

Abstract
Δ9-Tetrahydrocannabinol (THC) and cannabidiol (CBD) are the most prevalent biologically active constituents of Cannabis sativa. THC is the prototypic cannabinoid CB1 receptor agonist and is psychoactive and analgesic. CBD is also analgesic, but it is not a CB1 receptor agonist. Low voltage-activated T-type calcium channels, encoded by the CaV3 gene family, regulate the excitability of many cells, including neurons involved in nociceptive processing. We examined the effects of THC and CBD on human CaV3 channels stably expressed in human embryonic kidney 293 cells and T-type channels in mouse sensory neurons using whole-cell, patch clamp recordings. At moderately hyperpolarized potentials, THC and CBD inhibited peak CaV3.1 and CaV3.2 currents with IC50 values of ∼1 μm but were less potent on CaV3.3 channels. THC and CBD inhibited sensory neuron T-type channels by about 45% at 1 μm. However, in recordings made from a holding potential of -70 mV, 100 nm THC or CBD inhibited more than 50% of the peak CaV3.1 current. THC and CBD produced a significant hyperpolarizing shift in the steady state inactivation potentials for each of the CaV3 channels, which accounts for inhibition of channel currents. Additionally, THC caused a modest hyperpolarizing shift in the activation of CaV3.1 and CaV3.2. THC but not CBD slowed CaV3.1 and CaV3.2 deactivation and inactivation kinetics. Thus, THC and CBD inhibit CaV3 channels at pharmacologically relevant concentrations. However, THC, but not CBD, may also increase the amount of calcium entry following T-type channel activation by stabilizing open states of the channel.
Cannabis sativa has a long history of medicinal and social use (1). It is taken regularly by ∼5-8% of the adults in developed countries (2, 3) and by up to 20% of those suffering neurological conditions such as multiple sclerosis, epilepsy, and chronic pain (4-6). Since the isolation of the major psychologically active constituent of C. sativa, Δ9-tetrahydrocannabinol (THC)4 (7), more than 60 other compounds with biological activity have been identified (8). These include cannabidiol (CBD) (9), the most abundant biologically active compound after THC in the plant. The widespread use of cannabis for self-medication and social purposes and the potential of its constituents as new therapeutic agents make it important that the molecular targets for THC and CBD are well defined.
Most of the effects of THC are likely to occur through actions on G protein-coupled CB1 and CB2 cannabinoid receptors (10, 11) but CBD is an inverse agonist (CB2) or weak antagonist (CB1) at these receptors (12). When administered systemically, CB1 agonists cause a classic “tetrad” of behavioral effects in rodents: hypothermia, catalepsy, hypolocomotion, and antinociception (13). However, THC has non-CB receptor-mediated effects in animals including anti-nociceptive effects in the tail-flick assay of thermal nociception in CB1 receptor knock-out mice (14). Potential non-CB1/CB2 receptor sites of THC action (reviewed in Ref. 15) include GPR55 (16), the ionotropic 5-HT3 receptor (17), and the ion channels TRPA1 and TRPV2 (18, 19).
CBD lacks psychotropic activity (20, 21) but has anti-nociceptive (22) and anticonvulsant activity (23) and disrupts sleep (24); these effects are mediated in the central nervous system. The degree to which CBD action at CB receptors mediates its in vivo effects remains to be established. Other molecular effects of CBD are reviewed in Ref. 15 and include antagonism of GPR55 (16) and the putative abnormal cannabidiol receptor (25) and weak agonist activity at TRPV1 (21).
T-type Ca2+ channels are a family of voltage-gated Ca2+ channels with distinctive biophysical characteristics and widespread expression in neuronal and other tissue (26). Most notably, T-type channels open at membrane potentials significantly more negative than high voltage-activated N-, P/Q-, and R-type channels and inactivate relatively rapidly compared with L-, N-, and P/Q-type channels. Because T-type channels open at potentials between the resting membrane potential and the threshold potential for action potential firing, these channels are involved in a wide variety of physiological processes, including low-threshold calcium spiking, cardiac pacemaker activity, and modulation of neuronal excitability (26). Interestingly, important roles for T-type calcium channels in the regulation of nociception, epilepsy, and sleep have been proposed (27-32).
Three genes encode the pore forming α-subunits of the T-type calcium channels, CaV3.1, CaV3.2, and CaV3.3 (formerly α1G, α1H, α1I) (26). These channels are acutely inhibited by the endogenous cannabinoid N-arachidonoyl ethanolamine (anandamide) through a non-CB receptor-mediated mechanism (33). However, previous studies have reported conflicting data about the effectiveness of THC as an inhibitor of T-type ICa (33, 34). In NG-108 neuroblastoma cells, a high concentration of THC (30 μm) strongly inhibits the T-type ICa (34), but THC (10 μm) was reported not to affect recombinant CaV3.2 channels expressed in human embryonic kidney (HEK293) cells (33). In this study we have examined the effects of THC and CBD on the human CaV3 channel subtypes expressed in HEK293 cells. Both THC and CBD inhibited all three subtypes; but in addition, THC had unique and complex effects on CaV3.1 and CaV3.2 currents. The actions of THC and CBD at T-type calcium channels may be responsible for some of the non-CB receptor-mediated biological actions of phytocannabinoids.
EXPERIMENTAL PROCEDURES
Cell Culture and Transfection—HEK293 cells were grown in Dulbecco's modified Eagle's medium supplemented with 100 units of penicillin, 100 μg of streptomycin, and 10% fetal bovine serum or donor bovine serum (Invitrogen). HEK293 cells were stably transfected with plasmids containing cDNA for CaV3.1 (GenBank™ accession number AF190860) (35), CaV3.2 (GenBank™ accession number AF051946 (36), or CaV3.3 (GenBank™ accession number AF393329) (37) using Lipofectamine reagent according to manufacturer's instructions (Invitrogen). The stably transfected cell lines were cultivated in Dulbecco's modified Eagle's medium supplemented with 100 units of penicillin, 100 μg of streptomycin, 10% fetal bovine serum, or donor bovine serum and 1 mg/ml G418 (Invitrogen). Untransfected HEK293 cells did not express detectable ICa.
Isolation of Sensory Neurons—Adult mouse trigeminal ganglion neurons were isolated in a protocol modified from that described previously (38). All procedures were approved by the Royal North Hospital Animal Care and Ethics Committee. Briefly, male C57Bl6 mice, at least 8 weeks old, were anesthetized with isofluorane and decapitated, and the trigeminal ganglia were removed. The ganglia were placed in a modified HEPES-buffered saline (mHBS) containing (in mm): 130 NaCl, 2.5 KCl, 1.8 CaCl2, 10 MgCl2. 10 HEPES, 10 glucose (pH to 7.3 with NaOH, osmolarity = 330 ± 5 mosmol). The ganglia were minced with iridectomy scissors and incubated in mHBS containing 20 units of ml-1 papain for 25 min at 37 °C. The reaction was stopped with mHBS containing 1 mg ml-1 bovine serum albumin and 1 mg ml-1 trypsin inhibitor (type II-O). The tissue was then washed with mHBS, and cells were released by gentle trituration through fire-polished Pasteur pipettes. Cells were plated onto tissue culture, dishes, and used within 8 h of isolation.
