

Abstract
Beta-amyloid protein (Aβ) is thought to be responsible for neuronal apoptosis in Alzheimer’s disease (AD). Paradoxically, Aβ can also promote neurogenesis, both in vitro and in vivo, by inducing neural progenitor cells (NPCs) to differentiate into neurons. However, mechanisms of Aβ-induced neurogenesis are unknown. Here we examined the role of DNA polymerase-β (DNA pol-β), a DNA repair enzyme that is required for proper neurogenesis during brain development and is also responsible for Aβ-induced neuronal apoptosis. In neurospheres obtained from the adult mouse subventricular zone (SVZ), the knockdown of DNA pol-β or its pharmacological blockade showed that the enzyme functioned both to repress proliferation of early nestin+ progenitor cells and to promote the maturation of TuJ-1+ neuronal cells. In neurospheres challenged with oligomers of synthetic Aβ42, the expression levels of DNA pol-β were rapidly increased. DNA pol-β knockdown prevented the Aβ42-promoted differentiation of nestin+ progenitor cells into nestin+/Dlx-2+ neuroblasts. Moreover, when neurospheres were seeded to allow full differentiation of their elements, blockade of DNA pol-β prevented Aβ42-induced differentiation of progenitors into MAP-2+ neurons. Thus, our data demonstrate that Aβ42 arrests the proliferation of a subpopulation of nestin+ cells via the induction of DNA pol-β, thereby allowing for their differentiation toward the neuronal lineage. Our findings reveal a novel role of DNA pol-β in Aβ42-induced neurogenesis and identify DNA pol-β as a key mechanistic link between the neurogenic effect of Aβ42 on NPCs and the pro-apoptotic effect of Aβ42 on mature neurons.
Keywords: DNA polymerase-β, β-amyloid, neurogenesis, neurosphere, neural stem cell, Alzheimer’s disease
Introduction
Evidence for increased neurogenesis exists in the hippocampus and subventricular zone (SVZ) of Alzheimer’s disease (AD) patients (Ziabreva et al., 2004). Enhanced neurogenesis has also been described in β-amyloid protein precursor (APP) transgenic mice before the appearance of other AD-related pathological features (Lopez-Toledano et al., 2007), suggesting that it might be an important component of AD pathophysiology. Studies in vitro and in vivo have demonstrated that β-amyloid (Aβ), which is thought to be responsible for neuronal loss in AD, is able to drive the differentiation of neural progenitor cells (NPCs) toward neurons (Calafiore et al., 2006; Lopez-Toledano et al., 2004). Aβ may induce neurogenesis early in the pathogenesis of AD, thereby resulting in exhaustion of the neural stem cell pool later in life of AD patients (Lopez-Toledano et al., 2010).
Mechanisms underlying the neurogenic effect of Aβ are unknown. Understanding the mechanisms of Aβ-induced neurogenesis might be relevant for exploiting the regenerative potential of the AD brain. Mature neurons respond to Aβ by initiating cell cycle re-entry that leads to Aβ-induced neuronal apoptosis (Morishima et al., 2001; Herrup et al., 2004). Specifically, it is the ectopic S phase that triggers the death of mature neurons (Copani et al., 1999). Neurons treated with Aβ undergo an unusual type of de novo DNA replication, largely mediated by the repair enzyme DNA polymerase-β (DNA pol-β), as they lack the major replicative enzyme, DNA pol-α (Copani et al., 2002). Knockdown of DNA pol-β prevents Aβ-induced DNA replication and neuronal death (Copani et al., 2002, 2006).
In NPCs, which express a canonical repertoire of replicative enzymes, DNA pol-β might instead function as a DNA repair enzyme involved in non-homologous end-joining (Reichenberger et al., 1998) and gene recombination. In DNA pol-β deficient mice, neuronal apoptosis occurs within the developmental period of neurogenesis (Sugo et al., 2000). Interestingly, when apoptosis is prevented by the concomitant deficiency of the proapoptotic factor p53, mice still display serious brain abnormalities (Sugo et al., 2004), suggesting that DNA pol-β is required for neuronal differentiation.
In the present study, by using NPCs derived from the adult mouse SVZ, we examine the hypothesis that the induction of DNA pol-β plays an important role in the neurogenic effect of Aβ. Our results reveals a novel role of DNA pol-β in Aβ-induced neurogenesis, and identify DNA pol-β as a mechanistic link between the neurogenic effect of Aβ42 on NPCs and the pro-apoptotic effect of Aβ42 on mature neurons.
Materials and Methods
Aβ peptide preparation
Aβ42 was purchased from Bachem Distribution Service. Aβ42 was prepared as previously described (Giuffrida et al., 2009). Briefly, Aβ42 peptide was resuspended in dimethyl sulfoxide (DMSO) to 5 mM and then diluted to 100 μM in ice-cold Dulbecco’s modified Eagle’s medium-F12 (DMEM-F12, Gibco). The suspension was allowed to oligomerize overnight at 4°C and used to the final concentration of 1 μM in the presence of the ionotropic glutamate receptor antagonists MK-801 (1 μM) and DNQX (30 μM) to avoid the participation of the endogenous glutamatergic mechanism in subsequent experiments (Copani et al 1999). Control experiments were conducted in the same conditions except for the addition of the peptide.
