

Abstract
The secretase BACE1 is fundamentally involved in the development of cerebral amyloid pathology in Alzheimer's disease (AD). It has not been studied so far to what extent BACE1 activity in cerebrospinal fluid (CSF) mirrors in vivo amyloid load in AD. We explored associations between CSF BACE1 activity and fibrillar amyloid pathology as measured by carbon-11-labelled Pittsburgh Compound B positron emission tomography ([11C]PIB PET). [11C]PIB and CSF studies were performed in 31 patients with AD. Voxel-based linear regression analysis revealed significant associations between CSF BACE1 activity and [11C]PIB tracer uptake in the bilateral parahippocampal region, the thalamus, and the pons. Our study provides evidence for a brain region-specific correlation between CSF BACE1 activity and in-vivo fibrillar amyloid pathology in AD. Associations were found in areas close to the brain ventricles, which may have important implications for the use of BACE1 in CSF as a marker for AD pathology and for antiamyloid treatment monitoring.
1. Introduction
The cerebrospinal fluid (CSF) is in close contact with the extracellular space of the central nervous system; therefore, pathological changes such as those due to neurodegeneration are reflected in CSF and alterations of biomarkers can yield important mechanistic and diagnostic information on disease processes. In Alzheimer's disease (AD), a growing number of CSF biomarkers are studied, some of which mirror important events in the pathogenic cascade. These markers include several indicators of changes related to the amyloid cascade such as amyloid β 42 (Aβ42), total tau and phosphorylated tau [1]. The amyloid hypothesis of AD argues a cerebral accumulation of amyloid as primary driver of AD pathogenesis, including amyloid plaque deposition, neurofibrillary tangle formation, synapse loss, and neuronal cell death [2]. The amyloid deposits of insoluble protein material mainly contain high levels of the 40 and 42 amino acid long Aβ, which is produced through the cleavage of a membrane-bound amyloid precursor protein (APP) by the β- and γ-secretases. The cleavage of APP by the main cerebral β-secretase (β-site APP-cleaving enzyme 1, BACE1) [3] occurs at the N-terminus of the Aβ-domain and results in secreted soluble APP β (sAPPβ) and a C-terminal fragment (C99) [4], which subsequently undergoes cleavage by γ-secretase producing Aβ. The crucial role of BACE1 in AD pathogenesis is highlighted by its increased enzyme activity and protein concentration in AD brains [5]. Several previous studies have shown that BACE1 activity can readily be detected in CSF with activity increases in mild cognitive impairment (MCI) and clinically diagnosable AD [6–8]. The relevance of BACE1 in the amyloid cascade is also underscored by its positive association with the CSF products of APP cleavage, including Aβ40 and sAPPβ [9]. Therefore, the value of BACE1 as a clinical biomarker for AD pathology and for anti-amyloid treatment effects is currently under debate. Further in-vivo studies are urgently needed in order to explore the association between CSF BACE1 activity and amyloid pathology in AD; modern imaging techniques might play an important role in this regard since they are able to provide information about clinically suspected cerebral pathology and its spatial distribution.
Recently, the carbon-11-labeled positron emission tomography (PET) tracer Pittsburgh's Compound B ([11C]PIB) became available as an in-vivo imaging tool for cerebral amyloid deposits. Although [11C]PIB binds to several types of cerebral Aβ, including plaque, nonplaque, and vascular deposits, it has only minimal affinity to other protein aggregates such as Lewy bodies or neurofibrillary tangles [10–13]. Studies on patients with AD have demonstrated an increased tracer uptake, even in very mild clinical stages. Also, a progression of uptake with advancing clinical disease severity has been discussed [14, 15]; however, it seems that the progression of tracer uptake reaches a plateau when AD becomes clinically diagnosable [16]. The reported correlations between [11C]PIB tracer uptake and CSF markers related to amyloid pathology such as Aβ42 further underscore the relevance of [11C]PIB PET imaging as an AD biomarker [17–22].