Electrophysiology—HEK293 cells expressing CaV3.1, CaV3.2, or CaV3.3 channels were recorded in the whole-cell configuration of the patch clamp method (39) at room temperature. Dishes were perfused with HEPES-buffered saline containing (in mm): 140 NaCl, 2.5 KCl, 2.5 CaCl2, 1 MgCl2. 10 HEPES, 10 glucose (pH to 7.3 with NaOH, osmolarity = 330 ± 5 mosmol). For recording CaV3.1 and CaV3.2 currents, cells were bathed in an external solution containing (in mm): 140 tetraethylammonium chloride, 2.5 CsCl, 10 HEPES, 10 glucose, 1 MgCl2, 5 CaCl2 (pH to 7.3 with CsOH, osmolarity = 330 ± 5 mosmol). To minimize rundown of CaV3.3 currents, 5 mm CaCl2 was replaced by 5 mm BaCl2, and the pipette solution was modified as outlined below. Recordings were made with fire-polished borosilicate glass pipettes with resistance ranging from 2 to 3 megohms when filled with an internal solution containing (in mm): 130 CsCl, 10 HEPES, 2 CaCl2, 10 EGTA, 5 MgATP (pH to 7.3 with CsOH, osmolarity = 285 ± 5 mosmol). For recording of CaV3.3 currents, 10 mm EGTA was replaced by 10 mm BAPTA, and the concentration of MgATP was reduced to 1 mm. A liquid junction potential of -6 mV was corrected for in all reported membrane potentials. Recordings were made with a HEKA EPC 10 amplifier with Patchmaster acquisition software (HEKA Elektronik) or an Axopatch 1D amplifier using pCLAMP software (Molecular Devices, Sunnyvale, CA). Data were sampled at 3-5 kHz, filtered at 2 kHz, and recorded on hard disk for later analysis. Series resistance ranged from 3 to 10 megohms and was compensated by 80% in all experiments. All currents were leak-subtracted using a P/8 protocol. Cells were exposed to drugs via flow pipes positioned ∼200 μm from the cell. Concentration-response curves were generated by fitting the data to a logistic equation in GraphPad Prism 4. Activation curves were generated by fitting the whole-cell conductance data to a Boltzmann sigmoidal function, Y = 1/(1 + e((V0.5 - Vm)/slope)). Inactivation curves were generated by fitting data to a Boltzmann sigmoidal function, Y = 1-1/(1 + e((V0.5 - Vm)/slope)). T-type calcium channels were recorded from sensory neurons using the same internal solution as used for CaV3.1 and CaV3.2 recordings; however, the external tetraethylammonium solution contained 2.5 mm Ca2+ and 1 mm Mg2+. Recordings were made from type 2 trigeminal ganglion neurons, identified as outlined previously (38).
Pharmacological Agents—THC was obtained from Sigma-Aldrich. Two different batches of THC were used and gave similar results. CBD was obtained from Tocris (Bristol, UK) and the National Measurement Institute of Australia. CBD from both sources gave similar results. AM251 was obtained from Tocris. Drugs were kept in concentrated stock solutions in ethanol (85-100 mm) and stored at -20 °C. Daily dilutions were made from these stocks; the final ethanol concentration in all solutions was 0.1%.
Statistics—Data are reported as the mean ± S.E. of at least 6 independent experiments. Statistical significance for comparing the V0.5 values of activation and inactivation was determined using unpaired t tests and comparing values of V0.5 calculated for individual experiments. Changes in the time constant of inactivation and deactivation were assessed with a two-way ANOVA.
RESULTS
Both THC and CBD inhibited CaV3 channels expressed in HEK293 cells. The effects of 1 μm THC and CBD on CaV3.1, CaV3.2, and CaV3.3 channel currents evoked by a step from -86 to -26 mV are illustrated in Fig. 1. CBD (1 μm) inhibited CaV3.1 by an average of 54 ± 1%, CaV3.2 channels by 59 ± 4%, and CaV3.3 channels 12 ± 4%, whereas THC (1 μm) inhibited CaV3.1 channels by an average of 23 ± 4%, CaV3.2 channels by 32 ± 3%, and CaV3.3 channels by 9 ± 1%. A 5-min wash produced a reversal of CBD inhibition of CaV3.1, CaV3.2, and CaV3.3 of 56 ± 5, 58 ± 9, and 63 ± 6%, respectively, and the effects of THC were reversed by 26 ± 3, 21 ± 9, and 29 ± 7%, respectively. The inhibition of CaV3 currents by THC and CBD was concentration-dependent, and currents were completely inhibited by 10-30 μm THC and CBD (Fig. 2, Table 1). The lower potency of THC and CBD on CaV3.3 was not due to the different recording conditions necessary to obtain stable CaV3.3 currents (see “Experimental Procedures”). When recorded using CaV3.3 solutions, THC (1 μm) inhibited CaV3.1 by 25 ± 3% (compared with 23 ± 4% in control), and CBD (1 μm) inhibited CaV3.1 by 53 ± 2% (compared with 54 ± 2% in control).
FIGURE 1.
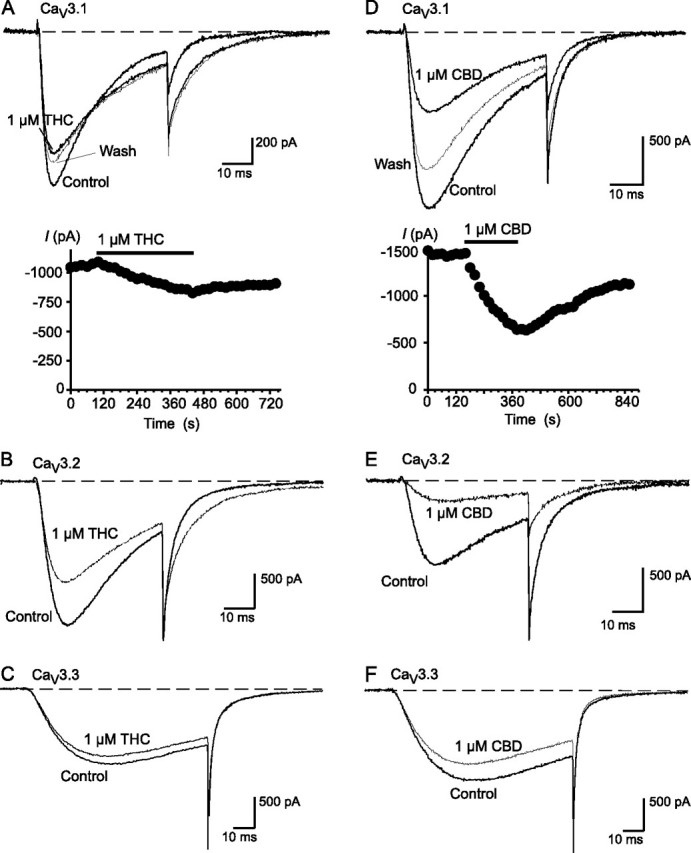
THC and CBD inhibit CaV3 calcium channels. Recordings of recombinant human CaV3 channels stably expressed in HEK293 cells were made as outlined under “Experimental Procedures.” Each trace represents the current elicited by a voltage step from -86 mV to -26 mV under control conditions and in the presence of 1 μm THC (A, CaV3.1; B, CaV3.2; C, CaV3.3) or CBD (D, CaV3.1; E, CaV3.2; F, CaV3.3). An example of the time course of inhibition and degree of reversibility for THC and CBD inhibition of CaV3.1 are illustrated in A and D, respectively. The data are representative of at least six cells for each experiment.
FIGURE 2.
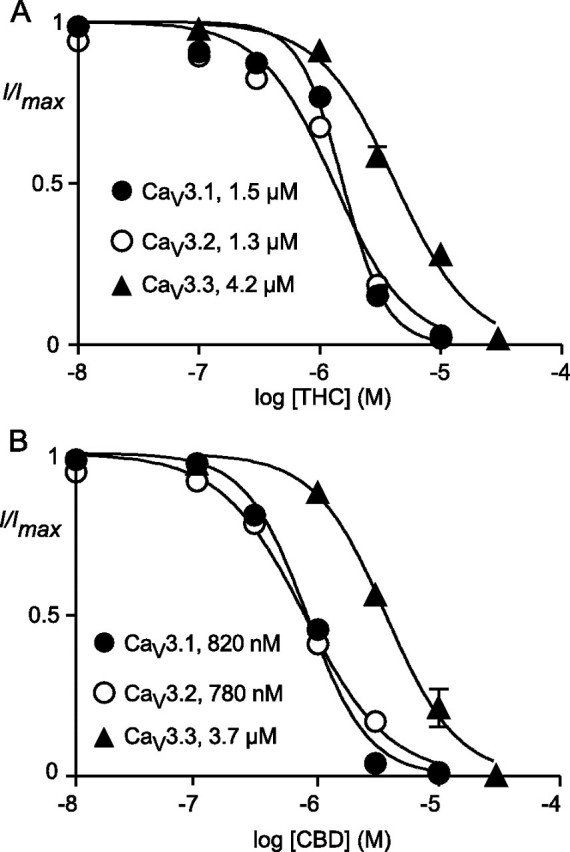
Concentration-response curve for the effect of THC (A) and CBD (B) on CaV3.1, CaV3.2, and CaV3.3 channels. Each point represents the mean ± S.E. of 6 cells and is presented as current remaining in the presence of drug compared with predrug current (I/Imax). One concentration of drug was applied per cell. The EC50 values reported are derived from the pEC50 values reported in Table 1.
TABLE 1.
