

Abstract
Intracellular protein aggregation is a common pathologic feature in neurodegenerative diseases such as Huntington' disease, amyotrophic lateral sclerosis and Parkinson' disease. Although progress towards understanding protein aggregation in vitro has been made, little of this knowledge has translated to patient therapy. Moreover, mechanisms controlling aggregate formation and catabolism in cellulo remain poorly understood. One limitation is the lack of tools to quantitatively monitor protein aggregation and disaggregation. Here, we developed a protein-aggregation reporter that uses huntingtin exon 1 containing 72 glutamines fused to the N-terminal end of firefly luciferase (httQ72-Luc). httQ72-Luc fails to aggregate unless seeded by a non-luciferase-containing polyglutamine (polyQ) protein such as Q80-cfp. Upon co-aggregation, httQ72-luc becomes insoluble and loses its enzymatic activity. Using httQ72-Luc with Q80(CFP/YFP) as seeds, we screened the Johns Hopkins Clinical Compound Library and identified leflunomide, a dihydroorotate dehydrogenase inhibitor with immunosuppressive and anti-psoriatic activities, as a novel drug that prevents polyQ aggregation. Leflunomide and its active metabolite teriflunomide inhibited protein aggregation independently of their known role in pyrimidine biosynthesis, since neither uridine treatment nor other pyrimidine biosynthesis inhibitors affected polyQ aggregation. Inducible cell line and cycloheximide-chase experiments indicate that these drugs prevent incorporation of expanded polyQ into an aggregate. This study demonstrates the usefulness of luciferase-based protein aggregate reporters for high-throughput screening applications. As current trials are under-way for teriflunomide in the treatment of multiple sclerosis, we propose that this drug be considered a possible therapeutic agent for polyQ diseases.
INTRODUCTION
Polyglutamine (polyQ) diseases comprise a heterogeneous group of neurological disorders that include spinobulbar muscular atrophy, Huntington disease, several spinocerebellar ataxias and dentatorubral pallidoluysian atrophy, which are characterized by intracellular protein aggregation and neuronal cell loss. In these diseases, an uninterrupted CAG trinucleotide repeat expansion in the coding sequences of specific genes causes the mutant protein to misfold, aggregate and trigger neurodegeneration by a gain-of-function mechanism (1,2). One polyQ disease, Huntington disease is progressive, autosomal dominant and characterized by personality changes, motor impairment and subcortical dementia (3,4). It is due to an expansion in the first exon of the IT15 gene coding for the protein huntingtin (5), a 3144 amino acid protein of unknown function. Huntington disease is pathologically characterized by degeneration of neurons in the striatum and subcortical regions. Patients with a CAG expansion >39 show a strict inverse correlation between the length of the polyQ-coding sequence and the age of onset with clinical severity directly correlating with the repeat number (4,6,7). Moreover, repeat length and concentration of the mutant protein dictate the propensity for huntingtin to aggregate in vivo and in vitro (8–11). Experiments with synthetic polyQ peptides and huntingtin exon 1 derivatives indicate that misfolding of an expanded polyQ monomer initiates aggregation in a nucleation-dependent process (12–14). Aggregation of bacterially expressed huntingtin fragments and synthetic polyQ peptides can be accelerated by adding aggregated polyQ seeds in vitro, supporting a nucleated growth assembly mechanism similar to amyloid formation (8,12,15,16). Therefore, aggregation of expanded huntingtin and other polyQ peptides can be ‘seeded’ by a polyQ aggregate (17).
An important property of expanded polyQ proteins is their capacity to incorporate both expanded and unexpanded polyQ proteins into the growing aggregate, which occurs through interactions with the polyQ domain (18–20). As a consequence, both homotypic and heterotypic aggregation of polyQ occurs in vitro and may underlie its neurotoxicity in vivo. Sequestration of heterologous proteins containing short stretches of polyQ, like CBP, has been proposed as a pathological mechanism of Huntington disease, as well as in other polyQ disorders (21–23). In addition, other polyQ proteins like TBP (24) and Brn-2 POU (25) are similarly sequestered into huntingtin aggregates in vivo where they lose their function and contribute to neurodegeneration. Therefore, non-aggregating, unexpanded polyQ-containing proteins can be forced to aggregate by aggregation-prone polyQ proteins by means of intercalation of polyQ into the β-strand sheets of bona fide aggregating proteins. This ‘seeding’ property of polyQ also takes place spontaneously in lower eukaryotes and presumably it plays a physiological role. Sup35 and Rnq1 are two prion-like, Q/N-rich proteins involved in metabolic adaptation of yeast to environmental conditions through self-aggregation. Expanded huntingtin or polyQ alone can induce the aggregation of Sup35 (26) and Rnq1 (27). In addition, replacing the Q/N-rich domain with polyQ in Sup35 allows polyQ-Sup35 prion propagation (28). Interestingly, Rnq1 inhibits polyQ aggregation (29,30) and Sup35 suppresses polyQ toxicity in Drosophila, indicating a complex interplay between Q/N-rich proteins and expanded polyQ proteins. On the other hand, mammalian Q/N-rich domain proteins TIA-1 (31), FUS (32,33) and TDP-43 (34,35) are also co-localized to polyQ aggregates in mammalian cells and in the human brain, indicating that ‘cross-seeding’ of prion-like and polyQ proteins is an evolutionarily conserved property of these self-aggregating proteins. Considering the high amount of Q/N-rich proteins coded in the human genome (36), the spectra of proteins potentially affected by polyQ aggregation in polyQ diseases increase enormously and efforts to understand homo- and hetero-polymeric aggregation/disaggregation of polyQ are of logical relevance.
Protein aggregation is a promising candidate target for disease intervention. Although there is still controversy about the role of intracellular polyQ aggregates as toxic or protective species (37), a decrease in polyQ aggregates correlates with reduced neurodegeneration in Drosophila and mouse models of Huntington disease (38–40). It is not surprising therefore that numerous in vitro and cell-based high-throughput initiatives have been developed in the past years to screen for small molecules that inhibit polyQ aggregation or cell toxicity (41–47). More recently, genetic screens with siRNA libraries have been developed to search for genes that modify the aggregation of polyQ proteins (48,49). Most of the cell-based screenings rely on aggregation changes of polyQ tagged to GFP or its derivatives. Recently, an improved method to quantitatively monitor polyQ aggregation by fluorescence resonance energy transfer (FRET) was described and adapted for high-throughput screening. Using this FRET-based assay, the authors identified a ROCK inhibitor, Y-27632, as a potent polyQ inhibitor in cellulo and in a Drosophila model of neurodegeneration (39,50). However, more robust and quantitative methods to assess protein-aggregation dynamics that are amenable to non-invasive, live imaging in cellulo and in vivo need to be generated.
The largest publicly accessible collection of existing drugs is the Johns Hopkins Clinical Compound Library (JHCCL), a collection of over 1500 drugs that includes drugs approved by the Food and Drug Administration (FDA) as well as drug candidates that have entered phase-II clinical trials. The JHCCL is intended to promote drug repurposing, i.e. finding new uses for existing drugs with known pharmacokinetics and side effects for accelerating drug discovery (51–53). Firefly luciferase has been previously used as an in vitro reporter to monitor co-translational protein folding of nascent polypeptides, using rabbit reticulocyte extracts, and it has also been used to monitor protein refolding after heat- or guanidinium-induced denaturation, where it regains enzymatic activity upon renaturation (54–56). To identify drugs that inhibit polyQ aggregation and are ready to use in clinical applications, we developed a novel aggregation-sensitive luciferase-based reporter to quantitate polyQ aggregation in cellulo, using an expanded huntingtin fragment (exon 1, httQ72-Luc), and screened the JHCCL collection.
RESULTS
Development of the httQ72-Luc protein aggregate reporter
We fused exon1 sequences of huntingtin to the N-terminus of firefly luciferase via a G/S linker to create httQ72-Luc. We included the 11-mer flexible linker (57) to minimize any potential steric hindrance imposed by FLuc on httQ72 self-assembly during aggregation (Fig. 1A). To test the aggregation state of this reporter in cell culture, we expressed httQ72-Luc in several cell lines and analyzed protein solubility by filter retardation. This assay consists of lysing cells and passing the lysate through a cellulose acetate filter with 0.2 μm pores, trapping aggregates which can be visualized with specific antibodies. Surprisingly, httQ72-Luc did not self-aggregate under the conditions tested. Similarly, we analyzed other luciferase-based constructs including Q80-Luc, Luc-Q80 and an androgen receptor fragment containing Q122, each of which failed to aggregate (data not shown). It is well established that polyQ proteins can ‘co-aggregate’ and that an aggregate prone polyQ can ‘seed’ an unexpanded, soluble polyQ protein. Therefore, we co-transfected httQ72-Luc with aggregation-prone, cyan fluorescent protein-tagged polyQ proteins httQ72-cfp (Q72-cfp) and pure Q80-cfp (Q80-cfp) or with a soluble, unexpanded polyQ as control (Q19-cfp) and tested the aggregation of the reporter by filter retardation assays in HEK-293 cells. httQ72-Luc is soluble when expressed with soluble polyQ but turns insoluble when co-expressed with aggregating polyQs (Fig. 1B). Aggregation of the httQ72-Luc reporter might be a general consequence of proteostasis impairment caused by aggregation of the expanded polyQ protein. As a control, Fluc solubility was also analyzed. In similar conditions, no aggregation of Fluc was observed, demonstrating that the aggregation of httQ72-Luc is inherent to the polyQ tract present in the reporter. In all conditions, the absence of protein in the filter is not a consequence of decreased expression of the reporter, as verified by western blot (Fig. 1B, right panel). To confirm that httQ72-Luc aggregation is seeded by expanded polyQ proteins, we performed luciferase immunostaining. Under normal conditions or when coexpressed with a soluble polyQ, httQ72-Luc reporter shows a widespread distribution in the cytoplasm. However, it becomes fully incorporated into a polyQ aggregate when coexpressed with Q80-cfp (Fig. 1C). On the contrary, Fluc remains soluble when co-expressed with either Q19-cfp or Q80-cfp, corroborating that httQ72-Luc is specifically trapped into polyQ aggregates. Together, these results indicate that httQ72-Luc aggregation requires seeding by an aggregate-prone expanded polyQ.
Figure 1.

Aggregation of httQ72-Luc reporter requires seeding by expanded polyQ. (A) Schematic representation of the httQ72-Luc reporter. The first 17 amino acids of the huntingtin protein and the flexible linker essential for proper luciferase folding are indicated in red and gray, respectively. (B) CFP-tagged polyQ proteins of different sizes (Q19-, Q72-, Q80-cfp) were co-transfected with httQ72-Luc or FLuc in HEK-293 cells. Forty-eight hours after transfection, lysates were prepared and protein aggregation analyzed by filter trap. Right panel: similar levels of expression in the same lysates were confirmed by western blot. (C) U2OS cells were co-transfected with indicated luciferase- and CFP-tagged constructs, and 48 h after transfection, luciferase staining was performed. httQ72-Luc but not FLuc was coaxed to aggregate into a polyQ aggregate. Scale bar, 20 μm.