In the present study, we quantitatively analyzed CSF BACE1 activity and [11C]PIB tracer uptake in 31 patients with probable AD, whose clinical diagnosis was supported by an AD typical metabolic pattern in their fluorine-18-labeled fluorodeoxyglucose ([18F]FDG) PET scans. Our main aim was to explore associations between [11C]PIB tracer uptake and BACE1 activity in CSF in order to provide evidence in support of BACE1 activity as an in-vivo biomarker for AD pathology.
2. Methods
2.1. Study Participants
Thirty-one patients with probable AD were recruited from the outpatient memory clinic of the Department of Psychiatry and Psychotherapy at the Technische Universität München. The clinical diagnosis was established by consensus of two experienced clinicians according to the NINCDS-ADRDA criteria. The clinical assessment included patient and proxy interviews, cognitive testing, physical examination, routine blood and CSF sampling, structural MRI, and [18F]FDG as well as [11C]PIB PET imaging of the brain. The psychometric workup was based on the Consortium to Establish a Registry for AD (CERAD) neuropsychological battery, which includes the Mini-Mental-State Examination (MMSE). Patients with mild to moderate dementia, who had a typical metabolic pattern for AD on [18F]FDG PET scans [23] were included in order to increase diagnostic accuracy. Patients were excluded from the study if they met criteria for other neurological or psychiatric disorders including Parkinson's disease, dementia with Lewy bodies, frontotemporal dementia, progressive supranuclear palsy, corticobasal degeneration, normal pressure hydrocephalus, depression, or alcohol dependence. All consecutive patients meeting the above in- and exclusion criteria were enrolled; [11C]PIB PET scans and BACE1 activity values were not used for diagnostic purposes. Written informed consent was obtained from all participants according to the 1975's Helsinki Declaration, and the study protocol was approved by the ethics committee of the Medical Faculty at the Technische Universität München.
2.2. Blood and CSF Assays
Serum and EDTA plasma samples for each subject were obtained by venous puncture for diagnostic purposes and genotyping. DNA was extracted from blood using standard procedures, and the apolipoprotein E (APOE) genotype was determined using polymerase chain reaction and restriction enzyme digestion according to an established standard protocol [24]. Patients were categorized as either APOE ε4 allele carriers or noncarriers for the present study. CSF (5–8 mL) was collected in sterile polypropylene tubes, using atraumatic cannulas placed in the L3/L4 or L4/L5 intervertebral space, and gently mixed. In the native CSF, determination of routine chemical parameters was performed. Subsequently, the CSF was centrifuged for 10 min at 4000 g, and aliquots of the remaining CSF supernatants were immediately frozen at −80°C for later determination of AD parameters. Tau and Aβ42 concentrations were measured in duplicate using enzyme-linked immunosorbent assay (Innotest Innogenetics, Zwijndrecht, Belgium) as described previously in detail [25].
BACE1 activity was measured using a time-resolved fluorescence activity assay based on SignalClimb technology (TruePoint Perkin Elmer, Turku, Finland) according to optimized manufacturer's instructions [6]. The synthetic TruePoint BACE1 substrate is a ten amino acid long peptide with a fluorescent europium (EU) chelate coupled to one end and a quencher of europium fluorescence (QSY7) coupled to the other end via lysine. The hydrolysis of the substrate's protein sequence CEVNLDAEFK by BACE1 results in a fluorescence signal proportional to the activity of BACE1. The fluorescence signal was measured at 37°C in a microplate reader using time-resolved fluorescence (FLUOstar Omega, BMG Labtech, Offenburg, Germany; excitation wavelength: 320 nm, emission wavelength: 615 nm) in black 96-well plates (Perkin Elmer, Turku, Finland) at a final volume of 27 μL, including 10 μL CSF, 2 μL DMSO, and 15 μL BACE1 substrate (0.80 nmol/mL). The continuous measurement of BACE1 was started immediately after adding the CSF sample; BACE1 activity was defined as the maximal activity within the first 30 min. Each sample was measured at least four times in order to verify reproducibility.
