

Abstract
The crystal structures of two ternary complexes of human spermine synthase (EC 2.5.1.22), one with 5′-methylthioadenosine and spermidine and the other with 5′-methylthioadenosine and spermine, have been solved. They show that the enzyme is a dimer of two identical subunits. Each monomer has three domains: a C-terminal domain, which contains the active site and is similar in structure to spermidine synthase; a central domain made up of four β-strands; and an N-terminal domain with remarkable structural similarity to S-adenosylmethionine decarboxylase, the enzyme that forms the aminopropyl donor substrate. Dimerization occurs mainly through interactions between the N-terminal domains. Deletion of the N-terminal domain led to a complete loss of spermine synthase activity, suggesting that dimerization may be required for activity. The structures provide an outline of the active site and a plausible model for catalysis. The active site is similar to those of spermidine synthases but has a larger substrate-binding pocket able to accommodate longer substrates. Two residues (Asp201 and Asp276) that are conserved in aminopropyltransferases appear to play a key part in the catalytic mechanism, and this role was supported by the results of site-directed mutagenesis. The spermine synthase·5′-methylthioadenosine structure provides a plausible explanation for the potent inhibition of the reaction by this product and the stronger inhibition of spermine synthase compared with spermidine synthase. An analysis to trace possible evolutionary origins of spermine synthase is also described.
Polyamines are essential for normal growth and development, and the polyamine biosynthetic pathway is an important target for the design of therapeutic agents (1, 2). The predominant polyamines in mammalian cells are spermidine (SPD)4 and spermine (SPM). These polyamines are made by the sequential addition of aminopropyl groups from decarboxylated S-adenosylmethionine (dcAdoMet). The addition of the first aminopropyl group to the diamine precursor putrescine is catalyzed by spermidine synthase (SpdSyn). The addition of a second aminopropyl group to the N-10 position5 of SPD to form SPM is catalyzed by spermine synthase (SpmSyn) (Fig. 1). The critical importance of polyamines is demonstrated by studies using mice with gene deletions of ornithine decarboxylase (3) or S-adenosylmethionine decarboxylase (AdoMetDC) (4), both of which are lethal at the earliest stages of embryonic development. No SpdSyn knock-out mice have been described, but studies in yeast show that this gene is required for viability and that SPD plays an essential role as a precursor of hypusine formed in the post-translational modification of eukaryotic initiation factor 5A (5). SpmSyn has received relatively little attention compared with the large number of publications describing ornithine decarboxylase, AdoMetDC, and SpdSyn (1, 6-8). The SpmSyn gene is not essential for growth of yeast (9). Mutants lacking the AdoMetDC gene, which cannot make SPD or SPM, grow normally when only SPD is provided in the media (10). However, in higher organisms, although not absolutely essential for life, SPM is required for normal development. Although cultured mouse cell lines with an inactivated SpmSyn gene grow at a normal rate in the complete absence of SPM (11-13), the loss of SPM has profound effects in mice. Male Gy mice, which have an X chromosome deletion that includes the SpmSyn gene, do survive. However, these mice, the tissues of which contain no SPM, are viable only on a B6C3H background and have a greatly reduced size, sterility, deafness, neurological abnormalities, and a very short life span (11, 14). Deletion of both of the genes, which were originally described as encoding SpmSyns in plants, also leads to developmental defects and sensitivity to drought but is not lethal (15-17). Recently, it has been demonstrated that one of these genes actually encodes thermospermine synthase (18). The relative roles of SPM and thermospermine in plant development are not well understood.
FIGURE 1.

General pathway for polyamine synthesis. ODC, ornithine decarboxylase.
An alteration in the human SpmSyn gene at Xp22.1 is the cause of Snyder-Robinson syndrome (19). This X-linked recessive condition causes mild-to-moderate mental retardation, hypotonia, cerebellar circuitry dysfunction, facial asymmetry, thin habitus, osteoporosis, and kyphoscoliosis. The mutation causes a splice variant of the SpmSyn gene that leads to the loss of exon 4 and inserts a premature stop codon, resulting in a truncated protein containing only the first 110 amino acids. SpmSyn activity in cells derived from Snyder-Robinson patients is reduced by 85-90%. This activity may be due to a small level of correct splicing.
