

Abstract
A series of C-6 alkyl, cycloalkyl, and aryl-9-(β-d-ribofuranosyl)purines were synthesized and their substrate activities with Escherichia coli purine nucleoside phosphorylase (E. coli PNP) were evaluated. (Ph3P)4Pd-mediated cross-coupling reactions of 6-chloro-9-(2,3,5-tri-O-acetyl-β-d-ribofuranosyl)-purine (6) with primary alkyl (Me, Et, n-Pr, n-Bu, isoBu) zinc halides followed by treatment with NH3/MeOH gave the corresponding 6-alkyl-9-(β-d-ribofuranosyl) purine derivatives 7–11, respectively, in good yields. Reactions of 6 with cycloalkyl(propyl, butyl, pentyl)zinc halides and aryl (phenyl, 2-thienyl)zinc halides gave under similar conditions the corresponding 6-cyclopropyl, cyclobutyl, cyclopentyl, phenyl, and thienyl -9-(β-d-ribofuranosyl)purine derivatives 12–16, respectively in high yields. E. coli PNP showed a high tolerance to the steric and hydrophobic environment at the 6-position of the synthesized purine ribonucleosides. Significant cytotoxic activity was observed for 8, 12, 15, and 16. Evaluation of 12 and 16 against human tumor xenografts in mice did not demonstrate any selective antitumor activity. In addition, 6-methyl-9-(β-d-arabinofuranosyl)purine (18) was prepared and evaluated.
Keywords: purine nucleoside phosphorylase, organozinc halides, cross-coupling reactions, 6-alkyl, cycloalkyl/aryl/heterocyclylpurine ribonucleosides
Introduction
Suicide gene therapy of cancer is an approach that is being evaluated as a potential treatment for solid tumors. We have developed a cancer gene therapy strategy that is based on the activation of a non-toxic purine nucleoside analog (prodrug) to a highly toxic purine analog by E. coli PNP selectively expressed in tumor cells [1–5]. E. coli PNP differs from human PNP in its ability to accept not only 6-oxopurine nucleosides, but also 6-aminopurine and certain adenine nucleoside analogs as substrates (Figure 1). This property has been used to cleave non-toxic adenine nucleoside analogs such as 9-(2-deoxy-β-D-ribofuranosyl)-6-methylpurine (MeP-dR, 1) and 2-fluoro-2′-deoxyadenosine (F-dAdo, 3), to the very toxic adenine analogs, 6-methylpurine (MeP, 2) and 2-fluoroadenine (F-Ade, 4) [6–8].
Figure 1.

One of our goals has been to get more information about the E. coli PNP substrate structural requirements. We have reported on the correlation between various modifications at the sugar moiety of adenine nucleoside analogs and the substrate activity with E. coli PNP [5]. Crystal structures of a number of complexes of E. coli PNP with various compounds of varied substrate activities such as adenosine, MeP-dR, F-dAdo, and 2-fluoro-9-(β-D-arabinofuranosyl) adenine (F-araA, 5) showed unique positioning of MeP-dR at the active site [9]. The nucleoside base moiety of MeP-dR was shown to be fitted into a hydrophobic pocket at the active site, resulting in a 2.6 Å shift of the sugar moiety of MeP-dR from the phosphate binding site when compared with adenosine. Although the binding of MeP-dR was significantly different from that of the natural substrate, it was still an excellent substrate for this enzyme [5]. It has been postulated that in the mechanism of the phosphorolysis reaction catalyzed by E. coli PNP, the glycosidic bond breaking occurs ahead of the phosphate bond formation and that the transition state has a considerable oxocarbenium character that is stabilized and subsequently attacked by one of the phosphate oxygens [10]. These observations suggested that an increase of the hydrophobic interaction at the C6 position would have a positive impact on the cleavage activity. If it is the case, an enhancement of the cleavage activity of certain poor substrates such as arabinofuranosyladenine analogs might be observed. Herein, we report on the synthesis of selected C-6 alkyl and arylpurine nucleosides, their cleavage activity by E. coli PNP, and their evaluations both in vitro and in vivo.
Results and Discussions
Chemistry
Transition metal catalyzed cross-couplings of organometallics [11] (such as arylmagnesium halides [12], alkyl/arylzinc halides [13], alkyl/aryltin [14], trialkylaluminium [15], alkylcuprates [16] reagents and arylboronic acids [17] with 6-halopurines and 6-halopurine nucleoside analogs) have been effectively used for C-C bond formations at the C-6 position of purine nucleosides. We have previously reported on the application of the palladium-mediated cross-coupling of methylzinc bromide with N9-protected-6-chloropurine and suitably protected 6-chloro (9-β-d-ribo- and deoxyribofuranosyl) purines for the synthesis of MeP and the corresponding nucleosides [18]. The mildness of the reaction conditions as well as the stability, safety, and the ease of the preparation of the organozinc reagents prompted us to utilize the same chemistry for the introduction of different carbon substituents at the C-6 position. Treatment of 6-chloro-9-(2,3,5-tri-O-acetyl-β-d-ribofuranosyl)purine (6) [19,20] with MeZnBr, EtZnBr, n-PrZnBr, n-BuZnBr, and isoBuZnBr in THF in the presence of ca. 0.05 equivalents of (Ph3P)4Pd at 55 °C, followed by treatment with NH3/MeOH gave the corresponding 6-alkyl-9-(β-d-ribofuranosyl) purines (7–11, Scheme 1, Table 1) [18] in good yields. Treatment of 6 with sec-alkylzinc halides under the same conditions, however gave mainly the corresponding 6-primary alkyl derivatives with the 6-sec-alkyl derivatives as minor products, which were not isolated. Cyclopropyl [22], cyclobutyl, and cyclopentylzinc halides were cross-coupled with 6 in the presence of (Ph3P)4Pd efficiently to give, after deprotection of the sugar hydroxyl groups, the corresponding 6-cycloalkylpurine ribonucleosides 12–14 in good yields (Table 1). Phenyl and 2-thienylzinc bromides were also cross-coupled with 6 under similar conditions to give after removal of the acetyl groups by NH3/MeOH treatment, 6-phenyl and 6-(2-thienyl)-9-(β-dribofuranosyl) purine derivatives (15) [12a,17c,27] and (16) [27,28,30], respectively, in good yields.
Scheme 1.

aReaction conditions: a) RZnX, (Ph3P)4Pd, THF, 55 °C; b) NH3, MeOH
Table 1.
Pd(PPh3)4 Catalyzed Cross-Coupling of 6 with organozinc halides followed by treatment with NH3/MeOH
| Entry | RZnX | Product, R | Yield % |
|---|---|---|---|
| 1 | MeZnBr | 7, CH3 | 95 |
| 2 | EtZnBr | 8, CH2CH3 | 82 |
| 3 | n-PrZnBr | 9, CH2CH2CH3 | 86 |
| 4 | n-BuZnBr | 10, CH2(CH2)2CH3 | 89 |
| 5 | isoBuZnBr | 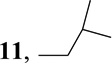 |
88 |
| 6 | cycloPrZnBr | 92 | |
| 7 | cycloBuZnBr | 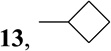 |
78 |
| 8 | cyclopentylZnBr | 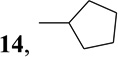 |
60 |
| 9 | PhZnBr | 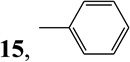 |
78 |
| 10 | 2-ThienylZnBr | 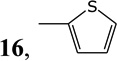 |
80 |
6-Methyl-9-(β-d-arabinofuranosyl)purine (18) was prepared by (Ph3P)4Pd catalyzed cross-coupling of 6-chloro- 9-(2,3,5-tri-O-acetyl-β-d-arabinofuranosyl)purine 17 [23] with MeZnBr under similar conditions. The arabinofuranosyl-6-chloropurine derivative 17 was prepared from 9-(β-d-arabinofuranosyl)-hypoxanthine in two steps with minor modification of the literature procedure [20,23]. Cross-coupling of 17 with CH3ZnBr followed by removal of the acetyl groups under the standard conditions furnished 18 in high yield (Scheme 2). NOE studies showed a distinct glycosidic torsional angle preference for the arabinofuranosyl purine derivatives 7 and 18 compared with the corresponding ribofuranosyl derivatives. Irradiation at the H-8 proton of 18 resulted in 2% and 5% NOE enhancements at the H-1′ and H-3′ signals, respectively. Irradiation at H-3′ gave enhancements of 4–5% and 2% of the signals at H-8 and at H-4′, respectively. These results are very similar to those reported for 9-(β-d-arabinofuranosyl) adenine [24] and suggest a more syn-conformational preference for 18.