2.3. Cerebral Imaging and Image Preprocessing
Structural MRI, [18F]FDG PET, and [11C]PIB PET scans of the brain were acquired according to published standard procedures [14] within four weeks after the initial clinical assessment. SPM8 software (Wellcome Functional Imaging Laboratory, London, UK) based on Matlab v7 (The Mathworks Inc, Natick, Mass USA) was used to preprocess and analyze the [11C]PIB PET data. For spatial transformation of [11C]PIB data, the individual standardized uptake value images (40–70 min p.i.) were coregistered to the corresponding high-resolution MRI scans and then normalized to the Montreal Neurological Institute space (MNI; bic.mni.mcgill.ca) with the warping parameters of the individual's MRI to obtain images comparable between patients. In order to adjust for interindividual differences in tracer uptake, a relative measure of [11C]PIB was obtained by calculating a cerebral to cerebellar vermis ratio as demonstrated previously [14]; briefly, the uptake in all pixels of the subjects' [11C]PIB scans was divided by the individual mean value in the cerebellum. The individual normalized images were then smoothed with a Gaussian kernel of 10 × 10 × 10 mm.
2.4. Statistical Analysis
Patient characteristics were analyzed in the Predictive Analytics Software package (PASW) v18 (The SPSS Inc., Chicago, Ill, USA). To explore the correlation between CSF BACE1 activity and [11C]PIB tracer uptake, a voxel-based linear regression analysis was performed within SPM8 with tracer uptake as the dependent variable and CSF BACE1 activity as the independent variable, adjusting for factors known to impact on [11C]PIB uptake and amyloid pathology including APOE genotype [14, 26], age [27], gender [28], and interval between PET scan and CSF sampling [14]. A significance threshold of P < 0.001 uncorrected for multiple comparisons was applied as in several previous studies using voxel-based multiple linear regression approaches [26].
3. Results
Patient characteristics are shown in Table 1. The average time interval between [11C]PIB PET scan and lumbar puncture was 29 ± 41 days. Men and carriers of an APOEε4 allele were slightly overrepresented in the sample. Visual inspection of the [18F]FDG PET scans demonstrated a typical pattern for AD in all patients, that is, a bilateral affection of temporal and/or parietal and/or posterior cingulate cortex [23]. The visual analysis of the [11C]PIB scans showed an AD-typical tracer uptake as previously described in all patients, including frontal and temporoparietal cortex, precuneus, posterior cingulate gyrus, and striatum, and less retention in sensorimotor, occipital, and cerebellar cortex [26]. No patient showed a [11C]PIB-negative scan. The CSF concentrations of tau and Aβ42 were in the expected ranges for this type of sample [25].
Table 1.
Description of the study sample.
| N | 31 |
|---|---|
| Male : female | 19 : 12 |
| Age, years* | 65.8 (7.86) |
| MMSE* | 23.9 (4.41) |
| CSF BACE1 activity, FU/μl* | 7793 (2003) |
| APOE ε4+ : ε4− | 17 : 14 |
| CSF tau (ng/L) | 635.58 (310.39) |
| CSF Aβ42 (ng/L) | 523.74 (116.79) |
*mean (SD), FU: fluorescence units.
The regression analysis with [11C]PIB tracer uptake as the dependent variable and CSF BACE1 activity as the independent variable, adjusted for covariates as described above, revealed significant positive associations in the bilateral parahippocampal region, the right thalamus, and the left pons (global maximum at MNI coordinates x/y/z 18/−24/−9, right parahippocampal gyrus, Brodmann area 35, P < 0.001 uncorrected). No other brain region showed a significant correlation between BACE1 activity in CSF and [11C]PIB tracer uptake (Table 2, Figures 1 and 2).
Table 2.