Inhibitory potency of phytocannbinoids on Cav3 channels
The potency of each drug was determined on the peak current of recombinant Cav3 channels stepped repetitively from −86 to −26 mV.
|
Channel |
pEC50 |
|
|---|---|---|
| Δ9-Tetrahydrocannabinol | Cannabidiol | |
| Cav3.1 | 5.81 ± 0.02 | 6.09 ± 0.01 |
| Cav3.2 | 5.88 ± 0.03 | 6.11 ± 0.02 |
| Cav3.3 | 5.37 ± 0.02 | 5.44 ± 0.03 |
The rate of inhibition of CaV3.1 by THC (3 μm) was dependent on the frequency at which currents were evoked, with inhibition occurring significantly faster when currents were evoked every 1 s than every 10 or 20 s (Fig. 3). The degree to which the channels were inhibited was not different at the different frequencies. If cells were not stepped at all during a 3-min THC application, there was no inhibition of the CaV3.1 current evoked by the first step, although the characteristic effect of THC on channel deactivation (e.g. Fig. 1) was fully developed (see below). In contrast, CBD inhibited CaV3.1 channels at the same rate whether currents were evoked at 1 or 0.05 Hz (Fig. 3). After a 3-min application of CBD during which the cells were not depolarized, the inhibition of the CaV3.1 current evoked by the first step (54 ± 9%) was similar to cells that had been continuously stepped. To confirm that CBD was interacting with a rested state of the channel, the experiment was repeated from a holding potential of -106 mV, where all CaV3.1 channels should be closed rather than inactivated. In this experiment the inhibition of the first step evoked after 3 min in CBD was 41 ± 4% of control.
FIGURE 3.
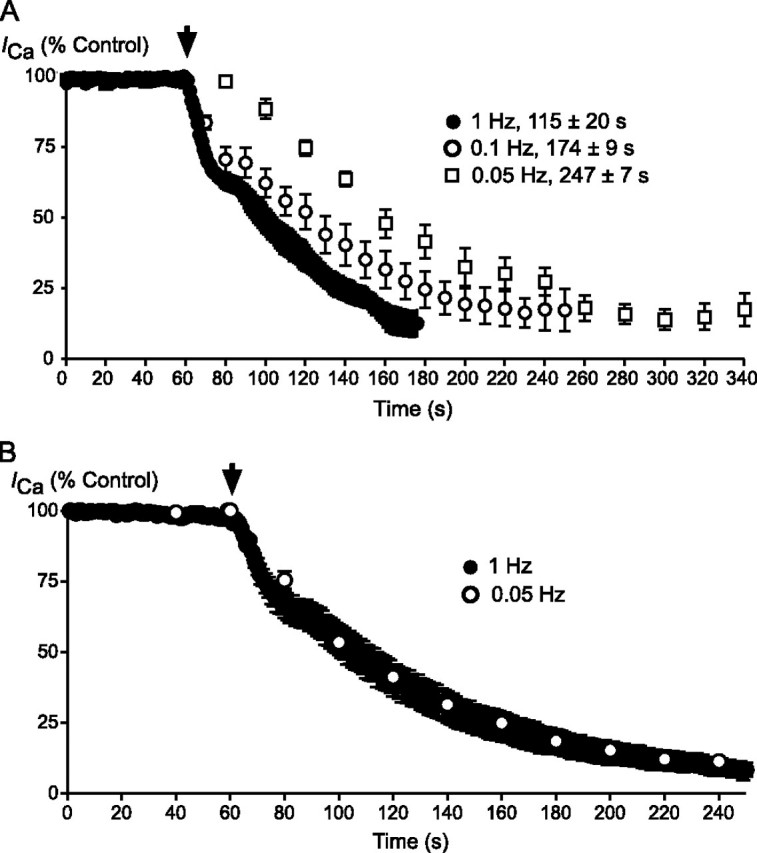
The onset of THC inhibition of CaV3.1 is use-dependent, and that of CBD is not. A, cells expressing CaV3.1 currents were stepped repetitively at 1, 0.1, and 0.05 Hz. 3 μm THC was superfused from the point indicated by the arrowhead. Each point represents the mean ± S.E. of 6 cells. The time to reach maximal inhibition is indicated on the figure; the times were significantly different at each frequency (one-way ANOVA, p < 0.05). B, cells expressing CaV3.1 currents were stepped repetitively at 1 and 0.05 Hz. 3 μm CBD was superfused from the point indicated by the arrowhead. Each point represents the mean ± S.E. of 6 cells; there was no difference in the time taken for CBD inhibition to reach equilibrium.
The effects of THC and CBD were strongly dependent on the resting membrane potential of the cells. When cells were held at -70 mV, CaV3.1 currents elicited by a repeated step to -26 mV at 1 Hz were inhibited 69 ± 3% by THC (100 nm) and 76 ± 3% by CBD (100 nm) (Fig. 4). This compares with inhibition CaV3.1 currents elicited by a step to -26 mV of 8 ± 2% for THC (1 μm) and 35 ± 10% for CBD (1 μm) when cells were held at -126 mV.
FIGURE 4.

THC and CBD inhibition of CaV3.1 is enhanced at less negative holding potentials. Cells expressing CaV3.1 were held at -70mV, a potential at which most channels would be inactivated, and currents were elicited by a step to -26 mV every 1 s. THC (100 nm (A)) and CBD (100 nm (B)) both inhibited CaV3.1 by more than 50% under these conditions. The left-hand panels illustrate the time courses of inhibition, with the data from 6 cells pooled for each drug. The right-hand panels show representative traces for THC and CBD inhibition of the small currents elicited from -70 mV.
Both THC and CBD inhibited the native T-type ICa of mouse trigeminal ganglion sensory neurons (Fig. 5). T-type currents were elicited with a test step from -80 to -40 mV. This test potential was chosen to avoid contamination of the T-type currents by high voltage-activated ICa. THC (1 μm) inhibited the T-type ICa by 42 ± 2%, and the inhibition by CBD (1 μm) was 44 ± 5%. To rule out the involvement of CB1 receptors in the effects of the phytocannabinoids we also tested them in the presence of the CB1 antagonist AM251 (40). AM251 (3 μm) inhibited the current evoked at -40 mV by 20 ± 2%; however, it did not diminish the effects of a subsequent co-application of THC (inhibition in AM251 was 42 ± 2%) or CBD (inhibition was 46 ± 2% in AM251). THC had no effect on the kinetics of channel inactivation from an open state or the kinetics of deactivation (Fig. 5).
FIGURE 5.

THC and CBD inhibit native T-type calcium channels in acutely isolated mouse trigeminal ganglion neurons. Cells were voltage-clamped at -80 mV and T-type current elicited by a step to -40 mV in order to minimize activation of native high voltage-activated channels.A, THC inhibition persisted in the presence of the CB1 antagonist AM251, which itself modestly inhibited the T-type currents. The inhibition by THC was not associated with a change in the kinetics of channel inactivation or deactivation, implying that these cells express predominantly CaV3.3. B, CBD also inhibited native T-type ICa in mouse trigeminal ganglion neurons. The time plots and traces are representative of at least 6 cells for each experiment.
To further describe the mechanisms by which THC and CBD inhibit CaV3 channels, we examined the effects of the drugs on channel activation and steady state inactivation. In activation experiments cells were held at -106 mV and stepped to potentials from -86 mV to +54 mV. In the presence of THC (1 μm), the V0.5 values for CaV3.1 and CaV3.2 were shifted to significantly more negative potentials (Fig. 6, Table 2), whereas the activation of CaV3.3 was unaffected by THC (3 μm). At -106 mV, steady state channel inactivation is minimal for CaV3.1 even in the presence of THC (see below), so the shift in activation produced by THC resulted in an increase in the absolute current amplitude at potentials below about -50 mV (Fig. 6). By contrast, CBD did not affect the activation of CaV3 channels (Fig. 7, Table 2).
FIGURE 6.

THC affects the activation and inactivation of CaV3 channels. A, current-voltage (I-V) relationship showing the activation of CaV3.1 from a holding potential of -106 mV in the absence and presence of 1 μm THC. The peak inward current amplitude is plotted. B, example traces from this experiment illustrating the effect of 1 μm THC at test potentials of -56 and -26 mV; current is enhanced at lower test potentials and inhibited at more depolarized potentials. The effects of THC on activation and steady state inactivation of CaV3 currents are illustrated: C, CaV3.1; D, CaV3.2; E, CaV3.3. Cells were voltage-clamped at -106 mV. For steady state inactivation, cells were voltage-clamped at the test potential for 2 s before currents were evoked by a step to -26 mV. For steady state inactivation, data are presented as conductance normalized to conductance at -26 mV; for activation curves data are normalized to the maximum conductance. The data are fitted with a Boltzmann equation; the effects of THC on activation and inactivation parameters are reported in Table 2. Each data point represents the mean ± S.E. of 6 cells.