Scheme 2.

aReaction conditions: a) CH3ZnBr, (Ph3P)4Pd, THF, 55 °C; b) NH3, MeOH
Biology
Substrate characteristics with E. coli PNP
The rate of cleavage of 100 µM adenosine (the natural substrate) and its analog, MeP-R were 398,000 and 84,000 nmoles/mg/h, respectively. The cleavage activities of 6-ethyl, n-propyl, n-butyl, isobutyl, cyclopropyl, cyclobutyl, and cyclopentylpurine ribonucleosides (8–14) by E. coli PNP were as good as the 6-methyl derivative (7) (Table 2). Surprisingly, a considerable cleavage activity was also observed with 6-phenyl and 6-thienyl ribonucleosides 15 and 16. These results show the tolerance of E. coli PNP to the steric and hydrophobic effects at the C-6 position of the purine nucleosides and reflect a good accommodation of these hydrophobic substituents in the enzyme’s active site. On the other hand, the observed poor substrate activity of 6-methyl-9-(β-D-arabinofuranosyl)purine (18) reflects the impact of the conformation around the glycosidic bond as a determinant factor in placing the molecule in the proper position at the active site. It is of interest that arabinofuranosyl-MeP (18) was much worse as a substrate than F-araA (5).
Table 2.
Substrate activities of 6-substituted purine nucleoside analogs with E. coli PNP
| Substrate | C6-substituent | Specific activity (nmoles/mg/hr) |
(N) |
|---|---|---|---|
| Adenosine* | amino | 398,000 | 5 |
| MeP-dR* | methyl | 461,000 | 10 |
| F-araA* | amino | 1,300 | 4 |
| 7 | methyl | 84,000 | 8 |
| 8 | ethyl | 69,000 | 3 |
| 9 | n-propyl | 72,000 | 3 |
| 10 | n-butyl | 86,000 | 3 |
| 11 | isobutyl | 79,000 | 3 |
| 12 | cyclopropyl | 51,000 | 3 |
| 13 | cyclobutyl | 63,000 | 3 |
| 14 | cyclopentyl | 42,000 | 3 |
| 15 | phenyl | 21,000 | 3 |
| 16 | thienyl | 9,900 | 2 |
| 18 | methyl | 14 | 2 |
Purified E. coli PNP (obtained from Dr. Steve Ealick, Cornell University, Ithaca, NY) was incubated at 25 °C with 100 µM of each nucleoside analog in the presence of 100 mM HEPES (pH 7.4), 50 mM phosphate (pH 7.4), 4% glycerol, 0.2 mM dithiothreitol, and an appropriate amount of enzyme to give a linear reaction. Samples were collected at various times after addition of substrate and the substrates and products were determined by monitoring UV absorbance as they eluted from a 150 × 4.6 mm, 5µm BDS hypersil C-18 column (Keystone Scientific Inc. Bellfonte, PA) using a 30-minute linear gradient of 5% to 50% acetonitrile in 50 mM NH3H2PO4 buffer pH 4.5 at a flow rate of 1 ml/minute. Each number is the average of at least 2 separate measurements (N).
From reference 5
Cytotoxicity
Because compound 7 is known to be very toxic to human cells, the 6-substituted purine nucleoside analogs were evaluated in a standard assay for cytotoxicity against CEM cells (Table 3). All of the compounds tested, except 11 and 18, were able to inhibit the growth of the CEM cells, but at greater concentrations than those required for compound 7. Because phosphorylation of compound 7 by adenosine kinase is required for its cytotoxicity, it is likely that this same activation is needed for the other compounds in Table 3. Thus, these results suggest that these compounds may be relatively poor substrates for this enzyme or that the phosphorylated metabolites are less active. Three of the most potent inhibitors (12,15,16) were also tested against a panel of solid tumor cell lines (Table 4), where they were generally quite active.
Table 3.
Inhibition of CEM cell growth by 6-substituted purine nucleoside analogs
| Substrate | C6-substituent | IC50 (µM) | (N) |
|---|---|---|---|
| 7 | methyl | 0.02 | 2 |
| 8 | ethyl | 0.72 | 3 |
| 9 | n-propyl | 6.8 | 3 |
| 10 | n-butyl | 49 | 2 |
| 11 | isobutyl | >130 | 2 |
| 12 | cyclopropyl | 1 | 3 |
| 13 | cyclobutyl | 7 | 2 |
| 14 | cyclopentyl | 16 | 2 |
| 15 | phenyl | 0.62 | 3 |
| 16 | thienyl | 0.09 | 3 |
| 18 | methyl | >130 | 2 |
CCRF-CEM cells (American Type Culture Collection) were incubated at 37 °C with various concentrations of the 6-substituted purine nucleoside analogs. Cell numbers were determined 72 hours after the addition of compound using a Coulter Counter and the amount of compound that resulted in 50% inhibition of cell growth was determined (IC50). Each number is the average of 2 or 3 separate measurements (N).
Table 4.
Inhibition of the growth of various solid tumor cell lines
| Cell line | IC50 of 6-substituted purine nucleoside analog (µM) | ||
|---|---|---|---|
| 12 | 15 | 16 | |
| SNB7 (CNS) | 33 | 31 | 10 |
| DLD-1 (colon) | 170 | 6 | 0.9 |
| NCI-H23 (lung) | 1 | 1.2 | 0.6 |
| ZR-75-1 (mammary) | 23 | 1.2 | 0.3 |
| LOXIMVI (melanoma) | >200 | 6.2 | 0.3 |
| PC-3 (prostate) | 67 | 1.2 | 0.9 |
| CAKI-1 (renal) | >200 | 31 | 30 |
The above cells were plated in 96-well microtiter plates and incubated with various concentrations of compound number 12, 15, or 16 at 37 °C. Cell viability was measured after 72 hours of continuous incubation with compound using the sulforhodamine B assay (absorbance read at 570 nm), and the concentration of compound that inhibited cell growth by 50% was determined. The results shown are the result of one experiment.
Because of the sensitivity of the NCI-H23 tumor cell line to these compounds, the in vivo efficacy of compounds 12 and 16 were determined. NCI-H23 tumors were grown on the flanks of nude mice. When the tumors reached approximately 200 mg, the animals were treated with 33, 50, or 75 mg/kg of compound 12 or 16 (given ip daily for 9 consecutive days). Neither compound at any dose had any effect of tumor growth. Seventy-five mg/kg of compound 12 killed 5 of 6 mice, whereas with compound 16 this dose caused a 14% decrease in weight. Therefore, at maximally tolerated doses neither compound exhibited selective antitumor activity in mice.
