

Abstract
Although cells can exit mitotic block aberrantly by mitotic slippage, they are prevented from becoming tetraploids by a p53-dependent postmitotic checkpoint. Intriguingly, disruption of the spindle-assembly checkpoint also compromises the postmitotic checkpoint. The precise mechanism of the interplay between these two pivotal checkpoints is not known. We found that after prolonged nocodazole exposure, the postmitotic checkpoint was facilitated by p53. We demonstrated that although disruption of the mitotic block by a MAD2-binding protein promoted slippage, it did not influence the activation of p53. Both p53 and its downstream target p21CIP1/WAF1 were activated at the same rate irrespective of whether the spindle-assembly checkpoint was enforced or not. The accelerated S phase entry, as reflected by the premature accumulation of cyclin E relative to the activation of p21CIP1/WAF1, is the reason for the uncoupling of the postmitotic checkpoint. In support of this hypothesis, forced premature mitotic exit with a specific CDK1 inhibitor triggered DNA replication without affecting the kinetics of p53 activation. Finally, replication after checkpoint bypass was boosted by elevating the level of cyclin E. These observations indicate that disruption of the spindle-assembly checkpoint does not directly influence p53 activation, but the shortening of the mitotic arrest allows cyclin E-CDK2 to be activated before the accumulation of p21CIP1/WAF1. These data underscore the critical relationship between the spindle-assembly checkpoint and the postmitotic checkpoint in safeguarding chromosomal stability.
Genome instability in the form of aneuploidy is commonly found in solid tumors. It has been suggested that mutations that lead to compromised spindle-assembly checkpoint may increase the chance of chromosome missegregation, leading to aneuploidy (1, 2). Tetraploidization is also believed to be a route to aneuploidy (3). Transient blocking of cytokinesis in p53-null mouse mammary epithelial cells generates tetraploids, which promotes aneuploidy and tumorigenesis (4). Another study reported that chromosome nondisjunction (both copies of a chromosome segregate to the same daughter cells) leads to binucleated tetraploids by promoting cleavage furrow regression; the tetraploid cells then become aneuploidy through further divisions (5). These and other studies provide strong evidence on the importance of polyploidization in tumorigenesis.
During mitosis, microtubules radiated from two opposite spindle poles and captured chromosomes by attaching to the kinetochores. Sister chromatid segregation is only initiated when all the chromosomes have achieved bipolar attachment to the mitotic spindles. The current paradigm states that unattached kinetochores or the absence of tension between paired kinetochores activates a surveillance mechanism called spindle-assembly checkpoint. The underlying mechanism of the checkpoint is not completely defined, but a growing body of evidence indicates that unattached kinetochores attract the components of the checkpoint machinery. This catalyzes the formation of diffusible mitotic checkpoint complexes (components include MAD2, BUBR1, and BUB3), which in turn inhibit APC/C-CDC20. This mechanism maintains high levels of active cyclin B1-CDK1 and locks the cell in prometaphase (reviewed in Ref. 6).
The mitotic block triggered by the spindle-assembly checkpoint is not permanent, and cells can exit mitosis aberrantly by a process known as mitotic slippage (also known as adaptation) (reviewed in Ref. 7). During mitotic slippage, cyclin B1-CDK1 is inactivated, and the cells enter G1 phase without first going through chromosome segregation and cytokinesis, yielding tetraploid G1 cells. DNA re-replication in these cells is prevented by a p53-dependent mechanism that arrests cells in postmitotic G1 phase, often referred to as the postmitotic checkpoint (8–13). The detailed molecular mechanism of this postmitotic checkpoint is unclear. It was previously believed that this checkpoint is triggered by the presence of tetraploid DNA content (14). More recent studies, however, refute this idea and indicate that there is no specific tetraploidy checkpoint that directly count DNA or centrosomes (reviewed in Ref. 15). Nevertheless, it is generally accepted that p53 does play an important role in preventing re-replication after mitotic slippage. For example, p53–/– mouse embryonic fibroblasts can spontaneously become tetraploids during culturing (16). Furthermore, tetraploids induced in mouse mammary epithelial cells by cytokinesis failure (4) or in human cells by virally induced cell fusion (17) can be propagated in the absence of p53. The G1 arrest of the postmitotic checkpoint is at least in part because of the cyclin-dependent kinase inhibitor p21CIP1/WAF1, a transcriptional target of p53 (10, 11, 18).
It is possible that other p53-independent mechanisms also contribute to the postmitotic checkpoint. Expression of HPV3 E6 oncoprotein, which targets p53 for degradation, can abolish the postmitotic checkpoint without affecting the spindle-assembly checkpoint. Interestingly, E6 mutant defective in p53 degradation can also partially induce polyploidy, suggesting the presence of a p53-independent postmitotic checkpoint mechanism (19). Moreover, proper function of the spindle-assembly checkpoint is required for the postmitotic G1 checkpoint (20). Vogel et al. (20) shows that spindle-assembly checkpoint-compromised HCT116 cells failed to arrest at the postmitotic checkpoint after nocodazole treatment.
What is the molecular mechanism underlying the linkage between the spindle-assembly checkpoint and the postmitotic checkpoint? We hypothesize that the spindle-assembly checkpoint may directly control the activation of the p53 pathway. Alternatively, the checkpoint may act independently of p53 activation, but a defective checkpoint may somehow desensitize the cell cycle to p53-mediated inhibition. In this study, we present observations that unequivocally show that the disruption of the spindle-assembly checkpoint does not influence the activation of p53. We found that after prolonged nocodazole exposure, the postmitotic checkpoint was facilitated by p53 and p21CIP1/WAF1 after mitotic slippage as well as after the cells were released into G1 phase. Curtailing the duration of the spindle-assembly checkpoint by either ectopic expression of MAD2L1BP or a CDK1 inhibitor abolished the postmitotic checkpoint. This was achieved through an acceleration of S phase entry, as reflected by the premature accumulation of cyclin E1, relative to the activation of p53/p21CIP1/WAF1. These results indicate that the spindle-assembly checkpoint does not directly control p53 activation, but the shortening of the mitotic arrest after uncoupling of the checkpoint allows cyclin E1-CDK2 to be activated before the accumulation of sufficient levels of p21CIP1/WAF1.