SpmSyn and SpdSyn are members of the family of aminopropyltransferases. These enzymes all transfer aminopropyl groups from dcAdoMet to amine acceptors. Representatives from four groups of this family have been characterized according to their preferred amine acceptor. In addition to SpdSyn (which utilizes putrescine) and SpmSyn (which adds the aminopropyl group to N-10 of SPD5) (reviewed in Refs. 20 and 21), aminopropylagmatine synthase uses agmatine (decarboxylated arginine) (22), and thermospermine synthase adds the aminopropyl group to N-1 of SPD5 (18). Structures have been determined by x-ray crystallography for a number of SpdSyns, including those from Thermotoga maritima (23), Caenorhabditis elegans (24), Plasmodium falciparum (25), Helicobacter pylori (26), and human (27), and for the aminopropylagmatine synthases from Thermus thermophilus (Protein Data Bank code 1UIR) (22) and Pyrococcus furiosus (28). A general mechanism for aminopropyl transfer has been proposed based on the human and T. maritima SpdSyn structures bound to substrates and inhibitors and the results of site-directed mutagenesis of key residues (27).
Although the crystal structures of the three mammalian enzymes responsible for the synthesis of SPD (ornithine decarboxylase (29), AdoMetDC (30), and SpdSyn (27)) have been determined and have provided much information on their substrate-binding specificity and mechanisms of catalysis and regulation, eukaryotic SpmSyns have so far eluded any attempts for the solution of a crystal structure. In this study, we describe the structure of the remaining enzyme in the mammalian polyamine biosynthetic pathway, human SpmSyn.
Here we report the crystal structures of two ternary complexes of human SpmSyn, one with the bound products 5′-methylthioadenosine (MTA) and SPM and the other one with the substrate SPD and the product MTA. Our studies show that human SpmSyn is a homodimer, with each monomer having three domains. The C-terminal domain, which contains the active site, adopts a classical AdoMet methyltransferase fold and exhibits structural similarity to known SpdSyns. Remarkably, the N-terminal domain, which forms the major part of the dimerization interface, shows a considerable structural similarity to the AdoMetDC-like fold. When expressed separately, both domains are enzymatically inactive and possess rigid tertiary structure, suggesting that human SpmSyn is a fusion protein of AdoMet methyltransferase-like and AdoMetDC-like folds. Key residues in the active site that allow catalysis and the ability to accommodate SPD as the preferred substrate were identified and confirmed by site-directed mutagenesis. The results of site-directed mutagenesis support a common mechanism for aminopropyltransferases.
EXPERIMENTAL PROCEDURES
Materials—[35S]dcAdoMet was synthesized enzymatically from l-[35S]methionine (PerkinElmer Life Sciences) (31). Unlabeled dcAdoMet was kindly provided by Professor A. Shirahata (Department of Biochemistry, Faculty of Pharmaceutical Sciences, Josai University, Saitama, Japan).
Expression, Purification, and Crystallization of Human SpmSyn—A DNA fragment encoding human SpmSyn was amplified by PCR and subcloned into the pET28a-LIC vector downstream of the polyhistidine coding region. Human SpmSyn was expressed in the Escherichia coli BL21-Codon Plus(DE3)-RIL strain (Stratagene) by the addition of 1 mm isopropyl 1-thio-β-d-galactopyranoside and incubated overnight at 15 °C. Harvested cells were resuspended in phosphate-buffered saline (pH 7.4) supplemented with 250 mm NaCl, 5 mm imidazole, 2 mm β-mercaptoethanol, 5% glycerol, and 1 mm phenylmethylsulfonyl fluoride. The cells were lysed by passing through a Microfluidizer (Microfluidics). The lysate was loaded onto a HiTrap chelating column (Amersham Biosciences) charged with Ni2+. The column was washed with 10 column volumes of 20 mm Tris-HCl (pH 8.0) containing 250 mm NaCl, 50 mm imidazole, and 5% glycerol, and the protein was eluted with elution buffer (20 mm Tris-HCl (pH 8.0), 250 mm NaCl, 250 mm imidazole, and 5% glycerol). The protein was loaded onto a Superdex 200 column (Amersham Biosciences) equilibrated with 20 mm Tris-HCl (pH 8.0) and 150 mm NaCl. Thrombin (Sigma) was added to combined fractions containing SpmSyn to remove the His tag. The protein was further purified to homogeneity by ion-exchange chromatography. The purification yield of SpmSyn was 2.3 mg/liter of culture.
Purified SpmSyn protein was crystallized in the presence of SPD and MTA or SPM and MTA using the hanging drop vapor diffusion method at 20 °C by mixing 1 μl of the protein solution (10 mg/ml) with 1 μl of the reservoir solution. The human SpmSyn·MTA·SPD complex (protein/MTA/SPD molar ratio of 1:5:10) was crystallized in 12% polyethylene glycol 20,000 and 0.1 m MES (pH 6.5). The human SpmSyn·MTA·SPM complex (protein/MTA/SPM molar ratio of 1:5:5) was crystallized in 18% polyethylene glycol 3,350, 0.1 m NaCl, and 0.1 m BisTris (pH 6.5). Crystals were soaked in the corresponding mother liquor supplemented with 20% glycerol as cryoprotectant before freezing in liquid nitrogen. Ligands were not added to the cryo solution to prevent possible over-occupancy of the active site of the enzyme.
