

Abstract
Mutations in the gene encoding the purine biosynthetic enzyme hypoxanthine–guanine phosphoribosyltransferase (HPRT) cause the intractable neurodevelopmental Lesch–Nyhan disease (LND) associated with aberrant development of brain dopamine pathways. In the current study, we have identified an increased expression of the microRNA miR181a in HPRT-deficient human dopaminergic SH-SY5Y neuroblastoma cells. Among the genes potentially regulated by miR181a are several known to be required for neural development, including Engrailed1 (En1), Engrailed2 (En2), Lmx1a and Brn2. We demonstrate that these genes are down-regulated in HPRT-deficient SH-SY5Y cells and that over-expression of miR181a significantly reduces endogenous expression of these genes and inhibits translation of luciferase plasmids bearing the En1/2 or Lmx1a 3′UTR miRNA-binding elements. Conversely, inhibition of miR181a increases the expression of these genes and enhances translation of luciferase constructs bearing the En1/2 and Lmx1a 3′UTR miRNA-binding sequences. We also demonstrate that key neurodevelopmental genes (e.g. Nurr1, Pitx3, Wnt1 and Mash1) known to be functional partners of Lmx1a and Brn2 are also markedly down-regulated in SH-SY5Y cells over-expressing miR181a and in HPRT-deficient cells. Our findings in SH-SY5Y cells demonstrate that HPRT deficiency is accompanied by dysregulation of some of the important pathways that regulate the development of dopaminergic neurons and dopamine pathways and that this defect is associated with and possibly due at least partly to aberrant expression of miR181a. Because aberrant expression of miR181a is not as apparent in HPRT-deficient LND fibroblasts, the relevance of the SH-SY5Y neuroblastoma cells to human disease remains to be proven. Nevertheless, we propose that these pleiotropic neurodevelopment effects of miR181a may play a role in the pathogenesis of LND.
INTRODUCTION
The housekeeping enzyme hypoxanthine–guanine phosphoribosyltransferase (HPRT) catalyzes a key step in the salvage of the purine bases hypoxanthine and guanine in the purine biosynthetic pathways of mammalian cells. Complete HPRT deficiency in humans causes the devastating and intractable Lesch–Nyhan disease (LND) characterized by hyperuricemia, dystonia and the hallmark aberrant neurobehavioral effect of self-mutilation (1–5). The HPRT gene is generally thought of as a ‘housekeeping’ gene and is expressed constitutively in most tissues, apparently without a high degree of developmental regulation. The abnormal neurological CNS phenotype of LND largely reflects aberrant basal ganglia function, including dopamine depletion and defective dopamine (DA) uptake (6,7). Unlike Parkinson disease, which also is associated with basal ganglia dopamine deficiency, the dopamine deficit in LND does not result from a major degree of DA neuronal degeneration since such cells are present in relatively normal numbers and have a relatively normal distribution in both human patients and the HPRT-knockout mouse (8,9). However, we have only a very incomplete understanding of the mechanisms by which the purine defects produce the neurological phenotype. Initial clues to the identity of genes aberrantly expressed in HPRT deficiency came from early gene expression studies in HPRT-deficient LND fibroblasts and in the striatum of HPRT-knockout mice (10), but those studies did not define responsible pathways for neural dysfunction.
More informative results were provided by recent studies demonstrating that HPRT deficiency and other kinds of purine metabolic derangement lead to dysregulated expression of transcription factors necessary for the development of dopaminergic neurons in mouse MN9D dopaminergic and human NT2 embryonic carcinoma cells undergoing differentiation in culture (11–13). In addition, a recent report identified transcriptional aberrations in HPRT-deficient human neural stem cells (14). We have also recently demonstrated that HPRT deficiency leads to dysregulation of key signaling pathways vital for early embryogenesis and neural development, such as Wnt-1 and presenilin (15). Even though these studies have identified some possible mechanisms responsible for neural aberrations in HPRT deficiency, a more thorough understanding of the neural dysfunction in HPRT deficiency will require a more detailed characterization of the complex interactions among other genetic networks involved in developmental gene regulation.
Toward that goal, we set out to determine whether dysregulated microRNA-mediated control mechanisms are responsible for aberrant expression of transcription factors necessary for dopaminergic (DA) neuron development. The expression and function of regulatory microRNAs has been extensively reviewed (16–20). MicroRNAs bind to sequences in the 3′UTR of many genes and regulate their expression largely by interfering with translation or (and) by initiating mRNA degradation. Many hundreds of microRNAs have been identified in mammalian systems (21–24) and because thousands of genes are known to contain exact or potential miRNA binding sites within their 3′UTR regions, a given miRNA can potentially regulate many, even hundreds of genes. This regulatory mechanism confers the enormous regulatory advantages by allowing coordinate regulation of multiple genes (25,26). The potentially pleiotropic effects of dysregulated miRNA expression may be particularly troublesome in the CNS in which genetic and cellular regulatory mechanisms are likely to be much more complicated and inter-dependent than in other organs. Indeed, aberrant miRNA expression has been reported in several CNS diseases, including schizophrenia, Alzheimer' disease and autism-related disorders (27–32).
In the current study, we have examined the microRNA expression profile in HPRT-deficient fibroblasts derived from patients with LND and HPRT-knockdown SH-SY5Y human neuroblastoma cells and we report that HPRT deficiency in these cells leads to dysregulation of miRNA-181a. We also provide evidence that abnormal expression of miR181a causes downstream abnormalities in the expression of multiple genes responsible for dopamine neuron development and differentiation. We postulate that dysregulation of miR181a may be responsible at least partly for the abnormal brain development in HPRT deficiency.
RESULTS
MicroRNA expression
In an initial preliminary study, we used miRNA array analysis (see Materials and Methods) to characterize miRNA expression in a single culture of primary classical LND fibroblasts compared with a normal HPRT+ human fibroblast culture [human dermal fibroblast (HDF), ScienCell Research Laboratories, Carlsbad, CA, USA]. Results from that preliminary examination (Table 1) identified several miRNAs that suggested altered expression of >1.5-fold (P < 0.05) in the LND cells compared with that of the normal control fibroblast culture, including mir-138-1*, miR181a, miR-601 and miR-765. Analysis by DAVID (database annotation visualization of integrated discovery) of the gene ontology (GO) terms of the potential target genes for those four miRNAs revealed that miR181a demonstrated the lowest P-value (4.1E − 03 Benjamini corrected) for the ‘neuro-related’ GO term (0048666). Using the combined microRNAs target gene databases generated by TargetCombo and TargetScan analysis methods (http://www.diana.pcbi.upen.edu/cgi-bin/TargetCombo.cgi; TargetScan.org), we identified approximately 1171 genes that contain 3′UTR potential miR181a-binding sites and that therefore constitute potential targets for miR181a regulation. Analysis by DAVID of those 1171 genes generated 364 gene clusters containing GO term categories (30,31) having similar biological significance. Supplementary Material, Table S2 indicates 47 genes that represent the cluster related to neurological processes with the GO term ‘neuron development’ (GO; 0048666). Of particular interest is the fact that potential target genes of miR181a include key transcription factors Lmx1a, En1, En2 and Pou3f2 (Brn2) that are known to be vital for the development of the dopaminergic neurons and dopaminergic pathways. Because our previous studies indicated dysregulated expression of these genes in differentiating MN9D neuroblastoma (12) and NT2 teratocarcinoma cells (11) and because of the potential for a regulatory role of miR181a for these genes, we selected miR181a for further analysis of HPRT-deficient human cells.
Table 1.
Partial list of microRNAs differentially expressed by at least 1.5-fold in primary fibroblast culture of a classical LND patient (HT) compared with control HDF
| MicroRNAs | Number of target genes | Number of clusters | P-value (neuro-related GO term) | P-value (corrected Benjamini) |
|---|---|---|---|---|
| mir-138-1* | 293 | 125 | 1.1E − 02 | 0.28 |
| miR181a | 841 | 278 | 4.4E − 05 | 4.1E − 03 |
| mir-601 | 54 | 17 | NA | NA |
| mir-765 | 183 | 71 | 0.71 | 1 |
Duplicate arrays were applied to each sample using an array platform containing three sub-array replicates (n = 6 for each patient). Each sub-array contained 1073 microRNA probes along with small nuclear RNA, mismatch and dye controls (see Materials and Methods). Target genes and clusters for the four microRNAs miR-138-1*, miR-181a, miR-601 and miR-765 were generated by TargetScan and DAVID.