EXPERIMENTAL PROCEDURES
Materials—All reagents were obtained from Sigma unless stated otherwise.
DNA Constructs and siRNA—MAD2L1BP cDNA (IMAGE 3945348) was amplified by PCR with the primers 5′GCCATGGCGGCGCCGGAGGCGG3′ and 5′CGGATCCTCACTCGCGGAAGCCTTT3′; the PCR product was cut with NcoI-BamHI and ligated into pUHD-P1 (21) to obtain FLAG-MAD2L1BP in pUHD-P1. A puromycin-resistant gene cassette was inserted into the BamHI site to create FLAG-MAD2L1BP in pUHD-P1/PUR. Plasmids for FLAG-p53 (22), cyclin E1 (23), and histone H2B-GFP (24) were constructed or obtained from sources as described previously. The cyclin E1 cDNA was amplified by PCR with the primers 5′GGAATTCATGAAGGAGGACGGCGG3′ and 5′GGAATTCTCACGCCATTTCCGG3′; the PCR product was cut with EcoRI and ligated into pUHD-P1 to obtain FLAG-cyclin E1 in pUHD-P1. An HPV E6-expressing construct was the generous gift from Dr. Nikki Harter (Case Western Reserve University, Cleveland, OH). Stealth siRNA targeting KIF11 (HSS105841) and control siRNA were obtained from Invitrogen.
Cell Culture—H1299 (non-small cell lung carcinoma), Hep3B (hepatocellular carcinoma), HeLa (cervical carcinoma), and HepG2 cells (hepatoblastoma) were obtained from the American Type Culture Collection (Manassas, VA). U2OS (osteosarcoma) Tet-On cell line was obtained from Clontech. Hep3B/p53 was a stable cell line with the expression of wild type p53 under doxycycline control (25) and was a gift from Dr. Paul Lai (Chinese University of Hong Kong, Hong Kong). p53+/+ and p53–/– mouse embryonic fibroblasts were prepared from embryos obtained from timed pregnant females at 15–16 days of gestation as described previously (26). Cells were grown in medium supplemented with calf serum or fetal bovine serum (Invitrogen) according to the supplier's instructions. Unless stated otherwise, cells were treated with the following reagents at the indicated final concentration: caspase inhibitor (benzyloxycarbonyl-VAD(OMe)-fluoromethyl ketone from Alexis, Lausen, Switzerland) (20 μm), doxycycline (1 μg/ml), nocodazole (0.1 μg/ml), puromycin (1 μg/ml), and RO3306 (Alexis) (10 μm). Cells were transfected with plasmids using the calcium phosphate precipitation method (27). Transfection of siRNA was carried out using Lipofectamine™ RNAiMAX (Invitrogen). Cell-free extracts were prepared as described previously (28). Double thymidine synchronization was performed as described previously (23). For generation of stable cell lines, U2OS cells were transfected with FLAG-MAD2L1BP in pUHD-P1/PUR and cultured in medium containing puromycin. After about 2 weeks of selection, individual colonies were isolated and propagated in the absence of puromycin.
Flow Cytometry—Propidium iodide staining and flow cytometry analysis were performed as described previously (29). Bromodeoxyuridine incorporation followed by flow cytometry analysis (24) and bivariate flow cytometry analysis (30) were performed as described previously.
Antibodies and Immunological Methods—Monoclonal antibodies E23 against cyclin A2 (31), A17 against CDK1 (30), YL1/2 against tubulin (21), M2 against FLAG (32), and polyclonal antibodies against CDK2 (30) were obtained from sources as described previously. Monoclonal antibodies GSN1 against cyclin B1 (sc-245), HD11 against cyclin D1 (sc-246), HE12 against cyclin E1 (sc-247), DO1 against p53 (sc-126), polyclonal antibodies against geminin (sc13015), phosphohistone H3Ser-10 (sc-8656R), KIF11 (sc-31643), p21CIP1/WAF1 (sc-397), and securin (sc-5839) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Monoclonal antibody AC-74 against β-actin was obtained from Sigma. Polyclonal antibodies against BUBR1 and phospho-histone H2AXSer-139 were obtained from Bethyl Laboratories (Montgomery, TX). Immunoblotting and immunoprecipitation were performed as described (28).
RESULTS
Activation of the p53-p21CIP1/WAF1 Axis during the Postmitotic Checkpoint—To define the relationship between mitotic slippage and p53 activation, HepG2 cells were cultured continuously with nocodazole for up to 72 h. The supplemental Fig. S1A shows that cyclin B1 and histone H3Ser-10 phosphorylation surged after addition of the drug. The hyper-phosphorylation of BUBR1 indicated the expected activation of the spindle-assembly checkpoint (33). Mitotic slippage occurred upon further incubation (48 h and 72 h), which was reflected by the return of cyclin B1 and phosphorylation of histone H3Ser-10 and BUBR1 to basal levels. The p53 pathway was strongly stimulated concurrently with the slippage, as indicated by the accumulation of both p53 and p21CIP1/WAF1. Flow cytometry analysis revealed that HepG2 cells display a 4N DNA content after slippage, indicating the cells were arrested in tetraploidy-G1 state (supplemental Fig. S1B). Similar results were obtained with another p53-containing cell line, U2OS (Fig. 1A). A notable cell line variation is that U2OS underwent massive cell death after prolonged nocodazole treatment, as indicated by the collapse of the cell cycle profile to sub-G1 population (Fig. 1B). This is in agreement with the previous findings that tetraploid cells arising because of mitotic slippage were prone to undergo BAX-mediated mitochondrial membrane permeabilization and subsequent apoptosis (34, 35). HepG2 also underwent nocodazole-induced apoptosis, albeit to a lesser extent, as indicated by the accumulation of sub-G1 population (supplemental Fig. S1B). Induction of γ-H2AX after nocodazole treatment in both cell lines (Fig. 1A and supplemental Fig. S1A) was probably because of apoptosis but not DNA damage (36, 37), because the γ-H2AX signal could be abolished by a caspase inhibitor (Fig. 1C).
FIGURE 1.