PolyQ aggregation can be qualitatively monitored by FRET in cell culture by expressing both CFP- and YFP-tagged polyQ proteins (39). In contrast, we hypothesized that luciferase activity is lost when it is forced to aggregate. To demonstrate this, HEK-293 cells were co-transfected with httQ72-Luc and different FRET pairs of polyQ ranging in size from 19 to 80 glutamines to confirm aggregation. A strict inverse correlation between luciferase activity and FRET was observed (Fig. 2A–C). As controls, single-point mutations in the first 17 amino acids of the httQ72-CFP/YFP pair that modify the aggregation of huntingtin were included (M.I.D., unpublished data). At the time point analyzed, a Lys-to-Arg mutation (Q72 K → R) slightly increases the aggregation of the reporter and a Met-to-Pro mutation (Q72 M → P) completely disrupts httQ72-CFP/YFP aggregation as indicated by FRET (Fig. 2B). A complete recovery in luciferase activity was observed in Q72 M → P-expressing cells, indicating that the reporter quantitatively senses the protein aggregate state of polyQ proteins (Fig. 2A). Increased dynamic range changes in luciferase compared with FRET in the Q72 K → R condition as well as various polyQ lengths indicated that httQ72-Luc was more sensitive than FRET to report polyQ aggregation. To verify the specificity of our reporters, we first demonstrated specificity for polyQ. We tested the effects of Q80(CFP/YFP) on FLuc alone and in a non-pathological length huntingtin reporter (httQ25-Luc). As expected for a polyQ-containing protein, Q80(CFP/YFP) decreased luciferase activity of httQ72-Luc and httQ25-Luc but not the Fluc reporter (Supplementary Material, Fig. S1A and B). Then, we wondered whether expression of another aggregation-prone protein known to interact with polyQ proteins might have an effect on httQ72-Luc. Fragments of TDP-43 aggregate and intercalate with expanded polyQ proteins by virtue of a prion-like Q/N-rich domain present in the C-terminal region of TDP-43 (34). We therefore tested whether C-terminal fragments of TDP-43 (p25) affected httQ72-Luc aggregation. Neither changes in httQ72-Luc activity nor co-localization was detected in p25-co-transfected cells (Supplementary Material, Fig. S1A and B). The change in luciferase activity was not due to alterations in transfection efficiency or changes in protein translation since a co-expressing renilla luciferase plasmid control did not change activity in the setting of Q19, Q35 or Q80 (Supplementary Material, Fig. S2).
Figure 2.

httQ72-Luc aggregates are inactive. (A–C) HEK-293 cells were co-transfected with httQ72-Luc and CFP/YFP-tagged polyQ of different sizes (Q19-, Q35-, Q72- and Q80(CFP/YFP)). Thirty-six hours after transfection, cells were split into sister 96-well plates for luciferase activity (A) or FRET (B) determinations. Single-point mutations of huntingtin that either increase (Q72K → R) or abolish (Q72M → P) httQ72-CFP aggregation were included. (C) A strict inverse correlation between luciferase activity and protein aggregation assessed by FRET was detected with a correlation coefficient of 0.9834. (D) HEK-293 cells were co-transfected with httQ72-Luc along with Q19-cfp or Q80-cfp. Forty-eight hours after transfection, live-cell BLI was performed. Fluorescence (CFP) and bright-field microscopy for the same field were recorded right before BLI. The arrow indicates a representative cell containing an httQ72-Luc/Q80-cfp co-aggregate devoid of luciferase activity. (E and F) HEK-293 cells were co-transfected as in (A), and 60 h later, lysates were prepared by sonication in the absence of detergents, spun for 10 min at 16 000g and total and cleared lysates assayed for luciferase and FRET.
To further demonstrate that luciferase activity was lost in the setting of aggregation, we analyzed luciferase activity in aggregate-containing cells by low-light microscopy. HEK-293 cells were co-transfected with equal amounts of httQ72-Luc and Q19-cfp or Q80-cfp-coding plasmids and cells visualized via live bioluminescence imaging (BLI) and fluorescence microscopy 24 h after transfection. With control Q19-cfp, luciferase activity parallels the localization of httQ72-Luc detected by immunostaining, with a widespread distribution in the cytosol and depletion from the nucleus (compare Fig. 2D with 1C). A loss in luciferase activity was detected in aggregate-containing cells, demonstrating that Q80-cfp-induced aggregation of httQ72-Luc caused a depletion of luciferase activity (Fig. 2C, arrow). It is known that polyQ aggregates are resistant to solubilization by both non-ionic and ionic detergents (58). To convincingly prove that luciferase activity comes from soluble species, we transfected cells as before and 1% TX-100 lysates were prepared and subjected to low-speed centrifugation (1000g, 10 min). Luciferase and FRET activities were then determined in both the lysate and the supernatants. Luciferase was detected in both the total lysate and the cleared supernatant. Conversely, FRET activity was completely lost after centrifugation, indicating that the luciferase activity and the aggregate are completely separable species and that luciferase activity comes from soluble, non-aggregated httQ72-Luc (Fig. 2E and F).
Screening of the JHCCL
We reasoned that any potential drug with disaggregating activity should increase luciferase activity and decrease FRET. We adapted the reporter system for high-throughput screening using luciferase and FRET as primary readouts in live cells. When using Q19- and Q80-FRET pairs to generate control-positive and -negative signals, the dynamic range of the reporter increased if we expressed the data as a ratio, i.e. luciferase/(FRET/donor), improving the calculated Z-factor (59) from 0.66 to 0.73. Therefore, we screened the JHCCL at a final concentration of 5 μm and scored the luciferase/(FRET/donor) ratio. A flow chart of the screening process is shown in Figure 3.
Figure 3.

Flow chart describing the screening steps. For the primary screen, 5 μm of each compound was tested in triplicate on separate plates. Zi scores were calculated for luciferase/(FRET/donor), and 20 drugs above a cutoff of 3 were pulled out. Fluorescent drugs that did not change luciferase activity were discarded. Zi scores were calculated for luciferase as before, and 10 primary hits identified. Dose–response studies with commercially available drugs were performed on hits showing >1.5-fold increase in luciferase activity in the primary screen. Of the 1560 compounds screened, 4 hits were obtained.
Library screen (27 drug plates) was completed in three sections by analyzing nine drug library plates in triplicate. For a section, we transfected HEK-293 cells in bulk as described in Materials and Methods and included positive and negative controls in every replica plate to monitor plate-to-plate variability. In order to maximize effects, we allowed aggregates to form 36 h before adding the compounds and treatments were carried out for 24 h. This approach should allow detecting both protein-aggregation inhibition and, more importantly, protein disaggregation. From the raw data, we noticed that the luciferase/(FRET/donor) ratio slightly decreased over time in each one of the sections, so we used a plate-by-plate Zi score analysis and set a cutoff of +3 for hit selection. We identified a set of 20 drugs that increased the luciferase/(FRET/donor) ratio (Supplementary Material, Fig. S3). Out of these drugs, only methylene blue and insulin fulfilled our initial criteria of increasing luciferase and decreasing FRET. Interestingly, these drugs had been previously implicated in polyQ aggregation inhibition and provided support that luciferase-based reporters could be used to monitor polyQ aggregation by high-throughput screening. Further analysis showed that ∼50% of the drugs were fluorescent in the cfp channel and did not change luciferase activity (Supplementary Material, Fig. S3), increasing the luciferase/FRET/donor ratio, artifactually indicating they were false positives. To identify potential hits that escaped the analysis, we repeated the Zi score analysis for luciferase changes only. We identified a total of nine drugs that increased luciferase activity. Of these, methylene blue (60,61) and nocodazole (62,63) had been previously described to participate in the metabolism of expanded polyQ proteins, including the aggregation of expanded N-terminal fragments of huntingtin (60,62,64). Insulin has also been demonstrated to decrease polyQ aggregation (64) but only met screening criteria when represented as a Luc/FRET ratio. We focused on drugs that increased luciferase activity by >50%, leaving four drugs to be validated in dose–response experiments: leflunomide, lansoprazole, piperine and nabumetone (Supplementary Material, Fig. S3). We performed dose–response experiments with these drugs using httQ72-Luc and Q80(CFP/YFP) and calculated the half maximal effective concentration (EC50) value for luciferase activity change after 48 h treatment. All of them increased luciferase activity >70%, with EC50 values in the low micromolar range (Table 1, Supplementary Material, Fig. S4).
Table 1.
Johns Hopkins Library screen
| Compound | Structure | CAS No. | EC50 (µm) | % Luc increase | Mechanism of action and clinical use |
|---|---|---|---|---|---|
| Leflunomide | 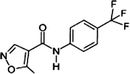 |
75706-12-6 | 3.10 | 173.2 | A pyrimidine biosynthesis inhibitor. Inhibits T- and B-lymohocyte proliferation. Prescribed in moderate-to-severe rheumatoid arthritis and psoriatic arthritis |
| Teriflunomide A77 1726 | 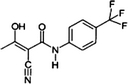 |
163451-81-8 | 1.84 | 206.4 | A DHODH inhibitor. Corresponds to leflunomide after ring opening in water solution or in the blood. Currently in Clinical Trial Phase III for the treatment of multiple sclerosis |
| Lansoprazole | 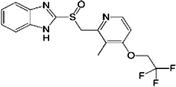 |
103577-45-3 | 5.30 | 74.4 | A K+/H+-ATPase antagonist. Inhibits both centrally and peripherally mediated gastric acid secretion. Prescribed to treat gastroesophageal reflux disease and in the treatment of active benign gastric ulcer |
| Piperine | 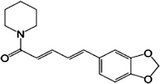 |
94-62-2 | 2.00 | 77.6 | A natural alkaloid extracted from black pepper. Possesses topical antibacterial activity but it is also in clinical trials for Alzheimer disease as it increases bioavailability of compounds like curcumin by inhibiting cytochrome P450 |
| Nabumetone | 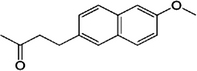 |
42924-53-8 | 1.54 | 98.2 | A napthaleneacetic acid-derivative nonsteroidal anti-inflammatory drug (NSAID). Prescribed to relieve pain, tenderness, swelling and stiffness caused by osteoarthritis and rheumatoid arthritis |
John Hopkins Library screening identifies leflunomide, lansoprazole, piperine and nabumetone as compounds able to increase reporter solubility. EC50 values were calculated based on luciferase activity only. Teriflunomide was not originally in the library but was included here because it is the active metabolite of leflunomide.
We pursued follow-up analysis of leflunomide for three reasons: (i) leflunomide is a pro-drug that is converted into its active metabolite teriflunomide in the blood (65), suggesting that higher effects in polyQ aggregation might be obtained with the actual active metabolite; (ii) leflunomide has been successfully used to suppress experimental autoimmune neuritis induced by myelin immunization, and hence it is neuroprotective in a central nervous system disease (66); and (iii) teriflunomide is a drug that is in current phase-III clinical trials for treatment of multiple sclerosis, an autoimmune, demyelinating disease that affects both brain and spinal cord, and has so far proved safe for CNS neurons (67,68).
Leflunomide and teriflunomide inhibit httQ72-Luc aggregation independently of pyrimidine biosynthesis inhibition
Leflunomide has a dose-dependent effect on polyQ aggregation inhibition at 24 h, using the same httQ72-Luc reporter in the presence of Q80(CFP/YFP). We observed a concentration-dependent increase in the luciferase activity of the reporter in cells treated for 24 h (Fig. 4A). As leflunomide is slowly converted into teriflunomide in solution, we repeated the same experiments using teriflunomide. A greater increase in luciferase activity over baseline was detected in cells treated with teriflunomide compared with leflunomide (130 versus 80% increase) at similar EC50 values of ∼0.5 and ∼1.1 μm. To rule out effects other than modification of the aggregation state, we repeated the same experiments with httQ72-Luc in the presence of the soluble Q19(CFP/YFP) FRET pair (Fig. 4B). httQ72-Luc does not aggregate or lose its activity in the presence of non-aggregating Q19(CFP/YFP). We detected a slight increase in the activity that could not be significantly adjusted to a sigmoidal fit under the experimental conditions. A similar behavior was detected when cells were treated for 48 h with leflunomide and teriflunomide, but with greater effects of 206 and 173% increase in luciferase activity, respectively (Table 1).