Experimental
1H NMR and 13C NMR spectra were recorded on a Nicolet NT 300 NB spectrometer operating at 300.635 MHz (1H) or 75.6 MHz (13C). Chemical shifts are expressed in parts per million from tetramethylsilane. The hydrogen-decoupled 13C-NMR spectra were assigned by comparison of the JCH values obtained from hydrogen-coupled 13C-NMR spectra. When necessary, selective hydrogen decoupling was performed in order to confirm the assignments. Ultraviolet absorption spectra were determined on Perkin-Elmer Lambda 19 spectrometer by dissolving each compound in methanol or water and diluting appropriately with 0.1 N HCl, pH 7 buffer, or 0.1 N NaOH. Values are in nanometers, and numbers in parentheses are extinction coefficients (ε × 10−3). Mass spectra were recorded on a Varian/MAT 311A double-focusing mass spectrometer in the fast atom bombardment (FAB) mode (glycerol matrix). CHN elemental analysis was carried out on Perkin-Elmer 2400 elemental analyzer. HPLC analysis was carried out on a Hewlett-Packard 1100 series liquid chromatograph with a Phenomenex Sphenclone 5 µM ODS (1) column (4.6 mm × 25 cm) with UV monitoring (254 nm). All flash column chromatography used 230–400 mesh silica gel from E. Merck. TLC was done on Analtech pre-coated (250 µm) silica gel (GF) plates.
6-Ethyl-9-(β-d-ribofuranosyl)purine (8)[25]
A solution of EtZnBr (1.2 mmoL) was generated by dropwise addition of ZnBr2 (1.13M, 1.1 mL, 1.2. mmol) in THF to 2M solution of EtMgBr (1.1 mmol, 0.3 mL) in THF (6 mL) for 1h at −78 °C. The solution was allowed to warm gradually to room temperature and then (Ph3P)4Pd (27 mg, 0.02 mmol) in THF (2 mL) was added to the mixture. A solution of compound 6 (0.197 g, 0.477 mmol) in dry THF (4 mL) was added and the mixture was heated under argon for 1h at 55 °C. The mixture was then cooled down to room temperature and quenched with saturated solution of NH4Cl. The solvent was concentrated under reduced pressure and the residue was partitioned between CHCl3 and H2O. The residue obtained by evaporation of the dried organic phase was dissolved in MeOH saturated with NH3 (15 mL) and kept overnight at room temperature. The solvent was evaporated and the residue was purified by a flash silica gel chromatography (elution with 5% EtOH in CHCl3) to give (0.11 g, 82%) 8 as a white solid, m.p. 98–100 °C, 1:1 H2O-EtOH (lit. [25] 104–106 °C): HPLC [99.5%; RT, 10.64 min, 0.01M NH4H2PO4: MeOH, 20 min linear gradient]; MS m/z 281 (M+1)+, UV λmax pH 1, 265.3 (7.4); pH 7, 260.6 (8.0); pH 13, 261.2 (8.1); 1H NMR (Me2SO-d6) δ 8.84 (1H, s, H-2, 1JC,H = 204.4 Hz), 8.76 (1H, s, H-8, 1JC,H = 214.3 Hz), 6.02 (1H, d, H-1′, J1′,2′ = 5.8 Hz), 5.54 (1H, d, 2′-OH, J = 5.9 Hz), 5.26 (1H, d, 3′-OH, J = 4.9 Hz), 5.14 (1H, t, 5′-OH, J = 5.6 Hz), 4.64 (1H, ddd, H-2′, J2′3′ = 4.7 Hz), 4.19 (1H, ddd, H-3′, J3′4′ = 3.4 Hz), 3.98 (1H, ddd, H-4′), 3.70 (1H, ddd, H-5′a, J4′,5′a = 3.7 Hz, J5′a,5′b = 12.1 Hz), 3.64 (1H, ddd, H-5′b, J4′,5′b = 4.4 Hz), 3.12 (2H, q, 6-CH2CH3), 1.35 (3H, t, 6-CH3); 13C NMR (Me2SO-d6) δ 162.64 (C-6), 151.78 (C-2), 150.22 (C-4), 143.97 (C-8), 132.19 (C-5), 87.59 (C-1′), 85.67 (C-4′), 73.58 (C-2′), 70.33 (C-3′), 61.30 (C-5′), 25.70 (6-CH2CH3), 12.20 (6-CH3); Anal. Calcd. for C12H16N4O4; C, 51.42; H, 5.75; N, 19.99. Found C, 51.22; H, 5.65; N 20.89.
6-n-Propyl-9-(β-d-ribofuranosyl)purine (9) [25]
A solution of (Ph3P)4Pd (25 mg, 0.02 mmol) in THF (1 mL) was added to a solution of n-PrZnCl [generated as above from a 2M solution n-PrMgCl (0.53 mL) and 1.13M solution ZnBr2 (1 mL) at −78 °C to r.t., for 1h] in THF (5 mL) at room temperature. A solution of 6 (0.175 g, 0.424 mmol) in THF (2 mL) was added and the mixture was heated for 5 h at 55 °C. The mixture was then cooled down to room temperature and quenched with saturated solution of NH4Cl. The solvent was concentrated under reduced pressure and the residue was partitioned between CHCl3 and H2O. The residue obtained by evaporation of the dried organic phase was dissolved in MeOH saturated with NH3 (10 mL) and stirred for 2 h at room temperature. The solvent was evaporated and the residue was purified by a flash silica gel chromatography (elution with 5% EtOH in CHCl3) to give (0.107 g, 86%) 9 as a pale yellow waxy solid, which was recrystallized from EtOH, m.p. 104–106 °C (lit. [25] foam): HPLC [99%; RT 12.64 min; 0.01 M NH4H2PO4: MeOH; 20 min linear gradient from 10–90%]; MS m/z 295 (M+1)+, UV λmax pH 1, 266.0 (8.0); pH 7, 261.2 (8.3); pH 13, 261.6 (8.2); 1H NMR (Me2SO-d6) δ 8.83 (1H, s, H-2), 8.76 (1H, s, H-8), 6.02 (1H, d, H-1′, J1′2′ = 5.9 Hz), 5.54 (1H, d, 2′-OH, J =6.0 Hz), 5.26 (1H, d, 3′-OH, J = 4.9 Hz), 5.14 (1H, t, 5′-OH, J = 5.6 Hz), 4.65 (1H, ddd, H-2′, J1′2′ = 4.7 Hz), 4.19 (1H, ddd, H-3′, J3′,4′ = 3.5 Hz), 3.98 (1H, ddd, H-4′), 3.70 (1H, ddd, H-5′a, J4′,5a′ = 3.7 Hz, J5′a,5′b = 12.1 Hz), 3.59 (1H, ddd, H-5′b, J4′,5′b = 4.1 Hz), 3.10 (2H, t, 6-CH2CH2CH3), 1.85 (2H, m, 6-CH2CH2CH3), 0.94 (3H, t, 6-CH2CH2CH3); 13C NMR (Me2SO-d6) δ 161.55 (C-6), 151.72 (C-2), 150.22 (C-4), 144.00 (C-8), 132.64 (C-5), 87.54 (C-1′), 85.68 (C-4′), 73.54 (C-2′), 70.34 (C-3′), 61.30 (C-5′), 34.29 (6-CH2CH2CH3), 20.96 (6-CH2CH2CH3), 13.78 (6-CH2CH2CH3). Anal. Calcd. for C13H18N4O4·0. 5 H2O: C, 51.48; H, 6.31; N, 18.47. Found: C, 51.82; H, 6.25; N, 18.37.