Prolonged nocodazole exposure induces slippage and activates p53. A, nocodazole (NOC) induces mitotic slippage and p53 activation. U2OS cells were exposed to nocodazole and harvested at the indicated time points. Cell-free extracts were prepared, and the expression of the indicated proteins was detected by immunoblotting. B, apoptosis is induced by prolonged exposure of nocodazole. U2OS cells were treated with nocodazole for 24 h before either buffer or caspase inhibitor was added. At the indicated time points, cells were harvested for flow cytometry analysis. The positions of 2N and 4N DNA content are indicated. The percentage of the sub-G1 population was indicated. C, inhibition of apoptosis does not affect mitotic slippage and p53 accumulation. U2OS cells were treated as in B. The cells were harvested at the indicated time points, and cell-free extracts were prepared. The expression of the indicated proteins was detected by immunoblotting.
To exclude the possibility that the cells did undergo another round of replication but subsequently became inviable, a caspase inhibitor was added with nocodazole to inhibit apoptosis. Fig. 1B shows that although the inhibitor significantly reduced cell death, no re-replication was detected. Importantly, neither mitotic slippage nor p53 activation was affected by the caspase inhibitor (Fig. 1C). When apoptosis was inhibited during slippage, both HepG2 and U2OS were arrested in the cell cycle with a 4N DNA content. The current paradigm states that replication after slippage is inhibited by p53. In agreement with this, re-replication can be detected after nocodazole and caspase inhibitor treatment in a p53-null background (supplemental Fig. S2).
The Activation of p53 Is Initiated during Mitotic Arrest and Continues after Mitotic Slippage—To delineate whether p53 is stabilized during mitosis or after slippage, the expression of p53 and histone H3Ser-10 phosphorylation was analyzed concurrently with flow cytometry (Fig. S3). The background level was established by excluding the primary antibodies as well as by using a p53-null cell line (H1299). At 24 h after nocodazole addition, p53 started to accumulate in both mitotic and nonmitotic cells. As expected, the number of mitotic cells reduced whereas the p53-expressing cells increased after longer exposure (36 h). A higher portion of the p53-expressing cells was found in the cells that had undergone slippage than in the remaining mitotic cells.
Ciciarello et al. (38) reported that p53 is expressed in lymphoblastoid cells and K562 erythroleukemic cells after a transient exposure to nocodazole, preventing G1 cells from progressing into S phase after the removal of nocodazole. To see if the relatively low level of p53 induced during mitotic block was sufficient to trigger a subsequent cell cycle arrest, cells were blocked transiently with nocodazole before replating in drug-free medium. This well established protocol prompted p53-negative cells such as HeLa (supplemental Fig. S4A) to progress through the cell cycle synchronously (note the periodic variation of cyclin E1, cyclin A2, and cyclin B1 through the cell cycle). By contrast, p53-positive cells such as HepG2 could not be synchronized effectively after activation of the spindle-assembly checkpoint. Although the majority of HepG2 cells displayed a 4N DNA content after exposure to nocodazole, only about half of the cells were released into 2N G1 phase (supplemental Fig. S4B). Furthermore, both 2N and 4N cells failed to enter S phase properly, with substantial delays in bromodeoxyuridine incorporation and cyclin E1 accumulation. Likewise, after an initial decrease, histone H3Ser-10 phosphorylation and cyclin B1 did not increase again during the course of the experiment. In accordance with the cell cycle arrest, we found that p53 and p21CIP1/WAF1 accumulated in both U2OS (Fig. 2A) and HepG2 (supplemental Fig. S4B) upon release from the spindle-assembly checkpoint. Interaction between p21CIP1/WAF1 and CDK2 was revealed by co-immunoprecipitation (Fig. 2B). Similar to the DNA re-replication in the continuous presence of nocodazole, replication after release from transient nocodazole block was also suppressed by the p53 network. Re-introduction of wild type p53 into the p53-null cell line Hep3B suppressed DNA replication after release from nocodazole block (supplemental Fig. S5). Similarly, disruption of p53 allows replication after nocodazole block release (supplemental Fig. S6).
FIGURE 2.

p53 is induced after release from transient nocodazole treatment. A, activation of the p53-p21CIP1/WAF1 pathway after release from mitotic block. U2OS cells were treated with nocodazole for 36 h and then released into fresh medium. Cells were harvested at the indicated time points and analyzed by immunoblotting. B, binding of p21CIP1/WAF1 to CDK2 after release from mitotic block. Protein expression in HepG2 cells cultured in control medium (lane 1), nocodazole (NOC)-containing medium for 24 h (lane 2), or at 12 h after release from a nocodazole block (lane 3) was analyzed by immunoblotting. The p21CIP1/WAF1 binding to CDK2 was detected by immunoprecipitation of CDK2 followed by immunoblotting for p21CIP1/WAF1. C, p53 is induced in both 2N and 4N cells. HepG2 cells were harvested at 12 h after release from a nocodazole block. The DNA content (propidium iodide, x axis) and p53 expression (fluorescein isothiocyanate, y axis) were analyzed by flow cytometry. The DNA profiles of p53-positive and -negative cells are also shown. Samples without primary antibody and asynchronously growing control cells serve as negative controls.
To distinguish whether p53 was expressed in G1 cells with 2N or 4N DNA contents, expression of p53 in individual cells was analyzed by flow cytometry. Fig. 2C shows that at 12 h after release from a nocodazole block, p53 was expressed in both 2N and 4N populations. Collectively, these data show that after a transient activation of the spindle-assembly checkpoint, the delay in both G1 and tetraploid G1 populations was accompanied by an accumulation of p53.
The Spindle-assembly Checkpoint Is Required for Postmitotic Cell Cycle Arrest but Not for p53-p21CIP1/WAF1 Activation—To determine the relationship between the spindle-assembly checkpoint and the postmitotic checkpoint, a U2OS cell line that conditionally expressed FLAG-tagged MAD2L1BP (also called MAD2-binding protein or p31comet), a potent inhibitor of MAD2 (39, 40), was generated (Fig. 3A). As expected, the spindle-assembly checkpoint was compromised in the presence of MAD2L1BP, as reflected by the lack of maintenance of histone H3Ser-10 phosphorylation upon nocodazole challenge. This is also supported by the absence of BUBR1 hyper-phosphorylation (see Fig. 4). Ectopically expressed MAD2L1BP triggered re-replication upon nocodazole challenge, indicating that MAD2L1BP also disrupted the postmitotic checkpoint (Fig. 3B). Intriguingly, both p53 and p21CIP1/WAF1 were still stimulated in MAD2L1BP-expressing cells. This surprising finding was verified when MAD2L1BP-expressing cells were compared with either the parental U2OS cells (Fig. 3A) or with the same U2OS/MAD2L1BP cells when the expression of the recombinant protein was suppressed by the absence of doxycycline (Fig. 3C). As a control, MAD2L1BP itself did not strongly stimulate the p53 pathway in the absence of nocodazole.