Data Collection and Structure Determination—X-ray diffraction data were collected at 100 K using a Rigaku FRE high brilliance x-ray generator with an R-AXIS IV detector. Data were processed using the HKL2000 suite (32). Structures of SpmSyn complexes were solved by molecular replacement using the program MOLREP (33). The crystal structure of human SpdSyn (Protein Data Bank code 2O06) (27) was used as the search model. ARP/wARP (34) was used for automatic model building. The graphic program COOT (35) was used for model building and visualization.
Assay of Aminopropyltransferase Activity—Activity was measured by following the production of [35S]MTA from [35S]dcAdoMet in 100 mm sodium phosphate buffer (pH 7.5) in the presence of SPD as the amine acceptor (31). Reactions were run for 30 min with an amount of enzyme that gave a linear rate of MTA production at 37 °C, and kinetic constants were determined using triplicate estimations at each concentration point, which covered an 8-fold range above and below the Km (27, 31). The results of the triplicate assays agreed within ±10%.
Production of SpmSyn Mutants—The human N-terminally truncated SpmSyn mutants were generated by PCR and subcloned into the pET28a-LIC vector. All site-directed mutants of human SpmSyn described in this work were generated by the QuikChange site-directed mutagenesis kit (Stratagene) using the pQE-hSpS plasmid (36) as a template. The entire coding sequence of each of the SpmSyn mutants was verified by DNA sequencing to ensure that no other mutations were introduced during PCR. All SpmSyn mutant proteins were purified as described above before assays.
Assay of AdoMetDC Activity—AdoMetDC was assayed by measuring the release of 14CO2 from S-adenosyl-l-[carboxy-14C]methionine (37) using human recombinant carboxyl-terminally His6-tagged AdoMetDC as described previously (38).
Secondary and Tertiary Structure Analysis—Thermo-melting profiling of all N-terminally truncated SpmSyn proteins was performed using an Aviv Model 215CD spectrophotometer. The proteins were heated to 95 °C and scanned at a fixed wavelength. The fluorescence spectra of the SpmSyn proteins were obtained on a SpectraMax Gemini XS (Molecular Devices) in the presence of different concentrations of urea in the protein samples.
RESULTS
Human SpmSyn Is a Dimer—To determine the three-dimensional structure of human SpmSyn, we generated a series of constructs encoding different lengths of the protein. The protein lacking the first 4 amino acids after removal of the His tag and in complex with either MTA and SPD or MTA and SPM produced crystals suitable for x-ray data collection. The structures of both ternary complexes, SpmSyn·MTA·SPD and SpmSyn·MTA·SPM, were solved by molecular replacement and refined to 1.95- and 2.45-Å resolution, respectively (Table 1). The SpmSyn·MTA·SPD complex was crystallized in space group P1 with four protein, four MTA, and four SPD molecules/asymmetric unit, whereas the SpmSyn·MTA·SPM complex was crystallized in space group P32 with four protein, four MTA, and four SPM molecules/asymmetric unit.
TABLE 1.