6-n-Butyl-9-(β-d-ribofuranosyl)purine (10) [25]
A solution of (Ph3P)4Pd (23 mg, 0.02 mmol) in THF (1 mL) was added to a solution of n-BuZnCl [generated as above from a 2M solution n-BuMgCl (0.5 mL) and 1.13M solution ZnBr2 (1 mL) at −78 °C to r.t., for 1h] in THF (5 mL) at room temperature. A solution of 6 (0.166 g, 0.4 mmol) in THF (2 mL) was added and the mixture was heated for 2 h at 55 °C. The mixture was then cooled down to room temperature and quenched with saturated solution of NH4Cl. The solvent was concentrated under reduced pressure and the residue was partitioned between CHCl3 and H2O. The residue obtained by evaporation of the dried organic phase was dissolved in MeOH saturated with NH3 (10 mL) and kept for 3 h at room temperature. The solvent was evaporated and the residue was purified by a flash silica gel chromatography (elution with 5% EtOH in CHCl3) to give (0.11 g, 89%) 10 as a pale yellow waxy solid, which was recrystallized from H2O-EtOH, m.p. 98–100 °C (lit. [25] foam): HPLC [99%; RT 12.64 min; 0.01M NH4H2PO4: MeOH; 20 min linear gradient from 10–90%]; MS m/z 309 (M+1)+, UV λmax pH 1, 267.0 (8.1); pH 7, 260.9 (8.4); pH 13, 261.1 (8.7); 1H NMR (Me2SO-d6) δ 8.83 (1H, s, H-2), 8.75 (1H, s, H-8), 6.03 (1H, d, H-1′, J1′,2′ = 5.9 Hz), 5.55 (1H, d, 2′-OH, J = 5.9 Hz), 5.27 (1H, d, 3′-OH, J = 4.8 Hz), 5.13 (1H, t, 5′-OH, J = 5.6 Hz), 4.64 (1H, ddd, H-2′, J2′,3′ = 5.0 Hz), 4.18 (1H, ddd, H-3′, J3′,4′ = 3.4 Hz), 4.00 (1H, ddd, H-4′), 3.69 (1H, ddd, H-5′a, J4′,5′a = 3.5 Hz, J5′a,5′b = 12.1 Hz), 3.59 (1H, ddd, H-5′b, J4′,5′b = 4.1 Hz), 3.12 (2H, t, 6-CH2CH2CH2CH3), 1.81 (2H, m, 6-CH2CH2CH2CH3), 1.33 (2H, m, 6-CH2CH2CH2CH3), 0.91 (3H, t, 6-CH2CH2CH2CH3); 13C NMR (Me2SO-d6) δ 161.76 (C-6), 151.72 (C-2), 150.21 (C-4), 143.98 (C-8), 132.56 (C-5), 87.53 (C-1′), 85.68 (C-4′), 73.54 (C-2′), 70.33 (C-3′), 61.30 (C-5′), 31.94 (6-CH2CH2CH2CH3), 29.74 (6-CH2CH2CH2CH3), 21.87 (6-CH2CH2CH2CH3), 13.64 (6-CH2CH2CH2CH3); Anal. Calcd. for C14H20N4O4: C, 54.53; H, 6.54; N, 18.17. Found: C, 54.17; H, 6.35; N, 18.04.
6-Isobutyl-9-(β-d-ribofuranosyl)purine (11)
A solution of (Ph3P)4Pd (73 mg, 0.06 mmol) in THF (2 mL) was added to a 0.5-M solution of isoBuZnCl (10 mL) in THF (5 mL) at room temperature. A solution of 6 (0.520 g, 1.26 mmol) in THF (5 mL) was added and the mixture was heated for 2 h at 55 °C. The mixture was then cooled down to room temperature and quenched with saturated solution of NH4Cl. The solvent was concentrated under reduced pressure and the residue was partitioned between CHCl3 and H2O. The residue obtained by evaporation of the dried organic phase was dissolved in MeOH saturated with NH3 (10 mL) and kept for 4 h at room temperature. The solvent was evaporated and the residue was purified by a flash silica gel chromatography (elution with 6% EtOH in CHCl3) to give (0.34 g, 88%) of a white solid: MS m/z 309 (M + 1)+; UV λmax pH 1, 266.8 (8.8); pH 7, 261.6 (9.2); pH 13, 261.7 (9.1); 1H NMR (Me2SO-d6) δ 8.84 (1H, s, H-2), 8.75 (1H, s, H-8), 6.02 (1H, d, H-1′, J1′,2′ = 5.8 Hz), 5.52 (1H, d, 2′-OH, J = 6.0 Hz), 5.24 (1H, d, 3′-OH, J = 4.8 Hz), 5.12 (1H, t, 5′-OH, J = 5.2 Hz), 4.67 (1H, ddd, H-2′, J2′,3′ =5.0 Hz), 4.19 (1H, ddd, H-3′, J3′,4′ = 3.4 Hz), 3.98 (1H, ddd, H-4′), 3.69 (1H, ddd, H-5′a, J4′,5′a = 4.0 Hz, J5′a,5′b = 11.9 Hz), 3.57 (1H, ddd, H-5′b, J4′,5′b = 4,1 Hz), 2.97 (2H, d, 6-CH2CH(CH3)2), 2.34 (1H, m, 6-CH2CH(CH3)2), 0.93 and 0.90 (3H, d, 6-CH2CH(CH3)2); 13C NMR (Me2SO-d6) δ 161.02 (C-6), 151.69 (C-2), 150.28 (C-4), 144.08 (C-8), 133.01 (C-5), 87.50 (C-1′), 85.72 (C-4′), 73.54 (C-2′), 70.37 (C-3′), 61.33 (C-5′), 41.34 (6-CH2CH(CH3)2), 27.69 (6-CH2CH(CH3)2), 22.49 and 22.47 (6-CH2CH(CH3)2); Anal. Calcd. for C14H20N4O4: C, 54.54; H, 6.54; N, 18.17. Found: C, 54.58; H, 6.32; N, 18.06.
6-Cyclopropyl-9-(β-d-ribofuranosyl)purine (12)
A mixture of magnesium turnings (74 mg, 3.0 mmol) and cyclopropyl bromide (0.24 mL, 3.02 mmol) in anhydrous THF (6 mL) was heated for 1 h at 60 °C until complete dissolution of the magnesium. The solution was cooled to room temperature and was treated with 1.13M solution ZnBr2 (2.7 mL, 3.0 mmol) and the resulting white suspension was stirred for 1h at room temperature. (Ph3P)4Pd (50 mg, 0.04 mmol) in THF (1 mL) was added to the mixture, followed by the addition of 6 (0.22 g, 0.53 mmol) in THF (2 mL) and the mixture was heated for 4 h at 45 °C. The mixture was then cooled down to room temperature and quenched with saturated solution of NH4Cl. The solvent was concentrated under reduced pressure and the residue was partitioned between CHCl3 and H2O. The residue obtained by evaporation of the dried organic phase was dissolved in MeOH saturated with NH3 (10 mL) and kept for 2 h at room temperature. The solvent was evaporated and the residue was purified by a flash silica gel chromatography (elution with 7% EtOH in CHCl3 to give (0.148 g, 92%) as a pale yellow solid which was crystallized from MeOH-heptane, m.p. 154–156 °C (lit. [26] 158–160 °C); HPLC [99.8%; RT 12.62 min; 0.01M NH4H2PO4: MeOH; 20 min linear gradient from 10–90%]; MS m/z 293.1 (M+1)+; UV λmax pH 1, 276.5 (12.5); pH 7, 265.3 (12.4); pH 13, 265.6 (12.7); 1H NMR (Me2SO -d6) δ. 8.74 (1H, s, H-2), 8.72 (1H, s, H-8), 6.00 (1H, d, H-1′, J1′,2′ = 5.9 Hz), 5.52 (1H, d, 2′-OH, J = 5.9 Hz), 5.25 (1H, d, 3′-OH, J = 5.1 Hz), 5.15 (1H, dd, 5′-OH, J5′a′,5′-OH = 5.0 Hz, J5′b′,5′-OH = 6.1Hz), 4.61 (1H, ddd, H-2′, J2′,3′ = 4.9 Hz), 4.17 (1H, ddd, H-3′, J3′,4′ = 3.5 Hz), 3.99 (1H, ddd, H-4′), 3.70 (1H, ddd, H-5′a, J4′,5′a = 4.0 Hz, J5′,5′b = 12.1 Hz), 3.58 (1H, dd, H-5′b, J4′5′b = 4.1 Hz), 2.70 (1H, m, 6-cycloPr-CH), 1.27-1.23 (4H, m, 6-cycloPr-CH2-CH2); 13C NMR (Me2SO-d6) δ 162.88 (C-6), 151.85 (C-2), 149.54 (C-4), 143.73 (C-8), 132.14 (C-5), 87.59 (C-1′), 85.62 (C-4′), 73.58 (C-2′), 70.27 (C-3′), 61.26 (C-5′), 12.69 (6-cycloPr-CH), 10.94 (6-cycloPr-CHCH2CH2); Anal. Calcd. for C13H16N4O4: C, 53.42; H, 5.52; N, 19.18. Found: C, 53.30; H, 5.23; N 19.20.