Figure 4.

Leflunomide and its active metabolite teriflunomide increase luciferase activity of aggregated httQ72-Luc in a dose–response manner. HEK-293 cells were transfected with httQ72-Luc along with Q80(CFP/YFP) seeds (A) or Q19(CFP/YFP) seeds (B) and split into 96-well plates 24 h after transfection. After 12 h recovery, cells were treated with 0.001, 0.01, 0.1, 1, 5 or 10 μm of leflunomide or teriflunomide and incubated for additional 24 h. Cells were washed, lysed and luciferase activity was determined. Luciferase activity is plotted as percentage to DMSO control for each drug set.
Teriflunomide directly binds to and inhibits the mitochondrial enzyme dihydroorotate dehydrogenase (DHODH), which catalyzes the conversion of dihydroorotate to orotate, the only redox reaction in the de novo biosynthesis of uracil-mono-phosphate (UMP), the precursor of all pyrimidine nucleotides (69–72). We wondered whether the effects of leflunomide/teriflunomide on httQ72-Luc aggregation depended on its role as a pyrimidine biosynthesis inhibitor. Effects that rely on the continuous biosynthesis of pyrimidine nucleotides are impaired by teriflunomide treatment and can be rescued by supplementing the media with uridine (72–74). We repeated the same experiments with 10 μm teriflunomide in the presence of saturating doses of uridine (100 μm). Uridine supplementation failed to alter teriflunomide effects, either at 24 h or at 48 h treatment, suggesting that inhibition of pyrimidine biosynthesis was not the mechanism of action of teriflunomide (Fig. 5A). To further demonstrate this, we used a set of anti-pyrimidine drugs. Acivicin and 6-azauridine are inhibitors of carbamoyl phosphate synthase and OMP-decarboxylase, the first and last steps in the biosynthesis of UMP, respectively (75), and the natural napthoquinone lapachol is an uncompetitive inhibitor also targeting DHODH (76). Notably, acivicin, 6-azauridine or the closest mimetic inhibitor to teriflunomide, lapachol, did not change the activity of the reporter after 24 or 48 h treatment (Fig. 5B). These results demonstrate that leflunomide and teriflunomide inhibit httQ72-Luc aggregation and that pyrimidine biosynthesis was not the mechanism of action of leflunomide/teriflunomide but represented an off-target effect of this drug instead.
Figure 5.

Leflunomide and teriflunomide increase httQ72-Luc solubility independently of pyrimidine biosynthesis inhibition. (A) HEK-293 cells were transfected with httQ72-Luc and Q80-CFP/YFP seeds and were split into 96-well plates 24 h later. After 12 h recovery, cells were treated with 100 μm teriflunomide or DMSO along with 100 μm uridine or vehicle. Luciferase activity was determined 24 or 48 h later and plotted as percentage to corresponding DMSO controls. (B) HEK-293 cells were transfected with httQ72-Luc along with Q19-CFP/YFP seeds or Q80-CFP/YFP seeds and were split into 96-well plates 24 h later. After 12 h recovery, cells were treated with 10 or 100 μm of the anti-pyrimidine drugs acivicin, 6-azauridine or lapachol, corresponding vehicles or teriflunomide for additional 24 or 48 h. Cells were washed and lysed and luciferase activity was determined. Luciferase activity is plotted as percentage to vehicle controls for each time set, as indicated.
Leflunomide and teriflunomide decrease polyQ aggregate size
Since leflunomide/teriflunomide did not affect FRET values, in the primary screen or the validation process (data not shown), we favored the hypothesis that these drugs interfered with the incorporation of httQ72-Luc into an aggregate. To test this, we evaluated the size and number of polyQ aggregates in cells treated with leflunomide/teriflunomide. The number of Q80-cfp and httQ72-cfp aggregates did not change with drug treatment, but they were smaller and more fragmented (Fig. 6A and B). To quantitate this, CFP immunofluorescence pictures were captured from HEK-293 cells transfected with httQ72-cfp or Q80-cfp and treated as before. Images were analyzed using the ImageJ software and the distribution of relative particle size was plotted as histogram plots. Leflunomide and teriflunomide treatment shifted the histogram distribution to the left in cells transfected with Q80-cfp, indicating that polyQ aggregates were smaller at the expense of the formation of bigger aggregates under the conditions tested (Fig. 6C). We defined a 300 pixel cutoff and similarly analyzed size distribution of aggregates using Q80-cfp. Leflunomide and teriflunomide decreased the size of aggregates in both httQ72-cfp- and Q80-cfp-transfected cells (Fig. 6D).
Figure 6.

Leflunomide and its active metabolite teriflunomide decrease polyQ aggregate size. (A) Representative CFP images of HEK-293 cells transiently transfected with httQ72-CFP or Q80-CFP and treated with 100 μm leflunomide, 100 μm teriflunomide or DMSO vehicle for 48 h. (B) Aggregates per cell in Q80-CFP transfected and treated cells as in (A) were counted in 10 independent fields from 2 independent experiments. (C) Images from experiments as in (A) using Q80-CFP were analyzed for particle size using ImageJ as described in Materials and Methods. Frequency distributions were plotted as histograms using 25pixel2 bins. Smaller aggregates (<300 pixel2) are more frequent in teriflunomide (red bars)- and leflunomide (white bars)-treated cells compared with vehicle (blue bars). Conversely, a decrease in aggregate size by leflunomide/teriflunomide was detected after a 300 pixel2 cutoff (horizontal bar). (D) Leflunomide and teriflunomide treatment decreases the size of both httQ72-CFP and Q80-CFP aggregates. **P < 0.01 and ***P < 0.005.
We reasoned that a decrease in polyQ aggregate size may also be associated with a decrease in the cytotoxicity associated with expanded polyQ expression. To test this, we expressed CFP control, Q35-CFP, httQ72-CFP and Q80-CFP in HEK-293 cells for 24h and then treated them with vehicle or 0.1, 1, 10 or 100 μm teriflunomide for an additional 48h. Consistent with previous studies, there was an increase in cell death as measured by LDH release in cells containing aggregated httQ72-CFP and Q80-CFP (45). However, the addition of teriflunomide at concentrations known to decrease aggregate size did not significantly alter cell viability (Supplementary Material, Fig. S5). These data confirm that teriflunomide is not toxic to cells at the concentrations used and any effect on protein aggregation is not a reflection of changes due to cell toxicity. However, these data do not support an effect for terfilunomide on improving polyQ toxicity under these conditions.
Leflunomide and teriflunomide inhibit polyQ aggregation by blocking recruitment into polyQ aggregates
To verify that decreased aggregate size and increased luciferase activity from the httQ72-Luc reporter correlated with increased solubility of polyQ proteins, we tested the aggregation of httQ72-cfp and Q80-cfp reporters by filter trap. HEK-293 cells were transiently transfected with httQ72-cfp and treated with 100 μm leflunomide/teriflunomide for 48 h starting 12 h after transfection. Despite similar levels of expression in the lysate, leflunomide and teriflunomide decreased the amount of aggregated httQ72-cfp and Q80-cfp (Fig. 7A), indicating that leflunomide/teriflunomide modified aggregation of polyQ independently of protein context. We next hypothesized that leflunomide/teriflunomide may inhibit incorporation of polyQ into an aggregate rather than disaggregating previously formed aggregates. To test this, we repeated the above experiments with an inducible cell line, Tet-ON U2OS[Q80-cfp], that expresses Q80-cfp upon induction with tetracycline (77). Cells grown in the presence of tetracycline for 3 days were re-plated in tetracycline-free media and treated either with 100 μm teriflunomide or with vehicle. This group represents teriflunomide effect on preformed aggregates (−Tet). Alternatively, similar cells were treated with 100 μm teriflunomide or vehicle in the presence of tetracycline. This group represents teriflunomide effect on a growing aggregate (+Tet). Cells were cultured for additional 48 h and filter retardation assays were performed as before. Teriflunomide increased solubility of Q80-cfp in the ‘growing aggregate’ setting but not in cells with preformed Q80-cfp aggregates (Fig. 7B).
Figure 7.

Teriflunomide blocks pure polyQ and expanded huntingtin aggregation by impeding incorporation into a polyQ aggregate. (A) HEK-293 cells were transiently transfected with httQ72-cfp or polyQ80-cfp and treated with 100 μm teriflunomide for 48 h. Lysates were prepared by sonication in the absence of detergents, and 20 μg of proteins were analyzed by filter trap using anti-GFP antibodies. Right panel: a representative western blot from three independent experiments to confirm similar levels of expression. (B) Tet-ON U2OS[Q80-cfp] cells were initially grown in the presence (−Tet) or absence (+Tet) of tetracycline for 3 days to preform aggregates and then replated in Tet-free or Tet-containing media to de novo formation of aggregates, respectively. Twelve hours after replating, cells were treated with 100 μm teriflunomide for 48 h and filter trap assays were done as before. Note that teriflunomide does not disaggregate preformed polyQ aggregates. (C) HEK-293 cells were co-transfected with httQ72-Luc and the indicated CFP/YFP-tagged polyQ proteins to seed the aggregation. Twenty-four hours after transfection, cells were split into 96-well plates and 12 h later, cells were treated with 100 μm teriflunomide for 24 h. Staggered CHX chase experiments were then performed for 0, 2 or 8 h in the presence of the drugs. Luciferase activity is represented as percentage to vehicle-only treatment at each time point. *P < 0.05, t-test (n = 8, from two independent experiments).
To further demonstrate that teriflunomide inhibited incorporation of polyQ into a growing aggregate, we performed cycloheximide (CHX)-chase experiments in the presence or the absence of teriflunomide. We hypothesized that, upon CHX treatment, a drop in luciferase activity in Q80-cfp-expressing cells compared with Q19-cfp-expressing cells would differentially reflect the incorporation of httQ72-Luc into an aggregate. The luciferase activity of httQ72-Luc in the presence of Q19-cfp remained constant in a time-window of 8 h CHX, indicating that httQ72-Luc is relatively long-lived. On the contrary, when httQ72-Luc was co-expressed with Q80-cfp, a 25% decrease in luciferase activity was detected after 8 h CHX treatment (Fig. 7C). Teriflunomide treatment restores luciferase activity at 2 and 8 h in the presence of CHX, by blocking incorporation into an existing aggregate. Together with the results obtained from the inducible Q80-cfp cell line, these experiments strongly suggest that teriflunomide interfered with the dynamic formation of polyQ aggregates by preventing the incorporation of httQ72-Luc into a growing polyQ aggregate.