FIGURE 3.

Cells expressing MAD2L1BP lack the postmitotic checkpoint but still express high level of p53. A, p53 and p21CIP1/WAF1 are activated by nocodazole (NOC) in MAD2L1BP-expressing cells. Parental U2OS and U2OS/MAD2L1BP cells were treated with doxycycline (Dox) together with either control buffer or nocodazole for 48 h. Cell-free extracts were prepared, and the indicated proteins were detected by immunoblotting. Uniform loading of lysates was confirmed by CDK2. B, cells expressing MAD2L1BP lack the postmitotic checkpoint. Parental U2OS and U2OS/MAD2L1BP cells were treated as described in A. The cells were harvested and processed for flow cytometry analysis. C, MAD2L1BP/U2OS cells were either mock-treated or treated with doxycycline and nocodazole for 48 h as indicated. Cell-free extracts were prepared, and the indicated proteins were detected by immunoblotting.
FIGURE 4.

The rate of p21CIP1/WAF1 accumulation relative to mitotic slippage regulates the postmitotic checkpoint. A, kinetics of mitotic slippage after the expression of MAD2L1BP. U2OS/MAD2L1BP cells were treated with thymidine, together with either buffer or doxycycline (Dox), for 16 h. Cells were released from the thymidine block and cultured continuously with or without doxycycline. Nocodazole was added to activate the spindle-assembly checkpoint. The cells were harvested at the indicated time points and processed for immunoblotting for the indicated proteins. Uniform loading of lysates was confirmed by immunoblotting for actin. B, kinetics of re-replication after the expression of MAD2L1BP. Cells were treated exactly as in A. The cells were harvested for flow cytometry analysis at the indicated time points.
To exclude the possibility that the p53-p21CIP1/WAF1 pathway was stimulated by checkpoint-independent effects of nocodazole, the spindle-assembly checkpoint was also activated using siRNA targeting the kinesin-like protein KIF11 (also called Eg5) (41). Similar to nocodazole treatment, knockdown of KIF11 promoted re-replication in MAD2L1BP-expressing cells but not in the parental U2OS cells (supplemental Fig. S7A). Moreover, KIF11 siRNA stimulated the expression of p53 and p21CIP1/WAF1 in both cell lines (supplemental Fig. S7B). Taken together, these data indicate that the postmitotic checkpoint also becomes impaired when the spindle-assembly checkpoint is disrupted. However, both p53 and p21CIP1/WAF1 were induced even in the absence of the postmitotic checkpoint.
The Constant Rate of Activation of the p53-p21CIP1/WAF1 Pathway Underlines the Relationship between the Spindle-assembly Checkpoint and the Postmitotic Checkpoint—To obtain a more detailed relationship between the spindle-assembly checkpoint and p53 activation, MAD2L1BP-expressing cells were first synchronized with thymidine, then released into the cell cycle, and treated with nocodazole at the same time. Fig. 4 shows that control cells were trapped in mitosis from t = 18 h, with robust phosphorylation of histone H3Ser-10. The activation of the spindle-assembly checkpoint was confirmed by the upward mobility shift of BUBR1 and the stabilization of anaphase-promoting complex/cyclosome substrates (cyclin B1, securin, and geminin). Mitotic slippage, characterized by reversal of mitotic events through dephosphorylation (histone H3Ser-10 and BUBR1) and proteolysis (cyclin B1, securin, and geminin), occurred at around t = 36 h. Cyclin E1, the mediator for G1-S transition, accumulated only after slippage. In marked contrast, cells expressing MAD2L1BP rapidly underwent mitotic slippage. Destruction of anaphase-promoting complex/cyclosome substrates occurred at t = 24 h, at least 12 h ahead of control cells. Flow cytometry analysis confirmed that although control cells were trapped with a 4N DNA content after slippage, MAD2L1BP-expressing cells readily re-replicated their DNA (Fig. 4B).
To see if the kinetics of p53 activation was affected by MAD2L1BP, the expression of both p53 and p21CIP1/WAF1 was examined in the same samples. Interestingly, the onset of nocodazole-induced accumulation of p53 and p21CIP1/WAF1 was identical in the presence or absence of MAD2L1BP (Fig. 4A). However, the postmitotic checkpoint was ineffective in MAD2L1BP-expressing cells because they entered S phase before p21CIP1/WAF1 accumulated to any substantial level. In agreement with this idea, cyclin E1 accumulated at t = 30 h in the presence of MAD2L1BP, well before p21CIP1/WAF1 was activated (Fig. 4A lane 15). In contrast, slippage and cyclin E1 accumulation occurred slower in control cells (t = 42 h, Fig. 4A, lane 8), when both p53 and p21CIP1/WAF1 had already stockpiled. Thus although the p53-p21CIP1/WAF1 pathway was activated at a constant rate after the introduction of nocodazole, cells lacking the spindle-assembly checkpoint entered G1 phase prematurely, allowing them to become resistant to the postmitotic checkpoint.
If the above hypothesis is correct, we expected that cells undergoing re-replication should not express high level of p21CIP1/WAF1. To test this idea, we examined the abundance of p21CIP1/WAF1 in individual cells in the MAD2L1BP re-replication model. Fig. 5A shows that p21CIP1/WAF1 was expressed at a relatively low level at the onset of re-replication (at 24 h). In particular, re-replication occurred only in cells with low p21CIP1/WAF1 expression (DNA contents between 4N and 8N). At a later time point, although cells that were re-replicating still expressed low levels of p21CIP1/WAF1, those that completed re-replication (at 8N content) possessed higher levels of p21CIP1/WAF1. These analyses further support the model that with a defective spindle-assembly checkpoint, p21CIP1/WAF1 is not activated in a timely manner to properly enforce the postmitotic checkpoint.
FIGURE 5.