Crystallography data and refinement statistics
|
SpmSyn·MTA·SPM |
SpmSyn·MTA·SPD |
|
|---|---|---|
| Protein Data Bank code | 3C6M | 3C6K |
| Data collection | ||
| Space group | P32 | P1 |
| Cell dimensions | ||
| a, b, c (Å) | 88.26, 88.26, 197.67 | 43.39, 74.03, 143.23 |
| α, β, γ | 90°, 90°, 120° | 94.2°, 92.9°, 107.1° |
| Resolution (highest resolution shell; Å) | 50.00-2.45 (2.54-2.45) | 50.00-1.95 (2.02-1.95) |
| Measured reflections | 293,197 | 416,665 |
| Unique reflections | 61,292 | 115,647 |
| Rmerge (%) | 12.5 (58.7) | 10.7 (37.9) |
| I/σI | 7.6 (2.6) | 6.8 (2.7) |
| Completeness (%) | 97.2 (93.4) | 94.3 (66.9) |
|
Redundancy
|
4.8 (4.4)
|
3.6 (2.9)
|
| Refinement | ||
| Resolution (Å) | 35.65-2.45 | 40.56-1.95 |
| No. reflections (test set) | 58,198 (3,045) | 109,886 (5,758) |
| Rwork/Rfree (%) | 20.9/27.1 | 19.8/25.7 |
| No. atoms | ||
| Protein | 9,958 | 10,972 |
| Cofactor | 136 | 120 |
| Water | 300 | 1,164 |
| 〈B〉-factors (Å2) | 33.3 | 23.3 |
| Protein | 32.7 | 22.6 |
| Cofactor | 21.0 | 16.2 |
| Water | 27.7 | 30.8 |
| r.m.s.d. | ||
| Bond length (Å) | 0.013 | 0.017 |
| Bond angle | 1.48° | 1.67° |
| Ramachandran plot % residues | ||
| Favored | 90.8 | 90.9 |
| Additionally allowed | 8.9 | 8.8 |
| Generously allowed | 0.3 | 0.3 |
| Disallowed | None | None |
Human SpmSyn, a protein with a molecular mass of 41 kDa, exists as a dimer in solution. The protein stays as a dimer even when the protein concentration is lowered to ∼2 μg/ml (supplemental Fig. S1), suggesting a strong dimerization mode. In both crystal structures, the protein exists as a dimer. It dimerizes mainly through the N-terminal domains (Fig. 2, A and B). After dimerization, ∼16% of the total surface area for each subunit is buried in both SpmSyn·MTA·SPD and SpmSyn·MTA·SPM structures. Of the buried surface area, ∼6% was from the central domain, ∼13% was from the C-terminal domain, and the rest was from the N-terminal domain. The dimerization interface is very extensive and includes sets of hydrophobic interactions (65.4% of the interface interaction) and hydrogen bonding interactions (34.6%). Human SpmSyn Monomer Is a Fusion of AdoMetDC- and AdoMet Methyltransferase-like Folds—The overall structures of the monomers of each ternary complex of human SpmSyn are identical. All four molecules can be superimposed with a root mean square deviation (r.m.s.d.) of ∼0.3 Å for all C-α atoms. Human SpmSyn consists of three domains (Fig. 2, C and D). The N-terminal domain (amino acids 1-117) includes seven β-strands and two α-helices. β-Strands 1-5 of the N-terminal domain are aligned in antiparallel fashion, whereas short β-strands 6 and 7 are aligned head to tail and extend the five-stranded antiparallel β-sheet, which is packed from one side against two α-helices (α1 and α2). The N-terminal domain is connected by a long loop (amino acids 118-137) to the central β-strand domain (amino acids 138-172), consisting of four antiparallel β-strands (β8-β11) that serve as a lid for the C-terminal catalytic domain (amino acids 173-366). The C-terminal domain consists of a central, mainly parallel, seven-stranded β-sheet (β12-β18) that is sandwiched by three α-helices on each face (α3-α5 and α6-α8) and one helix (α9) inserted between β-strands 17 and 18. The order of the β-strands is 14-13-12-15-16-18-17, where all strands, except β18, are in parallel orientation; this embellishment is analogous to the canonical AdoMet methyltransferase fold (39).
FIGURE 2.
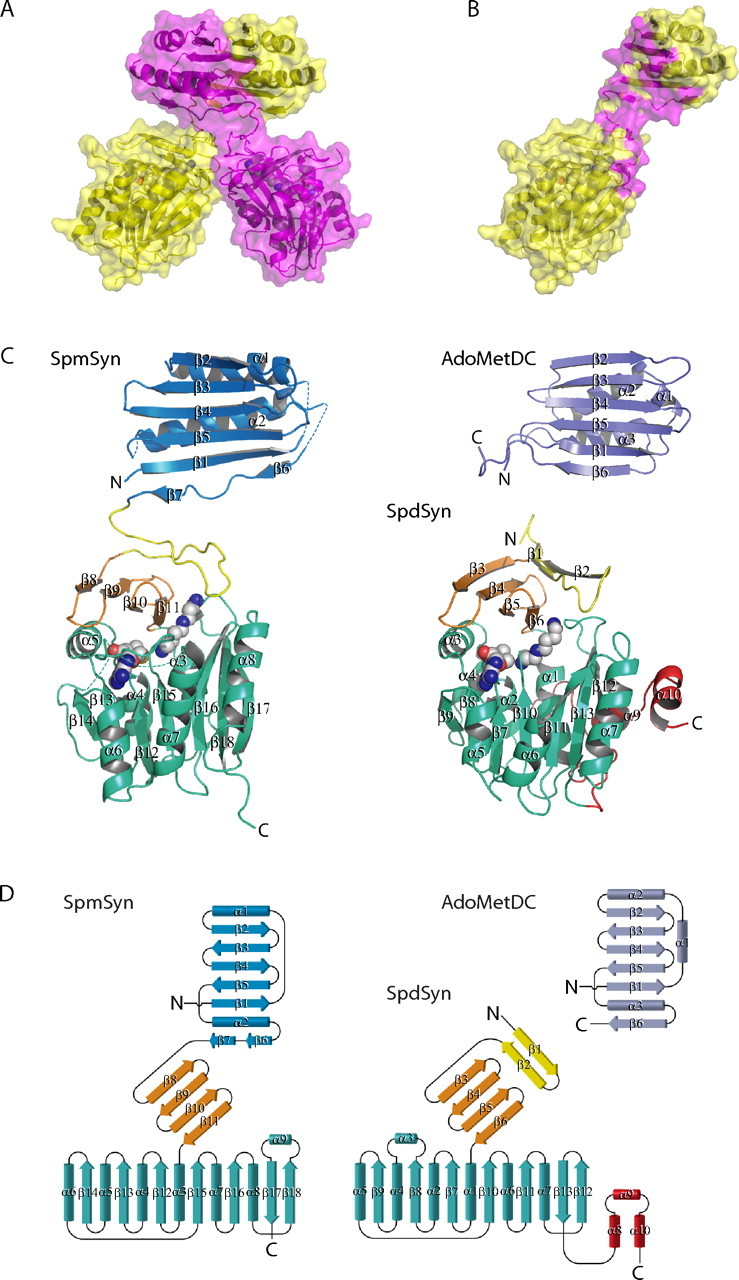