TABLE 2.
Effects of phytocannabinoids on the parameters of steady state activation and inaction of Cav3 channels
Cells expressing recombinant Cav3 channels were voltage-clamped at −106 mV and then stepped to potentials above −76 mV (activation) or stepped for 2 s to potentials between −126 and −36 mV before stepping to the test potential of −26 mV. The resulting peak currents were fitted to a Boltzmann equation. Changes in the voltage for half-activation/inactivation (V0.5) of the curves are reported below. “No drug” represents time-dependent changes under our recording conditions. Curves for THC are illustrated in Fig. 4; curves for CBD are illustrated in Fig. 5. *, p < 0.05; **, p < 0.01 from control.
|
Drug |
Cav |
Change in V0.5 |
|
|---|---|---|---|
| Activation | Inactivation | ||
| mV | |||
| 1 μm THC | 3.1 | −7 ± 2** | −8 ± 2** |
| 1 μm THC | 3.2 | −5 ± 2* | −9 ± 2** |
| 3 μm THC | 3.3 | −1 ± 2 | −11 ± 3** |
| 1 μm CBD | 3.1 | −2 ± 2 | −17 ± 7** |
| 1 μm CBD | 3.2 | −4 ± 5 | −10 ± 5** |
| 3 μm CBD | 3.3 | −2 ± 2 | −12 ± 4** |
| No drug | 3.1 | −1 ± 1 | −2 ± 3 |
| No drug | 3.2 | 1 ± 2 | −2 ± 2 |
| No drug | 3.3 | −2 ± 2 | −2 ± 2 |
FIGURE 7.
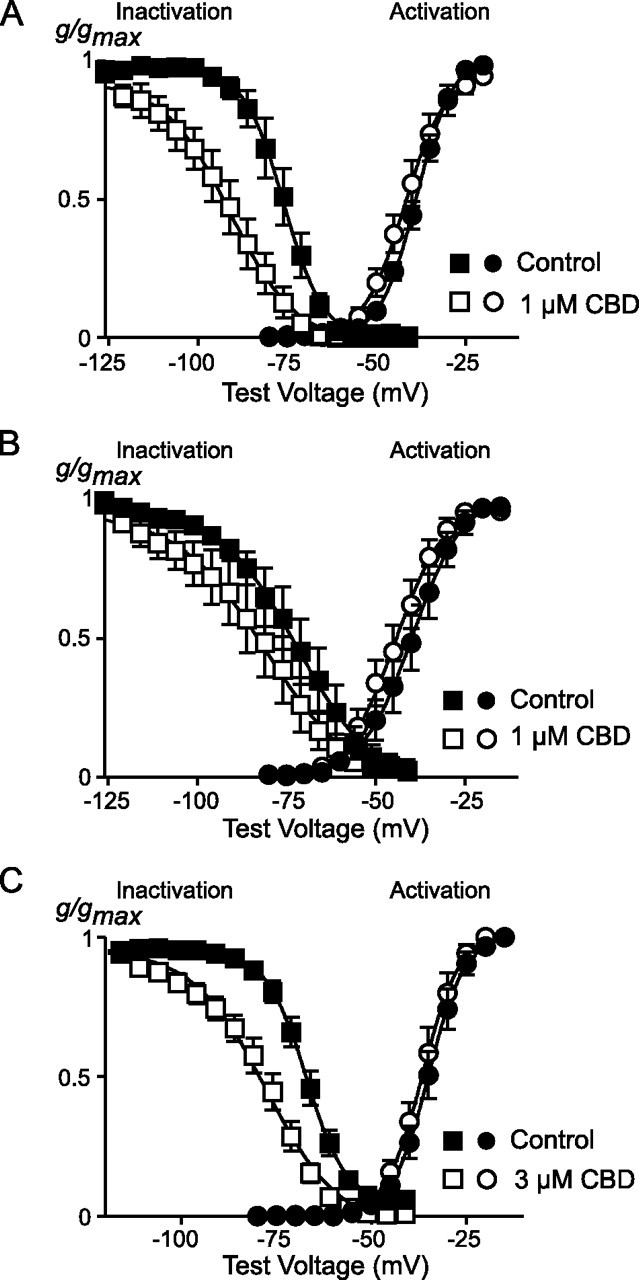
CBD affects the inactivation but not activation of CaV3 channels. The effects of CBD on activation and steady state inactivation of CaV3 currents are illustrated: i, CaV3.1; B, CaV3.2; C, CaV3.3. Cells were voltage-clamped at -106 mV. For steady state inactivation, cells were voltage-clamped at the test potential for 2 s before currents were evoked by a step to -26 mV. For steady state inactivation, data are presented as conductance normalized to conductance at -26 mV; for activation curves data are normalized to the maximum conductance. The data are fitted with a Boltzmann equation; the effects of CBD on activation and inactivation parameters are reported in Table 2. Each data point represents the mean ± S.E. of 6 cells.
The effects of THC and CBD on steady state inactivation were examined by holding cells at -106 mV, where inactivation is absent, and then applying a 2-s prepulse to potentials between -126 and -36 mV before a test step to -26 mV. In the presence of either THC or CBD, the potential at which half the channels were inactivated was shifted to significantly more negative potentials (Figs. 6 and 7, Table 1). An interaction of THC and CBD with inactivated state(s) of the CaV3 channels was further examined by determining the time course of recovery from open state inactivation at -106 mV in the presence and absence of the drugs (illustrated for CaV3.1 in Fig. 8). In these experiments cells were held at -106 mV, stepped to -26 mV for 70 ms (CaV3.1 and CaV3.2) or 350 ms (CaV3.3) to inactivate the channels, and then retested with 10-ms (CaV3.1 and CaV3.2)-or 50-ms (CaV3.3)-long steps at 0, 10, 20, 40, 80, 160, 320, 640, 1280, and 2560 ms after the end of the initial depolarization. THC and CBD significantly slowed the t½ for recovery from open state inactivation for each of the channels (Table 3).
FIGURE 8.
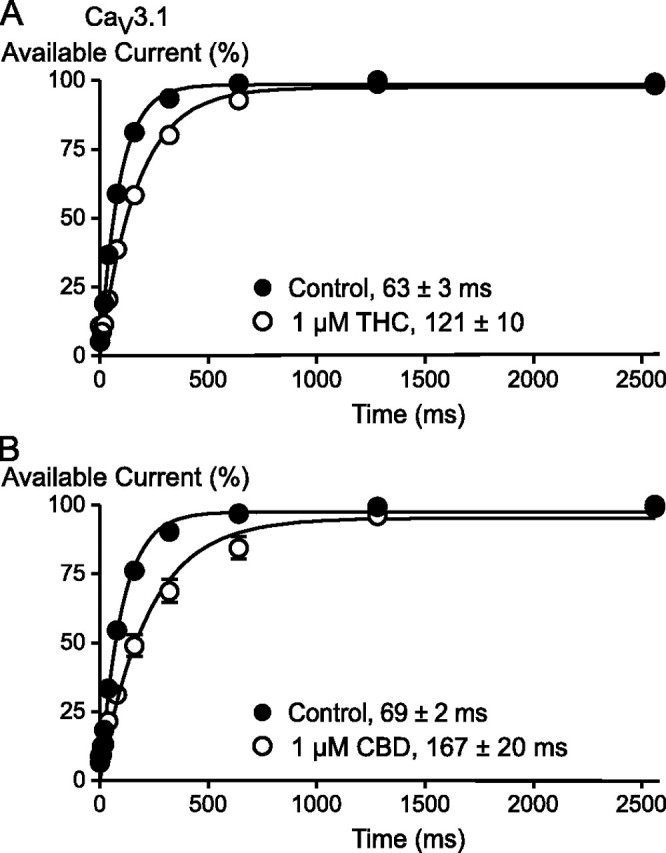
THC and CBD slow the recovery of CaV3.1 channels from inactivation. This effect is illustrated for THC (A) and CBD (B). Cells were voltage-clamped at -86 mV, stepped to -26 mV for 70 ms, and then retested with 10-ms steps to -26 mV at 10, 20, 40, 80, 160, 320, 640, 1280, and 2560 ms later. Each point represents the mean ± S.E. of 6 cells. Data were fitted with a single exponential function, and the half-time for recovery is shown. The half-time was significantly slowed (p < 0.01) by both CBD and THC.