DISCUSSION
In this work, we developed a sensitive and quantitative luciferase-based assay to assess expanded polyQ aggregation in cellulo, demonstrated its utility as a screening tool and identified leflunomide and its active metabolite teriflunomide as a novel class of polyQ aggregation inhibitors. Luciferase-based reporters that utilize firefly luciferase to directly assess protein aggregation in cellulo have not been reported. To date, luciferase refolding experiments have been utilized in vitro but only with the recombinant protein and never fused to an aggregation-prone domain (54,55). In vivo, a luciferase-based reporter has been described by Prusiner and co-workers (78,79), but protein aggregation is monitored indirectly as activation of the GFAP promoter in protein-aggregation environments such as the deposition of amyloid in APP transgenic mice or prion infection. Although it allows to non-invasively image dynamic processes in the brain, the reporter is an indicator of glial activation rather than protein aggregation itself. Another recently developed reporter to monitor protein aggregation uses a split Gaussia luciferase reporter fused to Aβ40/42 to monitor Aβ oligomer formation (80). The split luci-Aβ/ferase-Aβ reporter detects low-molecular-weight soluble oligomeric species of up to 24–36 subunits and allowed the confirmation that Aβ oligomer assembly is an intracellular event in route through the secretory pathway, with homopolymeric species of Aβ40 or Aβ42 being preferentially formed. However, whether this approach allows monitoring protein aggregation into higher molecular weight species like the insoluble aggregates found in Huntington and other polyQ diseases remains to be shown.
Our luciferase reporter is unique in assessing protein aggregation and offers advantages over other polyQ reporters. It distinguishes between soluble and insoluble species since luciferase signal is detected only when the protein is not in an aggregate. Therefore, it directly assesses protein aggregation by means of luciferase activity loss. Protein aggregates are in a dynamic equilibrium with soluble species and clearance of both soluble species and aggregates dictates their balance. When combined to FRET-based reporters, one can readily measure both the soluble and the insoluble protein components via plate reading or live imaging, allowing to simultaneously monitor both aggregate and, presumably, soluble intermediate fate in a single condition. We performed several direct comparisons between FRET-based reporters and our luciferase/FRET-coupled reporters and found that both reporters were able to quantify the aggregation state of expanded polyQ-containing proteins. However, with regard to a high-throughput screening approach, luciferase/FRET-coupled reporters had a superior Z-factor compared with FRET alone. In addition, many of the drugs identified had no effect on FRET but did have an effect on luciferase activity, suggesting that luciferase-based reporters may have broader utility than FRET-based reporters alone. Both assays are specific for polyQ aggregates, easy to use and cheap; however, the ability to measure aggregation and disaggregation in the same cell is an advance.
A novel class of drugs for polyQ inhibition
The results obtained in the present screening show that polyQ luciferase-based reporters can detect polyQ aggregate inhibitors that are missed by FRET-based reporters. Our screen was designed to identify drugs that assisted disassembly of preformed aggregates. Therefore, drugs were added 36 h after polyQ aggregates formed. We were unable to identify disaggregating drugs, as no hit simultaneously increased luciferase and decreased FRET to the levels of the Q19-seeds, i.e. with the soluble httQ72-Luc reporter. However, we did identify methylene blue as a drug that correspondingly modified those parameters (Supplementary Material, Fig. S3). Methylene blue is a phenothiazine which plays a role in polyQ aggregation. It modulates Hsp70 activity, promoting the degradation of aggregates of expanded androgen receptor fragments in cell culture (61) and increasing the solubility of Htt exon 1 Q103 fused to GFP in zebrafish embryos (60). Methylene blue has also been shown to reduce the aggregation of tau (81,82), Aβ (83,84) and TDP-43 (85) in cellular models and it also decreases tau burden in vivo (81).
We identified leflunomide and validated both leflunomide and its active metabolite teriflunomide as novel polyQ aggregate inhibitors. Both compounds increased luciferase activity of the httQ72-Luc reporter in a dose–response manner and only in the presence of polyQ aggregates. As expected, teriflunomide showed increased potency towards polyQ aggregate inhibition compared with leflunomide. This suggested that the mechanism of the action of leflunomide relies on its active metabolite teriflunomide, a DHODH inhibitor (72,74). However, we demonstrated that pyrimidine biosynthesis is not the mechanism of the action of teriflunomide. How leflunomide and teriflunomide inhibit polyQ aggregation is not clear at present. We suggest that teriflunomide blocks incorporation of soluble httQ72-luc into an existing aggregate. Because our methodology added leflunomide or teriflunomide to ‘preformed aggregates’, it appears that teriflunomide only makes aggregates smaller; however, Figure 7B evaluates the effect of teriflunomide on cells with growing aggregates versus non-growing aggregates, utilizing tetracycline-inducible cell lines. Teriflunomide is effective only on growing aggregates and did not change the aggregation state or disaggregate a preformed aggregate. We believe that teriflunomide blocks the incorporation of soluble polyQ into an aggregate and is therefore a polyQ aggregate inhibitor.
Other off-targets effects for teriflunomide, that fail to be rescued by uridine, have been previously described. These include (i) inhibition of tyrosine phosphorylation in Jurkat and CTLL-2 cells (86), (ii) inhibition of MAPK and p56lck leading to the blockade of NF-κB activation upon TNF stimulation in Jurkat cells (87), (iii) inhibition of JNK activation in cellular and animal models of acetaminophen-induced hepatotoxicity (88,89) and (iv) inhibition of the pro-survival pathways PDK1/Akt/GSK-3β leading to cell apoptosis in umbilical cord blood-derived mast cells (90). Of these, only JNK and MAPK inhibition were achieved at doses comparable with the ones used in the present study. The remaining off-target effects for teriflunomide are achieved only at very high concentrations (>50 μm) and therefore might not be mechanistically related to our findings. Interestingly, both MAPK and JNK signaling are induced by expanded polyQ and therefore might constitute an attractive target to further study (91,92). It is important to note that the cytotoxicity of aggregated httQ72-CFP or Q80-CFP in HEK-293 cells was not worsened by teriflunomide at doses capable of reducing aggregate burden. However, equally important, the cytotoxicity of polyQ expression was not rescued either. Whether leflunomide and teriflunomide are neuroprotective under conditions of polyQ toxicity remains to be assessed.
httQ72-Luc to study protein aggregate dynamics
We provide evidence that the cellular mechanism of protein aggregate inhibition by teriflunomide involves a blockade of incorporation of the reporter into a growing aggregate in vivo. Specifically, (i) maximal effect of teriflunomide on polyQ aggregation was observed only when the drug was added to cells that simultaneously express Q80-cfp, and not in cells with preformed aggregates, and (ii) CHX-chase experiments in the insoluble setting indicate that the decrease in luciferase activity can be rescued by teriflunomide treatment to levels similar to those of the soluble reporter (Fig. 7C). The interpretation of these experiments is consistent with a model where the mechanism of the action of the drug is not by disaggregating preformed aggregates but instead preventing polyQ incorporation into a growing aggregate. As a result, aggregate size of expanded polyQ decreased upon treatment and correlated with increased solubility of the protein aggregate (Fig. 7A). This model also agrees with the initial screen since leflunomide was identified based on its change in luciferase activity and not FRET. Our CHX-chase experiments allowed us to monitor the kinetics of the incorporation of httQ72-Luc into a Q80-cfp aggregate in cellulo. Assuming that the half-life of httQ72-Luc is similar in the soluble, non-aggregated phase of both Q19- and Q80-cfp-expressing cells, and considering that the luciferase activity arises only from cells expressing the soluble reporter (Fig. 2C–E), luciferase activity loss should reflect sequestration into an aggregate in that short CHX-chase kinetics. A 25% incorporation of httQ72-Luc into an aggregate in 8 h treatment (Fig. 7C) is in discrepancy with the fast appearance of aggregates in a cell (∼100min) (31,93). It is possible that recruitment kinetics are much slower than the homopolymeric aggregation process, but it is also possible that a decrease in luciferase activity under CHX-chase conditions represents the non-synchronized appearance of protein aggregates in the population of transfected cells. Regardless of which mechanism takes place, teriflunomide prevented luciferase activity loss induced by Q80-seeds, indicating that the drug prevented de novo incorporation into a preformed aggregate under the experimental conditions tested. This result was consistent with the fact that teriflunomide had an effect only when the treatment is co-temporal with Q80-cfp expression but not when Q80-cfp was already aggregated (Fig. 7B), validating the use of CHX-chase to monitor aggregate dynamics in cellulo.
Although httQ72-Luc aggregation is specific to expanded polyQ seeds, this reporter system can be conveniently modified to model heterologous protein sequestration into a polyQ aggregate similar to what occurs in Huntington disease with unexpanded polyQ proteins like CBP and prion-like proteins (94). Moreover, this method might be easily adapted to other protein aggregates as well. We foresee additional applications for httQ72-Luc and other luciferase-based protein aggregate reporters. By using inducible promoters, for example, protein disaggregation could be efficiently monitored at the cellular level. Starting with a highly aggregated reporter, an increase in luciferase activity might reflect the fraction of reporter being dissociated from the aggregate and should quantitatively reflect protein disaggregation. On the other hand, the responsiveness observed for polyQ-induced aggregation of both expanded and unexpanded huntingtin-luciferase makes the system ideal to also test different polyQ and Q/N-rich reporters, and CBP-Luc and TDP-43-Luc reporters to monitor heterologous recruitment into polyQ aggregates are under construction to evaluate this. In brain, non-invasive monitoring of protein aggregation has been shown feasible using the indirect reporter GFAP-Luc (78,79). We believe that adapting the httQ72-Luc reporter will provide an invaluable tool to monitor protein-aggregation dynamics in vivo using animal models for expanded huntingtin, like the R6/2 mice (95).
Teriflunomide as a CNS safe drug for the treatment of polyQ diseases
The importance of leflunomide/teriflunomide as a novel drug to prevent polyQ aggregation is emphasized by recent clinical data showing that teriflunomide has proved so far safe for the treatment of multiple sclerosis, a CNS autoimmune disease. In 1998, leflunomide was approved for the treatment of rheumatoid arthritis by the FDA and constitutes the latest antirheumatic disease-modifying drug and the first to receive the indication of retarding the structural damage of rheumatoid arthritis. Its strong ability to inhibit T-cell proliferation in autoimmune diseases like rheumatoid arthritis and myasthenia gravis led Jung and co-workers (66,96) to evaluate the use of this drug in another model, Experimental Autoimmune Neuritis (EAN). This animal model for multiple sclerosis involves the immunization of mice with myelin to evoke an immune response to myelin in both the spinal cord and peripheral nerves. The group found that teriflunomide suppressed EAN and proved to be an immunomodulatory agent to both peripheral and central milieaus. Neuroprotection was also observed in the Dark Agouti rat model of Experimental Autoimmune Encephalomyelitis, a similar model where myelin degeneration occurs deep in the CNS from spinal cord to the cerebellum. Teriflunomide at doses of 10 mg/kg decreased both demyelination and axonal loss in the posterior column of the spinal cord from 85 to 6% and from 67 to 4%, respectively (97). Although the actual bioavailability of teriflunomide in the CNS is still not clear, the effects presumably depend on the inhibition of de novo proliferation of activated T-cells migrating into the CNS through the blood–brain barrier. A phase-II study for the safety and efficacy of teriflunomide in multiple sclerosis with relapses has been recently developed (68). From this study, it seems that teriflunomide is well tolerated and effectively reduces MRI lesions, and recently reported phase-III TEMSO clinical trial confirm the initial findings (98). Considering that the safety profile of leflunomide in the treatment of patients with rheumatic arthritis is well known, after a clinical development program and more than 10 years of post-marketing surveillance, it is quite possible that teriflunomide will be approved for multiple sclerosis soon. The Johns Hopkins initiative for re-purposing drugs of clinical use allowed us to identify teriflunomide as a potential drug to decrease polyQ aggregates in vitro. This is the first report to attribute a role of leflunomide and teriflunomide as regulators of cellular protein aggregation. Considering that teriflunomide might be considered useful for treating a CNS disease and pilot studies in humans demonstrate safety, we propose that this drug be considered a potential drug for the treatment of polyQ diseases like Huntington.