HPRT knockdown in SH-SY5Y cells
To avoid the expected variability of miR181a expression in different human LND fibroblast samples and to focus more on a cell type more likely to be directly informative of functions relevant to neurogenesis and to brain development than a dermal fibroblast, we examined miR181a expression in normal and HPRT-knockdown derivatives of human SH-SY5Y cells, a model system used for studies of neurogenesis and differentiation (33). For HPRT knockdown, we used a retrovirus (retrosh2hprt) that expresses a short hairpin targeted to HPRT that we have shown in previous studies to produce a highly efficient reduction of HPRT expression in human fibroblasts (15). In that study, we reported that knockdown of HPRT with a retrovirus vector results in a reduction of ∼90% in the transcription of HPRT compared with transduction with an identically designed knockdown retrovirus vector targeted to luciferase (retroshlux). Figure 1A demonstrates that knockdown with the same vectors in SH-SY5Y neuroblastoma cells results in an almost complete (>98%) loss of HPRT gene expression (Fig. 1A). Other studies demonstrated that the knockdown cells also showed a marked but less complete reduction of HPRT expression by western immunoblot analysis (Supplementary Material, Fig. S1). In our earlier study, we demonstrated a similar phenomenon of a greater degree of transcriptional down-regulation than reduced HPRT protein levels in HPRT-knockdown cells (15).
Figure 1.

(A) Knockdown of HPRT expression in SH-SY5Y cells by infection with retrovirus (retrosh2hprt) expressing an shRNA targeted to HPRT (closed bar) compared with vector retroshlux expressing an shRNA targeted to luciferase (open bar). (B) Quantitative determination of expression level of miRNA-181a in SH-SY5Y cells transduced with control luciferase vector retroshlux (open bar) and HPRT-knockdown vector retrosh2hprt (closed bars). Bars represent mean ± SEM of triplicate PCR measurements for cDNA preparations from WT-Lux and HPRT-KD cells (n = 6, *P < 0.05, t-test).
miR181a over-expression in HPRT-deficient SH-SY5Y cells
Figure 1B illustrates the change in the expression of miR181a in SH-SY5Y cells in which HPRT had been transduced with the retrovirus short hairpin RNA (shRNA) vector targeted to HPRT (retrosh2hprt) compared with those infected with the control anti-luciferase vector retroshlux. The >98% knockdown of HPRT gene expression in SH-SY5Y cells determined by quantitative PCR (qPCR) methods resulted in an ∼3-fold over-expression of miR181a (P < 0.05, t-test) (Fig. 1B).
Down-regulated expression of potential miR181a target genes in HPRT-knockdown SH-SY5Y cells
To characterize the effect of HPRT deficiency on expression of the potential miR181a targets genes En1, En2, Lmx1a and Brn2 in SH-SY5Y cells, we performed qPCR and western blot analysis of their expression in parent and HPRT-knockdown cells (Fig. 2). Figure 2A indicates that expression of all four of these genes is significantly down-regulated (P<0.05, t-test) in HPRT-deficient SH-SY5Y cells compared with wild-type cells. Figure 2B and C demonstrates results of the western blot analysis for the same genes and indicates that the reduced expression of these genes shown by qPCR assays is paralleled by reduced expression of the gene products. There is good correlation between qualitative (Fig. 2B) and quantitative analyses (Fig. 2C) of the western blots.
Figure 2.

(A) qPCR assay of expression of miR181a neurodevelopmental target genes En1, En2, Lmx1a and Brn2 in wild-type (open bar) and HPRT-deficient (closed bars) SH-SY5Y cells. HPRT deficiency leads to robust down-regulation of En1, En2, Lmx1a and Brn2. Bars represent mean ± SEM of normalized mRNA levels. cDNA samples for each gene were measured in triplicate and repeated once (*P < 0.05, t-test, n = 6). (B) Representative western blot analysis of expression of miR181a target genes En, Lmx1a and Brn2 in wild-type cells transduced with control luciferase knockdown vector retroshlux (left lane) and HPRT-deficient SH-SY5Y cells transduced with retrosh2hprt (right lane). (C) Densitometric analysis of the relative protein amounts of En1, Lmx1a and Brn2 in wild-type (open bar) and HPRT-deficient (closed bar) SH-SY5Y cells. Measurements were carried out in duplicate for each sample (n = 2, *P < 0.05, t-test). Results indicate that HPRT knockdown in SH-SY5Y cells significantly reduces expression of En1, En2, Brn2 and Lmx1a as measured by both qPCR and western blotting methods.
Over-expression of miR181a in SH-SY5Y cells
For further documentation of regulation of target genes En1, En2, Lmx1a and Brn2 by miR181a, we used retrovirus vectors to over-express GFP alone (vector MDH-PGK-GFP) or to co-express GFP and mR181a (vector MDH1-hmiR181a-PGK-GFP) in native SH-SY5Y cells. For further analysis, GFP-positive cells were enriched by fluorescence-activated cells sorting (FACS), as described in Materials and Methods, to produce a population of GFP-positive cells of >99% homogeneity (data not presented). Figure 3A demonstrates that cells transduced with the vector MDH1-hmiR181a-PGK-GFP demonstrated a >10-fold increase in miR181a expression compared with cells transduced with the MDH1-PGK-GFP vector expressing GFP alone. SH-SY5Y cells over-expressing the transduced miR181a also showed a significant down-regulated expression of target genes En1, En2, Lmx1a, and Brn2, as measured by qPCR analysis (Fig. 3B, P < 0.05, t-test). To determine the functional consequences of transcriptional down-regulation of these miR181a target genes, we used western blot methods to determine the level of the protein products in SH-SY5Y cells over-expressing transduced miR181a. Figure 3C and D illustrates protein expression level of En1, Lmx1a and Brn2 after transduction with the retrovirus vectors expressing either GFP only (vector MDH1-PGK-GFP) or co-expressing GFP and miR181a (MDH1-hmiR181a-PGK-GFP). Expression of En1, Lmx1a and Brn2 is markedly reduced qualitatively (Fig. 3C) and quantitatively (Fig. 3D) in cells over-expressing miR181a from the vector MDH1-hmiR181a-PGK-GFP compared with the expression found in cells infected with the GFP-expressing vector MDH1-PGK-GFP.
Figure 3.

(A) qPCR demonstration of the mean over-expression of miR181a in SH-SY5Y cells transduced with retroviral vector MDH1-hmiR181a-PGK-GFP co-expressing GFP and miR181a (closed bars) compared with cells transduced with MDH-PGK-GFP expressing GFP alone (opened bars). Error bars indicate triplicate measurements repeated once for each sample normalized to U6. The asterisk denotes significant difference between the samples (n = 6, *P < 0.05, t-test). (B) qPCR determination of mean mRNA levels of putative miR181a target genes En1, En2, Lmx1a and Brn2 in cells expressing GFP alone (opened bars) and GFP together with miR181a (closed bars). Error bars represent ±SEM of triplicate measurements repeated once of mean mRNA levels normalized to TBP. Asterisks denote significant differences (n = 6, *P < 0.05, t-test). (C) Western blot analysis of the translation of En1, Lmx1a and Brn2 genes in cells infected with a retroviral vector MDH-PGK-GFP encoding GFP alone (left lane) and in cells infected with retroviral vector MDH1-hmiR181a-PGK-GFP co-expressing GFP and miR181a (right lane). (D) Densitometry analysis of the mean protein levels of En1/2 and Lmx1a in SH-SY5Y cells expressing GFP alone (opened bars) or GFP together with miR181a (closed bars). Samples were measured in triplicate and error bars represent ±SEM and asterisks denote significance (n = 3, *P < 0.05, t-test).
Anti-miR181a inhibitor up-regulates En1, En2, Lmx1a and Brm2 gene expression
To test the possible causal role of miR181a over-expression in down-regulating En1, En2, Lmx1a and Brn2 expression, we examined the effect of miR181a inhibition on the expression of En1, En2, Lmx1a and Brn2, using either a specific anti-miR181a inhibitor or the miR-ZIP™ lentivirus-based anti-microRNAs transduction (see Materials and Methods). Figure 4A demonstrates that exposure of cells to 20 µm of anti-miR181a results in a reduced expression of miR181a compared with the control inactive (scrambled) inhibitor and with an associated increased expression of the miR181a target genes En1, En2, Lmx1a and Brn2. Treatment of cells with the miR-ZIP lentivirus-based anti-microRNAs leads to enhanced protein expression of the miR181a target genes at both the mRNA and protein levels (Fig. 4B–D).
Figure 4.