TABLE 3.
Effects of phytocannabinoids on recovery from open channel inactivation of Cav3 channels
Cells expressing recombinant Cav3 channels were voltage-clamped at −86 mV, stepped to −26 mV for 70 ms (Cav3.1 and Cav3.2) or 350 ms (Cav3.3) to inactivate the channels, and then retested with steps to −26 mV at 20, 40, 80, 160, 320, 640, 1280, and 2560 ms after the end of the initial depolarization. Recovery curves for Cav3.1 in the presence of THC and CBD are illustrated in Fig. 6. *, p < 0.05; **, p < 0.001 from control.
|
Drug |
CaV |
Recovery from inactivation |
|
|---|---|---|---|
| Control | In drug | ||
| ms | |||
| 1 μm THC | 3.1 | 63 ± 3 | 121 ± 10** |
| 1 μm THC | 3.2 | 224 ± 10 | 430 ± 7** |
| 3 μm THC | 3.3 | 211 ± 5 | 410 ± 20** |
| 1 μm CBD | 3.1 | 69 ± 2 | 167 ± 20** |
| 1 μm CBD | 3.2 | 230 ± 10 | 384 ± 58** |
| 3 μm CBD | 3.3 | 184 ± 4 | 436 ± 27** |
THC had significant effects on CaV3.1 and CaV3.2 currents that were not shared by CBD, as is clear from the traces illustrated in Fig. 1. Most notably, the deactivation of channel currents following repolarization was dramatically slowed in the presence of THC, and the inactivation of the channel from an open state was also slowed. THC did not affect the deactivation or open state inactivation of CaV3.3. We analyzed the effects of THC on CaV3.1 and CaV3.2 in more detail by examining activation, inactivation from open states, and deactivation of these channels in the presence of THC.
THC (1 μm) did not affect the time to peak of CaV3.1 or CaV3.2 channels at any potential (data not shown). The effect of THC on the time constant of open state channel inactivation was studied using 300-ms-long test steps to potentials between -56 and +39 mV from a holding potential of -106 mV or -86 mV (Fig. 9). THC (1 μm) produced a significant slowing of channel inactivation (ANOVA, p < 0.01) in both channel types, from both holding potentials (Fig. 9), although the effects were more pronounced at the more depolarized test potentials. THC also produced a significant slowing of channel inactivation from an open state in the experiments in which cells were held at -70 mV and stepped to -26 mV (Fig. 4). The time constant for inactivation in control conditions was 11 ± 1 ms; in the presence of THC (100 nm) it was 14 ± 1.3 ms (n = 6, p = 0.03, paired t test). The time constant for activation of the currents under these conditions was unaffected by THC (2.1 ± 0.2 ms in control versus 2.1 ± 0.3 ms in THC).
FIGURE 9.
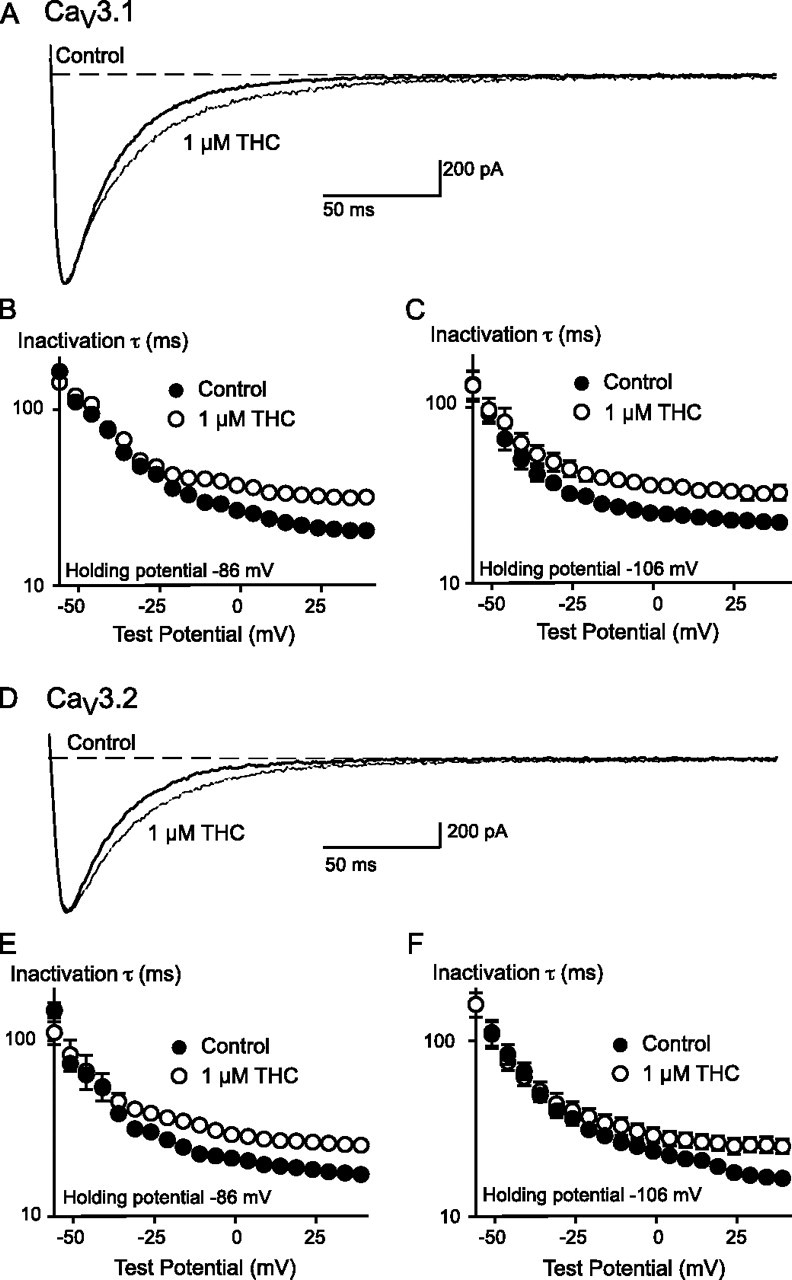
THC slows the inactivation from an open state of both CaV3.1 and CaV3.2. The effect of 1 μm THC on CaV3.1 inactivation is illustrated in A. the cell was voltage-clamped at -106 mV and stepped to 4 mV. The amplitude of the trace in the presence of THC was normalized to the control trace to allow ready comparison of the inactivation kinetics. The time constants of inactivation for CaV3.1 channels at holding potentials of -86 mV (B) and -106 mV (C) are also illustrated. The effect of 1 μm THC on CaV3.2 inactivation is illustrated in B. The cell was voltage-clamped at -106 mV and stepped to 4 mV. The amplitude of the trace in the presence of THC was normalized to the control trace to allow a ready comparison of the inactivation kinetics. The time constants of inactivation for CaV3.2 channels at holding potentials of -86 mV (C) and -106 mV (D) are also illustrated. Each point represents the mean ± S.E. of 6 cells. In the presence of THC the time constants for inactivation were significantly different from control for both channels at either holding potential (ANOVA, p < 0.05).
THC had a significant effect on the deactivation of CaV3.1 and CaV3.2 channels. Interestingly, unlike the THC inhibition of the peak current amplitude, the effect of THC in slowing deactivation of the tail currents was not use-dependent and appeared very rapidly upon THC superfusion (Fig. 10). When CaV3.1 currents were evoked every second, the increase in the time constant of channel decay produced by THC (3 μm) was close to maximal after about 5 s, a time when the inhibition of the peak current was only about 10%. The most striking illustration of the independence of the effects of THC on tail currents from those on peak current was seen when cells were depolarized every 20 s; the first step after THC application in this condition showed a maximal prolongation of the tail current but no effect on the peak amplitude of the current (Fig. 10). The pEC50 for THC slowing of the tail current decay for CaV3.1 was 5.97 ± 0.23 (∼1 μm), which was similar to the effect of THC on peak current amplitude. The effect of THC on the deactivation kinetics of CaV3.1 and CaV3.2 channels at a wide range of potentials is illustrated in Fig. 11. In the presence of THC, deactivation was significantly slowed (ANOVA, p < 0.05) for both CaV3.1 and CaV3.2. THC slowed the deactivation kinetics regardless of the test potential used to open the CaV3 channels (data not shown).