Neurosphere preparation and treatments
Neurosphere cultures were obtained from CD1 adult male mice. Brains were removed and placed in PIPES buffer (Fisher). After dissecting the lateral walls of the lateral ventricle, cell suspensions were grown in DMEM-F12 supplemented 10 mg/ml albumin, 5.2 μg/ml insulin, 0.63 ng/ml progesterone, 100 μg/ml transferin, 16.11 μg/ml putrescine, 2 mM glutamine, 0.5% glucose, 1% bovine serum albumin (BSA) ( all from Sigma, St. Louis, MO), 20 ng/ml EGF and 5 ng/ml of bFGF (both from Invitrogen, Carlsbad, CA) in 25-cm2 flasks. Neurospheres that had been grown for 12-15 DIV were passaged and transferred into 35-mm dishes. When necessary for differentiation studies, neurospheres were plated onto dishes pre-treated with poly-ornithine (20 μg/ml) and laminin (10 μg/ml) without EGF. The base analog 2′,3′-dideoxycytidine (ddC, 100 μM) was used as a selective inhibitor of DNA pol-β (Copani et al., 1999, 2002, 2006).
Use of oligonucleotide antisenses and shRNAs of DNA pol-β
The following “end-capped” phosphothioate oligonucleotide antisenses (obtained from MWG-Biotech) were used: pol β-As, 5′-tacttgtggatcgcctggct-3′; pol β-Sn, 5′-agccaggcgatccacaagta-3′ (bases 95-114 of the rat mRNA sequence NM017141). Cultures were treated with oligonucleotides (1.5 μM) 16 h before the addition of Aβ. DNA pol-β shRNAs cloned into pLKO.1 were purchased from Open Biosystem. The DNA pol-β shRNAs (TRCN0000077250 and TRCN0000077248), which that showed the highest levels of DNA pol-β knockdown among five plasmids we tested, were used in this study. To establish stable expression, we infected the cells with a lentivirus-based shRNA expresser, and cells were selected with puromycin. An shRNA encoded for a scrambled sequence was used as a negative control.
Fluorescence-activated cell sorting (FACS) analysis and immunofluorescence staining
Neurospheres were dissociated by incubation with 0.25% trypsin and collected by low-speed centrifugation after the addition of 20 % fetal bovine serum (FBS). Cellular pellets were fixed in 70% ethanol and treated with RNAse (100 μg/ml) for 1 h before propidium iodide staining (50 μg/ml for 10 min). Samples were simultaneously analyzed for cell cycle and apoptosis. DNA content and ploidy were assessed by using a CyAn flow cytometer, and cell-cycle distribution profiles were analyzed with the Multicycle AV software program. Apoptotic NPCs were scored from the area of hypoploid DNA preceding the G0/G1 DNA peak.
For FACS analysis, suspended cells, prepared as described above, were fixed in 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, and then stained overnight at 4°C with the following antibodies: mouse anti-nestin monoclonal antibody (Chemicon, 1 μg/ml), mouse anti-GFAP polyclonal antibody (Lab Vision, 1:500), mouse anti-PSA-NCAM (DHSB, 1:500), or rabbit anti-Dlx-2 polyclonal antibody (Chemicon, 1:400). The following secondary antibodies were used: Alexa Fluor 633 anti-mouse IgG (Invitrogen, 1:1000) for the detection of nestin, GFAP and PSA-NCAM, and Alexa Fluor 488 anti-rabbit IgG (Invitrogen, 1:1000) for the detection of Dlx-2.
Immunoblotting
Western blot analysis was carried out on total neurosphere protein extracts. Neurospheres were washed in phosphate-buffered saline (PBS) and resuspended in RIPA lysis buffer (Boston BioProducts) with a protease inhibitor cocktail (Roche) added. The cell suspension was sonicated and protein concentration was determined using bicinchoninic acid assay (Sigma-Aldrich). Electrophoresis was performed in SDS-polyacrylamide gel, using 40 μg of proteins per lane. The following primary antibodies were used: mouse monoclonal anti-nestin (Santa Cruz Biotech, 1:400), rabbit polyclonal anti-GFAP (Chemicon, 1:500 ), rabbit polyclonal anti-TuJ-1 (Covance, 1:1000), rabbit polyclonal anti-MAP-2 (Chemicon, 1:300), mouse monoclonal anti-DNA pol-β (Lab Vision, 1:200), rabbit polyclonal anti-Dlx-2 (Chemicon, 1:500), mouse monoclonal anti-Bax (Santa Cruz Biotech, 1:200), rabbit polyclonal anti-p21 (Santa Cruz Biotech, 1:200), and mouse monoclonal anti-GAPDH (Santa Cruz Biotech, 1:1000).