MATERIALS AND METHODS
Cell culture
HEK-293 cells and U2OS cells were grown in DMEM containing 10% FBS and 2 mm l-glutamine. Stable U2OS TRex cells for the inducible expression of polyQ80-CFP were grown in DMEM containing 10% FBS, 2 mm l-glutamine, 50 μg/ml hygromycin B and 65 μg/ml zeocin and induced with 1 μg/ml tetracycline as indicated.
DNA constructs
DNA constructs for Fluc, Q19-cfp, Q19-yfp, Q35-cfp, Q35-yfp, httQ72-cfp, httQ72-yfp, Q80-cfp and Q80-yfp were previously described (34,77,99). p25-cfp was a generous gift of Dr Robert H. Baloh (Washington University). httQ72-Luc was constructed by PCR cloning using the primers CTG CAG TCG ACG GTA CCG CGG ATG GCG ACC CTG GAA AAG CTG ATG AAG G (sense) and CAG AAC TGG AAC CTC CCG AGG ATC CGA ATT CAG ATC CAG GTC GGT GCA GA (antisense). A megaprimer of 474 bp was created by PCR using httQ72-cfp as a template (99), gel-purified and 350 ng used to reconstitute 50 ng of Q19-Luc using site-directed mutagenesis XL kit. The resulting plasmid contained the huntingtin exon 1 sequences and luciferase joined by a GSSGGSSSSGG flexible linker, as expected. However, the Q19 domain was not efficiently removed from the mother plasmid when the swapping reaction occurred, resulting in an in-frame insertion of 19 glutamines N-terminally to httQ72-Luc and we called this construct Q19httQ72-Luc. The extra Q19 domain was efficiently removed by an XhoI–SalI digestion, resulting in the plasmid httQ72-Luc. The new Kozik sequence is ctcgacGGTACCGCGGG (joint sequences indicated in bold lowercase). Similarly, httQ25-Luc was created using httQ25-CFP as a template. To create L221K monomeric mutants of CFP and YFP (numbering to GFP) in Q19- and Q80-tagged constructs, site-directed mutagenesis experiments were performed using primers as described before. All constructs used in this study were verified by sequencing in both strands, and they are all based on pECFP vector backbones (KanR, Clontech).
Luciferase+FRET polyQ aggregate reporter
HEK-293 cells were plated in 12-well plates the day before transfection at 3 × 105/well. DNA complexes were prepared the next day using 350 ng of CFP-tagged vector, 750 ng of YFP-tagged vector, 132 ng of httQ72-Luc and 3.5 μl of Fugene 6 in 50 μl of total volume as recommended by the manufacturer, but diluting the DNAs in DMEM before adding Fugene 6. After 30min of complex formation, mixtures were added dropwise to each well. For compensation controls, 600 ng of Q19-cfp or Q19-yfp and 1.8 μl of Fugene 6 in 50 μl of total volume in DMEM were used. Thirty-six hours after transfection, cells were tripsinized with 600 μl of tripsin and incubated for 2 min at 37°C. Growing media were then added to a final volume of 2 ml and 100 μl (1: 20) and were seeded in quadruplicate into sister cultures in 96-well clear-bottom white (Costar 3903) and 96-well clear-bottom black (Costar 3603) plates for luciferase and FRET determinations, respectively. For drug treatments, drugs were added in 10 μl (final volume 110 μl) 12 h after seeding and cells incubated for additional 24 h. For experiments involving longer time incubations, cells were identically treated but split 1:30 in the 96-well plates. After drug treatment, cells were washed twice in PBS and lysed for 30 min with 20 μl of Reporter Lysis Buffer 1× (Promega, Madison, WI, USA) for luciferase determination or fixed in PFA 4% for FRET determinations as described. Luciferase was measured using the Luciferase Assay System and a GloMax-Multi Detection System (Promega), and CFP, YFP and FRET were determined in an Infinite M1000 plate reader (Tecan Group Ltd, Männedorf, Switzerland). Luciferase activity was background-subtracted from mock-transfected cells. The CFP, YFP and FRET signals were first background-subtracted from mock-transfected cells and the FRET value was corrected for bleedthrough and crossover excitation according to: FRET/donor = [SMPL435/527 − X·(SMPL485/527)]/SMPL435/485, where X = YFP435/527/YFP485/527.
Immunocytochemistry and live BLI
U2OS cells were seeded in glass cover slips in 12-well plates the day before transfection at 5 × 105/well. Cells were transfected with 0.2 μg of luciferase-tagged and 0.2 μg of cfp-tagged constructs using Fugene 6 in a 1:3 ratio, as recommended by the manufacturer. After 48 h, cells were rinsed in cold PBS, fixed in 4% paraformaldehyde for 20 min at room temperature, washed again in PBS and permeabilized in PBS. After 48 h, cells were rinsed in cold PBS, fixed in 4% paraformaldehyde for 20 min at room temperature, washed again in PBS and permeabilized in PBS. After three washes with blocking solution, cells were incubated with Alexa 555 anti-goat antibodies (Invitrogen, 1:2000) for 30 min at 37°C, washed again and imaged. For BLI, HEK-293 cells were transfected in six-well plates exactly as for filter trap experiments and split into individual 35 mm dishes 12 h after transfection. Twenty-four hours later, cells were imaged in a low-light microscope as follows: cells were washed twice with PBS and incubated with 1 ml of luciferin reagent (150 μg/ml d-luciferin in PBS) for 15min. A field containing cells with both aggregated and soluble Q80-cfp was identified using fluorescence microscopy and imaged for both bright field and CFP. Then, cells were quickly imaged for bioluminescence with a cooled CCD camera using 2 min as the capture time.
Detergent fractionation experiments
The day before transfection, HEK-293 cells were plated in six-well plates (two wells per condition) at 8 × 105/well or in 12-well plates at 3 × 105/well for compensation controls. DNA complexes were prepared as before for CFP and YFP compensation controls or were scaled up 10 times for transfection into six-well plate (250 μl of complex per well). Cells were washed and harvested by scrapping the cells in PBS. Consolidated pellets were resuspended in 400 µl of lysis buffer (PBS + 1% Triton X-100 and protease inhibitor cocktail) and lysed by sonication as described before for filter trap assays. Total protein from the lysate was determined in each sample according to the Bradford method, and samples were diluted to 1 μg/μl with lysis buffer. An amount of 300 μl of diluted samples were centrifuged at 1000g for 10 min at 4°C. A total of 250 µl of the supernatant was removed for luciferase and FRET measurements in quadruplicate using 25 μl per well in 96-well plates, as before. Lysates were diluted prior to FRET determinations by adding 75 μl of lysis buffer per well.
Screening of the JHCCL
The JHCCL comprises a collection of 1937 FDA-approved drugs and 750 drugs that were either approved for use abroad or undergoing phase-II clinical trials as of 2006. Daughter plates were created in duplicate from the 384-well parent microtiter plates that contained the compounds at concentrations of 1 mm, using robotic liquid-handling system available through the High-Throughput Screening Core at Washington University School of Medicine. The library screen was completed in three sections using a Beckman Coulter Core robotics system, including an FX liquid handler, controlled by the Sagian graphical method development tool (SAMI scheduling software). For each one of the sections, HEK-293 cells were plated in 10 × 10 cm2 plates at 8 × 106 the day before transfection. Each plate was transfected with 5.25 μg of Q80-CFP, 11.25 μg of Q80-YFP, 1.98 μg of httQ72-Luc and 52.5 μl of Fugene 6 in 750 μl total volume as recommended by the manufacturer but diluting DNAs in DMEM first. Compensation controls (Q19-cfp and Q19-yfp) and controls to qualify the degree of aggregation of httQ72-Luc (Q19-, Q35-, Q72-, Q80(CFP/YFP) FRET pairs) were also included in every plate, and were obtained by transfecting 2 wells of a 12-well plate as indicated above. Thirty-six hours after transfection, cells were detached from the 10 cm plate by tripsin treatment 5 min at 37°C and brought to 30 ml in growing media. Cells were then combined and plated (100 μl) in each one of 96-well clear-bottom black plates (Costar 3603), with control cells seeded 1:20 at the edge columns. The plates were settled at room temperature for 30 min and then placed overnight in 90% humidity, 5% CO2 37°C incubators. The next day, library drugs were diluted 1:40 in growing media and then one-fifth of the media removed from the cell plates and replenished with the diluted drugs. Cells were incubated for exactly 24 h, carefully washed twice with DPBS (Invitrogen) and then plates were measured for CFP, YFP and FRET using a Perkin-Elmer EnVision Xcite 2102 Multilabel Reader (Perkin Elmer). Fifty microliters of DPBS was removed from the cells and 50 μl of d-luciferin (300 μg/ml) in DPBS was added. After 20min incubation at room temperature, luminescence was recorded using the same instrumentation. Finally, cell viability was assessed by adding 10 μl of Alamar Blue and reading the plates 10min later in a BMG FLUOstar Optima plate reader (Perkin Elmer).
Dose–response experiments
All drugs were purchased from Sigma-Aldrich, with the following exceptions: acivicin (Santa Cruz Biotechnologies), 6-azauridine (Chromadex), teriflunomide (A77 1726, Calbiochem). Commercially available drugs were prepared as 50 mm stocks in DMSO. Serial dilutions were prepared using liquid-handling robotics to different concentrations of 0.002, 0.01, 0.02, 0.1, 0.2, 1 and 2 mm. HEK-293 cells were transfected and treated with drugs as during the screening with the following modifications: each 10 cm plate was brought to 45 ml of growing media before seeding and the luciferase activity was measured 48 h after drug treatments. Cell lysates were prepared as before using the Luciferase Assay System kit (Promega) and luciferase activity measured using a GloMax®-Multi detection system (Promega). EC50 values from dose–response experiments were calculated using the Prism software and reported as average value from the best sigmoidal fit (GraphPad, Inc.).
Filter trap experiments
HEK-293 cells were seeded in six-well plates at 6 × 105/well and grown overnight. Cells were then co-transfected with 0.5 μg of httQ72-Luc (or Fluc) + 0.5 μg of CFP-tagged constructs or transfected with 1 μg of CFP-tagged polyQ alone using Fugene 6 in a 1:3 ratio as recommended by the manufacturer. For drug treatment experiments, cells were treated with 100 μm leflunomide, 100 μm teriflunomide or DMSO vehicle 12 h after transfection. For the experiments using the inducible Tet-ON U2OS[Q80-cfp] cell line, cells were plated in 60 mm dishes and grown in the presence or absence of tetracycline for 3 days. Cells were then split 1:2 in tetracycline-free media and treated with 100 μm teriflunomide or DMSO in the presence or absence of tetracycline of 1 μg/ml. After 48 h expression or treatment with drugs, cells were washed twice in PBS, scrapped and pelleted. Cell pellets were resuspended in 400 μl of PBS with 1 mm PMSF (PBS/PMSF) and sonicated with 10 pulses in an ultrasonic homogenizer model Omni-Ruptor 250 (Omni, Kennesaw, GA, USA) with 25% power and 10% pulser settings and lysates subjected to filter trap as before. Briefly, cell lysates were normalized to 0.2 mg/ml protein in PBS/PMSF and diluted in PBS/2% SDS buffer (1.6% w/v final concentration SDS). Twenty or 5 μg of cell lysate were immediately applied to a pre-wetted cellulose acetate 0.2 μm filter using a dot blot device (Bio-Rad). After two washes with 0.5 ml of PBS/2% SDS buffer, the membrane was incubated for 1 h in blocking buffer (5% milk in PBS containing 0.05% Tween 20) with gentle rocking at room temperature. The membrane was then incubated with anti-GFP antibodies (Sigma, 1:3000) or anti-Luciferase antibodies (Promega, 1:1000) in blocking buffer overnight, washed four times for 10 min with washing buffer (PBS with 0.05% Tween 20) and incubated with secondary antibodies in blocking buffer (1:5000) for 2–4 h at room temperature. The membranes were washed seven times, and proteins trapped in the filter were visualized using ECL reagent (GE Healthcare).