The postmitotic checkpoint following the uncoupling of the spindle-assembly checkpoint is restored by expression of exogenous p53. A, cells undergoing re-replication do not express high level of p21CIP1/WAF1. U2OS/MAD2L1BP cells were treated with doxycycline (Dox) and nocodazole (NOC). At the indicated time points, cells were harvested for bivariate analysis for p21CIP1/WAF1 and DNA. Controls lacking primary antibody or without nocodazole treatment were used to set the p21CIP1/WAF1 background expression level. The DNA profiles of cells with high and low p21CIP1/WAF1 expression levels for cells treated with nocodazole for 48 h are also shown. Immunoblotting was also performed to confirm the expression of the indicated proteins. B, increased expression of p53 restores the postmitotic checkpoint in U2OS/MAD2L1BP cells. Cells were transfected with a blank vector or a plasmid expressing FLAG-p53. A plasmid expressing GFP-tagged histone H2B was co-transfected to serve as a transfection marker. MAD2L1BP was turned on with doxycycline together with treatment of either buffer or nocodazole for 48 h. The cell cycle profiles of the transfected cells (GFP-positive) were analyzed by flow cytometry. Total lysates were also prepared, and the expression of FLAG-tagged p53 and MAD2L1BP was confirmed by immunoblotting. C, inhibition of p53 attenuates the post-mitotic checkpoint in U2OS cells. Cells were transfected with a blank vector or a plasmid expressing HPV E6. A plasmid expressing GFP-tagged histone H2B was co-transfected to serve as a transfection marker. The cells were then treated with either buffer or nocodazole for 48 h, and the cell cycle profiles of the transfected cells were analyzed by flow cytometry.
Another prediction of the above hypothesis is that an increase of the basal level of p53 should strengthen the postmitotic checkpoint even when the spindle-assembly checkpoint is prematurely silenced. To test this, U2OS/MAD2L1BP cells were transfected with plasmids expressing FLAG-tagged p53 prior to nocodazole challenge. Fig. 5B shows that the control MAD2L1BP-expressing cells underwent re-replication just as before. In contrast, cells co-expressing FLAG-p53 were significantly more resistant to re-replication under identical conditions. In a converse experiment, the postmitotic checkpoint in the parental U2OS cells (with intact spindle-assembly checkpoint) was attenuated when p53 function was disrupted by the expression of HPV E6 protein (Fig. 5C). Taken together, these results indicate that the abundance of p53 and p21CIP1/WAF1 after mitotic slippage is the salient factor for the postmitotic checkpoint, which in turn is determined by the duration of the spindle-assembly checkpoint-mediated arrest.
Forced Mitotic Slippage by Inactivation of Cyclin B1-CDK1 Uncouples the p53-mediated Postmitotic Checkpoint—The above conclusions are predominantly based on the disruption of the spindle-assembly checkpoint with MAD2L1BP. It can be argued that the proper activation of the spindle-assembly checkpoint is required to generate the dependence of the postmitotic checkpoint on p53-p21CIP1/WAF1. To exclude this possibility, cells were first trapped in mitosis with nocodazole to allow the spindle-assembly checkpoint to be fully activated. Mitotic slippage was induced artificially by addition of a specific CDK1 inhibitor called RO3306 (42). A schematic diagram of the experimental design is shown in Fig. 6A. As expected, cells that were cultured continuously with nocodazole (48 h) displayed a 4N DNA content (Fig. 6B). In contrast, punctuation of the nocodazole block with a pulse of RO3306 resulted in re-replication. These data suggested that inhibition of cyclin B1-CDK1 induced premature slippage and compromised the function of the postmitotic checkpoint. In support of this interpretation, whereas histone H3Ser-10 phosphorylation was maintained up to 30 h in control cells, it was rapidly dephosphorylated after addition of RO3306 (Fig. 6C, compare lanes 3 and 7). Similarly, anaphase-promoting complex/cyclosome was turned on because its substrates such as cyclin B1, securin, and geminin were rapidly destroyed. In agreement with the premature slippage and re-replication, cyclin E1 accumulated shortly following RO3306 treatment. Despite the eventual activation of p53 and p21CIP1/WAF1 (in fact they accumulated to higher levels in the presence of RO3306), their levels were relatively low at the time of slippage (Fig. 6C, lanes 7 and 8). Thus similar to the conclusion reached by disrupting the spindle-assembly checkpoint with MAD2L1BP, these results indicate that premature slippage leads to postmitotic checkpoint failure chiefly because the p53-p21CIP1/WAF1 pathway did not have sufficient time to activate and overcome cyclin E1-CDK2.
FIGURE 6.
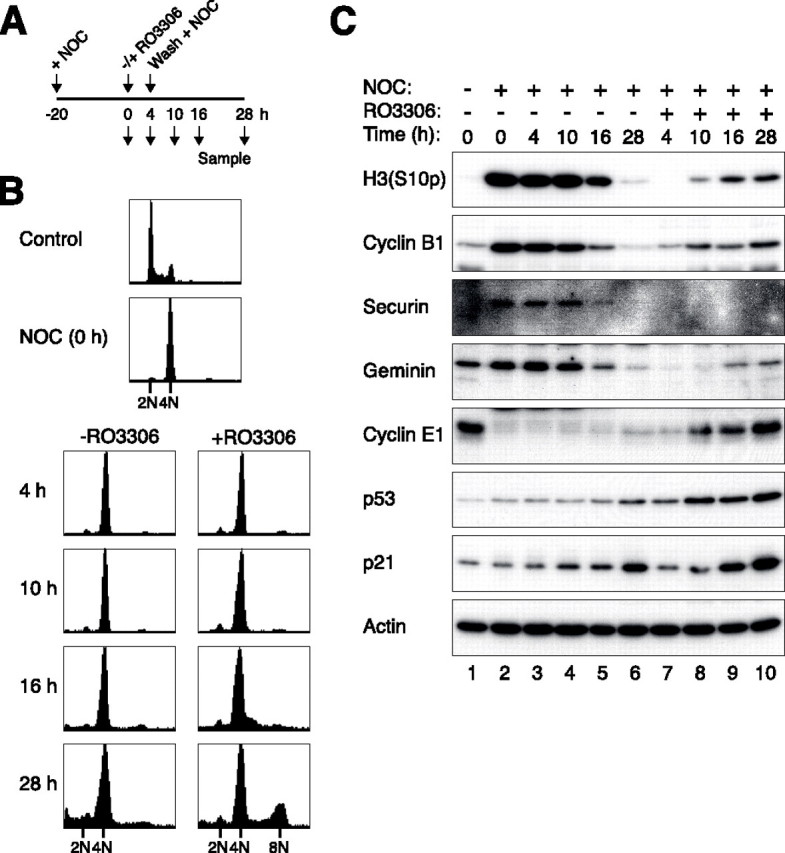
Forced mitotic slippage by an inhibitor of cyclin B1-CDK1 uncouples the postmitotic checkpoint. A, schematic diagram of experimental design. U2OS cells were treated with nocodazole (NOC) for 20 h before either buffer or RO3306 was added. After 4 h, the cells were washed and cultured in fresh medium supplemented with nocodazole. Samples were taken at the indicated time points for flow cytometry and immunoblotting analysis. B, RO3306 induces re-replication in nocodazole-blocked cells. Cells were treated as described in A and were subjected to flow cytometry analysis. C, RO3306-induced slippage activates p53 and p21CIP1/WAF1. Cells were treated as described in A. Cell-free extracts were prepared, and the indicated proteins were detected by immunoblotting.