Results
Aβ-induced neurogenesis and the effect of cell cycle arrest on neurosphere cultures from the SVZ
We previously reported that Aβ42 induced neural progenitors of the adult mouse SVZ to acquire a neuronal phenotype (Calafiore et al 2006), using the highly polysialated form of NCAM (PSA-NCAM) as a marker of neuronal lineage cells. We noted that, in suspended neurospheres derived from the SVZ, Aβ42-induced neurogenesis was coupled with an increased expression of the cyclin-dependent kinase (CDK) inhibitors, p21 and p27kip1, which promoted cell cycle arrest in the G1 phase (Calafiore et al 2006). This observation raised a fundamental question as to whether Aβ-induced neurogenesis is due to cell cycle arrest of the SVZ NPCs or direct induction of NPC differentiation into the neuronal lineage. Thus, we first examined whether cell cycle arrest per se, in the absence of Aβ42, would be sufficient to promote the differentiation of NPCs into the neuronal lineage. We compared cells treated with and without a commonly used cell cycle blocker, aphidicolin (Aph) (Jackman et al., 2001; Sheaff et al., 1991). Secondary neurospheres were exposed to 5 μM Aph at 3 days after expansion, and then were allowed to grow for 48 h (Fig. 1A). Under this condition, FACS analysis indicated that Aph modified the cell cycle distribution by arresting about 13% of the cells in the G1/S phases (Fig. 1B). Phase contrast microscopic image of cultures at Day 5 showed a dramatic reduction of the neurosphere size in cultures treated with Aph compared with controls (Fig. 1C). To quantify the effects of Aph treatment on death of NPCs, we assessed the levels of apoptosis by cytofluorimetric analysis (Darzynkiewicz et al., 1992; Lindsten et al, 2003). No change in apoptosis was detected in the Aph-treated group (Fig. 1D). These results indicated that NPC proliferation was effectively arrested in the G1/S phases and under this condition no toxicity was observed. Western blot analysis showed an increased expression of p21 and p27kip1 in neurospheres treated with Aph (Fig. 1D), whereas no changes were observed in the expression levels of Bax, a pro-apoptotic marker (Fig.1D).
Figure 1.
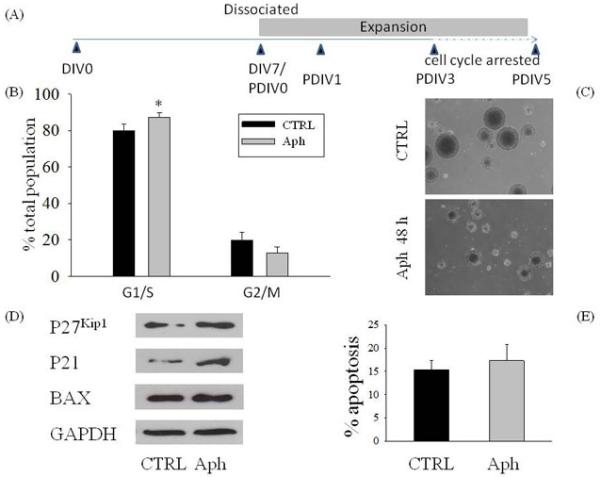
Effect of cell cycle arrest of neurospheres induced by aphidicolin in the presence or absence of Aβ42 on neural progenitor cells (NPCs). Schematic drawing of the experimental protocol (A). Neurospheres were grown as described in the Materials and Methods section. After 7 days in vitro (DIV 7), neurospheres were passaged and NPCs were allowed to form secondary neurospheres. Three days after the first passage (PDIV 3), neurospheres were treated with 5 μM of the cell cycle inhibitor aphidicolin (Aph) for 48 h. (C) Phase contrast microscopy of NPCs grown in vitro with or without Aph. Cell cycle distribution (B) and apoptosis (E) of neurospheres analyzed by flow cytometry 48 h after treatment with Aph (5 μM). Values are mean ± SEM of 5 different determinations. *p<0.05 (ANOVA followed by Tukey post hoc test). CTRL: control. (D) Representative blots of total protein extracts showing the increased expression of the cell cycle inhibitor proteins p21 and p27 after treatment with Aph, and no difference in the pro-apoptotic protein Bax.
We then used nestin as a marker to quantify NPCs, Dlx-2 as an early marker of differentiation into the neuronal lineage, and PSA-NCAM as a marker of neuroblasts (Moraes et al., 2009). Neurospheres were analyzed by immunofluorescence staining (Fig. 2A), and the percentage of immunopositive cells for different markers were scored by FACS analysis. In the control group, neurospheres consisted mainly of nestin+ cells (Fig 2A, 2C). When Aβ42 (1 μM) was added for 24 h, NPCs showed a 20% increase in the percentage of nestin+ cells (Fig. 2B, 2C), and entirely accounted for the population of cells also expressing Dlx-2 (nestin+/Dlx-2+) (Fig. 2C). We also found an increased percentage of Dlx-2+ cells that also stained for PSA-NCAM+ in neurospheres treated with Aβ42 (Fig. 2D). Dlx-2/PSA-NCAM labeled neuroblasts were virtually absent in control cultures (Fig. 2D). In contrast to Aβ treatments, addition of Aph to cells cultures neither increased the number of nestin+ cells (Fig. 2B, 2C), nor promoted the differentiation of nestin+ cells toward the neuronal lineage (Fig.2C, 2D) compared with the control group. Similar to Aβ42, Aph did not alter the percentage of cells expressing both nestin and GFAP (Fig. 2E).
Figure 2.