Structure of SpmSyn and structural homology to AdoMetDC and SpdSyn. A, dimer of SpmSyn. The two chains are in yellow and magenta. The enzyme is shown in schematic and surface representations; the bound molecules of SPD and MTA are shown in sphere representation, with carbon atoms in gray. B, monomer of SpmSyn with the dimerization interface in magenta. C, structures of the SpmSyn monomer, the SpdSyn monomer (Protein Data Bank code 2O06), and AdoMetDC (Protein Data Bank code 1TLU) shown in schematic representation. The N-terminal, central, and C-terminal domains in SpmSyn are in blue, orange, and greencyan, respectively. The loop connecting the N-terminal and central domains is in yellow. AdoMetDC is in purple. The SpdSyn domain color coding is the same as for SpmSyn, except for the first two β-strands, which structurally align with long interdomain loop of SpmSyn, are in yellow, and three C-terminal helices, which are absent in SpmSyn, are in red. SPD and MTA bound to SpmSyn and SpdSyn are shown in sphere representation, with carbon atoms in gray. The figures were generated using the program PyMOL (DeLano Scientific, San Carlos, CA). D, topology diagram for SpmSyn, SpdSyn, and AdoMetDC. All of the secondary structure elements are labeled. All of the domains in each molecule are colored as described for C.
Dali analysis (40) of the monomer of human SpmSyn did not identify any structural homologs. Analysis of the individual domains revealed that, remarkably, the N-terminal domain of human SpmSyn has significant structural homology to the AdoMetDC proenzyme from T. maritima (Protein Data Bank code 1TLU; Z-score of 10.6) (41) and human AdoMetDC (Protein Data Bank code 1JEN; Z-score of 9.4) (42) (Fig. 2, C and D). All of the C-α atoms of human SpmSyn N-terminal domain can be superimposed with T. maritima AdoMetDC with an r.m.s.d. of 2.8 Å and with human AdoMetDC with an r.m.s.d. of 3.0 Å. The structure formed by the central and C-terminal domains of human SpmSyn is very similar to that of human SpdSyn (Protein Data Bank code 2O07; Z-score of 22.0) (27), and the two structures can be superimposed with an r.m.s.d. of 1.9 Å for all C-α atoms (Fig. 2, C and D). A multiple sequence alignment of representative members of the SpmSyn family combined with secondary structure elements of human SpmSyn is shown in Fig. 3.
FIGURE 3.

Multiple sequence alignment of representative members of SpmSyn family. Identical residues are colored in red in the alignment. Secondary structure elements of human SpmSyn were assigned by the PROCHECK program (58) and are shown above the sequences and labeled: the helices are shown as cylinders, and the strands are shown as arrows. The three domains in human SpmSyn are colored in blue, orange, and greencyan. Catalytic residues are labeled with asterisks; the residues interacting with SPD and SPM are labeled with orange arrowheads; and the residues involved in MTA binding are labeled with blue arrowheads. The alignment was generated using ClustalW (59) and assisted with hand fittings. The sequences shown are from human (Homo sapiens; NP_004586), chicken (Gallus gallus; NP_001025974), zebrafish (Danio rerio; NP_571831), fruit fly (Drosophila melanogaster; NP_729798), mosquito (Anopheles gambiae; XP_315341), bee (Apis mellifera; XP_393567), sea urchin (S. purpuratus; XP_789223, 25 amino acids truncated from the C terminus), sea anemone (N. vectensis; XP_001636780, N terminus extended 50 amino acids using expressed sequence tag DV091768), M. brevicollis (Joint Genome Institute protein ID 30201), A. thaliana (NP_568785), and Saccharomyces cerevisiae (AAC19368).