6-Cyclobutyl-9-(β-d-ribofuranosyl)purine (13)
A mixture of magnesium turnings (45 mg, 1.8 mmol) and cyclobutyl bromide (0.25 mg, 1.85 mmol) in anhydrous THF (5 mL) was heated for 3 h at 60 °C until complete dissolution of the magnesium. The solution was cooled to −78 °C and treated ZnBr2 (1.13M, 1.6 mL, 1.8 mmol) in THF and the resulting white suspension was warmed gradually to room temperature and was stirred further for 1h at room temperature. (Ph3P)4Pd (27 mg) in THF (1 mL) was added, followed by addition of 3 (0.15 g, 0.36 mmol) in THF (2 mL) and the mixture was heated for 4 h at 55 °C. The mixture was then cooled down to room temperature and quenched with saturated solution of NH4Cl. The solvent was concentrated under reduced pressure and the residue was partitioned between CHCl3 and H2O. The residue obtained by evaporation of the dried organic phase was dissolved in MeOH saturated with NH3 (10 mL) and kept overnight at room temperature. The solvent was evaporated and the residue was purified by a flash silica gel chromatography (elution with 5% MeOH in CHCl3) to give (88 mg, 78%) as a pale yellow solid which was crystallized from EtOH in hexanes, m.p. 82–84 °C; HPLC [98%; RT 13.70 min; 0.01M NH4H2PO4: MeOH; 20 min linear gradient from 10–90%]; MS m/z 307 (M+1)+; UV λmax pH 1, 270.2 (9.9); pH 7, 263.1 (10.0); pH 13, 263.3 (10.0); 1H NMR (Me2SO-d6) δ 8.89 (1H, s, H-2), 8.74 (1H, s, H-8), 6.03 (1H, d, H-1′, J1′,2′ = 5.7 Hz), 5.52 (1H, d, 2′-OH, J = 5.2 Hz), 5.26 (1H, d, 3′-OH, J = 4.8 Hz), 5.14 (1H, t, 5′-OH, J = 5.6 Hz), 4.64 (1H, ddd, H-2′, J2′,3′ = 4.9 Hz), 4.29-4.18 (2H, m, 6-cycloBu; CHCH2CH2CH2 and H-3′), 4.01 (1H, ddd, H-4′, J3′,4′ = 3.2 Hz), 3.72 (1H, ddd, H-5′a, J4′,5′a = 3.5 Hz, J5′a,5′b = 12.1 Hz), 3.60 (1H, dd, H-5′b, J4′,5′b = 4.1 Hz), 2.60-2.47 (2H, m, 6-cycloBu CHCHaCHbCH2), 2.41-2.31 (2H, m, 6-cycloBu CHCHaCHbCH2), 2.20-1.93 (2H, m, HCHaCHbCH2); 13C NMR (Me2SO-d6) δ 163.14 (C-6), 151.87 (C-2), 150.33 (C-4), 143.90 (C-8), 131.51 (C-5), 87.64 (C-1′), 85.68 (C-4′), 73.64 (C-2′), 70.33 (C-3′), 61.31 (C-5′), 36.77 (6-cycloBu CHCH2CH2CH2), 26.96 (6-cycloBu CHCH2CH2CH2), 26.88 (6-cycloBu CHCH2CH2CH2), 18.17 (6-cycloBu CHCH2CH2CH2); Anal. Calcd. for C14H18N4O4 ∙0.5 H2O : C, 53.33; H, 6.07; N, 17.77. Found: C, 53.00; H, 5.95; N, 17.53.
6-Cyclopentyl-9-(β-d-ribofuranosyl)purine (14)
A solution of (Ph3P)4Pd (46 mg, 0.04 mmol) in THF (1.5 mL) was added to a solution of cyclopentyl ZnCl [generated as above from a 2M Et2O solution cyclopentyl MgCl (0.5 mL) and 1.13M THF solution ZnBr2 (1 mL) at −78 °C to r.t., for 1h] in THF (5 mL) at room temperature. A solution of 3 (0.224 g, 0.543 mmol) in THF (3 mL) was added and the mixture was heated for 45 min. at 55 °C. The mixture was then cooled down to room temperature and quenched with saturated solution of NH4Cl. The solvent was concentrated under reduced pressure and the residue was partitioned between CHCl3 and H2O. The residue obtained by evaporation of the dried organic phase was dissolved in MeOH saturated with NH3 (10 mL) and kept overnight at room temperature. The solvent was evaporated and the residue was purified by a flash silica gel chromatography (elution with 7% EtOH in CHCl3) to give (0.1 g, 60%) 14 as a pale yellow foam: HPLC [99%; RT 12.64 min; 0.01M NH4H2PO4: MeOH; 20 min linear gradient from 10–90%]; MS m/z 321.2 (M+1)+; UV λmax pH 1, 238.2 (6.4); pH 7, 262.2 (6.4); pH 13, 262.2 (6.4); 1H NMR (Me2SO-d6) δ 8.83 (1H, s, H-2), 8.74 (1H, s, H-8), 6.01 (1H, d, H-1′, J1′,2′. = 5.7 Hz), 5.53 (1H, d, 2′-OH, J = 5.6 Hz), 5.25 (1H, d, 3′-OH, J = 4.6 Hz), 5.13 (1H, t, 5′-OH, J = 5.5 Hz), 4.64 (1H,ddd, H-2′, J2′,3′ = 4.4 Hz), 4.18 (1H, ddd, H-3′, J3′,4′ = 3.1 Hz), 4.02 (1H, ddd, H-4′), 3.79 (1H, m, 6-CHCH2CH2CH2CH2), 3.68 (1H, ddd, H-5′a, J4′,5′a = 3.6 Hz, J5′a,5′b = 12.0 Hz), 3.56 (1H, dd, H-5′b, J4′,5′b = 4.6 Hz), 2.07-1.69 (8H, m, 6-CHCH2CH2CH2CH2), 13C NMR (Me2SO-d6) δ 165.59 (C-6), 152.36 (C-2), 150.76 (C-4), 144.36 (C-8), 132.57 (C-5), 87.59 (C-1′), 85.69 (C-4′), 73.57 (C-2′), 70.37 (C-3′), 61.33 (C-5′), 42.01 (6-CHCH2CH2CH2CH2), 32.09 and 32.06 (6-CHCH2CH2CH2CH2), 25.81 (6-CHCH2CH2CH2CH2); Anal. Calcd. for C15H20N4O4 ∙0.4 H2O : C, 54.96; H, 6.40; N, 17.17. Found: C, 55.11; H, 6.34; N, 16.84.