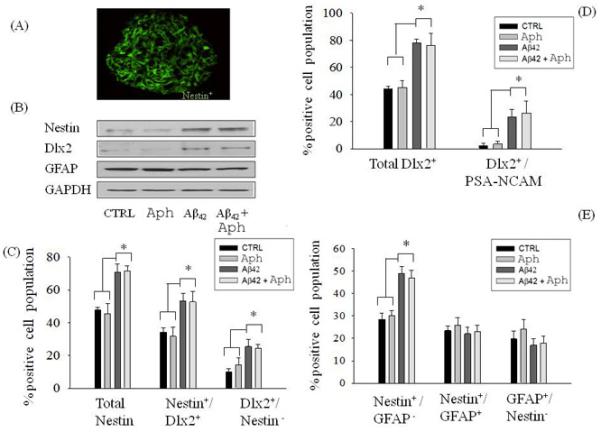
NPC differentiation in the presence or absence of Aβ42 was not affected by pharmacological blockade of the cell cycle. Confocal image of neurospheres showing immunoreactive nestin+ cells (A). Neurospheres from the adult mouse SVZ were treated with Aph (5 μM) for 48 h in the presence or absence of Aβ42 (1 μM), and expression levels of nestin, Dlx-2 and GFAP were analyzed in total protein extracts (B). The percentage of immunopositive cells for the different lineage markers were scored by FACS analysis (C-E). Values are expressed as percentage of total cell population and are mean ± SEM of 5 different determinations from 3 independent experiments. *p<0.05 (ANOVA followed by the Tukey post hoc test).
The effect of Aβ42 on DNA pol-β expression and the role of DNA pol-β in NPCs in SVZ neurospheres
The evidence that Aph did not mimic and did not potentiate the neurogenic effect of Aβ in SVZ neurospheres (Fig. 2B-D) indicated that cell cycle inhibition per se was not sufficient to account for Aβ-induced neurogenesis, and that Aβ may exert a direct neurogenic effect on NPCs. Copani et al. (1999) reported that, in post-mitotic neurons, Aβ42 re-activated an aberrant cell cycle by engaging the base excision repair enzyme, DNA polymerase β (DNA pol-β), within a non-canonical pathway of neuronal DNA replication. Interestingly, DNA pol-β was shown to be required for proper neurogenesis during embryonic development (Sugo et al., 2000). We wondered whether Aβ42-induced neurogenesis could be dependent on the activation of DNA pol-β in neurospheres. Virtually all the cells within the neurospheres constitutively expressed DNA pol-β (Fig. 3A-B), which was further increased (approximately 3 folds) following exposure to Aβ42 (1 μM) for 24 h (Fig. 3B). To gain insights into the role of DNA pol-β in neurogenesis, we carried out the short hairpin RNA (shRNA)-mediated silencing of the enzyme. The shRNA expression dramatically reduced the expression levels of DNA pol-β in total protein extract from NPCs (Fig. 3D). Twenty four hours after shRNA had been delivered through lentivirus, neurospheres were analyzed for both cell cycle distribution and apoptosis. FACS analysis indicated that DNA pol-β knockdown by shRNA significantly increased the percentage of cells entering S phase, an effect that was shared by the pharmacological inhibitor of DNA pol-β, 2′,3′-dideoxycytidine (ddC) (Copeland et al., 1992) (Fig. 3E). Basal levels of apoptosis were analyzed, but no statistically significant differences were observed between groups (Fig. 3C). Taken together, these data indicated that DNA pol-β could act as a repressor of proliferation of NPC neurospheres, consistent with previous data obtained with treatment of neurospheres with end-capped phosphothioate antisenses for DNA pol-β (1.5 μM for 16 h) (Copani et al. 2002). Both the antisense-induced knockdown of DNA pol-β and its pharmacological blockade with ddC induced a significantly increase of nestin protein levels at 24 h after treatment (Fig. 4A). This effect resulted from the increased number of early NPCs expressing nestin, but not GFAP or Dlx-2 (Fig. 4B), suggesting that DNA pol-β could restrain the proliferation of early NPCs in suspended neurospheres under basal conditions.
Figure 3.
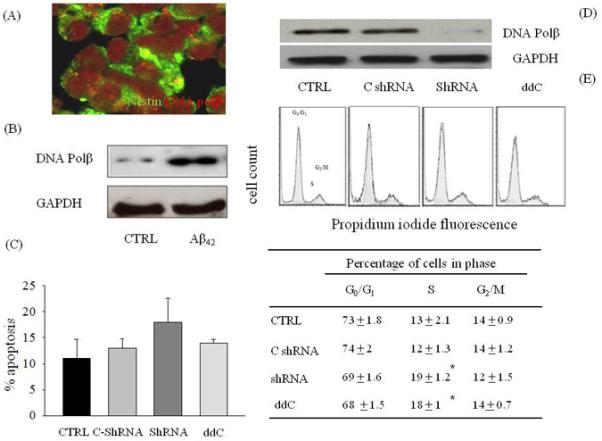
DNA pol-β acted as a repressor of proliferation in suspended neurospheres. (A) Confocal laser scanning microscopy of neurospheres. Cells were stained for nestin in green and DNA pol β in red. (B) Representative immunoblots of DNA pol-β in protein extracts from neurospheres exposed to 1 μM of Aβ42 for 24 h. Cell cycle distribution profiles (E) and extent of apoptosis (C) in NPCs expressing either a DNA pol-β shRNA or a control shRNA encoding a scramble sequence (C-shRNA) (D). Some neurospheres were treated with 2′,3′-dideoxycitidine (ddC, 100 μM), a selective inhibitor of DNA pol-β, for 24 h. In E, *p<0.05 vs. the respective control (CTRL, C-shRNA). Values are mean ± SEM of 3 different determinations from 5 independent cultures. For each determination, 50,000 cells were analyzed by cytofluorimetric analysis.