To address the functional role of the dimerization of SpmSyn, we generated a series of deletion mutants of human SpmSyn including the N-terminal domain (amino acids 1-128) and the central and C-terminal domains (amino acids 130-366) and tested their structural properties and enzymatic activity (Fig. 4). Deletions of all or part of the N-terminal domain led to the protein existing as a monomer as determined by gel filtration analysis (data not shown) and to virtually complete loss of activity (Fig. 4A). This is consistent with the concept that the dimerization is required for the activity of the enzyme. Neither the N-terminal domain alone nor the central and C-terminal domain fragment alone had any significant SpmSyn activity. The importance of the C-terminal domain is shown by the result from the truncation of the protein at position 346 (mutant C347Stop); removing the last 20 residues led to a complete loss of activity. A smaller truncation of only 9 residues (mutant Y358Stop) had a smaller effect but still reduced activity by ∼75%.
FIGURE 4.
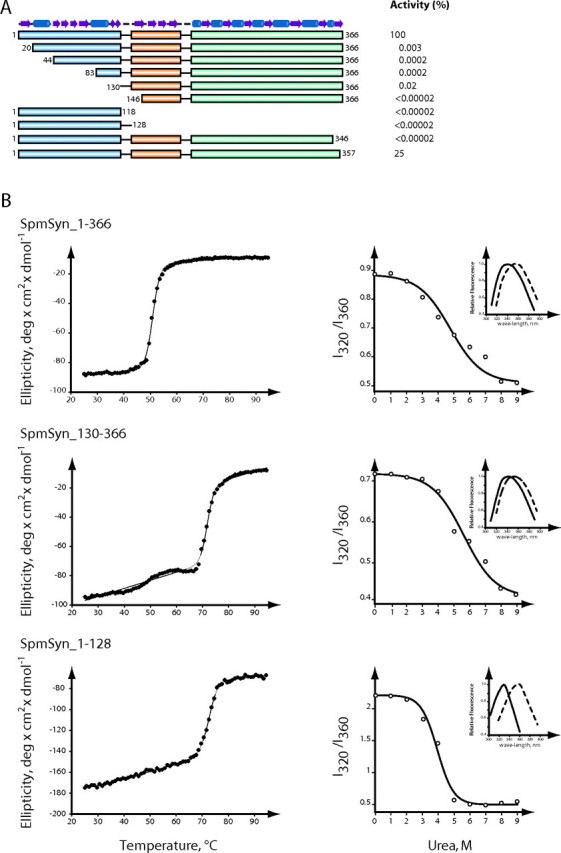
Enzymatic activity and tertiary structure of SpmSyn fragments. A, activity of each truncated protein shown as the percentage of the activity of the full-length enzyme. The three domains are colored in blue, orange, and greencyan. The secondary structure elements of SpmSyn are shown as cylinders (for helices) and arrows (for strands) on the top.B, analysis of the tertiary structure of full-length SpmSyn and its domains. The left panels show the thermo-melting profiles of full-length SpmSyn and its fragments. The ellipticity was measured at 220 nm. The right panels show the dependence of fluorescence spectra (I320/I360) of human SpmSyn and its fragments in the presence of different concentrations of urea. Insets show the fluorescence spectra in native buffer (20 mm Tris-HCl (pH 7.5) and 150 mm NaCl; solid lines) and in the presence of 9 m urea (dashed lines). The boundary of each construct is labeled on top of the profile. deg, degrees.
It is known that rigid tertiary structure in globular proteins is characterized by cooperative transition upon unfolding of proteins by increasing temperature or denaturing chemicals (43, 44). We observed cooperative transition of unfolding of both the N-terminal domain (amino acids 1-128) and central and C-terminal domain (amino acids 130-366) fragments upon melting by temperature monitored by ellipticity at 220 nm, as well as urea-induced unfolding monitored by tryptophan fluorescence spectra (Fig. 4B). Thus, both of these fragments have rigid tertiary structures, supporting the idea that human SpmSyn is a fusion of two proteins.