6-Phenyl-9-(β-d-ribofuranosyl)purine (1512) [a,17c,27]
A solution of PhZnBr (16.94 mmoL) was generated by dropwise addition of 1.13M solution of ZnBr2 (15 mL) in THF to 3M solution of PhMgBr (19.64 mmol, 5.64 mL) in THF (75 mL) at −0 °C for 1h. After the solution was allowed to warm to room temperature, a solution of (Ph3P)4Pd (0.5 g, 0.4 mmoL) in THF (10 mL) was added to it. A solution of compound 6 (3.55 g, 8.6 mmol) in dry THF (10 mL) was then added and the mixture was heated under argon for 4 h at 55 °C. The mixture was then cooled down to room temperature and quenched with saturated solution of NH4Cl. The mixture was then cooled down to room temperature and quenched with saturated solution of NH4Cl. The solvent was concentrated under reduced pressure and the residue was partitioned between CHCl3 and H2O. The residue obtained by evaporation of the dried organic phase was dissolved in MeOH saturated with NH3 (30 mL) and kept overnight at room temperature. The solvent was evaporated and the residue was purified by a flash silica gel chromatography (elution with 6% MeOH in CHCl3) to give (2.2 g, 78%) of 15 as a white solid with was crystallized from EtOH/toluene, m.p. 224–226 °C (lit. [17c] 228–230 °C): MS m/z 329 (M+1)+; UV λmax pH 1, 303.0 (18.3); pH 7, 288.4 (18.7); pH 13, 288.4 (18.4); 1H NMR (Me2SO-d6) δ 9.03 (1H, s, H-2), 8.92 (1H, s, H-8), 8.83-8.80 (2H, m, 6-Ph), 7.65-7.58 (3H, m, 6-Ph), 6.11 (1H, d, H-1′, J1′,2′ = 5.6 Hz), 5.60 (1H, d, 2′-OH, J = 5.9 Hz), 5.28 (1H, d, 3′-OH, J = 4.9 Hz), 5.16 (1H, t, 5′-OH, J = 5.5 Hz), 4.67 (1H, ddd, H-2′, J2′,3′ = 4.8 Hz), 4.23 (1H, ddd, H-3′, J3′,4′ = 3.9 Hz), 4.00 (1H, ddd, H-4′), 3.75 (1H, ddd, H-5′a, J4′,5′a = 3.6 Hz, J5′a,5′b = 11.9 Hz), 3.63 (1H, ddd, H-5′b, J4′,5′b = 4.1 Hz); 13C NMR (Me2SO -d6) δ 157.17(C-6 or C-4), 152.93 (C-4 or C-6), 151.85 (C-2), 144.86 (C-8), 135.19 (ipso C-6Ph), 131.08 (para C-6Ph), 130.82 (C-5), 129.33 and 128.63 (meta and ortho C-6Ph), 87.64 (C-1′), 85.63 (C-4′), 73.72 (C-2′), 70.21 (C-3′), 61.17 (C-5′); Anal. Calcd. for C16H16N4O4 ∙ 0.2 H2O: C, 57.85; H, 4.98; N, 16.94. Found: C, 57.78; H, 4.81; N, 16.99.
6-(2-Thienyl)-9-(β-d-ribofuranosyl)]purine (16) [27,28,30]
A mixture of magnesium turnings (466 mg, 19.2 mmol) and 2-thienyl bromide (1.8 mL, 19.2 mmol) in anhydrous THF (5 mL) was stirred under argon for 3 h at 37 °C. The resulting red color solution was cooled to 0 °C and treated with 1M THF solution of ZnBr2 (19.2 mL) and the thick suspension was stirred further for 1h at room temperature. (Ph3P)4Pd (277 mg, 0.24 mmol) in THF (5 mL) was added followed by the addition of a solution of 6 (2 g, 4.85 mmol) in THF (20 mL) and the mixture was heated for 2 h at 45 °C. The mixture was then cooled down to room temperature and quenched with saturated solution of NH4Cl. The solvent was concentrated under reduced pressure and the residue was partitioned between CHCl3 and H2O. The residue obtained by evaporation of the dried organic phase was dissolved in MeOH saturated with NH3 (20 mL) and kept overnight at room temperature. The solvent was evaporated and the residue was purified by a flash silica gel chromatography (elution with 5% MeOH in CHCl3) to give (1.3 g, 89%) as a yellow solid; MS m/z 307 (M+1)+; UV λmax pH 1, 338.8 (19.3); pH 7, 325.2 (24.1); pH 13, 324.8 (24.7); 1H NMR (Me2SO-d6) δ 8.91 (1H, s, H-2), 8.87 (1H, s, H-8), 8.84 (1H, dd, 6-C-S-CHCHCH, J = 1.1 Hz, J = 3.7 Hz), 7.94 (1H, dd, 6-C-S-CHCHCH, J = 1.1, J = 5.0 Hz), 7.36 (1H, dd, 6-C-S-CHCHCH, J = 5.0, J = 3.7 Hz), 6.06 (1H, d, H-1′, J1′2′ = 5.5 Hz), 5.57 (1H, d, 2′-OH, J = 5.9 Hz), 5.05 (1H, d, 3′-OH, J = 5.6 Hz), 5.15 (1H, t, 5′-OH, J = 5.5 Hz), 4.64 (1H, ddd, H-2′, J2′,3′ = 5.1 Hz), 4.21 (1H, ddd, H-3′, J3′,4′ = 3.2 Hz), 4.00 (1H, dd, H-4′), 3.71 (1H, ddd, H-5′a, J4′,5′a = 3.7, J5′a,5′b = 11.9 Hz), 3.60 (1H, dd, H-5′b, J4′,5′b = 4.6 Hz); Anal. Calcd. for C14H14N4O4S ∙ 0.3 H2O: C, 49.49; H, 4.33; N, 16.49. Found: C, 49.40; H, 4.03; N, 16.27.
6-Chloro-9-(tri-O-acetyl-β-d-arabinofuranosyl)purine (17) [23,30]
To a solution of 23 (1 g, 2.66 mmol) in anhydrous CHCl3 (25 mL) was added N,N-dimethylformamide (0.2 mL) and SOCl2 (4.5 mL, 30 mmol) dropwise over 10 min. The mixture was heated for 4 h at reflux temperature, then cooled down to room temperature and the solvent was evaporated. The residue was dissolved in EtOAc (50 mL) and neutralized with cold aqueous NaHCO3 solution. The organic phase was washed with H2O, dried over (MgSO4) and evaporated. The residue was purified by a flash silica gel chromatography (elution with; 1% MeOH in CHCl3) to give (1.05 g, 96%) of 17 as a colorless foam: MS m/z 413 (M+1)+; UV λmax pH 1, 263.2; pH 7, 263.2; pH 13, 261.6; 1H NMR (CDCl3) δ 8.78 (1H, s, H-2), 8.34 (1H, s, H-8), 6.66 (1H, d, H-1′, J1′,2′ = 4.6 Hz), 5.55 (1H, dd, H-2′, J2′,3′ = 3.1 Hz), 5.47 (1H, dd, H-3′, J3′,4′ = 4.5 Hz), 4.51 (1H, dd, H-5′a, J4′.5′a = 5.8 Hz, J5′a,5′b = 12.0 Hz), 4.49 (1H, dd, H-5′ b, J4′,5′ b = 4.4 Hz), 4.41 (1H, dt, H-4′), 2.19 (3H, s, Ac), 2.15 (3H, s, Ac), 1.90 (3H, s, Ac).