CHX chase experiments
HEK-293 cells were co-transfected with httQ72-Luc and Q19(CFP/YFP) (soluble setting) or Q80(CFP/YFP) (insoluble setting) in 12-well plates as before. Sixteen hours after transfection, cells were plated 1:30 in 96-well clear-bottom white (Costar 3903) plates. The next day, the cells were treated with 100 μm teriflunomide or DMSO vehicle for 40 h and staggered CHX chase experiments were performed by incubation with CHX or vehicle (ethanol) for 2 or 8 h at 37°C. Where necessary, teriflunomide was included to keep the concentration of the drug constant during the CHX chase. After completion of a 48 h total treatment with teriflunomide, luciferase activity was determined using the Luciferase Assay System kit and results plotted as % luciferase activity to the vehicle control at each time point.
Quantitation of polyQ aggregate size
HEK-293 cells from filter trap experiments were washed in PBS, and live images were acquired with a Cool Snap ES camera (Photometrics) mounted on a Nikon Eclipse TE-300 microscope, controlled using the MetaMorph software (Molecular Devices). CFP channel images were analyzed with the ImageJ software using the ‘analyze particles’ command with a threshold of 180–255 and particles >50 pixel2 in area were considered for histogram representations. Frequency was calculated for 25 pixel2 bin increments or a cutoff of 300 pixel2 was arbitrarily set.
Statistical analysis
Statistical significance (t-test) from at least three independent experiments performed either in triplicate or in quadruplicate is shown. *P< 0.05, **P < 0.01, ***P< 0.005.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
Conflict of Interest statement. The authors state no conflict of interest.
FUNDING
This work was supported by the National Institutes of Health (AG031867 to C.C.W., P50 CA94056 to D.P.-W.) and the Hope Center for Neurological Disorders, Washington University in St Louis (pilot grant to C.C.W. and D.P.-W.).
Supplementary Material
REFERENCES
- 1.La Spada A.R., Wilson E.M., Lubahn D.B., Harding A.E., Fischbeck K.H. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. 1991;352:77–79. doi: 10.1038/352077a0. [DOI] [PubMed] [Google Scholar]
- 2.Shao J., Diamond M.I. Polyglutamine diseases: emerging concepts in pathogenesis and therapy. Hum. Mol. Genet. 2007;16:R115–R123. doi: 10.1093/hmg/ddm213. Spec No. 2. [DOI] [PubMed] [Google Scholar]
- 3.Caine E.D., Shoulson I. Psychiatric syndromes in Huntington's disease. Am. J. Psychiatry. 1983;140:728–733. doi: 10.1176/ajp.140.6.728. [DOI] [PubMed] [Google Scholar]
- 4.Walker F.O. Huntington's disease. Lancet. 2007;369:218–228. doi: 10.1016/S0140-6736(07)60111-1. [DOI] [PubMed] [Google Scholar]
- 5.MacDonald M.E., Ambrose C.M., Duyao M.P., Myers R.H., Lin C., Srinidhi L., Barnes G., Taylor S.A., James M., Groot N., et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 6.Stine O.C., Pleasant N., Franz M.L., Abbott M.H., Folstein S.E., Ross C.A. Correlation between the onset age of Huntington's disease and length of the trinucleotide repeat in IT-15. Hum. Mol. Genet. 1993;2:1547–1549. doi: 10.1093/hmg/2.10.1547. [DOI] [PubMed] [Google Scholar]
- 7.Rubinsztein D.C., Leggo J., Coles R., Almqvist E., Biancalana V., Cassiman J.J., Chotai K., Connarty M., Crauford D., Curtis A., et al. Phenotypic characterization of individuals with 30–40 CAG repeats in the Huntington disease (HD) gene reveals HD cases with 36 repeats and apparently normal elderly individuals with 36–39 repeats. Am. J. Hum Genet. 1996;59:16–22. [PMC free article] [PubMed] [Google Scholar]
- 8.Scherzinger E., Sittler A., Schweiger K., Heiser V., Lurz R., Hasenbank R., Bates G.P., Lehrach H., Wanker E.E. Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: implications for Huntington's disease pathology. Proc. Natl Acad. Sci. USA. 1999;96:4604–4609. doi: 10.1073/pnas.96.8.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Narain Y., Wyttenbach A., Rankin J., Furlong R.A., Rubinsztein D.C. A molecular investigation of true dominance in Huntington's disease. J. Med. Genet. 1999;36:739–746. doi: 10.1136/jmg.36.10.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Graham R.K., Slow E.J., Deng Y., Bissada N., Lu G., Pearson J., Shehadeh J., Leavitt B.R., Raymond L.A., Hayden M.R. Levels of mutant huntingtin influence the phenotypic severity of Huntington disease in YAC128 mouse models. Neurobiol. Dis. 2006;21:444–455. doi: 10.1016/j.nbd.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 11.Colby D.W., Cassady J.P., Lin G.C., Ingram V.M., Wittrup K.D. Stochastic kinetics of intracellular huntingtin aggregate formation. Nat. Chem. Biol. 2006;2:319–323. doi: 10.1038/nchembio792. [DOI] [PubMed] [Google Scholar]
- 12.Chen S., Ferrone F.A., Wetzel R. Huntington's disease age-of-onset linked to polyglutamine aggregation nucleation. Proc. Natl Acad. Sci. USA. 2002;99:11884–11889. doi: 10.1073/pnas.182276099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nagai Y., Inui T., Popiel H.A., Fujikake N., Hasegawa K., Urade Y., Goto Y., Naiki H., Toda T. A toxic monomeric conformer of the polyglutamine protein. Nat. Struct. Mol. Biol. 2007;14:332–340. doi: 10.1038/nsmb1215. [DOI] [PubMed] [Google Scholar]
- 14.Thakur A.K., Jayaraman M., Mishra R., Thakur M., Chellgren V.M., Byeon I.J., Anjum D.H., Kodali R., Creamer T.P., Conway J.F., et al. Polyglutamine disruption of the huntingtin exon 1 N terminus triggers a complex aggregation mechanism. Nat. Struct. Mol. Biol. 2009;16:380–389. doi: 10.1038/nsmb.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen S., Berthelier V., Hamilton J.B., O'Nuallain B., Wetzel R. Amyloid-like features of polyglutamine aggregates and their assembly kinetics. Biochemistry. 2002;41:7391–7399. doi: 10.1021/bi011772q. [DOI] [PubMed] [Google Scholar]
- 16.Scherzinger E., Lurz R., Turmaine M., Mangiarini L., Hollenbach B., Hasenbank R., Bates G.P., Davies S.W., Lehrach H., Wanker E.E. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell. 1997;90:549–558. doi: 10.1016/s0092-8674(00)80514-0. [DOI] [PubMed] [Google Scholar]
- 17.Williams A.J., Paulson H.L. Polyglutamine neurodegeneration: protein misfolding revisited. Trends Neurosci. 2008;31:521–528. doi: 10.1016/j.tins.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perez M.K., Paulson H.L., Pendse S.J., Saionz S.J., Bonini N.M., Pittman R.N. Recruitment and the role of nuclear localization in polyglutamine-mediated aggregation. J. Cell. Biol. 1998;143:1457–1470. doi: 10.1083/jcb.143.6.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kazantsev A., Preisinger E., Dranovsky A., Goldgaber D., Housman D. Insoluble detergent-resistant aggregates form between pathological and nonpathological lengths of polyglutamine in mammalian cells. Proc. Natl Acad. Sci. USA. 1999;96:11404–11409. doi: 10.1073/pnas.96.20.11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen S., Berthelier V., Yang W., Wetzel R. Polyglutamine aggregation behavior in vitro supports a recruitment mechanism of cytotoxicity. J. Mol. Biol. 2001;311:173–182. doi: 10.1006/jmbi.2001.4850. [DOI] [PubMed] [Google Scholar]
- 21.McCampbell A., Taylor J.P., Taye A.A., Robitschek J., Li M., Walcott J., Merry D., Chai Y., Paulson H., Sobue G., et al. CREB-binding protein sequestration by expanded polyglutamine. Hum. Mol. Genet. 2000;9:2197–2202. doi: 10.1093/hmg/9.14.2197. [DOI] [PubMed] [Google Scholar]
- 22.Nucifora F.C., Jr, Sasaki M., Peters M.F., Huang H., Cooper J.K., Yamada M., Takahashi H., Tsuji S., Troncoso J., Dawson V.L., et al. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science. 2001;291:2423–2428. doi: 10.1126/science.1056784. [DOI] [PubMed] [Google Scholar]
- 23.Steffan J.S., Kazantsev A., Spasic-Boskovic O., Greenwald M., Zhu Y.Z., Gohler H., Wanker E.E., Bates G.P., Housman D.E., Thompson L.M. The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl Acad. Sci. USA. 2000;97:6763–6768. doi: 10.1073/pnas.100110097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang C.C., Faber P.W., Persichetti F., Mittal V., Vonsattel J.P., MacDonald M.E., Gusella J.F. Amyloid formation by mutant huntingtin: threshold, progressivity and recruitment of normal polyglutamine proteins. Somat. Cell Mol. Genet. 1998;24:217–233. doi: 10.1023/b:scam.0000007124.19463.e5. [DOI] [PubMed] [Google Scholar]
- 25.Yamanaka T., Tosaki A., Miyazaki H., Kurosawa M., Furukawa Y., Yamada M., Nukina N. Mutant huntingtin fragment selectively suppresses Brn-2 POU domain transcription factor to mediate hypothalamic cell dysfunction. Hum. Mol. Genet. 2010;19:2099–2112. doi: 10.1093/hmg/ddq087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Derkatch I.L., Uptain S.M., Outeiro T.F., Krishnan R., Lindquist S.L., Liebman S.W. Effects of Q/N-rich, polyQ, and non-polyQ amyloids on the de novo formation of the [PSI+] prion in yeast and aggregation of Sup35 in vitro. Proc. Natl Acad. Sci. USA. 2004;101:12934–12939. doi: 10.1073/pnas.0404968101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Urakov V.N., Vishnevskaya A.B., Alexandrov I.M., Kushnirov V.V., Smirnov V.N., Ter-Avanesyan M.D. Interdependence of amyloid formation in yeast: implications for polyglutamine disorders and biological functions. Prion. 2010;4:45–52. doi: 10.4161/pri.4.1.11074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DePace A.H., Santoso A., Hillner P., Weissman J.S. A critical role for amino-terminal glutamine/asparagine repeats in the formation and propagation of a yeast prion. Cell. 1998;93:1241–1252. doi: 10.1016/s0092-8674(00)81467-1. [DOI] [PubMed] [Google Scholar]