FIGURE 10.
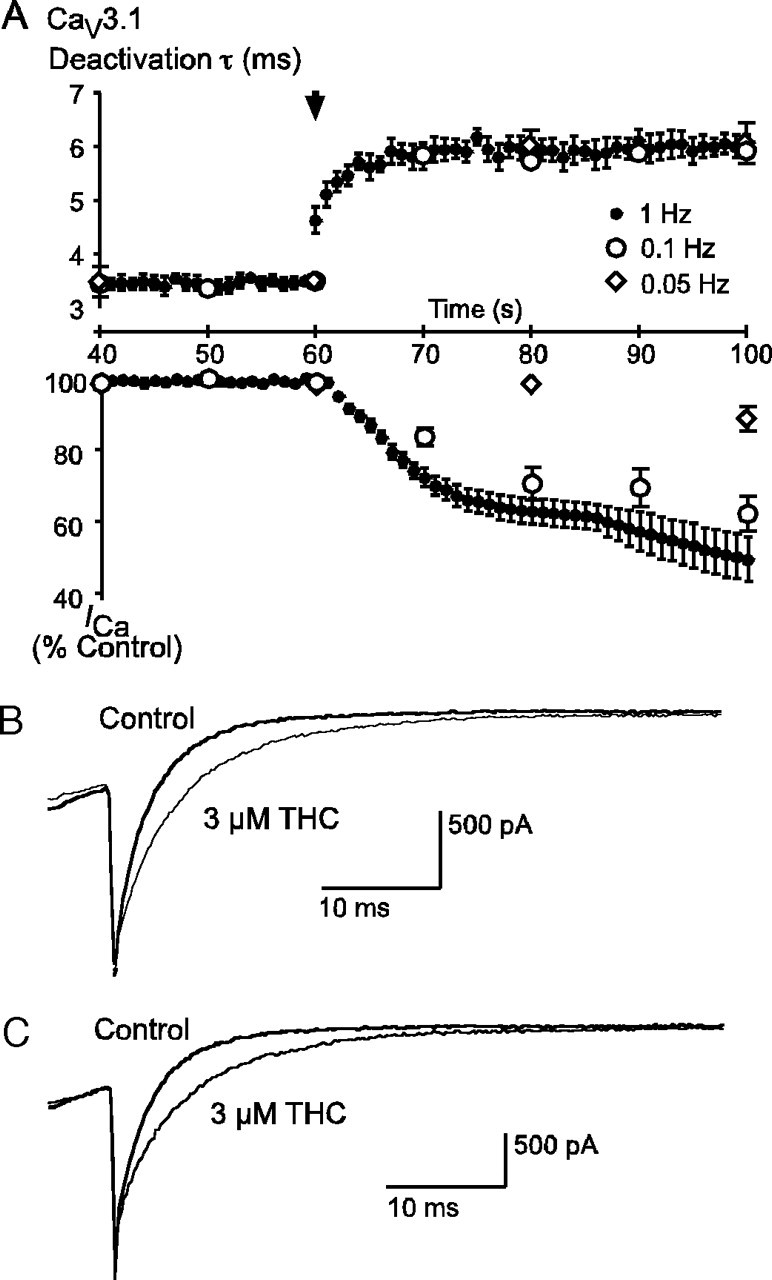
THC slows the deactivation of CaV3.1. A, the time course of 3 μm THC slowing of channel deactivation is compared with the THC inhibition of the peak current. Cells were stepped repetitively from -86 to -26 mV at 1, 0.1, and 0.05 Hz. The beginning of THC perfusion is indicated by the arrow-head. The time constant for channel deactivation following repolarization is compared with peak current amplitude, and the onset of the effect on channel deactivation is uncoupled from inhibition of the peak current and happens much more quickly. Each point represents the mean ± S.E. of 6 cells. Example traces illustrate the identical effect of 3 μm THC on tail currents at 5 s after application during 1-Hz stimulation (B) and 20 s after application during 0.05-Hz stimulation (C).
FIGURE 11.

THC slows deactivation of CaV3 channels at all test potentials. The effect of 1 μm THC on CaV3.1 deactivation is illustrated in A. The cell was voltage-clamped at -106 mV and stepped to +34 mV. The trace in the presence of THC was normalized to amplitude of the tail current of the control trace to allow ready comparison of the inactivation kinetics. B, the constant of deactivation of CaV3.1 channels at various potentials from a holding potential of -106 mV. The effect of 1 μm THC on CaV3.2 deactivation is illustrated in C. The cell was voltage-clamped at -106 mV and stepped to +34 mV. The trace in the presence of THC was normalized to the amplitude of the tail current of the control trace to allow ready comparison of the inactivation kinetics. D, the constant of deactivation of CaV3.2 channels at various potentials from a holding potential of -106 mV. Each point represents the mean ± S.E. of 6 cells. In the presence of THC, the time constants for deactivation were significantly different from the control for both channels (ANOVA, p < 0.05).
To assess the involvement of G proteins in THC and CBD inhibition of CaV3.1 in HEK293 cells, the effects of the drugs were reassessed with either 0.3 mm GTP or 1.2 mm GDPβS, a blocker of G protein activation, included in the internal pipette solution. The inhibition of CaV3.1 by both THC (1 μm, 24 ± 2% with GDPβS compared with 23 ± 2% with GTP) and CBD (55 ± 4% with GDPβS compared with 54 ± 3% with GTP) was unchanged by inhibiting G protein activation. In parallel experiments we reported previously that 1.2 mm GDPβS largely blocked μ-opioid receptor-mediated inhibition of high voltage-activated ICa in sensory neurons (41).
DISCUSSION
This study investigated the effect of the phytocannabinoids THC and CBD on T-type calcium channels and found that both the CB receptor agonist THC and CB receptor-inactive CBD inhibit recombinant human CaV3 channels and native mouse T-type currents. Both THC and CBD shift steady state inactivation of the channels to more negative potentials, which has the effect of reducing the number of channels that can open when the cell is depolarized. These effects on steady state inactivation most likely represent a major mechanism by which THC and CBD inhibit CaV3 channels, as neither drug slowed channel activation or accelerated the decay of the currents after opening, which might be indicative of an open channel block. The effects of CBD and THC on steady state channel inactivation are similar to those of many other pharmacological inhibitors of CaV3 channels (42) including the endocannabinoid anandamide, although anandamide also accelerates open channel inactivation (33), which is something neither phytocannabinoid did.
Although both CBD and THC caused a hyperpolarizing shift in CaV3 channel activation, several aspects of the acute inhibition by the drugs differed. In particular, the inhibitory effect of THC on CaV3.1 was completely use-dependent and also strongly dependent on the voltage at which the cells were held, with inhibition at a minimum when currents were evoked from strongly hyperpolarized membrane potentials. By contrast CBD still strongly inhibited CaV3.1 evoked by steps from holding potentials of -126 mV, and channel inhibition developed fully during superfusion of CBD onto cells held continuously at -106 mV. These data indicate that CBD, but not THC, interacts significantly with closed states of CaV3.1 channels.
THC had two effects on T-type ICa that we believe are unique for an organic modulator of these channels. The most dramatic of these is a substantial slowing of CaV3.1 and CaV3.2 (but not CaV3.3) channel deactivation following repolarization, which was readily evident at concentrations of THC below those that substantially inhibited the peak channel current. THC also strongly slowed the decline of CaV3.1 currents after activation, presumably by inhibiting the channel transition from an open to an inactivated state. This apparent stabilization of open channels was not seen with CBD and has only been observed previously for Hg2+ modulation of CaV3.1 (43) and Zn2+ modulation of CaV3.3 (44). The overall effects of Zn2+ on CaV3.3 are very similar to those of THC on CaV3.1 and CaV3.2. Zn2+ inhibits open channel inactivation and channel deactivation without affecting channel activation kinetics, and this is accompanied by a hyperpolarizing shift in steady state activation and inactivation. In contrast, Zn2+ potently inhibits CaV3.2 without dramatically changing macroscopic channel kinetics, similar to the effects of CBD on all CaV3 channels and THC on CaV3.3 (44). Zn2+ inhibition of CaV3.1 is associated with changes in activation and inactivation but not deactivation (44). Thus, it seems that common regulatory mechanisms exist for all 3 CaV3 channels, but they are differently accessed by THC and Zn2+ in different CaV3 subtypes. Zn2+is likely to act at no less than two different sites on CaV3 channels, and our data suggest a similar situation for THC. THC and CBD share a very similar structure but have quite distinct effects on CaV3 channels, making them intriguing candidates for lead molecules in probing the functional domains of these channels.