6-Methyl-9-(β-d-arabinofuranosyl)purine (18)
A solution of (Ph3P)4Pd (36 mg, 0.03 mmol) in THF (1 mL) was added to a solution of CH3ZnBr (1 mmol, generated as above) in THF (5 mL) at room temperature. A solution of 17 (0.167 g, 0.39 mmol) in THF (3 mL) was added at room temperature and the mixture was stirred for 5 h at 55 °C. After an aqueous work up, the residue obtained by evaporation of the dried organic phase was dissolved in MeOH saturated with NH3 (10 mL) and stirred for 3 h at room temperature. The solvent was evaporated and the residue was purified by silica gel chromatography (elution with 6% EtOH in CHCl3) to give (83 mg, 78%) of 18 as a colorless solid that was crystallized from hot ethanol, m.p. 220–222 °C; MS m/z 267.1 (M+1)+; UV λmax pH 1, 263.6 (7.4); pH 7, 260.3 (8.0); pH 13, 261.0 (8.3); 1H NMR (Me2SO-d6) δ 8.77 (1H, s, H-2), 8.56 (1H, s, H-8), 6.38 (1H, d, H-1′, J1′,2′ = 5.1 Hz), 5.66 (1H, br s, 2′-OH), 5.58 (1H, d, 3′-OH, J = 4.4 Hz), 5.11 (1H, br t, 5′-OH), 4.27 (1H, m, H-2′, J2′,3′ = 5.2 Hz), 4.20 (1H,ddd, H-3′, J3′,4′ = 5.2 Hz), 3.82 (1H, ddd, H-4′), 3.72-3.63 (2H, m, H-5′a,b), 2.72 (1H, s, 6-CH3); NOE: Irradiation at H-1′ an enhancements of 2%, 1–2% and 10% were observed at H-8, at H-4′ and H-2′, respectively. Irradiation at H-3′ gave enhancements of 4–5% and 2% of the signals at H-8 and at H-4′, respectively. 13C NMR (Me2SO-d6) δ 157.69 (C-6), 151.51 (C-2, 1JCH = 203.3 Hz), 150.09 (C-4), 144.55 (C-8, 1JCH = 215.5 Hz), 132.11 (C-5), 84.20 (C-4′), 83.71 (C-1′, 1JCH = 164.8 Hz), 75.64 (C-2′), 74.67 (C-3′), 60.65 (C-5′), 19.01 (6-CH3); Anal. Calcd. for C11H14N4O4; C 49.62, H 5.30, N 21.04; found C 49.45, H 5.15, N 21.00.
Acknowledgements
This investigation was supported by a National Cooperative Drug Discovery Grant (U19CA67763) from the National Cancer Institute. We thank M.D. Richardson, and J.C. Bearden of the Molecular Spectroscopy Laboratory of Southern Research Institute for analytical and spectral data and S. Campbell for HPLC analyses. We are grateful to M. Kirk, University of Alabama at Birmingham Comprehensive Cancer Center Shared Mass Spectrometry Facility, for supplying some of the mass spectral data. Special thanks are due to Dr. Omar Moukha-chafiq for technical assistance.
Abbreviations
- E. coli PNP
Escherichia coli purine nucleoside phosphorylase
- MeP-dR
9-(2-deoxy-β-d-ribofuranosyl)-6-methylpurine
- F-dAdo
2-fluoro-2′-deoxyadenosine
- F-araA
9-(β-d-arabinofuranosyl)-2-fluoroadenine
- MeP
6-methylpurine
- F-Ade
2-fluoroadenine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sorscher EJ, Peng S, Bebok Z, Allan PW, Bennett LL, Jr, Parker WB. Tumor Cell Bystander Killing in Colonic Carcinoma Utilizing the Escherichia Coli DeoD Gene to Generate Toxic Purines. Gene Ther. 1994;1:233–238. [PubMed] [Google Scholar]
- 2.Parker WB, King SA, Allan PW, Bennett LL, Jr, Secrist JA, III, Montgomery JA, Gilbert KS, Waud WR, Wells AH, Gillespie GY, Sorscher EJ. In Vivo Gene Therapy of Cancer with E. coli Purine Nucleoside Phosphorylase. Hum. Gene Ther. 1997;8:1637–1644. doi: 10.1089/hum.1997.8.14-1637. [DOI] [PubMed] [Google Scholar]
- 3.Hughes BW, King SA, Allan PW, Parker WB, Sorscher EJ. Cell to Cell Contact is Not Required for Bystander Cell Killing by Escherichia coli Purine Nucleoside Phosphorylase. J. Biol. Chem. 1998;273:2322–2328. doi: 10.1074/jbc.273.4.2322. [DOI] [PubMed] [Google Scholar]
- 4.Hughes BW, Wells AH, Bebok Z, Gadi VK, Garver RI, Jr, Parker WB, Sorscher EJ. Bystander Killing of Melanoma Cells Using the Human Tyrosinase Promoter to Express the Escherichia coli Purine Nucleoside Phosphorylase Gene. Cancer Res. 1995;55:3339–3345. [PubMed] [Google Scholar]
- 5.Secrist JA, III, Parker WB, Allan PW, Bennett LL, Jr, Waud WR, Truss JW, Fowler AT, Montgomery JA, Ealick SE, Wells AH, Gillespie GY, Gadi VK, Sorscher EJ. Gene Therapy of Cancer: Activation of Nucleoside Prodrugs with E. coli Purine Nucleoside Phosphorylase. Nucleosides Nucleotides. 1999;18:745–757. doi: 10.1080/15257779908041562. [DOI] [PubMed] [Google Scholar]
- 6.Bennett LL, Jr, Vail MH, Chumley S, Montgomery JA. Activity of Adenosine Analogs Against a Cell Culture Line Resistant to 2-Fluoroadenine. Biochem. Pharmacol. 1966;15:1719–1728. [Google Scholar]
- 7.a) Philips FS, Sternberg SS, Hamilton L, Clarke DA. The Toxic Effects of 6-Mercaptopurine and Related Compounds. Ann. N.Y. Acad. Sci. 1954;60:283–296. doi: 10.1111/j.1749-6632.1954.tb40019.x. [DOI] [PubMed] [Google Scholar]; b) Clarke DA, Philips FS, Sternberg SS, Stock CC. Effects of 6-Mercaptopurine and Analogs on Experimental Tumors. ibid. 1954;60:235–243. doi: 10.1111/j.1749-6632.1954.tb40014.x. [DOI] [PubMed] [Google Scholar]
- 8.Montgomery JA, Hewson K. Analogs of 6-Methyl-9-(β-D-ribofuranosylpurine. J. Med. Chem. 1968;11:48–52. doi: 10.1021/jm00307a010. [DOI] [PubMed] [Google Scholar]
- 9. Unpublished results. [Google Scholar]
- 10.Erion MD, Stoeckler JD, Guida WC, Walter RL, Ealick SE. Purine Nucleoside Phosphorylase. 2. Catalytic Mechanism. Biochemistry. 1997;36:11735–11748. doi: 10.1021/bi961970v. [DOI] [PubMed] [Google Scholar]
- 11.For a general review, please see: Boudier A, Bromm LO, Lotz M, Knochel P. New Applications of Polyfunctional Organometallic Compounds in Organic Synthesis. Angew. Chem. Int. Ed. 2000;39:4414–4435.
- 12.For examples on magnesium chemistry, please see: Bergstrom DE, Reddy PA. Synthesis of 6-Alkyl and 6-Aryl Substituted 9-(β-D-ribofuranosyl Purines Via The Nickel Catalyzed Coupling of Grignard Reagents to 2′,3′,5′-Tris-O-(t-butyldimethylsilysl)-9-(β-D-ribofuranosyl-6-chloropurine. Tetrahedron Lett. 1982;23:4191–4194. Estep KG, Josef KA, Bacon ER, Carabateas PM, Rumney S, IV, Pilling GM, Krafte DS, Volberg WA, Dillon K, Dugrenier N, Briggs GM, Cannif PC, Gorczyca WP, Stankus GP, Ezrin AM. Synthesis and Structure-Activity Relationships of 6-Heterocyclic-Substituted Purines as Inactivation Modifiers of Cardiac Sodium Channels. J. Med. Chem. 1995;38:2582–2595. doi: 10.1021/jm00014a011.