- 29.Meriin A.B., Zhang X., He X., Newnam G.P., Chernoff Y.O., Sherman M.Y. Huntington toxicity in yeast model depends on polyglutamine aggregation mediated by a prion-like protein Rnq1. J. Cell Biol. 2002;157:997–1004. doi: 10.1083/jcb.200112104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Osherovich L.Z., Weissman J.S. Multiple Gln/Asn-rich prion domains confer susceptibility to induction of the yeast [PSI(+)] prion. Cell. 2001;106:183–194. doi: 10.1016/s0092-8674(01)00440-8. [DOI] [PubMed] [Google Scholar]
- 31.Furukawa Y., Kaneko K., Matsumoto G., Kurosawa M., Nukina N. Cross-seeding fibrillation of Q/N-rich proteins offers new pathomechanism of polyglutamine diseases. J. Neurosci. 2009;29:5153–5162. doi: 10.1523/JNEUROSCI.0783-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Doi H., Koyano S., Suzuki Y., Nukina N., Kuroiwa Y. The RNA-binding protein FUS/TLS is a common aggregate-interacting protein in polyglutamine diseases. Neurosci. Res. 2010;66:131–133. doi: 10.1016/j.neures.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 33.Doi H., Okamura K., Bauer P.O., Furukawa Y., Shimizu H., Kurosawa M., Machida Y., Miyazaki H., Mitsui K., Kuroiwa Y., et al. RNA-binding protein TLS is a major nuclear aggregate-interacting protein in huntingtin exon 1 with expanded polyglutamine-expressing cells. J. Biol. Chem. 2008;283:6489–6500. doi: 10.1074/jbc.M705306200. [DOI] [PubMed] [Google Scholar]
- 34.Fuentealba R.A., Udan M., Bell S., Wegorzewska I., Shao J., Diamond M.I., Weihl C.C., Baloh R.H. Interaction with polyglutamine aggregates reveals a Q/N-rich domain in TDP-43. J. Biol. Chem. 2010;285:26304–26314. doi: 10.1074/jbc.M110.125039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwab C., Arai T., Hasegawa M., Yu S., McGeer P.L. Colocalization of transactivation-responsive DNA-binding protein 43 and huntingtin in inclusions of Huntington disease. J. Neuropathol. Exp. Neurol. 2008;67:1159–1165. doi: 10.1097/NEN.0b013e31818e8951. [DOI] [PubMed] [Google Scholar]
- 36.Michelitsch M.D., Weissman J.S. A census of glutamine/asparagine-rich regions: implications for their conserved function and the prediction of novel prions. Proc. Natl Acad. Sci. USA. 2000;97:11910–11915. doi: 10.1073/pnas.97.22.11910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takahashi T., Katada S., Onodera O. Polyglutamine diseases: Where does toxicity come from? What is toxicity? Where are we going? J. Mol. Cell Biol. 2010;2:180–191. doi: 10.1093/jmcb/mjq005. [DOI] [PubMed] [Google Scholar]
- 38.Kazantsev A., Walker H.A., Slepko N., Bear J.E., Preisinger E., Steffan J.S., Zhu Y.Z., Gertler F.B., Housman D.E., Marsh J.L., et al. A bivalent Huntingtin binding peptide suppresses polyglutamine aggregation and pathogenesis in Drosophila. Nat. Genet. 2002;30:367–376. doi: 10.1038/ng864. [DOI] [PubMed] [Google Scholar]
- 39.Pollitt S.K., Pallos J., Shao J., Desai U.A., Ma A.A., Thompson L.M., Marsh J.L., Diamond M.I. A rapid cellular FRET assay of polyglutamine aggregation identifies a novel inhibitor. Neuron. 2003;40:685–694. doi: 10.1016/s0896-6273(03)00697-4. [DOI] [PubMed] [Google Scholar]
- 40.Tanaka M., Machida Y., Niu S., Ikeda T., Jana N.R., Doi H., Kurosawa M., Nekooki M., Nukina N. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat. Med. 2004;10:148–154. doi: 10.1038/nm985. [DOI] [PubMed] [Google Scholar]
- 41.Heiser V., Engemann S., Brocker W., Dunkel I., Boeddrich A., Waelter S., Nordhoff E., Lurz R., Schugardt N., Rautenberg S., et al. Identification of benzothiazoles as potential polyglutamine aggregation inhibitors of Huntington's disease by using an automated filter retardation assay. Proc. Natl Acad. Sci. USA. 2002;99(Suppl. 4):16400–16406. doi: 10.1073/pnas.182426599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang J., Gines S., MacDonald M.E., Gusella J.F. Reversal of a full-length mutant huntingtin neuronal cell phenotype by chemical inhibitors of polyglutamine-mediated aggregation. BMC Neurosci. 2005;6:1. doi: 10.1186/1471-2202-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aiken C.T., Tobin A.J., Schweitzer E.S. A cell-based screen for drugs to treat Huntington's disease. Neurobiol. Dis. 2004;16:546–555. doi: 10.1016/j.nbd.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 44.Coufal M., Maxwell M.M., Russel D.E., Amore A.M., Altmann S.M., Hollingsworth Z.R., Young A.B., Housman D.E., Kazantsev A.G. Discovery of a novel small-molecule targeting selective clearance of mutant huntingtin fragments. J. Biomol. Screen. 2007;12:351–360. doi: 10.1177/1087057107299428. [DOI] [PubMed] [Google Scholar]
- 45.Piccioni F., Roman B.R., Fischbeck K.H., Taylor J.P. A screen for drugs that protect against the cytotoxicity of polyglutamine-expanded androgen receptor. Hum. Mol. Genet. 2004;13:437–446. doi: 10.1093/hmg/ddh045. [DOI] [PubMed] [Google Scholar]
- 46.Ferrington D.A., Husom A.D., Thompson L.V. Altered proteasome structure, function, and oxidation in aged muscle. FASEB J. 2005;19:644–646. doi: 10.1096/fj.04-2578fje. [DOI] [PubMed] [Google Scholar]
- 47.Zhang X., Smith D.L., Meriin A.B., Engemann S., Russel D.E., Roark M., Washington S.L., Maxwell M.M., Marsh J.L., Thompson L.M., et al. A potent small molecule inhibits polyglutamine aggregation in Huntington's disease neurons and suppresses neurodegeneration in vivo. Proc. Natl Acad. Sci. USA. 2005;102:892–897. doi: 10.1073/pnas.0408936102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Doumanis J., Wada K., Kino Y., Moore A.W., Nukina N. RNAi screening in Drosophila cells identifies new modifiers of mutant huntingtin aggregation. PLoS One. 2009;4:e7275. doi: 10.1371/journal.pone.0007275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang S., Binari R., Zhou R., Perrimon N. A genomewide RNA interference screen for modifiers of aggregates formation by mutant Huntingtin in Drosophila. Genetics. 2010;184:1165–1179. doi: 10.1534/genetics.109.112516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Desai U.A., Pallos J., Ma A.A., Stockwell B.R., Thompson L.M., Marsh J.L., Diamond M.I. Biologically active molecules that reduce polyglutamine aggregation and toxicity. Hum. Mol. Genet. 2006;15:2114–2124. doi: 10.1093/hmg/ddl135. [DOI] [PubMed] [Google Scholar]
- 51.Ashburn T.T., Thor K.B. Drug repositioning: identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004;3:673–683. doi: 10.1038/nrd1468. [DOI] [PubMed] [Google Scholar]
- 52.Chong C.R., Chen X., Shi L., Liu J.O., Sullivan D.J., Jr A clinical drug library screen identifies astemizole as an antimalarial agent. Nat. Chem. Biol. 2006;2:415–416. doi: 10.1038/nchembio806. [DOI] [PubMed] [Google Scholar]
- 53.Chong C.R., Sullivan D.J., Jr New uses for old drugs. Nature. 2007;448:645–646. doi: 10.1038/448645a. [DOI] [PubMed] [Google Scholar]
- 54.Hendrick J.P., Langer T., Davis T.A., Hartl F.U., Wiedmann M. Control of folding and membrane translocation by binding of the chaperone DnaJ to nascent polypeptides. Proc. Natl Acad. Sci. USA. 1993;90:10216–10220. doi: 10.1073/pnas.90.21.10216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schroder H., Langer T., Hartl F.U., Bukau B. DnaK, DnaJ and GrpE form a cellular chaperone machinery capable of repairing heat-induced protein damage. EMBO J. 1993;12:4137–4144. doi: 10.1002/j.1460-2075.1993.tb06097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Buchberger A., Schroder H., Buttner M., Valencia A., Bukau B. A conserved loop in the ATPase domain of the DnaK chaperone is essential for stable binding of GrpE. Nat. Struct. Biol. 1994;1:95–101. doi: 10.1038/nsb0294-95. [DOI] [PubMed] [Google Scholar]
- 57.Gross S., Piwnica-Worms D. Real-time imaging of ligand-induced IKK activation in intact cells and in living mice. Nat. Methods. 2005;2:607–614. doi: 10.1038/nmeth779. [DOI] [PubMed] [Google Scholar]
- 58.Li M., Chevalier-Larsen E.S., Merry D.E., Diamond M.I. Soluble androgen receptor oligomers underlie pathology in a mouse model of spinobulbar muscular atrophy. J. Biol. Chem. 2007;282:3157–3164. doi: 10.1074/jbc.M609972200. [DOI] [PubMed] [Google Scholar]
- 59.Zhang J.H., Chung T.D., Oldenburg K.R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 60.van Bebber F., Paquet D., Hruscha A., Schmid B., Haass C. Methylene blue fails to inhibit Tau and polyglutamine protein dependent toxicity in zebrafish. Neurobiol. Dis. 2010;39:265–271. doi: 10.1016/j.nbd.2010.03.023. [DOI] [PubMed] [Google Scholar]
- 61.Wang A.M., Morishima Y., Clapp K.M., Peng H.M., Pratt W.B., Gestwicki J.E., Osawa Y., Lieberman A.P. Inhibition of hsp70 by methylene blue affects signaling protein function and ubiquitination and modulates polyglutamine protein degradation. J. Biol. Chem. 2010;285:15714–15723. doi: 10.1074/jbc.M109.098806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Webb J.L., Ravikumar B., Rubinsztein D.C. Microtubule disruption inhibits autophagosome-lysosome fusion: implications for studying the roles of aggresomes in polyglutamine diseases. Int. J. Biochem. Cell Biol. 2004;36:2541–2550. doi: 10.1016/j.biocel.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 63.Shimohata T., Sato A., Burke J.R., Strittmatter W.J., Tsuji S., Onodera O. Expanded polyglutamine stretches form an ‘aggresome. Neurosci. Lett. 2002;323:215–218. doi: 10.1016/s0304-3940(02)00162-3. [DOI] [PubMed] [Google Scholar]
