [Google Scholar]
- 64.Matikainen, S., Sareneva, T., Ronni, T., Lehtonen, A., Koskinen, P. J., and Julkunen, I. (1999) Blood 93 1980–1991 [PubMed] [Google Scholar]
- 65.Stout, B. A., Bates, M. E., Liu, L. Y., Farrington, N. N., and Bertics, P. J. (2004) J. Immunol. 173 6409–6417 [DOI] [PubMed] [Google Scholar]
- 66.Horinaga, M., Okita, H., Nakashima, J., Kanao, K., Sakamoto, M., and Murai, M. (2005) Urology 66 671–675 [DOI] [PubMed] [Google Scholar]
- 67.Huang, H. F., Murphy, T. F., Shu, P., Barton, A. B., and Barton, B. E. (2005) Mol. Cancer 4 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gao, L., Zhang, L., Hu, J., Li, F., Shao, Y., Zhao, D., Kalvakolanu, D. V., Kopecko, D. J., Zhao, X., and Xu, D. Q. (2005) Clin. Cancer Res. 11 6333–6341 [DOI] [PubMed] [Google Scholar]
- 69.Nakahara, C., Nakamura, K., Yamanaka, N., Baba, E., Wada, M., Matsunaga, H., Noshiro, H., Tanaka, M., Morisaki, T., and Katano, M. (2003) Clin. Cancer Res. 9 5409–5416 [PubMed] [Google Scholar]
- 70.Domingo-Domenech, J., Oliva, C., Rovira, A., Codony-Servat, J., Bosch, M., Filella, X., Montagut, C., Tapia, M., Campas, C., Dang, L., Rolfe, M., Ross, J. S., Gascon, P., Albanell, J., and Mellado, B. (2006) Clin. Cancer Res. 12 5578–5586 [DOI] [PubMed] [Google Scholar]
- 71.Li, J., Minnich, D. J., Camp, E. R., Brank, A., Mackay, S. L., and Hochwald, S. N. (2006) J. Surg. Res. 132 112–120 [DOI] [PubMed] [Google Scholar]
- 72.Hammerman, P. S., Fox, C. J., Cinalli, R. M., Xu, A., Wagner, J. D., Lindsten, T., and Thompson, C. B. (2004) Cancer Res. 64 8341–8348 [DOI] [PubMed] [Google Scholar]
- 73.Yan, B., Wang, H., Kon, T., and Li, C. Y. (2006) Braz. J. Med. Biol. Res. 39 169–176 [DOI] [PubMed] [Google Scholar]
- 74.Calabrese, E. J., Bachmann, K. A., Bailer, A. J., Bolger, P. M., Borak, J., Cai, L., Cedergreen, N., Cherian, M. G., Chiueh, C. C., Clarkson, T. W., Cook, R. R., Diamond, D. M., Doolittle, D. J., Dorato, M. A., Duke, S. O., Feinendegen, L., Gardner, D. E., Hart, R. W., Hastings, K. L., Hayes, A. W., Hoffmann, G. R., Ives, J. A., Jaworowski, Z., Johnson, T. E., Jonas, W. B., Kaminski, N. E., Keller, J. G., Klaunig, J. E., Knudsen, T. B., Kozumbo, W. J., Lettieri, T., Liu, S. Z., Maisseu, A., Maynard, K. I., Masoro, E. J., McClellan, R. O., Mehendale, H. M., Mothersill, C., Newlin, D. B., Nigg, H. N., Oehme, F. W., Phalen, R. F., Philbert, M. A., Rattan, S. I., Riviere, J. E., Rodricks, J., Sapolsky, R. M., Scott, B. R., Seymour, C., Sinclair, D. A., Smith-Sonneborn, J., Snow, E. T., Spear, L., Stevenson, D. E., Thomas, Y., Tubiana, M., Williams, G. M., and Mattson, M. P. (2007) Toxicol. Appl. Pharmacol. 222 122–128 [DOI] [PubMed] [Google Scholar]
- 75.Liu, G., Gong, P., Bernstein, L. R., Bi, Y., Gong, S., and Cai, L. (2007) Crit. Rev. Toxicol. 37 587–605 [DOI] [PubMed] [Google Scholar]
- 76.Calabrese, E. J. (2005) Crit. Rev. Toxicol. 35 463–582 [DOI] [PubMed] [Google Scholar]
- 77.Lechanteur, C., Jacobs, N., Greimers, R., Benoit, V., Deregowski, V., Chariot, A., Merville, M. P., and Bours, V. (2005) Oncogene 24 1788–1793 [DOI] [PubMed] [Google Scholar]
- 78.Yang, P., He, X. Q., Peng, L., Li, A. P., Wang, X. R., Zhou, J. W., and Liu, Q. Z. (2007) J. Toxicol. Environ. Health A 70 976–983 [DOI] [PubMed] [Google Scholar]
- 79.Bullock, A. N., Debreczeni, J. E., Fedorov, O. Y., Nelson, A., Marsden, B. D., and Knapp, S. (2005) J. Med. Chem. 48 7604–7614 [DOI] [PubMed] [Google Scholar]
- 80.Holder, S., Lilly, M., and Brown, M. L. (2007) Bioorg. Med. Chem. 15 6463–6473 [DOI] [PubMed] [Google Scholar]
- 81.Pogacic, V., Bullock, A. N., Fedorov, O., Filippakopoulos, P., Gasser, C., Biondi, A., Meyer-Monard, S., Knapp, S., and Schwaller, J. (2007) Cancer Res. 67 6916–6924 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.