Effect of miR181a inhibition on expression of miR181a and its target genes. (A) qPCR measurement of mean levels of miR181a in SH-SY5Y cells transfected either with 20 µm anti-miR181a inhibitor (closed bar) or with 20 µm anti-miR181a negative control (opened bar). Error bars represent ±SEM and the asterisk denotes significance (n = 4, *P < 0.05, t-test). (B) qPCR analysis of the expression of En1, En2, Lmx1a and Brn2 in SH-SY5Y cells expressing control anti-miR control (open bar) and in cells expressing anti-miRNA inhibitor of human miR181a (closed bar). Statistical analysis is as in (A). (C) Western blot analysis of En1 and Lmx1a proteins in cells infected with a lentivirus expressing a control (scramble) inhibitor for miR-Zip vector (left lane) and in cells infected with lentivirus miR-ZIP expressing miR-Zip-181a inhibitor (right lane). (D) Mean densitometric measurements of the levels of En1, Lmx1a and Brn2 proteins in SH-SY5Y cells expressing a control inhibitor (open bars) or miR-ZIP-181a inhibitor (closed bar) expressed from the miR-Zip lentivirus vector. Error bars represent ±SEM (n = 2, *P < 0.05, t-test).
miR181a interacts with 3′UTR regions of En1/2 and Lmx1a
To determine whether the effect of miR181a is an authentic miRNA effect and is mediated by the expected miRNA mechanism of interaction with the 3′UTR miRNA-binding sites of target genes, we used an established luciferase reporter assay to test the interaction of miR181a with luciferase-expressing plasmids containing exactly matched wild-type or mutated 3′UTR En1/2 or Lmx1a-binding sites (Fig. 5A). Luciferase plasmids pMIR-report-CMV-3′UTR (Ambion) containing the exact or mutated 3′UTR-binding sites were co-transfected into HEK 293 cells with plasmid constructs expressing the retrovirus vectors MDH1-PGK-GFP or MDH1-hmir-181A-PGK-GFP (Fig. 5B) or with anti-miR-control or anti-miR-181a (Fig. 5C). Normalized luciferase expression was analyzed 48h post-transfection. Results were analyzed by one-way ANOVA and post hoc Tukey t-tests. The bars in Figure 5B represent ratios of luciferase expression in cells transfected with MDH1-PGK-GFP (not expressing miR181a) to luciferases activity in cells transfected with MDH1-hmiR181a-PGK (expressing miR181a). Luciferase expression was reduced only in the case of transfection of authentic 3′UTR-binding site sequences (EN or Lmx1a, filled bars) but not of a mutated 3′UTR sequence (EN-mu or Lmx1a-mu, shaded bars). For both En1/2 and Lmx1a, luciferase expression is significantly reduced (filled bars) by miR181a expression in cells transfected with wild-type 3′UTR sequences compared with plasmids containing mutated 3′UTR sequences of En (ANOVA, P = 0.03) and Lmx1a (ANOVA, P = 0.02). In contrast, luciferase expression from both the En1/2 and Lmx1a constructs is increased (Fig. 5C) in the presence of the control inactive miR181a En inhibitor (ANOVA P = 0.04) and Lmx1a (ANOVA = 0.02) (Ambion). We conclude from these data that miR181a acts on En1/2 and Lmx1a gene expression by interacting with the 3′UTR binding sites of the genes, as expected for typical microRNAs.
Figure 5.

Interaction of miR181a with target genes. (A) Partial sequence alignment of miR181a with potential bindings sites in 3′UTR regions of En1/2 and Lmx1a mRNA. Asterisks indicate positions of base pairing. (B) Mean luciferase activities in 293 cells transfected with either MDH1-PGK-GFP expressing GFP alone or plasmid MDH1-hmiR181a-PGK co-expressing GFP and miR181a. After 48 h, the cells were transfected again with either the control luciferase plasmid pCMV-lux (CTL), pMIR plasmid containing native (closed bars) or mutated pMIR containing mutated 3′UTR miR-binding site sequences for En or Lmx1a (gray bars). Data were obtained from six independent measurements for each group and represent mean ± SEM of the ratio of luciferase expression in cells transfected with MDH1-PGK-GFP to luciferases activity in cells transfected with MDH1-hmiR181a-PGK. The results for each group were compared by one-way ANOVA. The overall ANOVA was significant for the changes in luciferase activity for engrailed and Lmx1a (F = 4.06, P = 0.03 and F = 4.54, P = 0.02, respectively). Post hoc Tukey t-tests indicate significant difference for En- versus En-mutated (P = 0.01) and Lmx1a- versus Lmx1a-mutated (P = 0.02). The asterisks represent statistical significance (n = 6). For both En and Lmx1a, luciferase activity is significantly reduced in cells transfected with wild-type 3′UTR sequences (filled bars) but not after transfection with plasmids containing mutated 3′UTR sequences (gray bars). (C) Mean luciferase activities in 293 cells transfected either with anti-miR181a-containing sequences complementary to miR181a or with scrambled anti-miR. As above, cells were super-transfected after 48 h with either the control luciferase plasmid pCMV-lux (CTL), pMIR plasmid containing native (closed bars) or mutated pMIR containing mutated 3′UTR miR-binding site sequences for En or Lmx1a (gray bars). Luciferase expression is expressed as ratios of activity in cells transfected with the scrambled anti-miR181a compared with expression in cells transfected with anti miR181a. Statistical analysis was performed as in (B).
HPRT-deficiency-induced dysregulation of miR181a is associated with aberrant expression of dopaminergic transcription factors
Lmx1a is necessary and sufficient for dopaminergic differentiation from neuronal precursor cells (34). In addition, the Brn2 protein has been reported to act synergistically with pro-neuronal proteins such as Mash1 to regulate several facets of the neuro-developmental program (35). We have previously demonstrated dysregulated Mash1 expression during in vitro neuronal differentiation of HPRT-deficient human embryonic carcinoma cells (11). Because our current studies show that Lmx1a and Brn2, both potential targets for miR181a regulation, are down-regulated in HPRT-deficient SH-SY5Y cells in association with over-expression of miR181a, we examined the expression of several known functional partners of Lmx1a for dopaminergic neurogenesis, including Nurr1, PitX3 and Wnt-1 (36) and the putative functional partner of Brn2, MashI, in HPRT-deficient SH-SY5Y cells. We used qPCR methods and western blot analysis to estimate expression of these genes in HPRT-deficient SH-SY5Y cells. Figure 6A demonstrates that HPRT-deficient SH-SY5Y cells demonstrate a marked decrease in expression of Mash1, Nurr1, Pixt3 and Wnt1 (P < 0.05, t-test) compared with HPRT+ cells. The reduced protein expression of Mash1, Nurr1, Pixt3 and Wnt1 in HPRT-deficient SH-SY5Y cells was confirmed by western blot analysis (Fig. 6B) and densitometric analysis of the western blot bands (Fig. 6C).
Figure 6.

Mean levels of expression of functional neuro-developmental partners of Brn2 (Mash1) and Lmx1 (Nurr1, Pixt3 and Wnt1) in wild-type (open bar) and HPRT-knockdown (closed bars) SH-SY5Y cells measured by qPCR analysis. (A) HPRT deficiency is associated with reduced expression of Mash1, Nurr1, Pitx3 and Wnt1. Error bars represent ±SEM of triplicate measurements of two independent experiments of normalized mRNA levels The asterisks represent statistical significance (n = 6, P < 0.05, t-test). (B) Western blot results of wild-type (WT-LUX) and HPRT-knockdown (HPRT-KD) SH-SY5Y cells demonstrating markedly reduced protein levels for all four genes in the HPRT-knockdown cells, consistent with qPCR results in (A). (C) Densitometric analysis of the western blot in (B), confirming reduced levels of all four proteins in HPRT-knockdown SH-SY5Y cells (n = 3, *P < 0.05, t-test).
Aberrant expression of Lmx1a and Brn2 functional partners
To clarify the possible role of dysregulated miR181a expression in the aberrant expression of the dopaminergic developmental factors Mash1, Nurr1, Pitx3 and Wnt-1, we examined their expression in SH-SY5Y cells over-expressing miR181a after transduction with the miR181a-expressing retrovirus vector MDH1-hmiR181a-PGK-GFP co-expressing GFP and miR181a, with lentivirus miR-ZIP vectors expressing anti-miR181a inhibitor to knock down miR181a expression or with the control lentivirus miR-ZIP vector encoding an inactive scrambled anti-miR sequence. Gene expression was measured by western immunoblotting assays, with cells transduced with the GFP-expressing retroviral vector MDH-PGK-GFP serving as control. Figure 7A and B demonstrates reduced protein expression of Mash1, Nurr1, Pitx3 and Wnt-1 in cells over-expressing miR181a (GFP-miR181a) compared with control cells (CTL-GFP). Figure 7C and D illustrates western blots for Mash1, Nurr1, Pitx3 and Wnt-1 expression in SH-SY5Y cells infected with miR-ZIP vector expressing anti-miR181a compared with the control lentivirus containing the inactive inhibitor (CTL-Scramble) and shows qualitatively (panel C) and quantitatively increased levels (filled bars, Fig. 7D) of all four proteins. These results support our working hypothesis that HPRT deficiency causes aberrant expression of the dopaminergic transcription factors LMX1a, En1/2 Mash1, Nurr1, Pixt3 and Wnt1 at least partly through dysregulated expression of miR181a.
Figure 7.