Finally, we tested if elevating the cyclin E1 level could boost re-replication after checkpoint bypass. After expressing FLAG-tagged cyclin E1, U2OS cells were treated with nocodazole followed by a period of RO3306 as before. Fig. 7 shows that in comparison with vector-transfected cells, re-replication was boosted by the increased levels of cyclin E1.
FIGURE 7.
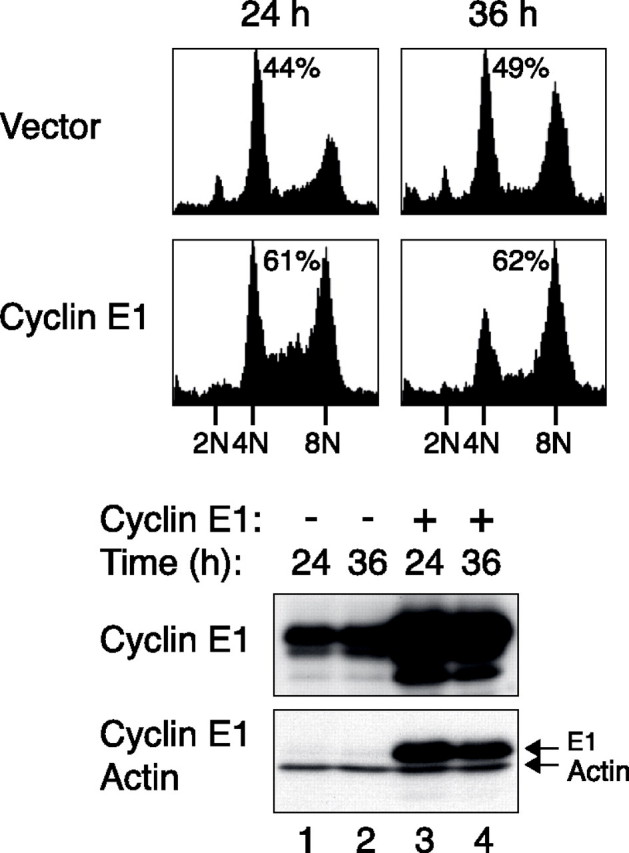
Re-replication after premature spindle-assembly checkpoint inactivation is promoted by increased expression of cyclin E1. U2OS cells were transfected with either a blank vector or a plasmid expressing FLAG-cyclin E1 (the expression of FLAG-cyclin E1 was turned on with doxycycline). A plasmid expressing GFP-tagged histone H2B was co-transfected to serve as a transfection marker. The cells were then treated with nocodazole for 20 h followed by addition of RO3306. After 4 h, the cells were washed and cultured in fresh medium supplemented with nocodazole. At 24 and 36 h, the cells were harvested, and the cell cycle profiles of the transfected cells (GFP-positive) were analyzed by flow cytometry. The percentage of cells with DNA contents greater than 4N is indicated. The expression of the recombinant cyclin E1 was confirmed by immunoblotting. Actin analysis confirmed constant loading of the extracts.
DISCUSSION
We demonstrated that p53 is stabilized and activated after prolonged activation of the spindle-assembly checkpoint. Activation of the p53 pathway was not limited to treatment with spindle poisons (Fig. 3) but also by knockdown of KIF11 (supplemental Fig. S7). Our results indicated that p53 starts to accumulate during the mitotic block and continues after mitotic slippage (supplemental Fig. S3). Slippage per se is not absolutely required for p53 activation, since cells that were released from the mitotic block into G1 also accumulated p53 (Fig. 2 and supplemental Fig. S4).
One of the most interesting recent findings is that spindle-assembly checkpoint-compromised cells can undergo DNA re-replication after mitotic slippage even if they possess functional p53 (20). This indicates that both functional spindle-assembly checkpoint and p53 are required for the postmitotic G1 checkpoint. To explain this, Vogel et al. (20) speculated that the transcriptional activity of p53 may be altered in spindle checkpoint-compromised cells. In disagreement with this idea, we found that the p53 downstream target p21CIP1/WAF1 was induced at the same rate in normal U2OS cells and in cells deficient in the spindle-assembly checkpoint (Fig. 4A). Importantly, this does not (as the data may have superficially suggested) imply an insignificant role of the p53-p21CIP1/WAF1 pathway in the postmitotic checkpoint. On the contrary, because the duration of the mitotic block was shortened in the absence of the spindle-assembly checkpoint, the cells expressed cyclin E and entered S phase before p21CIP1/WAF1 accumulated to any substantial level. These results also suggest that after the initiation of p53 stabilization, its accumulation rate is constant irrespective of the duration of the mitotic arrest or slippage.