Peak correlations between [11C]PIB tracer uptake and CSF BACE1 activity.
| Region | x | y | z | Z-score | Cluster |
|---|---|---|---|---|---|
| Parahippocampal gyrus (r), BA 35 | 18 | −24 | −9 | 3.91 | 733 |
| Parahippocampal gyrus (l), BA 28 | −24 | −24 | −6 | 3.58 | 372 |
| Parahippocampal gyrus (l), BA 35 | −14 | −13 | −28 | 3.51 | 235 |
| Thalamus (r) | 2 | −19 | 3 | 3.02 | 733 |
| Pons (r) | 8 | −20 | −38 | 3.12 | 495 |
Significant brain clusters were labeled in Talairach daemon software (talairach.org/Daemon.html) after conversion of MNI (bic.mni.mcgill.ca) to Talairach's coordinates using the Matlab function mni2tal (http://imaging.mrc-cbu.cam.ac.uk/imaging/MniTalairach); brain regions are indicated by Talairach's coordinates; cluster: extent of contiguous voxels within the cluster; r: right, l: left; BA: Brodmann's area.
Figure 1.
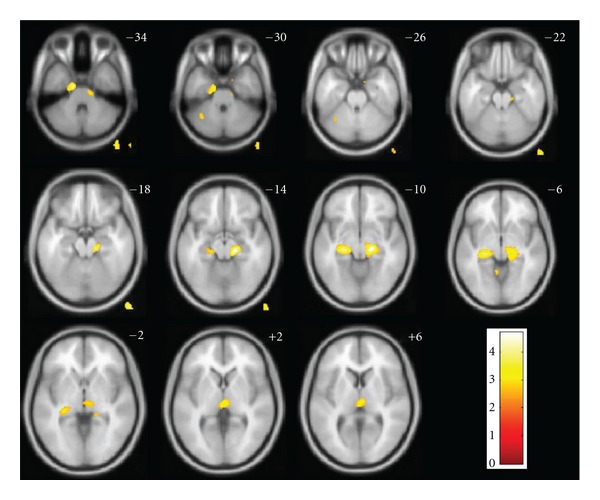
SPM8 maps of voxel-based correlations between [11C]PIB tracer uptake and CSF BACE1 activity. Anatomical localization as projected on axial sections of a normal T1-weighted MRI, spatially normalized into the MNI template at the given z coordinates in Talairach's space (P < 0.01 uncorrected for display purposes; voxels outside of the brain and in the cerebellum are artifacts due to this threshold); color bars represent Z-scores; images are displayed in neurological orientation (right side is right).
Figure 2.
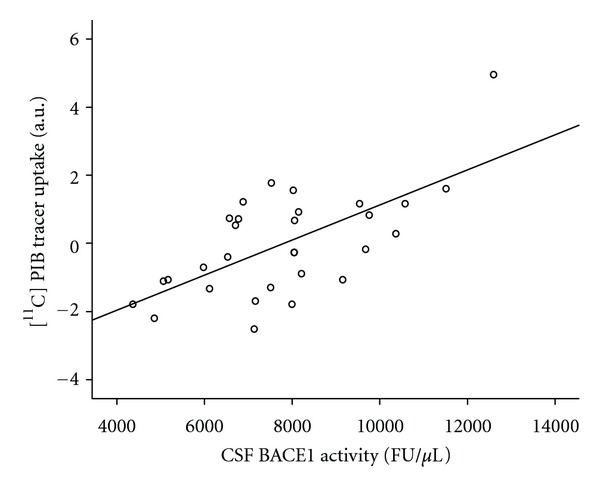
Linear regression analysis of fitted and adjusted [11C]PIB uptake and CSF BACE1 activity at the localization of the most significant cluster (Talairach's coordinates x/y/z 18/−24/−9, right parahippocampal gyrus, Brodmann's area 35), FU: fluorescence units.