Effect of miR181a on functional expression of target genes. (A) Western blot analysis of the expression of Mash1, Nurr1, Pitx3 and Wnt1 in cells infected with a retroviral vector MDH-PGK-GFP encoding GFP alone (CTL-GFP) or in cells infected with retroviral vector MDH1-hmiR181a-PGK-GFP co-expressing GFP and miR181a (GFP-miT-181a). (B) Densitometric analysis of the protein levels of Mash1, Nurr1, Pitx3 and Wnt1in SH-SY5Y cells expressing GFP alone (open bars) and co-expressing GFP and miR181a (closed bars). Error bars represent ±SEM (n = 3, *P < 0.05, t-test) and asterisks denote significant changes. The quantitation of protein levels supports qualitative results in (A). (C) Western blot analysis of Mash1, Nurr1, Pitx3 and Wnt1 protein expression in SH-SY5Y cells infected with a lentivirus miR-Zip vector encoding a scrambled anti-miR control (CTL-scramble) or with lentivirus miR-Zip vector expressing anti-miR181a inhibitor (anti-miR181a). (D) Densitometric quantitation of Mash1, Nurr1, Pitx3 and Wnt1 protein levels in SH-SY5Y cells transfected by the lentivirus anti-miR p-miR-ZIP (closed bars) or control (open bars) anti-Mir as described in Materials and Methods. Statistical analysis as in (B).
miR181a expression in LND fibroblasts
With the evidence for miR181a aberration in the SH-SY5Y neuroblastoma cell culture model system for HPRT deficiency, we used qPCR analysis to re-examine the possibility of miR181a involvement in LND (Fig. 8). We assayed miR181a expression in primary fibroblasts derived from six patients with the severe form of HPRT deficiency (LND), and six control patients (CTL) without HPRT deficiency. Figure 8 illustrates the miR181a expression in each of the two patient groups and demonstrates the variability of miR181a expression in cells derived from different LND patients. As tested by Student's t-test, miR181a expression in HPRT-deficient cells showed a non-significant relative elevation of 1.53 compared with control samples (P = 0.3).
Figure 8.

Variable miR181a expression in human fibroblasts derived from six control samples (CTL) and from six patients with full classical LND, all normalized to control mRNA (U6) (upper panel) and a comparison of the two groups (lower panel). The apparent up-regulation in LND samples by ∼1.53-fold is not statistically significant (P = 0.3), as evaluated by Student's t-test.
DISCUSSION
We and other groups have recently demonstrated that HPRT deficiency dysregulates the expression of a number transcription factors known to affect neuronal development (9–11). Because of the complex pleiotropic effects of HPRT deficiency on genetic regulatory systems, we conjectured that such broad effects could at least partially reflect the effects of dysregulated miRNA expression since such mechanisms would represent efficient methods for coordinate regulation of such widespread genetic functions. Toward the goal of examining the potential role of microRNA expression in the HPRT-deficiency phenotype, we initially used microarray methods to examine the microRNA expression profile in fibroblasts derived from a patient with the classic LND phenotype compared with a normal human fibroblast control cell and found preliminary evidence for aberrant expression of a number of miRNAs, one of which (miR181a) was found by DAVID to have numerous potential target genes involved in neurodevelopmental pathways that characterize human HPRT deficiency. Although this result provided useful clues to the role of HPRT expression in miRNA expression, our initial studies of miR181a expression in fibroblasts from several classical LND patients, patients with variant HPRT-deficiency phenotype and normal patient controls established only a minimal up-regulation of miR181a in human LND fibroblasts (Fig. 8), suggesting a more complex role of miR181a in HPRT deficiency in vivo in LND patients.
Studies in primary human fibroblasts of neurodevelopmental genes are unlikely to illuminate many of the mechanisms relevant to neural development, and we therefore suggest that a clarification in model systems of the connections between purine metabolism, HPRT deficiency, miRNA metabolism and neural development would be important for an understanding of this puzzling neurological disorder and in development of more specific approaches to its treatment. We therefore elected to examine a generally accepted model system for the study of dopamine-related neural defects in human neurodegenerative and neurodevelopmental disease; i.e. a more homogeneous population of HPRT-knockdown human SH-SY5Y neuroblastoma cells. Our current results present experimental evidence that the molecular, cellular and possibly the neurodevelopmental aberrations of HPRT deficiency are associated with aberrant microRNA expression and therefore suggest that miRNA dysregulation may play a role in the pathogenesis of LND. Although our results clearly point to miR181a as one such HPRT-regulated miRNA, it is likely that other miRNAs will also be found to play roles in a complex miRNA regulatory network that controls normal development of the neurotransmission pathways and that, in dysregulated form, contribute to the neurobehavioral phenotype in HPRT deficiency.
Because the degree of neurological dysfunction in HPRT-deficient LND patients is correlated with the degree of HPRT deficiency and because significant levels of residual HPRT enzyme activity protect against development of the neurological phenotype (37), an efficient HPRT knockdown would be required to allow firm conclusions to be drawn relevant to CNS development in such an in vitro system. Fortunately, we have achieved a knockdown of >98% with the retrovirus-expressed shRNA targeted to HPRT (Fig. 1), a change that leads to a significant over-expression of miR181a (Fig. 1).
Concomitant with the miR181a dysregulation in HPRT-deficient cells is the aberrant expression of potential miR181a target genes that are known to play important roles in dopaminergic and general neurogenesis. By DAVID analysis of the potential targets of miR181a generated by TargetCombo and TargetScan, we have identified clusters and GO categories that are possible targets of miR181a and that can represent plausible candidate contributors to the aberrant neurogenesis and defective function of the dopamine pathways that underlie aspects of the abnormal neural phenotype of HPRT deficiency LND (34–36,38–43). The presence of dysregulated miR181a target genes identified in the GO categories of developmental processes and nervous system development (Table 2) is consistent with our working hypothesis that the complex neurological phenotype in HPRT deficiency reflects dysfunction of complex interacting miRNA networks and pathways that affect many aspects of CNS development.
Table 2.
Cluster of GO terms pertaining to the term ‘neuronal process’ derived from miR181a target genes selected from the TargetCombo and TargetScan databases
| Term | Count | P-value |
|---|---|---|
| GO:0030182, neuron differentiation | 57 | 2.52E − 06 |
| GO:0048666, neuron development | 47 | 4.12E − 06 |
| GO:0048812, neuron projection morphogenesis | 34 | 5.89E − 06 |
| GO:0000904, cell morphogenesis involved in differentiation | 37 | 7.05E − 06 |
| GO:0048667, cell morphogenesis involved in neuron differentiation | 33 | 1.04E − 05 |
| GO:0007409, axonogenesis | 31 | 1.45E − 05 |
| GO:0000902, cell morphogenesis | 47 | 1.51E − 05 |
| GO:0048858, cell projection morphogenesis | 36 | 1.92E − 05 |
| GO:0031175, neuron projection development | 37 | 2.08E − 05 |
| GO:0032989, cellular component morphogenesis | 50 | 2.75E − 05 |
| GO:0030030, cell projection organization | 47 | 3.52E − 05 |
| GO:0032990, cell part morphogenesis | 36 | 4.89E − 05 |
| GO:0007411, axon guidance | 17 | 0.002103 |
Some of the target genes subsequently validated and analyzed for our studies are among the 47 genes in the GO term ‘neuron development’.
In these studies, we have focused on the candidate genes En1, En2, Lmx1a and Brn2 because of their known role in dopaminergic neurogenesis and neural pathway development and because of the previous demonstrations from our laboratory and others that several of them are aberrantly expressed in HPRT deficiency (11,12) in cell culture systems. Transcription factors En1, En2 and Lmx1a play important roles in early and late stages of the maturation and specification of dopaminergic neurons (34–36,38–43). The transcription factors En1/2 influence the development of DA neurons from the early to the late stage of differentiation (42,43). Lmx1a belongs to the family of LIM homeodomain transcription factors expressed in early progenitors and induces down-stream transcription factors that are necessary and sufficient to drive development of dopaminergic neurons from neuronal precursor cells. Lmx1a is essential and sufficient for induction and specification of the maturation of DA neurons via this developmental pathway (34,44,45). The transcription factor Brn2, unlike the other transcription factors cited above, is generally thought not to be implicated specifically in the development of DA neurons but rather belongs to a family of ATGCAAAT-binding transcription factors that have a more general role in neurogenesis (35). Interestingly, it has been recently reported that Brn2 is one of the three transcription factors needed for the direct trans-differentiation of fibroblasts to functional neurons (46).
We present evidence that the four transcription factor genes En1, En2, Lmx1a and Brn2 are all down-regulated in HPRT-deficient SH-SY5Y (Fig. 2). Interestingly, our present results with En1 and En2 dysregulated expression are not consistent with the results previously reported for MN9D cells, which described up-regulation of En1 and En2 in human LND fibroblasts and in HPRT-deficient MND9 mouse cells (12). We have not yet established the reasons for this discrepancy, but one possibility is that different cell types—some such as dermal fibroblasts very distant from neural differentiation pathways and others such as SH-SY5Y neuroblastoma cells that retain the capacity for neuronal differentiation. We expect that HPRT deficiency may interact with the different differentiation programs very differently in such diverse cells.