An essential but yet unanswered question is how p53 is activated after spindle disruption. A number of mechanisms have been proposed, but the process remains mysterious and controversial. It has been suggested that spindle disruption leads to DNA breaks, which triggers a DNA damage response in G1 (43). Indeed, many studies that addressed the postmitotic checkpoint are complicated by the subsequent realization that many drug treatments and synchronization procedures in fact generated DNA damage (reviewed in Ref. 15). We did not obtain evidence of DNA damage after nocodazole treatment or KIF11 knockdown. Although histone H2AXSer-139 is highly phosphorylated during prolonged mitotic block (Fig. 1A and supplemental Fig. S1A) or after release from the block (Fig. 2B), we believed that it was mainly a reflection of apoptosis rather than DNA damage. No significant amount of DNA strand breaks was detected by comet assays.4 Although γ-H2AX is known to be a classic DNA damage marker, previous reports have shown that γ-H2AX is induced after initiation of DNA fragmentation (44) and is required for DNA ladder formation (37). In agreement with these studies, we found that most apoptotic cells, but not mitotic and interphase cells, were stained positive for γ-H2AX after nocodazole treatment.4 Importantly, caspase inhibitors could abolish the nocodazole-induced γ-H2AX without affecting p53 activation (Fig. 1C).
In support of the role of DNA damage in the postmitotic checkpoint, ATM has also been suggested to be an activator of p53 after spindle disruption. Tritarelli et al. (45) proposed a model that during normal mitosis, p53Ser-15 is phosphorylated by ATM and is localized to the centrosomes, where dephosphorylation of p53Ser-15 takes place (45). Spindle disruption inhibits the p53-centrosome association (38), thus preventing the dephosphorylation of p53Ser-15. Thus AT cells, which are unable to phosphorylate p53Ser-15, do not undergo postmitotic arrest after nocodazole block and release (45). In disagreement with these findings, Vogel et al. (20) found that several components of the DNA damage checkpoint (ATM, ATR, CHK1, and CHK2) are not required for the nocodazole-induced p53 activation. We were also not able to detect p53Ser-15 phosphorylation after nocodazole treatment, and we found that caffeine (an ATM/ATR inhibitor) could neither prevent p53 activation nor the postmitotic checkpoint response.4
Several recent studies have also suggested that aberrant centrosomes may underlie the signals that activate p53 during the postmitotic checkpoint (46–48). In this connection, a p38 mitogen-activated protein kinase-dependent stress-response pathway is believed to lie upstream of p53 (46, 48). Another candidate for activating p53, LATS2, is also connected to the centrosomes. Nocodazole induces LATS2 translocation from centrosomes to the nucleus, where it binds and inhibits MDM2 (49). This allows p53 to accumulate, which in turn up-regulates LATS2 in a positive feedback loop. Given that subtle treatments of the cells or even the act of imaging may activate checkpoints specifically in tetraploid cells (46, 50), further work is required to ascertain whether these suggestions are true or not.
Supplementary Material
Acknowledgments
We thank Talha Arooz, Anita Lau, and Sandy Siu for technical support.
This work was supported in part by Research Grants Council Grants HKUST6123/04M, HKUST6415/05M, and HKUST6439/06 M (to R. Y. C. P.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S7.
Footnotes
The abbreviations used are: HPV, human papillomavirus; GFP, green fluorescent protein; siRNA, small interfering RNA; GFP, green fluorescent protein.
Y. W. Chan and R. Y. C. Poon, unpublished data.
References
- 1.Rajagopalan, H., Nowak, M. A., Vogelstein, B., and Lengauer, C. (2003) Nat. Rev. Cancer 3 695–701 [DOI] [PubMed] [Google Scholar]
- 2.Kops, G. J., Weaver, B. A., and Cleveland, D. W. (2005) Nat. Rev. Cancer 5 773–785 [DOI] [PubMed] [Google Scholar]
- 3.Ganem, N. J., Storchova, Z., and Pellman, D. (2007) Curr. Opin. Genet. Dev. 17 157–162 [DOI] [PubMed] [Google Scholar]
- 4.Fujiwara, T., Bandi, M., Nitta, M., Ivanova, E. V., Bronson, R. T., and Pellman, D. (2005) Nature 437 1043–1047 [DOI] [PubMed] [Google Scholar]
- 5.Shi, Q., and King, R. W. (2005) Nature 437 1038–1042 [DOI] [PubMed] [Google Scholar]
- 6.Musacchio, A., and Salmon, E. D. (2007) Nat. Rev. Mol. Cell Biol. 8 379–393 [DOI] [PubMed] [Google Scholar]
- 7.Weaver, B. A., and Cleveland, D. W. (2005) Cancer Cell 8 7–12 [DOI] [PubMed] [Google Scholar]
- 8.Cross, S. M., Sanchez, C. A., Morgan, C. A., Schimke, M. K., Ramel, S., Idzerda, R. L., Raskind, W. H., and Reid, B. J. (1995) Science 267 1353–1356 [DOI] [PubMed] [Google Scholar]
- 9.Di Leonardo, A., Khan, S. H., Linke, S. P., Greco, V., Seidita, G., and Wahl, G. M. (1997) Cancer Res. 57 1013–1019 [PubMed] [Google Scholar]
- 10.Khan, S. H., and Wahl, G. M. (1998) Cancer Res. 58 396–401 [PubMed] [Google Scholar]
- 11.Lanni, J. S., and Jacks, T. (1998) Mol. Cell. Biol. 18 1055–1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Casenghi, M., Mangiacasale, R., Tuynder, M., Caillet-Fauquet, P., Elhajouji, A., Lavia, P., Mousset, S., Kirsch-Volders, M., and Cundari, E. (1999) Exp. Cell Res. 250 339–350 [DOI] [PubMed] [Google Scholar]
- 13.Sablina, A. A., Agapova, L. S., Chumakov, P. M., and Kopnin, B. P. (1999) Cell Biol. Int. 23 323–334 [DOI] [PubMed] [Google Scholar]