- 13.For examples on zinc chemistry, please see: Gundersen L-L, Bakkestuen AK, Aasen AJ, Øverås H, Rise F. 6-Halopurines in Palladium-Catalyzed Coupling with Organotin and Organozinc Reagents. Tetrahedron. 1994;50:9743–9756. Gundersen L-L, Langli G, Rise F. Regioselective Pd-Mediated Coupling Between 2,6-Dichloropurines and Organometallic Reagents. Tetrahedron Lett. 1995;36:1945–1948. Stevenson TM, Prasad ASB, Citineni JR, Knochel P. Preparation of Zinc Organometallics Derived from Nucleosides and Nucleic Acid Bases and Pd(0) Catalyzed Coupling with Aryl Iodides. Tetrahedron Lett. 1996;37:8375–8378. Prasad ASB, Stevenson TM, Citineni JR, Nyzam V, Knochel P. Preparation and Reactions of New Zincated Nitrogen-Containing Heterocycles. Tetrahedron. 1997;53:7237–7254.
- 14.For examples on tin chemistry, please see: a) Reference 13a. Hocek M, Masojídková M, Holý A. Synthesis of Acyclic Nucleotide Analogues Derived from N-Substituted 6-(1-Aminoethyl)Purines Via 6-Acetylpurine Derivatives. Tetrahedron. 1997;53:2291–2302. Moriarty RM, Epa WR, Awasthi AK. Palladium Catalysed C-8 Allylation and Vinylation of Adenosine, 2′-Deoxyadenosine and 2′,3′-Dideoxyadenosine Nucleosides. Tetrahedron Lett. 1990;31:5877–5880. Sessler JL, Wang B, Harriman A. Long-Range Photoinduced Electron Transfer in an Associated but Non-covalently Linked Photosynthetic Model System. J. Amer. Chem. Soc. 1993;115:10418–10419. Van Aerschot AA, Mamos P, Weyns NJ, Ikeda S, De Clercq E, Herdewijn PA. Antiviral Activity of C-Alkylated Purine Nucleosides Obtained by Cross-Coupling with Tetraalkyltin Reagents. J. Med. Chem. 1993;36:2938–2942. doi: 10.1021/jm00072a013.
- 15.For an example on aluminum chemistry, please see: Hirota K, Kitade Y, Kanbe Y, Maki Y. Convenient Method for the Synthesis of C-Alkylated Purine Nucleosides: Palladium-Catalyzed Cross-Coupling Reaction of Halogenopurine Nucleosides with Trialkylaluminums. J. Org. Chem. 1992;57:5268–5270.
- 16.For an example on copper chemistry, please see: Dvořáková H, Dvořák D, Holý A. A, Coupling of 6-Chloropurines with Organocuprates Derived from Grignard Reagents: A Convenient Route to sec and tert 6-Alkylpurines. Tetrahedron Lett. 1996;37:1285–1288.
- 17.For examples on aryl boronic acids, please see: a) Reference 14b. Havelková M, Hocek M, Česnek M, Dvořák D. D, The Suzuki-Miyaura Cross-Coupling Reactions of 6-Halopurines with Boronic Acids Leading to 6-Aryl- and 6-Alkenylpurines. Synlett. 1999:1145–1147. Hocek M, Holý A, Votruba I, Dvořáková H. Synthesis and Cytostatic Activity of Substituted 6-Phenylpurine Bases and Nucleosides: Application of the Suzuki–Miyaura Cross-Coupling Reactions of 6-Chloropurine Derivatives with Phenylboronic Acids. J. Med. Chem. 2000;43:1817–1825. doi: 10.1021/jm991167+.
- 18.Hassan AEA, Abou-Elkhair RAI, Montgomery JA, Secrist JA., III Convenient Synthesis of 6-Methylpurine and Related Nucleosides, Nucleosides. Nucleotides & Nucleic Acids. 2000;19:1123–1134. doi: 10.1080/15257770008035035. [DOI] [PubMed] [Google Scholar]
- 19.Buck IM, Reese CB. An Unambiguous Synthesis of Adenylosuccinic Acid and its Constituent Nucleoside. J. Chem. Soc. Perkin Trans. 1990;1:2937–2942. [Google Scholar]
- 20.Robins MJ, Basom GL. Nucleic Acid Related Compounds. 8. Direct Conversion of 2′-Deoxyinosine to 6-Chloropurine 2′-Deoxyriboside and Selected 6-Substituted Deoxynucleosides and Their Evaluation as Substrates of Adenosine Deaminase. Can. J. Chem. 1973;51:3161–3169. [Google Scholar]
- 21.Giovannini R, Knochel P. Ni(II)-Catalyzed Cross-Coupling between Polyfunctional Arylzinc Derivatives and Primary Alkyl Iodides. J. Am. Chem. Soc. 1998;120:11186–11187. [Google Scholar]
- 22.Campbell JB, Jr, Firor JW, Davenport TW. Facile Palladium-Catalyzed Cross-Coupling of Monoorganozinc Halides with 3-Iodoanthranilonitriles. Synth. Commun. 1989;19:2265–2272. [Google Scholar]
- 23.Kotra LP, Manouilov KK, Cretton-Scott E, Sommadossi J-P, Boudinot FD, Schinazi RF, Chu CK. Synthesis, Biotransformation, and Pharmacokinetic Studies of 9-(β-D-Arabinofuranosyl)-6-azidopurine: A Prodrug for Ara-A Designed to Utilize the Azide Reduction Pathway. J. Med. Chem. 1996;39:5202–5207. doi: 10.1021/jm960339p. [DOI] [PubMed] [Google Scholar]
- 24.Rosemeyer H, Seela F. Configurational and Conformational Analysis of Regular and Modified Nucleosides by 1D-NOE Difference Spectroscopy. Nucleosides & Nucleotides. 1990;9:417–418. [Google Scholar]
- 25.Yamane A, Matsuda A, Ueda T. Reaction of 6-Methylsulfonylpurine Riboside with Carbon Nucleophiles and the Synthesis of 6-Alkylpurine Nucleosides (Nucleosides and Nucleotides. XXIX) Chem. Pharm. Bull. 1980;28:150–156. [Google Scholar]
- 26.Kuchar M, Pohl R, Klepetarova B, Votruba I, Hocek M. Synthesis of Substituted 6-Cyclopropylpurine Bases and Nucleosides by Cross-coupling reactions or Cyclopropanations. Org. Biomol. Chem. 2008:2377–2387. doi: 10.1039/b802833h. [DOI] [PubMed] [Google Scholar]
- 27.Gundersen L-L, Nissen-Meyer J, Spilsberg B. Synthesis and Antimycobacterial Activity of 6-Arylpurines: The Requirements for the N-9 Substituent in Active Antimycobacterial Purines. J. Med. Chem. 2002;45:1383–1386. doi: 10.1021/jm0110284. [DOI] [PubMed] [Google Scholar]
- 28.Hocek M, Holý A, Votruba I, Dvořáková H. Cytostatic 6-Arylpurine Nucleosides III. Synthesis and Structure-activity Relationship Study in Cytostatic Activity of 6-Aryl-, 6-Hetaryl- and 6-Benzylpurine Ribonucleosides. Collect. Czech. Chem. Commun. 2001;66:483–499. [Google Scholar]
- 29.Hocek M, Naus P, Pohl R, Votruba I, Furman PA, Tharnish PM, Otto MJ. Cytostatic 6-Arylpurine Nucleosides. 6. SAR in Anti-HCV and Cytostatic Activity of Extended Series of 6-Hetarylpurine Ribonucleosides. J. Med. Chem. 2005;48:5869–5873. doi: 10.1021/jm050335x. [DOI] [PubMed] [Google Scholar]
- 30.Kaneko M, Kimura M, Nishimura T, Shimizu B. Synthesis of N6- or 8-substituted 9-((β-D-Arabinofuranosyl) Adenines and Their Antiviral Activities Against Herpes Simplex and Vaccinia Viruses. Chem. Pharm. Bull. 1977;25:2482–2489. doi: 10.1248/cpb.25.2482. [DOI] [PubMed] [Google Scholar]