Potential mechanisms for the probable causal rather than mere coincidental role of aberrant expression of miR181a in dysregulating expression of vital neural developmental transcription factor genes in HPRT-knockdown cells are suggested by the reduced expression of the Engrailed1/2, Lmx1a and Brn2 in response to over-expression of an exogenous retrovirally transduced miR181a in wild-type SH-SY5Y cells and by reversal of the effect by an anti-microRNA hairpin for miR181a (Fig. 4). These results are consistent with a known regulatory effect of the miRNAs on the target genes, either by inhibiting translation or by degrading mRNA through the interaction with 3′UTR miRNA-binding site in the target genes (16–19).
Since the transcription factors Lmx1a and Brn2 are potential targets of miR181a and are markedly down-regulated in HPRT-deficient SH-SY5Y cells, we hypothesized that their functional partners Wnt1, Nurr1, Pitx3 and Mash1 may also be dysregulated in HPRT deficiency. It has been reported that Lmx1a regulates Wnt1 expression during dopaminergic neuronal differentiation and that Lmx1a directly binds the promoter elements and regulates expression of Nurr1 and Pitx3 (36). The transcription factor Brn2 has been shown to cooperate during neurogenesis with pro-neural basic helix-loop-helix transcription factors such as Mash 1 through cooperative binding of a conserved DNA motif (35). Our present study demonstrates that HPRT deficiency in SH-SY5Y cells dysregulates expression of each of these secondary partners of Lmx1a and Brn2 and that over-expression of miR181a in SH-SY5Y cells leads to decreased protein expression of each of the Lmx1a and Brn2 downstream targets Nurr1 Wnt1, Pitx3 and Mash1. Conversely, as expected, miR181a knockdown leads to their increased protein expression (Fig. 7). These data underscore the pleiotropic regulatory effects that a single dysregulated miRNA such miR181a can have directly or indirectly on key neuro-developmental genes (Fig. 9).
Figure 9.

Schematic working model of the potential cascade of pleiotropic effects of HPRT deficiency and miR181a expression on key neuro-developmental dopaminergic regulatory genes. We propose that HPRT deficiency causes an underlying purine aberration that in turn affects expression of a number of microRNAs, with particular emphasis in this study to miR181a. The aberrant expression of miR181a takes part in the down-regulation of the expression of genes such as En1, En2, Lmx1a and Brn2, which, in turn, down-regulates expression of Lmx1a and Brn2 effector genes Nurr1, Pitx3, Wnt1 and Mash1. Our working model suggests that this cascade of defects leads to aberrant development of dopaminergic neurogenesis and defective development of dopaminergic signaling pathways in HPRT-deficient cells and may thereby contribute to the neuropathology of the HPRT-deficiency phenotype in LND.
We propose that HPRT-deficiency contributes to the regulation of dopaminergic neuron development and to the function in the mammalian CNS and that one of its principal mechanisms of action involves miRNA expression, including among others, dysregulation of miR181a. It also seems likely that the neurological phenotype caused by HPRT deficiency in the human at least partly reflects coordinated dysregulation of a number of miRNAs and transcription factor genes required for correct development of DA neurons and dopamine pathways. A detailed validation and understanding of targets of disparate miRNAs as well as the mechanism by which they bring about the dystonia and behavioral and the cognitive defects in LND awaits further study. There is still a paucity of detailed information on the role of miRNAs and their target transcription factors in the development and function of the CNS and in neuropathology, but the present study suggests that HPRT expression may be a valuable model system and offers clues to the role of miRNA expression in the development of the mammalian CNS and in the development of neurodegenerative and neurodevelopmental disorders. Finally, the surprisingly powerful role of the classical purine metabolic ‘housekeeping’ gene HPRT in regulating vital neuro-developmental and regulatory mechanisms at least partly through microRNA-mediated mechanisms may help to identify common pathogenic mechanisms in LND and other neurodevelopmental and neurodegenerative disorders. Finally, we propose that detailed understanding of microRNA expression in the HPRT-deficiency phenotype may point to useful modulators of these pathways that may find a place in treatment of this presently completely intractable disease.
MATERIALS AND METHODS
Primers for PCR assays
All PCR primers used in the following studies are listed in Supplementary Material, Table S1.
Cells
Primary normal HDF cells obtained from ScienCell Research Laboratories were grown to ∼70% confluence in DMEM high-glucose medium supplemented with 10% fetal bovine serum (FBS) and 50 µg/ml penicillin/streptomycin (Invitrogen, Carlsbad, CA) in 5% CO2 atmosphere. Medium was changed every 2–3 days. For miRNA studies, we selected additional human LND and control fibroblasts including 15 primary fibroblast cultures derived from 5 normal HPRT-negative patients and 10 HPRT-deficient patients exhibiting HPRT enzymatic activity ranging from undetectable to ∼50% residual enzyme activity. Human SH-SY5Y cells were obtained from ATCC and were maintained in a 1:1 mixture of minimum essential medium and F12 medium (Gibco, Carlsbad, CA, USA) containing 10% FBS and 50 µg/ml penicillin/streptomycin (Invitrogen, Carlsbad, CA) in 5% CO2 atmosphere. Media were changed every 4–7 days.
HPRT and luciferase small hairpin oligoneucleotides and knockdown vectors
The shRNA sequences against the luciferase and HPRT genes were selected and prepared as previously described (9). VSV-G-pseudotyped retrovirus vectors expressing short hairpin oligonucleotides targeted against either HPRT (retrosh2hprt) or luciferase (retroshlux) were produced, isolated and titered on HT-1080 as previously described (47). The hairpin oligonucleotides were cloned into RNAi-Ready pSIREN Moloney leukemia virus-based retrovirus vectors that express shRNA from the human U6 promoter (Clonetech, Mountain View, CA, USA). Virus packaging was carried out in the packaging cell line GP-293 co-transfected with pSIREN vectors and pseudotyped with plasmid pCMV-G encoding the glycoprotein of vesicular stomatitis virus (48).
HPRT knockdown and selection of SH-SY5Y cells
SH-SY5Y cells were infected at a multiplicity of infection (MOI) of approximately 1 with the knockdown vectors retrosh2hprt or with control vector retroshlux. Infected cells were grown for 10 days in complete DMEM containing 3 µg/ml of puromycin. Bulk cultures were re-plated and maintained in DMEM without puromycin selection for an additional 7 days, after which cells were examined for HPRT expression by transfer to DMEM containing 250 µm 6-thioguanine (SIGMA). The cells infected with the HPRT-knockdown vector retrosh2hprt showed unimpeded growth and expansion in 6TG, whereas the control cells infected with retroshlux were unable to grow in 6-TG (data not shown). The HPRT-deficiency phenotype of the selected cells was confirmed using qPCR and western blot against HPRT (Fig. 1A & Fig. 1S).
Total RNA isolation and qPCR analysis
Total RNA was isolated using PureLink™ RNA Mini Kit for control and HPRT-deficient cells, according to the manufacturer's instructions (Invitrogen). RNA quantity, quality and integrity were evaluated using the RNA 6000 Labchip Kit on the Agilent 2100 Bioanalyzer (Agilent Technologies). An amount of 500 ng of the resulting RNA was used for a reverse transcription reaction using Qiagen miScript Reverse Transcription Kit according to the manufacturer's instructions (Qiagen, Hilden Germany). The synthesized cDNA was then used for qPCR analysis using a Qiagen miScript kit which permits quantification of mature miRNA as well as mRNA from the same cDNA sample.
All PCR primers used in the following studies are listed in Supplementary Material, Table S1. qPCR was carried out in the presence of primers (see Supplementary Material, Table S1) designed and prepared using the web-based software program, OligoPerfect (Invitrogen). PCR reactions were carried out twice in duplicate or triplicate, using the Opticon 2 system DNA engine (Bio-Rad, Hercules, CA, USA) in a total reaction volume of 20 µl in the presence of 200nm of each of the primers. Primers specific for the TATA box-binding protein (TBP) or the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were used as normalization controls. In order to verify the specificity of each amplicon, a melting curve analysis was included at the end of each run. PCR for microRNAs was carried out using U6 small RNA as an internal control. Differences of expression between control and knockdown groups were calculated by normalizing Ct values of the test genes or miRNAs to the Ct values of an endogenous control (U6 for miRNA and TBP or GAPDH for mRNA). The fold change or difference in mRNA expression was calculated using the equation 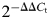 (49), or by normalizing all sample signals to signals corresponding to 100 ng of GAPDH, TBP or U6 RNA. PCR efficiency was verified for each PCR experiment by using normalization primers with serially diluted control cDNA. The statistical significance of variation of expression was assessed by comparing ΔCt of each group by one-way ANOVA or Student's t-test.