4. Discussion
APP cleavage by BACE1 is the rate-limiting step in the production of Aβ and the activity of BACE1 in CSF can readily be measured [29]; thus, CSF-based detection of BACE1 activity might be valuable in the early detection, differential diagnosis, and anti-amyloid treatment monitoring in AD. Our results not only confirm findings of previous studies that suggest an association between BACE1 activity and in-vivo amyloid pathology [5, 9] but also extend these earlier investigations by presenting evidence for a region-specific pattern of this relation, reporting a significant positive correlation between BACE1 activity in CSF and [11C]PIB tracer uptake in the parahippocampal region, the thalamus, and the pons.
Increased BACE1 expression, concentration, and activity in and around senile plaques have been reported in several studies in cognitively healthy elderly individuals and patients with AD [5, 30, 31]. BACE1 activity increases, and correlations with amyloid pathology were most consistently found not only in AD-vulnerable brain regions such as the temporal cortex, the hippocampal region, as well as the prefrontal cortex [32] but also in some less vulnerable structures in the diencephalon and brain stem including the thalamus and the pons [33, 34]. Furthermore, a coexpression of BACE1 and APP, which is a prerequisite for Aβ production, in the hippocampal region has been reported in APP/BACE1 transgenic mice [35–37]. The findings of these postmortem and animal model studies are corroborated by our in-vivo findings; however, associations between BACE1 activity in lumbar CSF and amyloid load in [11C]PIB PET scans were not found in some of the previously reported brain regions including the prefrontal cortex [31].
In line with our recent finding that the correlation between CSF Aβ42 and [11C]PIB tracer uptake is strongest in brain regions close to the ventricles [20], our present results may indicate that BACE1 activity measured in lumbar CSF primarily reflects amyloid pathology in structures bordering the ventricular system. This observation can possibly be explained by the fact that CSF is drained from the ventricles into the spinal space before a small part of it reaches cortical areas such as prefrontal cortex [38] as well as the fact that proteins from ventricle remote regions are drained via other ways including the perivascular interstitial fluid channels following the cortical arteries [39]. Therefore, BACE1 activity measured in lumbar CSF may predominantly correspond to the enzymatic activity located in specific brain structures. As an alternative explanation, brain regional BACE1 activity differences might actually exist in patients with clinically diagnosable AD, and the plateau phase of [11C]PIB uptake increase in this disease stage might further restrict the association to certain brain areas.
Some limitations of our study have to be mentioned. The patients were recruited from a specialized university center and may, therefore, not truly represent the whole population with AD. Furthermore, a modest number of patients were included and replication in larger samples would be helpful. An additional concern is the lack of pathologic confirmation of AD. However, the validity of present clinical diagnostic criteria compared with autopsy diagnoses has been reported to be very good in study cohorts recruited at specialized centers [40]; moreover, only patients with a typical metabolic pattern for AD on [18F]FDG PET scans were included. Finally, although our cross-sectional study demonstrated a correlation between BACE1 activity and in vivo amyloid load, longitudinal studies are necessary to determine the stability or variability of this association over time.
To conclude, our results point to an association between BACE1 activity in lumbar CSF and in-vivo fibrillar amyloid pathology in regions adjacent to the brain ventricular system. Thus, our data implicate that CSF BACE1 activity reflects AD pathology in particular brain regions, whereas [11C]PIB tracer uptake may map the entire cerebral amyloid load. This observation suggests that BACE1 activity in CSF can only be considered a peripheral measure for AD pathology in relation to certain brain regions. Our results support the notion that CSF analysis may represent an important tool for the detection of ongoing intracerebral pathology, but complementation with imaging procedures may allow the assessment of actual presence, quantity, and regional distribution of pathology in the brain. It is also important to note that the decision for or against CSF and imaging biomarkers in clinical routine and treatment trials will also depend on the willingness among clinicians to perform and the willingness of patients to undergo lumbar puncture on the one hand and access to cyclotrons and PET scanners as well as financial considerations on the other hand. Our findings encourage further studies in order to explore the value of BACE1 activity in CSF as a reliable, affordable, and easily accessible AD biomarker. However, our results also narrow the possible applications of CSF BACE1 activity due to its selective correlation with brain regional amyloid pathology.