Mixing of the fragments did not greatly alter the activity from that expected for the two pieces individually. For example, adding up to 10 μg of the N-terminal domain (amino acids 1-128) to 2 ng of the full-length protein had no inhibitory effect, and mixing up to 100 μg of the N-terminal domain (amino acids 1-118 or 1-128) with the central plus C-terminal domain fragment (amino acids 130-366 or 146-366) did not restore activity. The addition of any of the fragments including the N-terminal domain (amino acids 1-128) or the full-length protein to purified human AdoMetDC did not reduce the AdoMetDC activity, and the addition of AdoMetDC did not alter the SpmSyn activity of full-length SpmSyn. These results suggest there is no direct interaction between the two polyamine biosynthetic enzymes, but it is possible that interaction occurs only during folding of the nascent protein and may require co-translation. None of the SpmSyn constructs had any AdoMetDC activity (data not shown). This is consistent with the absence from the N-terminal domain of a serine that forms the precursor of the pyruvate cofactor of AdoMetDC (41, 45).
Active Site of Human SpmSyn—The active site of SpmSyn is clearly defined by the binding of MTA, SPD, and SPM (Fig. 5). It is formed between the central β-sheet domain, which forms a lid on top of the active site, and the C-terminal domain.
FIGURE 5.
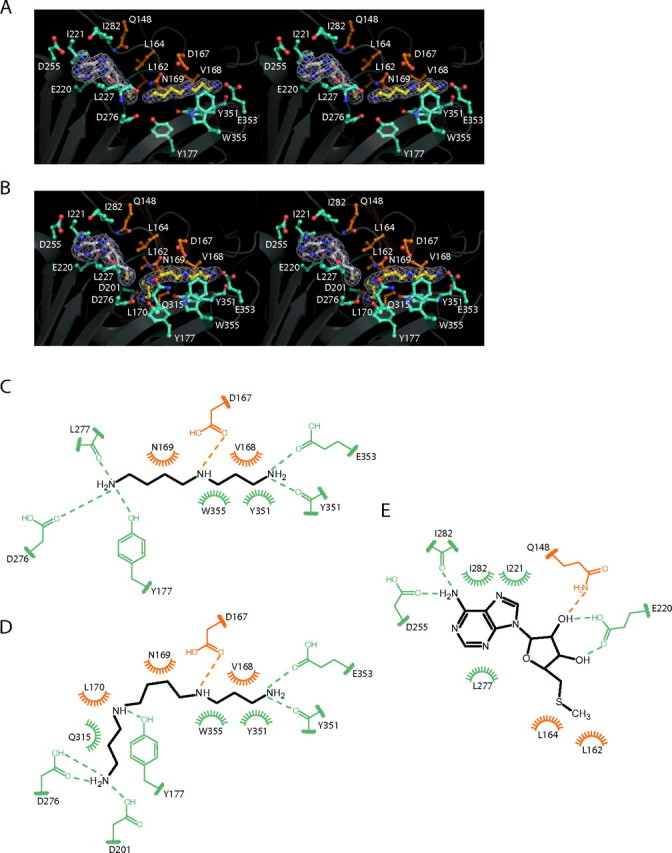
Active site of SpmSyn. A and B, stereo views of the active sites of SpmSyn in the SpmSyn·MTA·SPD and SpmSyn·MTA·SPM ternary complexes, respectively. The enzyme is shown in schematic representation. The MTA, SPD, and SPM molecules are shown in stick-and-ball representation, with carbon atoms in MTA in gray and carbon atoms in SPD and SPM in yellow. The 2mFo - DFc map of each ligand is shown in mesh (contoured to 1.0σ). All of the residues interacting with the ligands are shown in stick-and-ball representation. The carbon atoms of residues from the central domain of SpmSyn are in orange, whereas the carbon atoms of residues from the C-terminal domain are in greencyan. C-E, schematic illustrations of interactions between the activesite residues and SPD, SPM, and MTA, respectively. Dashed lines represent hydrogen bonds, and arcs represent hydrophobic interactions. The residues from the central and C-terminal domains are in orange and greencyan.
The adenine ring of MTA is placed between Ile221 and Ile282 on one side and Leu227 on the other side (Fig. 5, A, B, and E). N-6 of the MTA adenine forms hydrogen bonds with the side chain of Asp255 and the backbone carbonyl of Ile282. The ribose ring of MTA establishes hydrogen bonds with the side chains of Gln148 and Glu220. The CH3S moiety of MTA makes van der Waals contacts with Leu162 and Leu164, which are highly conserved in SpmSyns from different species (Fig. 3) and thus play an important role in orienting the reactive thiol alkane group for aminopropyl transfer. The MTA-binding site is next to the amine substrate-binding site, placing the sulfur atom of MTA close to the N-10 amino group of SPD, with a sulfur-nitrogen distance of 3.77 Å.