(49), or by normalizing all sample signals to signals corresponding to 100 ng of GAPDH, TBP or U6 RNA. PCR efficiency was verified for each PCR experiment by using normalization primers with serially diluted control cDNA. The statistical significance of variation of expression was assessed by comparing ΔCt of each group by one-way ANOVA or Student's t-test.
MicroRNA arrays and data analysis
Ncode™ human miRNA Microarray V3 (Invitrogen) was used for expression array analysis, using conditions recommended by the manufacturer (NCode human miRNA Microarray V3, http://invitrogen.com, catalog no. MIRH3-05). Briefly, this array consists of epoxide-coated glass slides, on which are printed 1073 probe sequences targeting all known mature miRNAs available in the miRbase Sequence Sanger database, Release 10.0 (http://microrna.sanger.ac.uk). The arrays also contain positive controls for hybridization validation using the multi-species miRNA array as well as Alexa Fluor Dye control probes for easy normalization of signal intensities and mismatch controls for screening 1 and 2 base sequence mismatched dye control. Each sample was processed in duplicate on arrays containing three sub-array replicates (n = 6 for each microRNA measurement).
An amount of 0.5–5 µg of RNA was labeled using the NCode Rapid miRNA Labeling System, which makes use of Alexa Fluor labeling technology (NCode Rapid miRNA Labeling System, http://invitrogen.com, catalog no. MIRLSRPD-20). First, poly(A) tailing of total RNA was carried out in 10× miRNA reaction buffer containing 25 mm MnCl2, 0.1 mm ATP in the presence of poly(A) polymerase, and incubated at 37° for 15 min. Subsequently, ligation of poly-tailed RNA was carried out according to the manufacturer's instructions in a reaction mixture containing 6× Alexa Fluor ligation Mix (Cy3 or Cy5), NCode dye normalization control and the ligase enzyme. The ligation cocktail was incubated in light-free conditions at room temperature for 30 min. Ligations were terminated by adding the NCode stop solution at room temperature. Following ligation, the labeled miRNA as well as the appropriate controls was mixed in 2× enhanced hybridization buffer (Invitrogen), incubated for 65°C for 10 min in the absence of light. The hybridization cocktail was then added to the array slide in the hybridization chamber (Corning) and the hybridization chamber was placed in an incubator at 52°C overnight (8–20 h). Following hybridization, arrays were removed from the chamber and the slides were washed twice in 2× SSC, 0.2% SDS washing buffer at room temperature and once in 0.2 SSC washing buffer also at room temperature. The slides were subsequently dried by centrifugation for 4 min at 600g at room temperature and scanned within 30 min after the final wash to avoid photo-bleaching using Genepix 4000B Scanner (Molecular Devices).
MicroRNA expression was analyzed in control and experimental groups using the NCode profiler software tool (NCode Profiler User Guide, http://invitrogen.com). Scanned arrays in the GenePix format were imported into the NCode profiler in which only positive data are used by the profiler algorithms. The NCode profiler analysis output file was formatted according to the manufacturer's recommendations (NCode Profiler User Guide, http://invitrogen.com). Positive fold changes indicate increased expression in HPRT-deficient cells, whereas negative fold changes indicate increased expression in control cells. The P-values were determined by bootstrapping the error distribution of the model fit of the data based on the formula [1/number of bootstraps + 2]. For ample details, see NCode Profiler User Guide, http://invitrogen.com.
Gene clustering and GO analysis
To identify target genes of miR181a and to determine their possible relevance to the aberrant neurodevelopmental phenotype of HPRT deficiency, we interrogated the miRNA database TargetCombo and TargetScan (http://www.diana.pcbi.upen.edu/cgi-bin/TargetCombo.cgi, TargetScan.org). TargetCombo is an open database which combines target predictions from the major database such as PicTar and Diana-microT. Data for annotation and visualization of integrated discovery (DAVID) functional classification clustering is a tool that organizes genes into functionally related clusters. Within each cluster are GO categories of genes having similar biological significance (50,51).
Preparation of miRNA-expressing retroviral vectors
The retroviral cassette MDH1-PGK-GFP for miRNA expression was a gift from C. Chen (School of Medicine, Stanford University) and has been previously described (52). Briefly, the cassette MDH1-PGK-GFP contains the human H1 promoter for the RNA component of RNase P and a murine phosphoglycerate-kinase (PGK) promoter driving the expression of GFP. To produce the miR181a-expressing derivative (MDH1-hmiR181a-PGK-GFP), we amplified human genomic DNA (Promega) using the primers 5′-TCTCGAGAATAATCTCTGCACAGGGAAGAGA-3′, and 5′-TGAATTCTGAAATGGCATAAAAATGCATAA-3′ followed by ligation into the Xho1 and EcoRI site of the MDH1-PGK-GFP retroviral cassette. The MDH1-PGK-GFP and MDH1-hmiR181a-PGK-GFP cassettes were used to produce VSV-G pseudotyped retrovirus vectors as published (47).
Generation of anti-miRNA expression lentivirus
Plasmids expressing anti-miR181a lentivirus p-miR-ZIP as well as a control anti-Mir against a scrambled sequence were obtained from SBI System Biosciences (Mountain View, CA, USA). The details of miR-zip construct and design are described at www.Systembio.com/downloads/Manual_miRZip-081208_web.pdf. p-miR-ZIP lentivirus consists of anti-sense microRNAs stably expressing an RNAi hairpin that expresses anti-microRNA activity. VSV-G pseudotyped lentivirus HIV-1-based vectors expressing the anti-miR181a hairpin as well as the control anti-miR hairpin were prepared using HEK 293 T and were subjected to the established triple transduction protocol as previously described (47,48).
FACS analysis
Prior to qPCR or western immunoblotting, cells transfected to express GFP were subjected to FACS sorting to prepare highly enriched GFP-positive cells. Cells infected with the retrovirus MDH1-PGK-GFP and MDH1-hmiR181a-PGK-GFP were collected in 1× PBS buffer containing 1% FCS and 5 mm EDTA at the density of 2.5 × 106 cells per milliliter and sorted using BD FACSARIA™ II from (BD Biosciences). Enriched GFP-producing cells were subsequently used for qPCR and western blot analysis.
Western blotting
Cells were cultured, lysed in six-well plates and the extracts were obtained using RIPA buffer with a protease inhibitor mixture (Santa Cruz, Inc.) (50 mm Tris–Cl, pH 8, 150 mm NaCl, 0.5% of deoxycholate, 0.1% SDS, 2 mm EDTA, 1% aprotinin, 1 mm PMSF, 1 mm sodium orthovanadate). The cell lysates were centrifuged at 15 000g at 4°C for 10 min. Protein concentration assays of supernatant were carried out by BCA assay (Pierce) and 10–20 µg of the lysates were then separated by reducing tricine–SDS–PAGE and the separated protein bands were transferred onto polyvinylidene fluoride (PVDF) membrane (Invitrogen) using Mini Trans-Blot Electrophoretic Transfer Cell (Bio-Rad). Blotted PVDF membranes were blocked by blocking solution containing 1% bovine serum albumin, 20 mm of Tris, 137 mm of NaCl and 0.1% of Tween-20 (Sigma-Aldrich) for 1h at room temperature. Immunodetection of primary antibodies was carried out overnight in 4°C, and signal amplification using horseradish peroxidase (HRP)-conjugated secondary antibody was performed at room temperature for 1h. The chemiluminescence reagent, SuperSignal West Pico (Thermo Scientific, Rockford, IL, USA), was used signal amplification reagent. X-ray films were developed by SRX101A film processor (Konica Minolta, Motosu-shi, Gifu-ken). All primary and secondary antibodies were diluted in blocking solution. The primary polyclonal goat, mouse or rabbit antibody against HPRT, Mash1, Nurr1, Pitx3 Wnt1 and β-actin were all obtained from Santa Cruz, Inc. and used at a dilution ranging from 1:100 to 1:500. The secondary IgG antibodies against the corresponding primary antibodies were labeled with HRP (from Santa Cruz) and used at the dilution of 1:20 000. Western blot signal was quantified using densitometry ImageJ software according to the protocol published at http://openwetware.org/wiki/Bitan:densitometry. Expression of β-actin was used as a loading normalization control.
MiR inhibition studies
SH-SY5Y or HEK 293 cells were cultured in 12-well dishes at ∼70% confluence, as described above, and transfected with 20 µm of a single-stranded (2′-O-methyl)-modified nucleic acid anti-miRNA inhibitor of human miR181a using siPORT NeoFX transfection agent (Ambion). The equivalent amount of anti-miRNA inhibitor control (scramble) (20 µm) provided by the manufacturer was also used on SH-SY5Y cells as control. SH-SY5Y or HEK 293 cells cultured in 12-well dishes at ∼70% confluence were also infected with miR-ZIP, a lentivirus vector expressing a short hairpin targeted against miR181a or a scrambled control sequence at MOI of 1. After 48 h, transfected and infected cells were harvested by trypsinization and the RNA was extracted for qPCR and western blot analysis as mentioned above.