Acknowledgments
The study was supported by the German Research Foundation (Deutsche Forschungsgemeinschaft, Grant numbers HE 4560/1-2, DR 445/3-1, DR 445/4-1), the Bund der Freunde der Technischen Universität München e.V. (Grant number 22592), and the Kommission für Klinische Forschung of the Klinikum rechts der Isar München (grant numbers B06-09, 8764151). The sponsors did not have any role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; and preparation, review, or approval of the paper. The authors report no conflicts of interest. The authors wish to thank Dorottya Ruisz for proofreading. T. Grimmer and P. Alexopoulos are authors who contributed equally.
References
- 1.Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nature Reviews Neurology. 2010;6(3):131–144. doi: 10.1038/nrneurol.2010.4. [DOI] [PubMed] [Google Scholar]
- 2.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 3.Cai H, Wang Y, McCarthy D, et al. BACE1 is the major β-secretase for generation of Aβ peptides by neurons. Nature Neuroscience. 2001;4(3):233–234. doi: 10.1038/85064. [DOI] [PubMed] [Google Scholar]
- 4.Vassar R, Bennett BD, Babu-Khan S, et al. β-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286(5440):735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- 5.Yang LB, Lindholm K, Yan R, et al. Elevated β-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nature Medicine. 2003;9(1):3–4. doi: 10.1038/nm0103-3. [DOI] [PubMed] [Google Scholar]
- 6.Holsinger RMD, Lee JS, Boyd A, Masters CL, Collins SJ. CSF BACE1 activity is increased in CJD and Alzheimer disease versus [corrected] other dementias. Neurology. 2006;67(4):710–712. doi: 10.1212/01.wnl.0000229925.52203.4c. [DOI] [PubMed] [Google Scholar]
- 7.Zhong Z, Ewers M, Teipel S, et al. Levels of β-secretase (BACE1) in cerebrospinal fluid as a predictor of risk in mild cognitive impairment. Archives of General Psychiatry. 2007;64(6):718–726. doi: 10.1001/archpsyc.64.6.718. [DOI] [PubMed] [Google Scholar]
- 8.Ewers M, Zhong Z, Bürger K, et al. Increased CSF-BACE 1 activity is associated with ApoE-ε4 genotype in subjects with mild cognitive impairment and Alzheimer’s disease. Brain. 2008;131(5):1252–1258. doi: 10.1093/brain/awn034. [DOI] [PubMed] [Google Scholar]
- 9.Zetterberg H, Andreasson U, Hansson O, et al. Elevated cerebrospinal fluid BACE1 activity in incipient Alzheimer disease. Archives of Neurology. 2008;65(8):1102–1107. doi: 10.1001/archneur.65.8.1102. [DOI] [PubMed] [Google Scholar]
- 10.Fodero-Tavoletti MT, Smith DP, McLean CA, et al. In vitro characterization of Pittsburgh compound-B binding to Lewy bodies. Journal of Neuroscience. 2007;27(39):10365–10371. doi: 10.1523/JNEUROSCI.0630-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lockhart A, Lamb JR, Osredkar T, et al. PIB is a non-specific imaging marker of amyloid-beta (Aβ) peptide-related cerebral amyloidosis. Brain. 2007;130(10):2607–2615. doi: 10.1093/brain/awm191. [DOI] [PubMed] [Google Scholar]