- 14.Andreassen, P. R., Lohez, O. D., Lacroix, F. B., and Margolis, R. L. (2001) Mol. Biol. Cell 12 1315–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ganem, N. J., and Pellman, D. (2007) Cell 131 437–440 [DOI] [PubMed] [Google Scholar]
- 16.Borel, F., Lohez, O. D., Lacroix, F. B., and Margolis, R. L. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 9819–9824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duelli, D. M., Hearn, S., Myers, M. P., and Lazebnik, Y. (2005) J. Cell Biol. 171 493–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stewart, Z. A., Leach, S. D., and Pietenpol, J. A. (1999) Mol. Cell. Biol. 19 205–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu, Y., Heilman, S. A., Illanes, D., Sluder, G., and Chen, J. J. (2007) Cancer Res. 67 2603–2610 [DOI] [PubMed] [Google Scholar]
- 20.Vogel, C., Kienitz, A., Hofmann, I., Muller, R., and Bastians, H. (2004) Oncogene 23 6845–6853 [DOI] [PubMed] [Google Scholar]
- 21.Yam, C. H., Ng, R. W., Siu, W. Y., Lau, A. W., and Poon, R. Y. C. (1999) Mol. Cell. Biol. 19 635–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chan, W. M., Siu, W. Y., Lau, A., and Poon, R. Y. C. (2004) Mol. Cell. Biol. 24 3536–3551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arooz, T., Yam, C. H., Siu, W. Y., Lau, A., Li, K. K., and Poon, R. Y. C. (2000) Biochemistry 39 9494–9501 [DOI] [PubMed] [Google Scholar]
- 24.Chow, J. P. H., Siu, W. Y., Ho, H. T. B., Ma, K. H. T., Ho, C. C., and Poon, R. Y. C. (2003) J. Biol. Chem. 278 40815–40828 [DOI] [PubMed] [Google Scholar]
- 25.Chi, T. Y., Chen, G. G., and Lai, P. B. (2005) Cancer Cell Int. 5 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Woo, R. A., and Poon, R. Y. C. (2004) Genes Dev. 18 1317–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ausubel, F., Brent, R., Kingston, R., Moore, D., Seidman, J., Smith, J., and Struhl, K. (1991) Current Protocols in Molecular Biology, John Wiley & Sons, Inc., New York
- 28.Poon, R. Y. C., Toyoshima, H., and Hunter, T. (1995) Mol. Biol. Cell 6 1197–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Siu, W. Y., Arooz, T., and Poon, R. Y. C. (1999) Exp. Cell Res. 250 131–141 [DOI] [PubMed] [Google Scholar]
- 30.Siu, W. Y., Lau, A., Arooz, T., Chow, J. P., Ho, H. T., and Poon, R. Y. C. (2004) Mol. Cancer Ther. 3 621–632 [PubMed] [Google Scholar]
- 31.Yam, C. H., Siu, W. Y., Kaganovich, D., Ruderman, J. V., and Poon, R. Y. C. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 497–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fung, T. K., Siu, W. Y., Yam, C. H., Lau, A., and Poon, R. Y. C. (2002) J. Biol. Chem. 277 35140–35149 [DOI] [PubMed] [Google Scholar]
- 33.Li, W., Lan, Z., Wu, H., Wu, S., Meadows, J., Chen, J., Zhu, V., and Dai, W. (1999) Cell Growth & Differ. 10 769–775 [PubMed] [Google Scholar]
- 34.Castedo, M., Coquelle, A., Vivet, S., Vitale, I., Kauffmann, A., Dessen, P., Pequignot, M. O., Casares, N., Valent, A., Mouhamad, S., Schmitt, E., Modjtahedi, N., Vainchenker, W., Zitvogel, L., Lazar, V., Garrido, C., and Kroemer, G. (2006) EMBO J. 25 2584–2595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tao, W., South, V. J., Zhang, Y., Davide, J. P., Farrell, L., Kohl, N. E., Sepp-Lorenzino, L., and Lobell, R. B. (2005) Cancer Cell 8 49–59 [DOI] [PubMed] [Google Scholar]
- 36.Rogakou, E. P., Pilch, D. R., Orr, A. H., Ivanova, V. S., and Bonner, W. M. (1998) J. Biol. Chem. 273 5858–5868 [DOI] [PubMed] [Google Scholar]
- 37.Lu, C., Zhu, F., Cho, Y. Y., Tang, F., Zykova, T., Ma, W. Y., Bode, A. M., and Dong, Z. (2006) Mol. Cell 23 121–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ciciarello, M., Mangiacasale, R., Casenghi, M., Zaira Limongi, M., D'Angelo, M., Soddu, S., Lavia, P., and Cundari, E. (2001) J. Biol. Chem. 276 19205–19213 [DOI] [PubMed] [Google Scholar]
- 39.Xia, G., Luo, X., Habu, T., Rizo, J., Matsumoto, T., and Yu, H. (2004) EMBO J. 23 3133–3143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Habu, T., Kim, S. H., Weinstein, J., and Matsumoto, T. (2002) EMBO J. 21 6419–6428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blangy, A., Lane, H. A., d'Herin, P., Harper, M., Kress, M., and Nigg, E. A. (1995) Cell 83 1159–1169 [DOI] [PubMed] [Google Scholar]
- 42.Vassilev, L. T., Tovar, C., Chen, S., Knezevic, D., Zhao, X., Sun, H., Heimbrook, D. C., and Chen, L. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 10660–10665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quignon, F., Rozier, L., Lachages, A. M., Bieth, A., Simili, M., and Debatisse, M. (2007) Oncogene 26 165–172 [DOI] [PubMed] [Google Scholar]
- 44.Rogakou, E. P., Nieves-Neira, W., Boon, C., Pommier, Y., and Bonner, W. M. (2000) J. Biol. Chem. 275 9390–9395 [DOI] [PubMed] [Google Scholar]
- 45.Tritarelli, A., Oricchio, E., Ciciarello, M., Mangiacasale, R., Palena, A., Lavia, P., Soddu, S., and Cundari, E. (2004) Mol. Biol. Cell 15 3751–3757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Uetake, Y., Loncarek, J., Nordberg, J. J., English, C. N., La Terra, S., Khodjakov, A., and Sluder, G. (2007) J. Cell Biol. 176 173–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Srsen, V., Gnadt, N., Dammermann, A., and Merdes, A. (2006) J. Cell Biol. 174 625–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mikule, K., Delaval, B., Kaldis, P., Jurcyzk, A., Hergert, P., and Doxsey, S. (2007) Nat. Cell Biol. 9 160–170 [DOI] [PubMed] [Google Scholar]
- 49.Aylon, Y., Michael, D., Shmueli, A., Yabuta, N., Nojima, H., and Oren, M. (2006) Genes Dev. 20 2687–2700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Uetake, Y., and Sluder, G. (2004) J. Cell Biol. 165 609–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.