Target gene luciferase reporter assays
HEK293 cells grown to ∼70% confluence were cultured as above in 12-well plates and co-transfected using SuperFect® transfection reagent (Qiagen) with either 1 µg of control MDH1-PGK-GFP or MDH1-hmiR181a-PGK1-GFP retroviral vectors along with 1 µg of standard luciferase vector driven by CMV promoter (PCMV-LUX, from Clontech) or the appropriate luciferase plasmid pMIR-report-CMV-3′UTR (Ambion) in addition to 0.5 µg of CMV-β-galactosidase vector for normalization of transfection efficiency (Ambion). Parallel transfection of HEK293 was also carried out using 20 µm of anti-miR scramble sequence (as control) or anti-miR181a along with 1 µg of standard luciferase vector driven by CMV promoter PCMV-LUX (no 3′UTR) or the appropriate luciferase plasmid pMIR-report-CMV-3′UTR in addition to 0.5 µg of CMV-β-galactosidase vector for normalization of transfection efficiency. Cells were harvested 48 h after transfection. Dual luminescence assay was performed using Dual-light® or Nova-bright® (from Ambion and Invitrogen, respectively). Data were presented as percent of luciferase activity relative to cells transfected with the control MDH1-PGK-GFP or control anti-MiR vectors.
Statistical analysis
Statistical analyses were carried out using KaleidaGraph graphing and data analysis software package (Synergy Software, Reading, PA, USA). The data are reported as mean ± standard error. For most analyses, Student's paired t-test was performed for control and experimental groups. One-way ANOVA with Tukey's post hoc test was also performed. For most analyses, statistical significance was set at P < 0.05.
Gene symbols
HUGO symbols: HPRT (HGPRT1), Engrailed 1 (EN1), Engrailed 2 (EN2), Lmx1a (LMX1A), Nurr1 (MR4A2), PitX3 (PITX3), Wnt1 (WNT1), Brn2 (POU3F2), Mash1 (HCVS), β-actin (ACTB), miR181a (MIR181A).
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
Conflict of Interest statement. None declared.
FUNDING
This work was supported by grant R24DK082840 from the National Institutes of Health, USA, and a grant from the Lesch–Nyhan Disease Children's Research Foundation.
Supplementary Material
ACKNOWLEDGEMENTS
The authors wish to thank Jorge Valencia and Adam Schiller of the UCSD Gene Chip Microarray Core of the Veterans Medical Research Foundation for their skillful help in the microRNA array analysis, Eric O'Connor of the UCSD Flow Cytometry and Imaging of Human Embryonic Stem Cell Core Facility for his help with FACS sorting analysis and Dr Kersi Pestonjamasp of the UCSD Moores Cancer Center Imaging Resources. We also thank Dr William Nyhan for fibroblast cultures from LND patients.
REFERENCES
- 1.Jinnah H.A., De Gregorio L., Harris J.C., Nyhan W.L., O'Neill J.P. The spectrum of inherited mutations causing HPRT deficiency: 75 new cases and a review of 196 previously reported cases. Mutat. Res. 2000;463:309–326. doi: 10.1016/s1383-5742(00)00052-1. [DOI] [PubMed] [Google Scholar]
- 2.Jinnah H.A., Friedmann T. Lesch–Nyhan disease and its variants. In: Scriver C.R., Beaudet A.L., Sly W.S., Valle D., editors. Metabolic and Molecular Bases of Inherited Disease. McGraw Hill; 2000. pp. 2537–2570. [Google Scholar]
- 3.Rosenbloom F.M., Kelley W.N., Miller J., Henderson J.F., Seegmiller J.E. Inherited disorder of purine metabolism. Correlation between central nervous system dysfunction and biochemical defects. JAMA. 1967;202:175–177. doi: 10.1001/jama.202.3.175. [DOI] [PubMed] [Google Scholar]
- 4.Watts R.W., Spellacy E., Gibbs D.A., Allsop J., McKeran R.O., Slavin G.E. Clinical, post-mortem, biochemical and therapeutic observations on the Lesch-Nyhan syndrome with particular reference to the neurological manifestations. Q. J. Med. 1982;51:43–47. [PubMed] [Google Scholar]
- 5.Visser J.E., Bär P.R., Jinnah H.A. Lesch-Nyhan disease and the basal ganglia. Brain Res. Rev. 2000;32:449–475. doi: 10.1016/s0165-0173(99)00094-6. Review. [DOI] [PubMed] [Google Scholar]
- 6.Jinnah H.A., Wojcik B.E., Hunt M., Narang N., Lee K.Y., Goldstein M., Wamsley J.K., Langlais P.J., Friedmann T. Dopamine deficiency in a genetic mouse model of Lesch-Nyhan disease. J. Neurosci. 1994;14:1164–1175. doi: 10.1523/JNEUROSCI.14-03-01164.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Egami K., Yitta S., Kasim S., Lewers J.C., Roberts R.C., Lehar M., Jinnah H.A. Basal ganglia dopamine loss due to defect in purine recycling. Neurobiol. Dis. 2007;26:396–407. doi: 10.1016/j.nbd.2007.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Del Bigio M.R., Halliday W.C. Multifocal atrophy of cerebellar internal granular neurons in Lesch-Nyhan disease: case reports and review. J. Neuropathol. Exp. Neurol. 2007;66:346–353. doi: 10.1097/nen.0b013e3180515319. [DOI] [PubMed] [Google Scholar]
- 9.Smith D.W., Friedmann T. Characterization of the dopamine defect in primary cultures of dopaminergic neurons from hypoxanthine phosphoribosyltransferase knockout mice. Mol. Ther. 2000;1:486–491. doi: 10.1006/mthe.2000.0057. [DOI] [PubMed] [Google Scholar]
- 10.Song S., Friedmann T. Tissue-specific aberrations of gene expression in HPRT-deficient mice: functional complexity in a monogenic disease? Mol. Ther. 2007;15:1432–1443. doi: 10.1038/sj.mt.6300199. [DOI] [PubMed] [Google Scholar]
- 11.Guibinga G.H., Hsu S., Friedmann T. Deficiency of the housekeeping gene hypoxanthine-guanine phosphoribosyltransferase (HPRT) dysregulates neurogenesis. Mol. Ther. 2010;18:54–62. doi: 10.1038/mt.2009.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ceballos-Picot I., Mockel L., Potier M.C., Dauphinot L., Shirley T.L., Torero-Ibad R., Fuchs J., Jinnah H.A. Hypoxanthine-guanine phosphoribosyl transferase regulates early developmental programming of dopamine neurons: implications for Lesch-Nyhan disease pathogenesis. Hum. Mol. Genet. 2009;18:2317–2327. doi: 10.1093/hmg/ddp164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Messina E., Micheli V., Giacomello A. Guanine nucleotide depletion induces differentiation and aberrant neurite outgrowth in human dopaminergic neuroblastoma lines: a model for basal ganglia dysfunction in Lesch-Nyhan disease. Neurosci. Lett. 2005;375:97–100. doi: 10.1016/j.neulet.2004.10.076. [DOI] [PubMed] [Google Scholar]
- 14.Cristini S., Navone S., Canzi L., Acerbi F., Ciusani E., Hladnik U., de Gemmis P., Alessandri G., Colombo A., Parati E., Invernici G. Human neural stem cells: a model system for the study of Lesch-Nyhan disease neurological aspects. Hum. Mol. Genet. 2010;19:1939–1950. doi: 10.1093/hmg/ddq072. [DOI] [PubMed] [Google Scholar]
- 15.Kang T.H., Guibinga G., Friedmann T. HPRT deficiency coordinately dysregulates canonical WNT and presenilin-1 signaling: a neuro-developmental regulatory role for a housekeeping gene? PLoS One. 2011;6:e16572. doi: 10.1371/journal.pone.0016572. doi:10.1371/journal.pone.oo16572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bushati N., Cohen S.M. MicroRNA functions. Annu. Rev. Cell Dev. Biol. 2007;23:175–205. doi: 10.1146/annurev.cellbio.23.090506.123406. [DOI] [PubMed] [Google Scholar]
- 17.Liu J. Control of protein synthesis and mRNA degradation by microRNAs. Curr. Opin. Cell Biol. 2008;20:214–221. doi: 10.1016/j.ceb.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 18.Siomi H., Siomi M.C. Posttranscriptional regulation of microRNA biogenesis in animals. Mol. Cell. 2010;38:323–332. doi: 10.1016/j.molcel.2010.03.013. [DOI] [PubMed] [Google Scholar]