- 12.Ikonomovic MD, Klunk WE, Abrahamson EE, et al. Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer’s disease. Brain. 2008;131(6):1630–1645. doi: 10.1093/brain/awn016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ye L, Velasco A, Fraser G, et al. In vitro high affinity α-synuclein binding sites for the amyloid imaging agent PIB are not matched by binding to Lewy bodies in postmortem human brain. Journal of Neurochemistry. 2008;105(4):1428–1437. doi: 10.1111/j.1471-4159.2008.05245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grimmer T, Tholen S, Yousefi BH, et al. Progression of cerebral amyloid load is associated with the apolipoprotein e ε4 genotype in Alzheimer's disease. Biological Psychiatry. 2010;68(10):879–884. doi: 10.1016/j.biopsych.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kadir A, Almkvist O, Forsberg A, et al. Dynamic changes in PET amyloid and FDG imaging at different stages of Alzheimer’s disease. doi: 10.1016/j.neurobiolaging.2010.06.015. Neurobiology of Aging. In press. [DOI] [PubMed] [Google Scholar]
- 16.Engler H, Forsberg A, Almkvist O, et al. Two-year follow-up of amyloid deposition in patients with Alzheimer’s disease. Brain. 2006;129(11):2856–2866. doi: 10.1093/brain/awl178. [DOI] [PubMed] [Google Scholar]
- 17.Forsberg A, Engler H, Almkvist O, et al. PET imaging of amyloid deposition in patients with mild cognitive impairment. Neurobiology of Aging. 2008;29(10):1456–1465. doi: 10.1016/j.neurobiolaging.2007.03.029. [DOI] [PubMed] [Google Scholar]
- 18.Fagan AM, Mintun MA, Mach RH, et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Annals of Neurology. 2006;59(3):512–519. doi: 10.1002/ana.20730. [DOI] [PubMed] [Google Scholar]
- 19.Forsberg A, Almkvist O, Engler H, Wall A, Långström B, Nordberg A. High PIB retention in Alzheimer’s disease is an early event with complex relationship with CSF biomarkers and functional parameters. Current Alzheimer Research. 2010;7(1):56–66. doi: 10.2174/156720510790274446. [DOI] [PubMed] [Google Scholar]
- 20.Grimmer T, Riemenschneider M, Förstl H, et al. Beta amyloid in Alzheimer’s disease: increased deposition in brain is reflected in reduced concentration in cerebrospinal fluid. Biological Psychiatry. 2009;65(11):927–934. doi: 10.1016/j.biopsych.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tolboom N, van der Flier WM, Yaqub M, et al. Relationship of cerebrospinal fluid markers to 11C-PiB and 18F-FDDNP binding. Journal of Nuclear Medicine. 2009;50(9):1464–1470. doi: 10.2967/jnumed.109.064360. [DOI] [PubMed] [Google Scholar]
- 22.Degerman Gunnarsson M, Lindau M, Wall A, et al. Pittsburgh compound-B and Alzheimer’s disease biomarkers in CSF, plasma and urine: an exploratory study. Dementia and Geriatric Cognitive Disorders. 2010;29(3):204–212. doi: 10.1159/000281832. [DOI] [PubMed] [Google Scholar]
- 23.Mosconi L, Tsui WH, Herholz K, et al. Multicenter standardized 18F-FDG PET diagnosis of mild cognitive impairment, Alzheimer’s disease, and other dementias. Journal of Nuclear Medicine. 2008;49(3):390–398. doi: 10.2967/jnumed.107.045385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wenham PR, Price WH, Blandell G. Apolipoprotein E genotyping by one-stage PCR. The Lancet. 1991;337(8750):1158–1159. doi: 10.1016/0140-6736(91)92823-k. [DOI] [PubMed] [Google Scholar]
- 25.Hulstaert F, Blennow K, Ivanoiu A, et al. Improved discrimination of AD patients using β-amyloid(1-42) and tau levels in CSF. Neurology. 1999;52(8):1555–1562. doi: 10.1212/wnl.52.8.1555. [DOI] [PubMed] [Google Scholar]
- 26.Drzezga A, Grimmer T, Henriksen G, et al. Effect of APOE genotype on amyloid plaque load and gray matter volume in Alzheimer disease. Neurology. 2009;72(17):1487–1494. doi: 10.1212/WNL.0b013e3181a2e8d0. [DOI] [PubMed] [Google Scholar]