Figure 4.
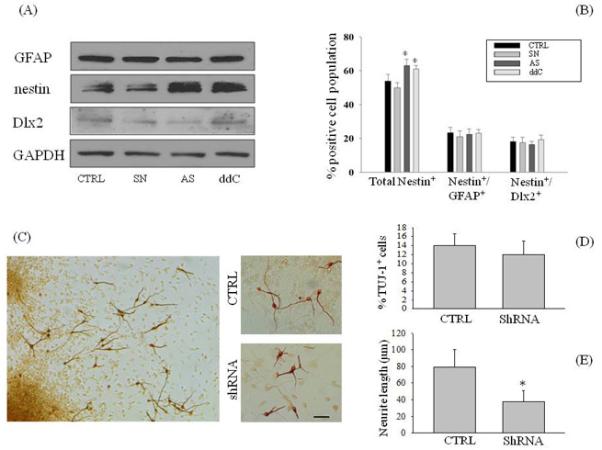
Knockdown of DNA pol-β altered the differentiation of NPCs. (A) Expression levels of GFAP, nestin and Dlx-2 in total protein extracts from neurospheres treated with DNA pol-ß antisenses (As) or senses (Sn), or treated with ddC (100 μM). GAPDH was used as a loading control. (B) The percentage of total nestin+ cells was increased in neurospheres treated with DNA pol-β antisenses (1.5 μM), but not the number of nestin+ cells co-expressing GFAP or Dlx-2. *p<0.05 vs the respective controls. (C) Neurospheres were plated on poly-L-ornithine and laminin coated dishes for 72 h in growing medium in the absence of EGF. Immature neurons, in which DNA pol-β had been silenced with shRNA, were immunostained with TUJ-1, and the number of immunopositive cells (D) as well as neurite length (E) were compared with the controls (CTRL). For each determination, 20 randomly selected fields from 3 independent cultures were analyzed. Values are mean ± SEM of 4 different determinations from 3 independent experiments. In E, *p< 0.05 (ANOVA followed by Tukey post hoc test) vs. the control group. Scale bar, 30 μm.
Neurospheres cultured onto laminin, in the absence of growth factors, consist of a mixture of precursor cells and differentiated neurons, astrocytes and oligodendrocytes (Suslov et al., 2002). Twenty four hours after the transfection with shRNA, neurospheres were plated onto poly-L-ornithine and laminin and allowed to differentiate for 3 days. We used an antibody against the β-tubulin III isoform (TuJ-1) to determine the percentage of immature neurons in culture. Although the silencing of DNA pol-β did not influence the percentage of TuJ-1+ cells as compared to controls (Fig. 4C-D), the assessment of neurite outgrowth revealed that neurons generated from shRNA-treated neurospheres showed a dramatic reduction of neurite length (38 ± 13 μm, n=157) compared to the control group (79 ± 21 μm, n= 168) (Fig 4C, 4E).
The role of DNA pol-β in Aβ42-induced neuronal differentiation of SVZ neuroshpheres
When Aβ42 was added in suspended neurospheres where DNA pol-β had been knocked down by antisenses, Aβ42 treatment failed to increase the number of cells immunoreactive for both nestin and Dlx-2 (Fig 5A, 5D) and a parallel increase of nestin-only immunopositive cells was observed (Fig 5D). These data indicated that DNA pol-β was engaged by Aβ42 to promote the differentiation of early nestin+ NPCs toward the neuronal lineage in free floating neurosphere cultures.
Figure 5.
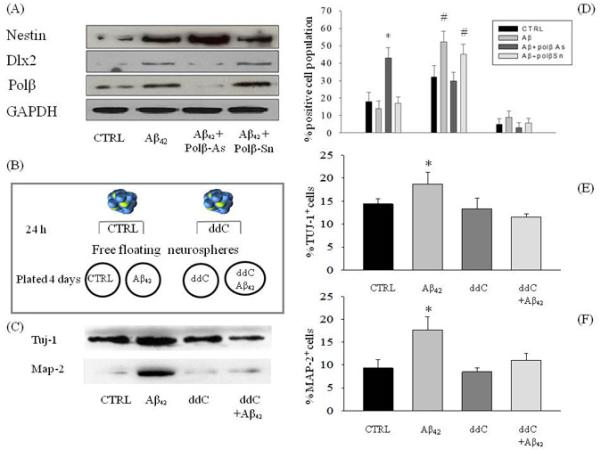
DNA pol-β blockade inhibited neuronal differentiation in neurospheres exposed to Aβ42. Cultures were treated with antisenses (pol β-As) or senses (pol β-Sn) (1.5 μM) for 16 h before the addition of 1 μM Aβ42. Representative immunoblots of nestin, Dlx-2 and DNA pol-β from neurospheres exposed to 1 μM of Aβ42, after antisense or sense treatment, are in (A). GAPDH was the loading control. (D) pol β-As prevented the expression of Dlx-2 in neurospheres exposed to Aβ42 for 24 h. * p< 0.05 vs. CTRL. # p< 0.05 vs CTRL and Aβ-As. (B) Suspended neurospheres were treated with ddC (100 μM) 24 h before plating. Plated neurospheres were maintained in cultures for 4 days either in the absence or presence of 1 μM Aβ42 and with or without ddC. (C) Representative immunoblots of MAP-2 and TuJ-1 in protein extracts from neurospheres treated as described in (B). The cell cycle inhibitor ddC prevented the increase in the total number of neurons (F) and neuroblasts (E) that was observed in neurospheres plated with Aβ42. Data represent mean ± SEM of 9 different determinations from 3 independent experiments. * p< 0.05 vs CTRL and # p<0.05 vs Aβ.