- 19.Cai Y., Yu X., Hu S., Yu J. A brief review on the mechanisms of miRNA regulation. Genomics, Proteomics Bioinformatics. 2009;7:147–154. doi: 10.1016/S1672-0229(08)60044-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo H., Ingolia N.T., Weissman J.S., Bartel D.P. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835–840. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thomas M., Lieberman J., Lal A. Desperately seeking microRNA targets. Nat. Struct. Mol. Biol. 2010;17:1169–1174. doi: 10.1038/nsmb.1921. [DOI] [PubMed] [Google Scholar]
- 22.Chitwood D.H., Timmermans M.C. Small RNAs are on the move. Nature. 2010;467:415–419. doi: 10.1038/nature09351. [DOI] [PubMed] [Google Scholar]
- 23.McManus M.T., Sharp P.A. Gene silencing in mammals by small interfering RNAs. Nat. Rev. Genet. 2002;3:737–747. doi: 10.1038/nrg908. [DOI] [PubMed] [Google Scholar]
- 24.Chua J.H., Armugam A., Jeyaseelan K. MicroRNAs: biogenesis, function and applications. Curr. Opin. Mol. Ther. 2009;11:189–199. [PubMed] [Google Scholar]
- 25.Stark A., Brennecke J., Bushati N., Russell R.B., Cohen S.M. Animal microRNAs confer robustness to gene expression and have a significant impact on 3′UTR evolution. Cell. 2005;123:1133–1146. doi: 10.1016/j.cell.2005.11.023. [DOI] [PubMed] [Google Scholar]
- 26.Le M.T., Xie H., Zhou B., Chia P.H., Rizk P., Um M., Udolph G., Yang H., Lim B., Lodish H.F. MicroRNA-125b promotes neuronal differentiation in human cells by repressing multiple targets. Mol. Cell Biol. 2009;229:5290–5305. doi: 10.1128/MCB.01694-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perron M.P., Provost P. Protein components of the microRNA pathway and human diseases. Methods Mol. Biol. 2009;487:369–385. doi: 10.1007/978-1-60327-547-7_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bushati N., Cohen S.M. MicroRNAs in neurodegeneration. Curr. Opin. Neurobiol. 2008;18:292–296. doi: 10.1016/j.conb.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 29.Miller B.H., Wahlestedt C. MicroRNA dysregulation in psychiatric disease. Brain Res. 2010;1338:89–99. doi: 10.1016/j.brainres.2010.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abu-Elneel K., Liu T., Gazzaniga F.S., Nishimura Y., Wall D.P., Geschwind D.H., Lao K., Kosik K.S. Heterogeneous dysregulation of microRNAs across the autism spectrum. Neurogenetics. 2008;9:153–161. doi: 10.1007/s10048-008-0133-5. [DOI] [PubMed] [Google Scholar]
- 31.Provost P. MicroRNAs as a molecular basis for mental retardation, Alzheimer's and prion diseases. Brain Res. 2010;1338:58–66. doi: 10.1016/j.brainres.2010.03.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beveridge N.J., Tooney P.A., Carroll A.P., Gardiner E., Bowden N., Scott R.J., Tran N., Dedova I., Cairns M.J. Dysregulation of miRNA 181b in the temporal cortex in schizophrenia. Hum. Mol. Genet. 2008;17:1156–1168. doi: 10.1093/hmg/ddn005. [DOI] [PubMed] [Google Scholar]
- 33.Constantinescu R., Constantinescu A.T., Reichmann H., Janetzky B. Neuronal differentiation and long-term culture of the human neuroblastoma line SH-SY5Y. J. Neural. Transm. 2007;72:17–28. doi: 10.1007/978-3-211-73574-9_3. [DOI] [PubMed] [Google Scholar]
- 34.Andersson E., Tryggvason U., Deng Q., Friling S., Alekseenko Z., Robert B., Perlmann T., Ericson J. Identification of intrinsic determinants of midbrain dopamine neurons. Cell. 2006;124:393–405. doi: 10.1016/j.cell.2005.10.037. [DOI] [PubMed] [Google Scholar]
- 35.Castro D.S., Skowronska-Krawczyk D., Armant O., Donaldson I.J., Parras C., Hunt C., Critchley J.A., Nguyen L., Gossler A., Gottgens B., Matter J.M., Guillemot F. Proneural bHLH and Brn proteins co-regulate a neurogenic program through cooperative binding to a conserved DNA motif. Dev. Cell. 2006;11:831–844. doi: 10.1016/j.devcel.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 36.Chung S., Leung A., Han B.S., Chang M.Y., Moon J.I., Kim C.H., Hong S., Pruszak J., Isacson O., Kim K.S. Wnt1-lmx1a forms a novel autoregulatory loop and controls midbrain dopaminergic differentiation synergistically with the SHH-FoxA2 pathway. Cell Stem Cell. 2009;5:646–658. doi: 10.1016/j.stem.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Page T., Nyhan W.L. The spectrum of HPRT deficiency: an update. Adv. Exp. Med. Biol. 1989;253A:129–133. doi: 10.1007/978-1-4684-5673-8_20. [DOI] [PubMed] [Google Scholar]
- 38.Burbach J.P., Smidt M.P. Molecular programming of stem cells into mesodiencephalic dopaminergic neurons. Trends Neurosci. 2006;29:601–603. doi: 10.1016/j.tins.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 39.Smidt M.P., Burbach J.P. How to make a mesodiencephalic dopaminergic neuron. Nat. Rev. Neurosci. 2007;8:21–32. doi: 10.1038/nrn2039. [DOI] [PubMed] [Google Scholar]
- 40.Ferri A.L., Lin W., Mavromatakis Y.E., Wang J.C., Sasaki H., Whitsett J.A., Ang S.L. Foxa1 and Foxa2 regulate multiple phases of midbrain dopaminergic neuron development in a dosage-dependent manner. Development. 2007;134:2761–2769. doi: 10.1242/dev.000141. [DOI] [PubMed] [Google Scholar]
- 41.Simon H.H., Alavian K.N. Transcriptional regulation of their survival: the Engrailed homeobox genes. Adv. Exp. Med. Biol. 2009;651:66–72. doi: 10.1007/978-1-4419-0322-8_6. [DOI] [PubMed] [Google Scholar]
- 42.Alavian K.N., Scholz C., Simon H.H. Transcriptional regulation of mesencephalic dopaminergic neurons: the full circle of life and death. Mov. Disord. 2008;23:319–328. doi: 10.1002/mds.21640. [DOI] [PubMed] [Google Scholar]
- 43.Sgadò P., Albéri L., Gherbassi D., Galasso S.L., Ramakers G.M., Alavian K.N., Smidt M.P., Dyck R.H., Simon H.H. Slow progressive degeneration of nigral dopaminergic neurons in postnatal Engrailed mutant mice. Proc. Natl Acad. Sci. USA. 2006;103:15242–15247. doi: 10.1073/pnas.0602116103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nakatani T., Kumai M., Mizuhara E., Minaki Y., Ono Y. Lmx1a and Lmx1b cooperate with Foxa2 to coordinate the specification of dopaminergic neurons and control of floor plate cell differentiation in the developing mesencephalon. Dev. Biol. 2010;339:101–113. doi: 10.1016/j.ydbio.2009.12.017. [DOI] [PubMed] [Google Scholar]
- 45.Lin W., Metzakopian E., Mavromatakis Y.E., Gao N., Balaskas N., Sasaki H., Briscoe J., Whitsett J.A., Goulding M., Kaestner K.H., Ang S.L. Foxa1 and Foxa2 function both upstream of and cooperatively with Lmx1a and Lmx1b in a feedforward loop promoting mesodiencephalic dopaminergic neuron development. Dev. Biol. 2009;333:386–396. doi: 10.1016/j.ydbio.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 46.Vierbuchen T., Ostermeier A., Pang Z.P., Kokubu Y., Sudhof T.C., Wernig M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature. 2010;463:1035–1041. doi: 10.1038/nature08797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guibinga G.H., Hall F.L., Gordon E.M., Ruoslahti E., Friedmann T. Ligand-modified vesicular stomatitis virus glycoprotein displays a temperature-sensitive intracellular trafficking and virus assembly phenotype. Mol. Ther. 2004;9:76–84. doi: 10.1016/j.ymthe.2003.09.018. [DOI] [PubMed] [Google Scholar]
- 48.Yee J.-K., Miyanohara A., LaPorte P., Bouic K., Burns J.C., Friedmann T. A general method for generation of high titer, pantropic retroviral vectors: highly efficient infection of primary hepatocytes. Proc. Natl Acad. Sci. USA. 1994;91:9564–9568. doi: 10.1073/pnas.91.20.9564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT. Method. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 50.Huang D.W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 51.Huang D.W., Sherman B.T., Lempicki R.A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Min H., Chen C.Z. Methods for analyzing microRNA expression and function during hematopoietic lineage differentiation. Methods Mol. Biol. 2006;342:209–227. doi: 10.1385/1-59745-123-1:209. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.