- 64.Yamamoto A., Cremona M.L., Rothman J.E. Autophagy-mediated clearance of huntingtin aggregates triggered by the insulin-signaling pathway. J. Cell Biol. 2006;172:719–731. doi: 10.1083/jcb.200510065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kalgutkar A.S., Nguyen H.T., Vaz A.D., Doan A., Dalvie D.K., McLeod D.G., Murray J.C. In vitro metabolism studies on the isoxazole ring scission in the anti-inflammatory agent lefluonomide to its active alpha-cyanoenol metabolite A771726: mechanistic similarities with the cytochrome P450-catalyzed dehydration of aldoximes. Drug Metab. Dispos. 2003;31:1240–1250. doi: 10.1124/dmd.31.10.1240. [DOI] [PubMed] [Google Scholar]
- 66.Korn T., Toyka K., Hartung H.P., Jung S. Suppression of experimental autoimmune neuritis by leflunomide. Brain. 2001;124:1791–1802. doi: 10.1093/brain/124.9.1791. [DOI] [PubMed] [Google Scholar]
- 67.Gold R., Wolinsky J.S. Pathophysiology of multiple sclerosis and the place of teriflunomide. Acta Neurol. Scand. 2011;124:75–84. doi: 10.1111/j.1600-0404.2010.01444.x. [DOI] [PubMed] [Google Scholar]
- 68.O'Connor P.W., Li D., Freedman M.S., Bar-Or A., Rice G.P., Confavreux C., Paty D.W., Stewart J.A., Scheyer R. A phase II study of the safety and efficacy of teriflunomide in multiple sclerosis with relapses. Neurology. 2006;66:894–900. doi: 10.1212/01.wnl.0000203121.04509.31. [DOI] [PubMed] [Google Scholar]
- 69.Davis J.P., Cain G.A., Pitts W.J., Magolda R.L., Copeland R.A. The immunosuppressive metabolite of leflunomide is a potent inhibitor of human dihydroorotate dehydrogenase. Biochemistry. 1996;35:1270–1273. doi: 10.1021/bi952168g. [DOI] [PubMed] [Google Scholar]
- 70.Greene S., Watanabe K., Braatz-Trulson J., Lou L. Inhibition of dihydroorotate dehydrogenase by the immunosuppressive agent leflunomide. Biochem. Pharmacol. 1995;50:861–867. doi: 10.1016/0006-2952(95)00255-x. [DOI] [PubMed] [Google Scholar]
- 71.Liu S., Neidhardt E.A., Grossman T.H., Ocain T., Clardy J. Structures of human dihydroorotate dehydrogenase in complex with antiproliferative agents. Structure. 2000;8:25–33. doi: 10.1016/s0969-2126(00)00077-0. [DOI] [PubMed] [Google Scholar]
- 72.Williamson R.A., Yea C.M., Robson P.A., Curnock A.P., Gadher S., Hambleton A.B., Woodward K., Bruneau J.M., Hambleton P., Moss D., et al. Dihydroorotate dehydrogenase is a high affinity binding protein for A77 1726 and mediator of a range of biological effects of the immunomodulatory compound. J. Biol. Chem. 1995;270:22467–22472. doi: 10.1074/jbc.270.38.22467. [DOI] [PubMed] [Google Scholar]
- 73.Cherwinski H.M., Cohn R.G., Cheung P., Webster D.J., Xu Y.Z., Caulfield J.P., Young J.M., Nakano G., Ransom J.T. The immunosuppressant leflunomide inhibits lymphocyte proliferation by inhibiting pyrimidine biosynthesis. J. Pharmacol. Exp. Ther. 1995;275:1043–1049. [PubMed] [Google Scholar]
- 74.Bruneau J.M., Yea C.M., Spinella-Jaegle S., Fudali C., Woodward K., Robson P.A., Sautes C., Westwood R., Kuo E.A., Williamson R.A., et al. Purification of human dihydro-orotate dehydrogenase and its inhibition by A77 1726, the active metabolite of leflunomide. Biochem. J. 1998;336:299–303. doi: 10.1042/bj3360299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Christopherson R.I., Lyons S.D., Wilson P.K. Inhibitors of de novo nucleotide biosynthesis as drugs. Acc. Chem. Res. 2002;35:961–971. doi: 10.1021/ar0000509. [DOI] [PubMed] [Google Scholar]
- 76.Knecht W., Henseling J., Loffler M. Kinetics of inhibition of human and rat dihydroorotate dehydrogenase by atovaquone, lawsone derivatives, brequinar sodium and polyporic acid. Chem. Biol. Interact. 2000;124:61–76. doi: 10.1016/s0009-2797(99)00144-1. [DOI] [PubMed] [Google Scholar]
- 77.Ju J.S., Miller S.E., Hanson P.I., Weihl C.C. Impaired protein aggregate handling and clearance underlie the pathogenesis of p97/VCP-associated disease. J. Biol. Chem. 2008;283:30289–30299. doi: 10.1074/jbc.M805517200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tamguney G., Francis K.P., Giles K., Lemus A., DeArmond S.J., Prusiner S.B. Measuring prions by bioluminescence imaging. Proc. Natl Acad. Sci. USA. 2009;106:15002–15006. doi: 10.1073/pnas.0907339106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Watts J.C., Giles K., Grillo S.K., Lemus A., DeArmond S.J., Prusiner S.B. Bioluminescence imaging of Abeta deposition in bigenic mouse models of Alzheimer's disease. Proc. Natl Acad. Sci. USA. 2011;108:2528–2533. doi: 10.1073/pnas.1019034108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hashimoto T., Adams K.W., Fan Z., McLean P.J., Hyman B.T. Characterization of oligomer formation of amyloid-{beta} peptide using a split-luciferase complementation assay. J. Biol. Chem. 2011;286:27081–27091. doi: 10.1074/jbc.M111.257378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.O'Leary J.C., III, Li Q., Marinec P., Blair L.J., Congdon E.E., Johnson A.G., Jinwal U.K., Koren J., III, Jones J.R., Kraft C., et al. Phenothiazine-mediated rescue of cognition in tau transgenic mice requires neuroprotection and reduced soluble tau burden. Mol. Neurodegener. 2010;5:45. doi: 10.1186/1750-1326-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Taniguchi S., Suzuki N., Masuda M., Hisanaga S., Iwatsubo T., Goedert M., Hasegawa M. Inhibition of heparin-induced tau filament formation by phenothiazines, polyphenols, and porphyrins. J. Biol. Chem. 2005;280:7614–7623. doi: 10.1074/jbc.M408714200. [DOI] [PubMed] [Google Scholar]
- 83.Medina D.X., Caccamo A., Oddo S. Methylene blue reduces abeta levels and rescues early cognitive deficit by increasing proteasome activity. Brain Pathol. 2011;21:140–149. doi: 10.1111/j.1750-3639.2010.00430.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Necula M., Breydo L., Milton S., Kayed R., van der Veer W.E., Tone P., Glabe C.G. Methylene blue inhibits amyloid Abeta oligomerization by promoting fibrillization. Biochemistry. 2007;46:8850–8860. doi: 10.1021/bi700411k. [DOI] [PubMed] [Google Scholar]
- 85.Yamashita M., Nonaka T., Arai T., Kametani F., Buchman V.L., Ninkina N., Bachurin S.O., Akiyama H., Goedert M., Hasegawa M. Methylene blue and dimebon inhibit aggregation of TDP-43 in cellular models. FEBS Lett. 2009;583:2419–2424. doi: 10.1016/j.febslet.2009.06.042. [DOI] [PubMed] [Google Scholar]
- 86.Xu X., Williams J.W., Bremer E.G., Finnegan A., Chong A.S. Inhibition of protein tyrosine phosphorylation in T cells by a novel immunosuppressive agent, leflunomide. J. Biol. Chem. 1995;270:12398–12403. doi: 10.1074/jbc.270.21.12398. [DOI] [PubMed] [Google Scholar]
- 87.Manna S.K., Aggarwal B.B. Immunosuppressive leflunomide metabolite (A77 1726) blocks TNF-dependent nuclear factor-kappa B activation and gene expression. J. Immunol. 1999;162:2095–2102. [PubMed] [Google Scholar]
- 88.Latchoumycandane C., Seah Q.M., Tan R.C., Sattabongkot J., Beerheide W., Boelsterli U.A. Leflunomide or A77 1726 protect from acetaminophen-induced cell injury through inhibition of JNK-mediated mitochondrial permeability transition in immortalized human hepatocytes. Toxicol. Appl. Pharmacol. 2006;217:125–133. doi: 10.1016/j.taap.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 89.Latchoumycandane C., Goh C.W., Ong M.M., Boelsterli U.A. Mitochondrial protection by the JNK inhibitor leflunomide rescues mice from acetaminophen-induced liver injury. Hepatology. 2007;45:412–421. doi: 10.1002/hep.21475. [DOI] [PubMed] [Google Scholar]
- 90.Sawamukai N., Saito K., Yamaoka K., Nakayamada S., Ra C., Tanaka Y. Leflunomide inhibits PDK1/Akt pathway and induces apoptosis of human mast cells. J. Immunol. 2007;179:6479–6484. doi: 10.4049/jimmunol.179.10.6479. [DOI] [PubMed] [Google Scholar]
- 91.Apostol B.L., Illes K., Pallos J., Bodai L., Wu J., Strand A., Schweitzer E.S., Olson J.M., Kazantsev A., Marsh J.L., et al. Mutant huntingtin alters MAPK signaling pathways in PC12 and striatal cells: ERK1/2 protects against mutant huntingtin-associated toxicity. Hum. Mol. Genet. 2006;15:273–285. doi: 10.1093/hmg/ddi443. [DOI] [PubMed] [Google Scholar]
- 92.Scappini E., Koh T.W., Martin N.P., O'Bryan J.P. Intersectin enhances huntingtin aggregation and neurodegeneration through activation of c-Jun-NH2-terminal kinase. Hum. Mol. Genet. 2007;16:1862–1871. doi: 10.1093/hmg/ddm134. [DOI] [PubMed] [Google Scholar]
- 93.Arrasate M., Mitra S., Schweitzer E.S., Segal M.R., Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- 94.Frost B., Diamond M.I. The expanding realm of prion phenomena in neurodegenerative disease. Prion. 2009;3:74–77. doi: 10.4161/pri.3.2.8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Crook Z.R., Housman D. Huntington's disease: can mice lead the way to treatment. Neuron. 2011;69:423–435. doi: 10.1016/j.neuron.2010.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Korn T., Magnus T., Toyka K., Jung S. Modulation of effector cell functions in experimental autoimmune encephalomyelitis by leflunomide—mechanisms independent of pyrimidine depletion. J. Leukoc. Biol. 2004;76:950–960. doi: 10.1189/jlb.0504308. [DOI] [PubMed] [Google Scholar]
- 97.Merrill J.E., Hanak S., Pu S.F., Liang J., Dang C., Iglesias-Bregna D., Harvey B., Zhu B., McMonagle-Strucko K. Teriflunomide reduces behavioral, electrophysiological, and histopathological deficits in the Dark Agouti rat model of experimental autoimmune encephalomyelitis. J. Neurol. 2009;256:89–103. doi: 10.1007/s00415-009-0075-3. [DOI] [PubMed] [Google Scholar]
- 98.Palmer A.M. Teriflunomide, an inhibitor of dihydroorotate dehydrogenase for the potential oral treatment of multiple sclerosis. Curr. Opin. Investig. Drugs. 2010;11:1313–1323. [PubMed] [Google Scholar]
- 99.Shao J., Welch W.J., Diprospero N.A., Diamond M.I. Phosphorylation of profilin by ROCK1 regulates polyglutamine aggregation. Mol. Cell. Biol. 2008;28:5196–5208. doi: 10.1128/MCB.00079-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}