Next, neurospheres were plated onto laminin and allowed to differentiate for 4 days in the absence or presence of Aβ42. Both TuJ-1+ cells and MAP-2+ cells, which correspond to more mature neurons, were found in control cultures (Fig. 5C, 5E-F). Exposure to Aβ42 resulted into an increased number of both TuJ-1+ cells and MAP-2+ cells (Fig. 5C, 5E-F), indicating that Aβ42 was able to promote the differentiation of NPCs into the neuronal lineage. Similar to shRNA-mediated silencing of DNA pol-β, pharmacological blockade of the enzyme with ddC (100 μM applied 24 h before and during plating) did not affect the total number of TuJ-1+ cells in control cultures (Fig. 5C, 5E). However, the presence of ddC completely prevented Aβ42-induced increase of both TuJ-1+ and MAP-2+ cells (Fig. 5C, 5E-F).
Discussion
Enhanced neurogenesis has been reported in AD patients (Ziebreva et al., 2004) and in animal models of AD, including the APP23 (Bondolfi e al., 2002), PDGF-APPSw-Ind (Jin e tal., 2004) and PDAPP mice (Donovan et al., 2006), in all of which the human Aβ peptide is overproduced. Jin and colleagues (2004) suggested that increased neurogenesis could be a compensatory response to disease manifestation, whereas Lopez-Toledano et al. (2007) proposed that Aβ itself drives neurogenesis in the early phases of AD, leading to the exhaustion of the NPC pool available for brain repair later in the neurodegenerative process of the disease. Our early work demonstrated that synthetic human Aβ42, under the conformation state that is usually toxic for adult neurons, promoted instead the differentiation of NPCs of the adult mouse SVZ toward the neuronal phenotype (Calafiore et al., 2006). Mechanisms underlying this phenomenon are unknown. In the present study, our data indicate that synthetic oligomers of Aβ42 drive the differentiation of nestin+ NPCs into MAP-2+ neurons, by engaging the DNA repair enzyme, DNA pol-β. High levels of DNA pol-β are found in the rat brain during embryonic ages (Nowak et al., 1990).
Neuronal differentiation may arise from genomic rearrangement factors that generate a certain type of DNA damage that is repairable by DNA pol-β (Sugo et al., 2004). However, the specific role of DNA pol-β in Aβ-induced neurogenesis has not been studied. We showed that DNA pol-β acted to arrest the proliferation of a subset of nestin+ progenitor cells, allowing them to acquire the early marker of the neuronal lineage, Dlx-2, in response to Aβ42. This effect appeared to be specific for DNA pol-β, since a generic arrest of NPC proliferation by aphidicolin, both in the absence and in the presence of Aβ42, did not result into neuronal differentiation. Although DNA pol-β was expressed virtually in all NPCs, it appeared that its pro-differentiation effects were restricted to a population of progenitors committed to the neuronal lineage by Aβ42. It is well known that NPCs are heterogeneous, and this heterogeneity is particularly likely in the NPC population in cultured neurospheres. The evidence that DNA pol-β per se was required for the maturation of TuJ-1+ cells suggested that the enzyme could serve multiple functions in cells belonging to the neuronal lineage. It was intriguing that DNA pol-β appeared to control proliferation and differentiation of nestin+ progenitors, but it was not required for cell survival. A hypothetic model of the DNA pol-β involvement in Aβ-induced neurogenesis is presented in Fig.6. This model is based on results from this study as well as those from our previous study (Calafiore et al., 2006). Our findings strengthen the molecular link between the function of DNA poly-β and the induction of neural differentiation.
Figure 6.

A model depicting the role for DNA pol-β in the neurogenic effect of Aβ. This hypothesis of neuronal differentiation driven by Aβ is based on the results from the present study and from Calafiore et al. (2006). Aβ42 arrests the proliferation of a subpopulation of nestin+ cells via the induction of DNA pol-β, thus allowing for their differentiation toward the neuronal phenotype. Aβ42 also promotes the appearance of nestin immunoreactivity in nestin−/Sox-2+ cells. The precise mechanisms underlying this effect remain to be further determined.