- 27.Rowe CC, Ellis KA, Rimajova M, et al. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiology of Aging. 2010;31(8):1275–1283. doi: 10.1016/j.neurobiolaging.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 28.Corder EH, Ghebremedhin E, Taylor MG, Thal DR, Ohm TG, Braak H. The biphasic relationship between regional brain senile plaque and neurofibrillary tangle distributions: modification by age, sex, and APOE polymorphism. Annals of the New York Academy of Sciences. 2004;1019:24–28. doi: 10.1196/annals.1297.005. [DOI] [PubMed] [Google Scholar]
- 29.Vassar R. BACE1: the β-secreiase enzyme in Alzheimer’s disease. Journal of Molecular Neuroscience. 2004;23(1-2):105–113. doi: 10.1385/JMN:23:1-2:105. [DOI] [PubMed] [Google Scholar]
- 30.Sun A, Koelsch G, Tang J, Bing G. Localization of β-secretase memapsin 2 in the brain of Alzheimer’s patients and normal aged controls. Experimental Neurology. 2002;175(1):10–22. doi: 10.1006/exnr.2002.7875. [DOI] [PubMed] [Google Scholar]
- 31.Leuba G, Wernli G, Vernay A, Kraftsik R, Mohajeri MH, Saini KD. Neuronal and nonneuronal quantitative BACE immunocytochemical expression in the entorhinohippocampal and frontal regions in Alzheimer’s disease. Dementia and Geriatric Cognitive Disorders. 2005;19(4):171–183. doi: 10.1159/000083496. [DOI] [PubMed] [Google Scholar]
- 32.Fukumoto H, Cheung BS, Hyman BT, Irizarry MC. β-secretase protein and activity are increased in the neocortex in Alzheimer disease. Archives of Neurology. 2002;59(9):1381–1389. doi: 10.1001/archneur.59.9.1381. [DOI] [PubMed] [Google Scholar]
- 33.Schmechel A, Strauss M, Schlicksupp A, et al. Human BACE forms dimers and colocalizes with APP. Journal of Biological Chemistry. 2004;279(38):39710–39717. doi: 10.1074/jbc.M402785200. [DOI] [PubMed] [Google Scholar]
- 34.Herzig MC, Paganetti P, Staufenbiel M, Jucker M. BACE1 and mutated presenilin-1 differently modulate Aβ40 and Aβ42 levels and cerebral amyloidosis in APPDutch transgenic mice. Neurodegenerative Diseases. 2007;4(2-3):127–135. doi: 10.1159/000101837. [DOI] [PubMed] [Google Scholar]
- 35.Irizarry MC, Locascio JJ, Hyman BT. β-site APP cleaving enzyme mRNA expression in APP transgenic mice: anatomical overlap with transgene expression and static levels with aging. The American Journal of Pathology. 2001;158(1):173–177. doi: 10.1016/s0002-9440(10)63955-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chiocco MJ, Kulnane LS, Younkin L, Younkin S, Evin G, Lamb BT. Altered amyloid-β metabolism and deposition in genomic-based β-secretaee transgenic mice. Journal of Biological Chemistry. 2004;279(50):52535–52542. doi: 10.1074/jbc.M409680200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chiocco MJ, Lamb BT. Spatial and temporal control of age-related APP processing in genomic-based β-secretase transgenic mice. Neurobiology of Aging. 2007;28(1):75–84. doi: 10.1016/j.neurobiolaging.2005.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brodbelt A, Stoodley M. CSF pathways: a review. British Journal of Neurosurgery. 2007;21(5):510–520. doi: 10.1080/02688690701447420. [DOI] [PubMed] [Google Scholar]
- 39.Zhang ET, Richards HK, Kida S, Weller RO. Directional and compartmentalised drainage of interstitial fluid and cerebrospinal fluid from the rat brain. Acta Neuropathologica. 1992;83(3):233–239. doi: 10.1007/BF00296784. [DOI] [PubMed] [Google Scholar]
- 40.Morris JC, McKeel DW, Fulling K, Torack RM, Berg L. Validation of clinical diagnostic criteria for Alzheimer’s disease. Annals of Neurology. 1988;24(1):17–22. doi: 10.1002/ana.410240105. [DOI] [PubMed] [Google Scholar]