THC and CBD both inhibited native mouse T-type ICa in acutely isolated trigeminal ganglion sensory neurons. The inhibition of the current was not associated with any obvious change in current inactivation from an open state or deactivation. The CaV3 subunits that are responsible for the T-type calcium currents in small trigeminal ganglion sensory neurons from mouse are unknown, although mRNA for all three genes is found in the ganglion.5 If mouse CaV3 channels are affected in a manner similar to human CaV3 channels, then our data suggest that CaV3.3 may be the main contributor to these currents, consistent with the previously reported nickel and cadmium sensitivity of the currents (38).
Our data with THC are consistent with the findings of Caulfield and Brown(34), who showed that a single high concentration of THC inhibits native T-type calcium channels in NG-108 cells, but is different from the findings of Chemin et al. (33), who reported that THC does not inhibit recombinant CaV3.2 channels expressed in HEK293 cells. There is no obvious explanation for the differences between the latter study and ours, but we note that THC is readily oxidized and relatively insoluble. We used THC from two different commercial batches and confirmed its activity at CB1 receptors in an assay of GABAergic (where GABA is γ-aminobutyric acid) synaptic transmission6 (45).
THC and CBD were unlikely to be exerting their effects via G proteins or G protein-coupled receptors. Most of our experiments were conducted without GTP in the internal solution, and inclusion of either the inhibitor of G protein activation, GDPβS, or GTP itself failed to affect the inhibition of CaV3.1 by submaximally effective concentrations of THC or CBD. Although G protein-independent signaling is well recognized for many G protein-coupled receptors, HEK293 cells are not thought to express CB1 or CB2 receptors (46), and although other G protein-coupled receptor targets of CBD have been proposed, none of them have been directly identified. The inhibition of native T-type ICa was not sensitive to the CB1 antagonist AM251, which like its structural analog SR141716A, also inhibited the T-type currents (33). The recently described cannabinoid receptor GPR55 is also expressed in sensory neurons (47). However, THC and AM251 are agonists at this receptor, and CBD is an antagonist (16), so the observation that all three drugs had similar effects on the native T-type currents suggests that these receptors were also not involved in the cannabinoid inhibition of T-type ICa seen in this study.
T-type calcium channels are involved in a range of physiological processes that THC and CBD are also known to affect. These include the peripheral transduction and central processing of noxious stimuli, sleep regulation, and epilepsy (27-32). Although any effects of CBD on these processes are likely to be independent of CB receptor activation, and thus potentially to involve inhibition of CaV3 channels, THC may act through several independent mechanisms to modulate the one physiological process. These mechanisms may be difficult to dissect, as the commonly used CB1 receptor antagonist SR141716A is also a T-type calcium channel blocker at CB receptor-relevant concentrations (33).
It is likely that concentrations of THC and CBD sufficient to affect T-type calcium channels significantly are reached following Cannabis ingestion in humans. When HEK293 cells were held at a typical physiological membrane potential of -70 mV, THC and CBD inhibited CaV3.1 channels by more than 50% at a concentration of 100 nm (∼30 ng/ml). There is limited information available on THC and CBD pharmacokinetics in humans, but smoking a 3.55% THC marijuana cigarette produces peak blood levels of about 750 nm (49). Most unregulated preparations of cannabis have higher concentrations of THC (50), and medical Cannabis preparations prescribed in Holland have standardized THC contents of up to 18% (3, 51). Postmortem THC concentrations in the human brain have been reported up to 20-30 ng/g, which is higher than in matched blood samples (52). In pigs, an animal model chosen for its similarity to humans, THC concentrations in the brain 30 min after administration of a typical human dose of THC were 50 ng/g (53). This evidence strongly suggests that T-type ICa channels are a likely to be affected by THC in many social and self-medicating users.
A dose of THC (0.3 mg/kg) that is close to the EC50 for producing the classic tetrad of behavioral signs in mice produces blood and brain levels of 100-300 nm (54), sufficient to affect native T-type calcium channels strongly. Commonly used doses of CBD (3-10 mg/kg) produce brain levels of about 200 nm and 3 μm, respectively (54). Even though there is no evidence that the cannabinoid behavioral tetrad is mediated by anything other than interactions at CB1 receptors, it has been pointed out that non-CB receptor-mediated effects of THC may have been missed because most investigators focus on processes known to be CB receptor-mediated (48). The lack of selective blockers of T-type ICa has made it difficult to study the role of these channels in vivo, and it is perhaps not surprising that there are no behavioral assays for T-type channel activity. The concentrations of THC required to inhibit T-type channels are similar to those required for inhibition of recombinant 5-HT3 receptors (16) and much lower than those required to activate other non-CB receptor effectors such as TRPA1 (EC50 > 10 μm (18)) or TRPV2 (EC50 43 μm (19)).
In summary, we have shown that the phytocannabinoids THC and CBD inhibit all three CaV3 subtypes as well as native T-type ICa. The CB1 agonist THC additionally causes slowing of both inactivation and deactivation of CaV3.1 and CaV3.2, unique actions that may have the effect of increasing calcium entry into neurons at moderate concentrations of THC. Actions on CaV3 channels may explain some of the pharmacological effects of THC and CBD that cannot be explained by CB receptor activation, and the two compounds are likely to be valuable tools for dissecting the structural features of these channels responsible for their unique electrophysiological signatures.
Acknowledgments
We thank Ed Perez-Reyes for his kind gift of CaV3.1, CaV3.2, and CaV3.3 cDNA and for ongoing comments on these studies.
This work was supported in part by National Health & Medical Research Council of Australia Project 402564 (to M. C.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: THC, Δ9-tetrahydrocannabinol; CBD, cannabidiol; HEK293, human embryonic kidney 293 cells; mHBS, modified HEPES-buffered saline; GDPβS, guanyl-5′-yl thiophosphate; ANOVA, analysis of variance.