DNA pol-β might be critically involved in the differentiation of neural progenitors for several reasons. First, DNA pol-β might be part of an endogenous “checkpoint system” aimed at regulating the quantity and quality of nestin+ progenitors via a cell cycle arrest. Second, DNA pol-β might function to maintain the integrity of genes (e.g., Dlx-2) to be expressed in certain progenitors (e.g., nestin+/Dlx-2+ cells), thus coordinating proliferation arrest with differentiation. Third, DNA pol-β might be required to repair oxidative base damage occurring in neuronal types actively undergoing axon pathfinding (Sugo et al., 2004). Consistent with this notion, our results indicated that the neurite outgrowth of Tuj-1+ cells was dependent on the presence of DNA pol-β. Although it appeared that constitutive DNA pol-β participated in neurogenesis, our results indicated that an increased expression of the enzyme per se, in the absence of the concomitant stimulation by Aβ42, would not be sufficient to fully promote neuronal differentiation.
Our findings reveal a novel role of DNA pol-β in Aβ42-induced neurogenesis, and also indentify DNA pol-β as a key mechanistic link between the neurogenic effect of Aβ42 on NPCs and the pro-apoptotic effect of Aβ42 on mature neurons. Because DNA pol-β is also involved in Aβ-mediated apoptosis of mature neurons (Copani et al., 2006), Aβ-induced neurons might undergo apoptosis once they reach the post-mitotic state. Thus, it is likely that Aβ-induced neurogenesis in AD is fated to be unproductive. Nevertheless, DNA pol-β could participate to the attempted neurogenesis in AD and under many other neuropathological conditions, such as brain ischemia, where the enzyme activity is expected to be increased as a result of the concomitant DNA damage that needs to be repaired.
Acknowledgements
This work was in part supported by grants from National Institutes of Health (R01NS061983 and R01ES015988 to W. D.), the National Multiple Sclerosis Society (to W. D.), Shriners Hospitals for Children (to W. D.), and Italian Ministry of University and Research (grant 105/04) (to A. C.).
Footnotes
The authors declare no conflicts of financial interest related to this work.
References
- Bondolfi L, Calhoun M, Ermini F, Kuhn HG, Wiederhold KH, Walker L, Staufenbiel M, Jucker M. Amyloid-associated neuron loss and gliogenesis in the neocortex of amyloid precursor protein transgenic mice. J Neurosci. 2002 Jan 15;22(2):515–22. doi: 10.1523/JNEUROSCI.22-02-00515.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calafiore M, Battaglia G, Zappalà A, Trovato-Salinaro E, Caraci F, Caruso M, Vancheri C, Sortino MA, Nicoletti F, Copani A. Progenitor cells from the adult mouse brain acquire a neuronal phenotype in response to beta-amyloid. Neurobiol Aging. 2006 Apr;27(4):606–13. doi: 10.1016/j.neurobiolaging.2005.03.019. [DOI] [PubMed] [Google Scholar]
- Copani A, Condorelli F, Caruso A, Vancheri C, Sala A, Giuffrida Stella AM, Canonico PL, Nicoletti F, Sortino MA. Mitotic signaling by beta-amyloid causes neuronal death. FASEB J. 1999 Dec;13(15):2225–34. [PubMed] [Google Scholar]
- Copani A, Sortino MA, Caricasole A, Chiechio S, Chisari M, Battaglia G, Giuffrida-Stella AM, Vancheri C, Nicoletti F. Erratic expression of DNA polymerases by beta-amyloid causes neuronal death. FASEB J. 2002 Dec;16(14):2006–8. doi: 10.1096/fj.02-0422fje. [DOI] [PubMed] [Google Scholar]
- Copani A, Hoozemans JJ, Caraci F, Calafiore M, Van Haastert ES, Veerhuis R, Rozemuller AJ, Aronica E, Sortino MA, Nicoletti F. DNA polymerase-beta is expressed early in neurons of Alzheimer’s disease brain and is loaded into DNA replication forks in neurons challenged with beta-amyloid. J Neurosci. 2006 Oct 25;26(43):10949–57. doi: 10.1523/JNEUROSCI.2793-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland WC, Chen MS, Wang TS. Human DNA polymerases alpha and beta are able to incorporate anti-HIV deoxynucleotides into DNA. J Biol Chem. 1992 Oct 25;267(30):21459–64. [PubMed] [Google Scholar]
- Darzynkiewicz Z, Bruno S, Del Bino G, Gorczyca W, Hotz MA, Lassota P, Traganos F. Features of apoptotic cells measured by flow cytometry. Cytometry. 1992;13:795–808. doi: 10.1002/cyto.990130802. [DOI] [PubMed] [Google Scholar]
- Donovan MH, Yazdani U, Norris RD, Games D, German DC, Eisch AJ. Decreased adult hippocampal neurogenesis in the PDAPP mouse model of Alzheimer’s disease. J Comp Neurol. 2006 Mar 1;495(1):70–83. doi: 10.1002/cne.20840. [DOI] [PubMed] [Google Scholar]
- Giuffrida ML, Caraci F, Pignataro B, Cataldo S, De Bona P, Bruno V, Molinaro G, Pappalardo G, Messina A, Palmigiano A, Garozzo D, Nicoletti F, Rizzarelli E, Copani A. Beta-amyloid monomers are neuroprotective. J Neurosci. 2009 Aug 26;29(34):10582–7. doi: 10.1523/JNEUROSCI.1736-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrup K, Neve R, Ackerman SL, Copani A. Divide and die: cell cycle events as triggers of nerve cell death. J Neurosci. 2004 Oct 20;24(42):9232–9. doi: 10.1523/JNEUROSCI.3347-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman J, O’Connor PM. Methods for synchronizing cells at specific stages of the cell cycle. Curr Protoc Cell Biol. 2001 May; doi: 10.1002/0471143030.cb0803s00. Chapter 8:Unit 8.3. [DOI] [PubMed] [Google Scholar]
- Jin K, Galvan V, Xie L, Mao XO, Gorostiza OF, Bredesen DE, Greenberg DA. Enhanced neurogenesis in Alzheimer’s disease transgenic (PDGF-APPSw,Ind) mice. Proc Natl Acad Sci U S A. 2004 Sep 7;101(36):13363–7. doi: 10.1073/pnas.0403678101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, Peel AL, Mao XO, Xie L, Cottrell BA, Henshall DC, Greenberg DA. Increased hippocampal neurogenesis in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2004 Jan 6;101(1):343–7. doi: 10.1073/pnas.2634794100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsten T, Golden JA, Zong WX, Minarcik J, Harris MH, Thompson CB. The proapoptotic activities of Bax and Bak limit the size of the neural stem cell pool. J Neurosci. 2003 Dec 3;23(35):11112–9. doi: 10.1523/JNEUROSCI.23-35-11112.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Toledano MA, Shelanski ML. Neurogenic effect of beta-amyloid peptide in the development of neural stem cells. J Neurosci. 2004 Jun 9;24(23):5439–44. doi: 10.1523/JNEUROSCI.0974-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Toledano MA, Shelanski ML. Increased neurogenesis in young transgenic mice overexpressing human APP(Sw,Ind) J Alzheimers Dis. 2007 Nov;12(3):229–40. doi: 10.3233/jad-2007-12304. [DOI] [PubMed] [Google Scholar]
- Lopez-Toledano MA, Ali Faghihi M, Patel NS, Wahlestedt C. Adult neurogenesis: a potential tool for early diagnosis in Alzheimer’s disease? J Alzheimers Dis. 2010;20(2):395–408. doi: 10.3233/JAD-2010-1388. [DOI] [PubMed] [Google Scholar]
- Moraes L, de Moraes Mello LE, Shimabukuro MK, de Castro Batista CM, Mendez-Otero R. Lack of association between PSA-NCAM expression and migration in the rostral migratory stream of a Huntington’s disease transgenic mouse model. Neuropathology. 2009 Apr;29(2):140–7. doi: 10.1111/j.1440-1789.2008.00959.x. [DOI] [PubMed] [Google Scholar]
- Morishima Y, Gotoh Y, Zieg J, Barrett T, Takano H, Flavell R, Davis RJ, Shirasaki Y, Greenberg ME. Beta-amyloid induces neuronal apoptosis via a mechanism that involves the c-Jun N-terminal kinase pathway and the induction of Fas ligand. J Neurosci. 2001 Oct 1;21(19):7551–60. doi: 10.1523/JNEUROSCI.21-19-07551.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichenberger S, Pfeiffer P. Cloning, purification and characterization of DNA polymerase beta from Xenopus laevis--studies on its potential role in DNA-end joining. Eur J Biochem. 1998 Jan 15;251(1-2):81–90. doi: 10.1046/j.1432-1327.1998.2510081.x. [DOI] [PubMed] [Google Scholar]
- Sheaff R, Ilsley D, Kuchta R. Mechanism of DNA polymerase alpha inhibition by aphidicolin. Biochemistry. 1991 Sep 3;30(35):8590–7. doi: 10.1021/bi00099a014. [DOI] [PubMed] [Google Scholar]
- Sugo N, Aratani Y, Nagashima Y, Kubota Y, Koyama H. Neonatal lethality with abnormal neurogenesis in mice deficient in DNA polymerase beta. EMBO J. 2000 Mar 15;19(6):1397–404. doi: 10.1093/emboj/19.6.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugo N, Niimi N, Aratani Y, Takiguchi-Hayashi K, Koyama H. p53 Deficiency rescues neuronal apoptosis but not differentiation in DNA polymerase beta-deficient mice. Mol Cell Biol. 2004 Nov;24(21):9470–7. doi: 10.1128/MCB.24.21.9470-9477.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suslov ON, Kukekov VG, Ignatova TN, Steindler DA. Neural stem cell heterogeneity demonstrated by molecular phenotyping of clonal neurospheres. Proc Natl Acad Sci U S A. 2002 Oct 29;99(22):14506–11. doi: 10.1073/pnas.212525299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziabreva I, Perry E, Perry R, Minger SL, Ekonomou A, Przyborski S, Ballard C. Altered neurogenesis in Alzheimer’s disease. J Psychosom Res. 2006 Sep;61(3):311–6. doi: 10.1016/j.jpsychores.2006.07.017. [DOI] [PubMed] [Google Scholar]