E. Johnson and M. Connor, unpublished observations.
C. Vaughan, personal communication.
References
- 1.xLemberger, L. (1980) Annu. Rev. Pharmacol. Toxicol. 20 151-172 [DOI] [PubMed] [Google Scholar]
- 2.Teesson, M., Baillie, A., Lynskey, M., Manor, B., and Degenhardt, L. (2006) Drug Alcohol Depend. 81 149-155 [DOI] [PubMed] [Google Scholar]
- 3.Gorter, R. W., Butorac, M., Cobian, E. P., and van der Sluis, W. (2005) Neurology 64 917-919 [DOI] [PubMed] [Google Scholar]
- 4.Ware, M. A., Doyle, C. R., Woods, R., Lynch, M. E., and Clark, A. J. (2003) Pain 102 211-216 [DOI] [PubMed] [Google Scholar]
- 5.Gross, D. W., Hamm, J., Ashworth, N. L., and Quigley, D. (2004) Neurology 62 2095-2097 [DOI] [PubMed] [Google Scholar]
- 6.Clark, A. J., Ware, M. A., Yazer, E., Murray, T. J., and Lynch, M. E. (2004) Neurology 62 2098-2100 [DOI] [PubMed] [Google Scholar]
- 7.Goani, Y., and Mechoulam, R. (1964) J. Am. Chem. Soc. 86 1646-1647 [Google Scholar]
- 8.Elsohly, M. A., and Slade, D. (2005) Life Sci. 78 539-548 [DOI] [PubMed] [Google Scholar]
- 9.Petrzilka, T., Haefliger, W., and Sikemeier, C. (1969) Helv. Chim. Acta 52 1102-1134 [DOI] [PubMed] [Google Scholar]
- 10.Matsuda, L. A., Lolait, S. J., Brownstein, M. J., Young, A. C., and Bonner, T. I. (1990) Nature 346 561-564 [DOI] [PubMed] [Google Scholar]
- 11.Munro, S., Thomas, K. L., and Abu-Shaar, M. (1993) Nature 365 61-65 [DOI] [PubMed] [Google Scholar]
- 12.Thomas, A., Baillie, G. L., Phillips, A. M., Razdan, R. K., Ross, R. A., and Pertwee, R. G. (2007) Br. J. Pharmacol. 150 613-623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Compton, D. R., Johnson, M. R., Melvin, L. S., and Martin B. R. (1992) J. Pharmacol. Exp. Ther. 260 201-209 [PubMed] [Google Scholar]
- 14.Zimmer, A., Zimmer, A. M., Hohmann, A. G., Herkenham, M., and Bonner, T. I. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 5780-5785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pertwee, R. G. (2008) Br. J. Pharmacol. 153 199-215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ryberg, E., Larsson, N., Sjogren, S., Hjorth, S., Hermansson, N.-O., Leonova, J., Elebring, T., Nilsson, K., Drmota, T., and Greasley, P. J. (2007) Br. J. Pharmacol. 152 1092-1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barann, M., Molderings, G., Bruss, M., Bonisch, H., Urban, B. W., and Gothert, M. (2002) Br. J. Pharmacol. 137 589-596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jordt, S. E., Bautista, D. M., Chaung, H. H., McKemy, D. D., Zygmunt, P. M., Hogestatt, E. D., Meng, I. D., and Julius, D. (2004) Nature 427 260-265 [DOI] [PubMed] [Google Scholar]
- 19.Neeper, M. P., Liu, Y., Hutchinson, T. L., Wang, Y., Flores, C. M., and Qin, N. (2007) J. Biol. Chem. 282 15894-15902 [DOI] [PubMed] [Google Scholar]
- 20.Perez-Reyes, M., Timmons, M. C., Davis, K. H., and Wall, E. M. (1973) Experientia (Basel) 29 1368-1369 [DOI] [PubMed] [Google Scholar]
- 21.Bisogno, T., Hanus, L., De Petrocellis, L., Tchilibon, S., Ponde, D. E., Brandi, I., Moriello, A. S., Davis, J. B., Mechoulam, R., and Di Marzo, V. (2001) Br. J. Pharmacol. 134 845-852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Costa, B., Trovato, A. E., Comelli, F., Giagnoni, G., and Colleoni, M. (2007) Eur. J. Pharmacol. 556 75-83 [DOI] [PubMed] [Google Scholar]
- 23.Karler, R., Cely, W., and Turkanis, S. A. (1974) Life Sci. 15 931-947 [DOI] [PubMed] [Google Scholar]
- 24.Murillo-Rodriguez, E., Millan-Aldaco, D., Palomero-Rivero, M., Mechoulam, R., and Drucker-Colin, R. (2006) FEBS Lett. 580 4337-4345 [DOI] [PubMed] [Google Scholar]
- 25.Jarai, Z., Wagner, J. A., Varga, K., Lake, K. D., Compton, D. R., Martin, B. R., Zimmer, A. M., Bonner, T. I., Buckley, N. E., Mezey, E., Razdan, R. K., Zimmer, A., and Kunos, G. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 14136-14141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perez-Reyes, E. (2003) Physiol. Rev. 83 117-161 [DOI] [PubMed] [Google Scholar]
- 27.Bourinet, E., Alloui, A., Monteil, A., Barrere, C., Couette, B., Poirot, O., Pages, A., McRory, J., Snutch, T. P., Eschalier, A., and Nargeot, J. (2005) EMBO J. 24 315-324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Todorovic, S. M., Jevtovic-Todorovic, V., Meyenburg, A., Mennerick, S., Perez-Reyes, E., Romano, C., Olney, J. W., and Zorumski, C. F. (2001) Neuron 31 75-85 [DOI] [PubMed] [Google Scholar]
- 29.Kim, D., Park, D., Choi, S., Lee, S., Sun, M., Kim, C., and Shin, H. S. (2003) Science 302 117-119 [DOI] [PubMed] [Google Scholar]
- 30.Anderson, M. P., Mochizuki, T., Xie, J., Fischler, W., Manger, J. P., Talley, E. M., Scammell, T. E., and Tonegawa, S. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 1743-1748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feinberg, I., Jones, R., Walker, J., Cavness, C., and Floyd, T. (1976) Clin. Pharmacol. Ther. 19 782-794 [DOI] [PubMed] [Google Scholar]
- 32.Tsakiridou, E., Bertollini, L., de Curtis, M., Avanzini, G., and Pape, H. C. (1995) J. Neurosci. 15 3110-3117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chemin, J., Monteil, A., Perez-Reyes, E., Nargeot, J., and Lory, P. (2001) EMBO J. 20 7033-7040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Caulfield, M. P., and Brown, D. A. (1992) Br. J. Pharmacol. 106 231-232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cribbs, L. L., Gomora, J. C., Daud, A. N., Lee, J. H., and Perez-Reyes, E. (2000) FEBS Lett. 466 54-58 [DOI] [PubMed] [Google Scholar]
- 36.Cribbs, L. L., Lee, J. H., Yang, J., Satin, J., Zhang, Y., Daud, A., Barclay, J., Williamson, M. P., Fox, M., Rees, M., and Perez-Reyes, E. (1998) Circ. Res. 83 103-109 [DOI] [PubMed] [Google Scholar]
- 37.Gomora, J. C., Murbartian, J., Arias, J. M., Lee, J.-H., and Perez-Reyes, E. (2002) Biophys. J. 83 229-241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Borgland, S. L., Connor, M., and Christie, M. J. (2001) J. Physiol. (Lond.) 536 35-47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hamill, O. P., Marty, A., Neher, E., Sakmann, B., and Sigworth, F. J. (1981) Pfluegers Arch. Eur. J. Physiol. 391 85-100 [DOI] [PubMed] [Google Scholar]
- 40.Lan, R., Liu, Q., Fan, P., Lin, S., Fernando, S. R., McCallion, D., Pertwee, R., and Makriyannis, A. (1999) J. Med. Chem. 42 769-776 [DOI] [PubMed] [Google Scholar]
- 41.Roberts, L. A., Ross, H. R., and Connor, M. (2008) Neuropharmacology 54 172-180 [DOI] [PubMed] [Google Scholar]
- 42.Heady, T. N., Gomora, J. C., Macdonald, T. L., and Perez-Reyes, E. (2001) Jpn. J. Pharmacol. 85 339-350 [DOI] [PubMed] [Google Scholar]
- 43.Tarabova, B., Kurejova, M., Sulova, Z., Drabova, M., and Lacinova, L. (2006) J. Pharmacol. Exp. Ther. 317 418-427 [DOI] [PubMed] [Google Scholar]
- 44.Traboulsie, A., Chemin, J., Chevalier, M., Quignard, J. F., Nargeot, J., and Lory, P. (2007) J. Physiol. (Lond.) 578 159-171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vaughan, C. W., Connor, M., Bagley, E. E., and Christie, M. J. (2000) Mol. Pharmacol. 57 288-295 [PubMed] [Google Scholar]
- 46.Tao, Q., and Abood, M. E. (1998) J. Pharmacol. Exp. Ther. 285 651-658 [PubMed] [Google Scholar]
- 47.Lauckner, J. E., Jensen J. B., Chen, H. Y., Lu, H. C., Hille, B., and Mackie, K. (2008) Proc. Natl. Acad. Sci. U. S. A. 105 2699-2704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Varvel, S. A., Bridgen, D. T., Tao, Q., Thomas, B. F., Martin, B. R., and Lichtman, A. H. (2005) J. Pharmacol. Exp. Ther. 314 329-337 [DOI] [PubMed] [Google Scholar]
- 49.Huestis, M. A., and Cone, E. J. (2004) J. Anal. Toxicol. 28 394-399 [DOI] [PubMed] [Google Scholar]
- 50.Hall, W., and Swift, W. (2000) Aust. N. Z. J. Public Health 24 503-508 [DOI] [PubMed] [Google Scholar]
- 51.Engels, F. K., de Jong, F. A., Mathijssen, R. H. J., Erkens, J. A., Herings, R. M., and Verweij, J. (2007) Eur. J. Cancer 43 2638-2644 [DOI] [PubMed] [Google Scholar]
- 52.Mura, P., Kintz, P., Dumestre, V., Raul, S., and Hauet, T. (2005) J. Anal. Toxicol. 29 842-843 [DOI] [PubMed] [Google Scholar]
- 53.Brunet, B., Doucet, C., Venisse, N., Hauet, T., Hebrard, W., Papet, Y., Mauco, G., and Mura, P. (2006) Forensic Sci. Int. 161 169-174 [DOI] [PubMed] [Google Scholar]
- 54.Varvel, S. A., Wiley, J. L., Yang, R., Bridgen, D. T., Long, K., Lichtman, A. H., and Martin, B. R. (2006) Psychopharmacology (Berl.) 186 226-234 [DOI] [PubMed] [Google Scholar]