SpmSyn is strongly inhibited by MTA with a Ki of ∼0.3 μm (21, 46-48). This inhibition does not have great importance in limiting SPM synthesis in vivo because MTA is normally rapidly degraded by MTA phosphorylase (49). However, inhibition of this enzyme allows MTA to accumulate with deleterious effects on polyamine content, and very potent MTA phosphorylase inhibitors are now in clinical development as anti-tumor agents (50). MTA is a much less potent inhibitor of SpdSyns with a Ki of 2-10 μm (21, 48, 51). Analysis of crystal structures of human SpdSyn (27) and SpmSyn provides an explanation for this difference. MTA from both structures super-imposes very well and has similar interaction networks in the SpdSyn and SpmSyn structures (Fig. 6A). However, hydrophobic surfaces involved in interaction with the adenine ring of MTA are very different (Fig. 6, B and C). The hydrophobic surface (in magenta) made by Ile221 and Leu282 in SpmSyn is larger than the corresponding one in SpdSyn (Ile125). On the other side of the adenine ring of MTA, the hydrophobic surface (in yellow) of SpdSyn made by Ile100 and Leu184 is shifted and partially covers the adenine ring of MTA, whereas in the SpmSyn structure, Leu227 is positioned to cover the adenine ring completely. Thus, MTA is a more potent inhibitor for SpmSyn than for SpdSyn due to extensive hydrophobic interaction with the adenine ring of MTA.
FIGURE 6.
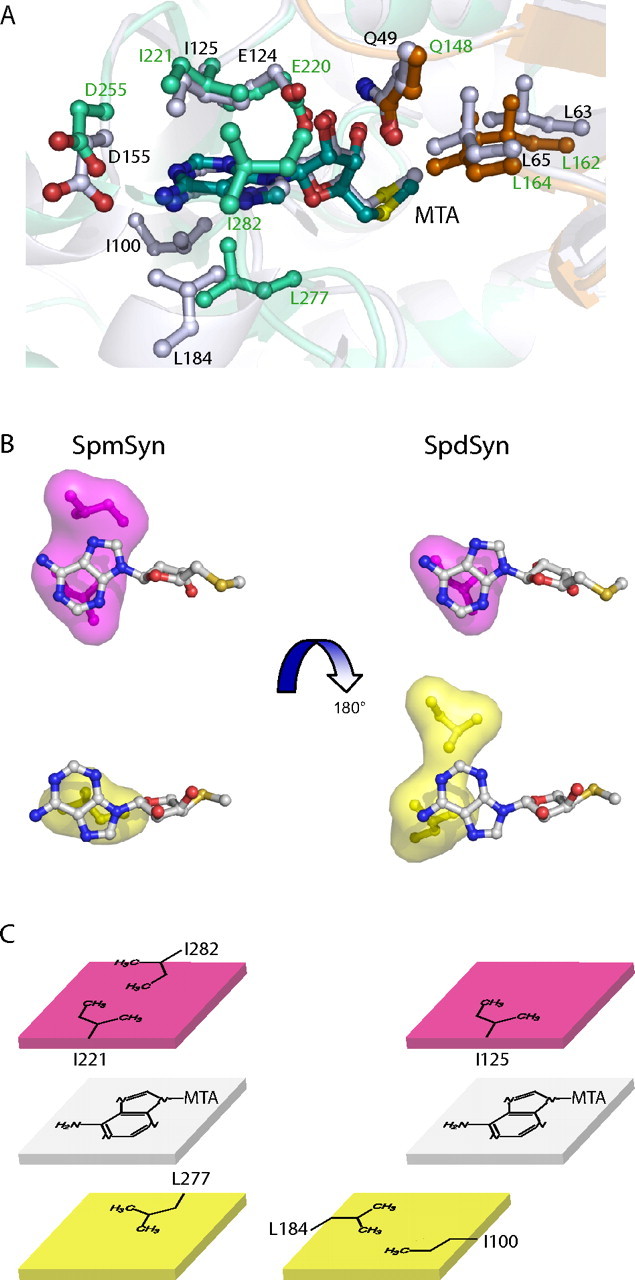
Structural basis for inhibition of SpmSyn by MTA. A, superimposition of MTA-binding sites in SpmSyn and SpdSyn. The structures of SpmSyn (Protein Data Bank code 3C6K) shown in greencyan and SpdSyn (Protein Data Bank code 2O0L) shown in gray are shown in schematic representation and superimposed. MTA and the residues interacting with MTA in both structures are shown in stick-and-ball representation, and the carbon atoms are in the same color as the corresponding structure, except for the three residues from the central domain of SpmSyn, which are in orange. B and C, illustrations of the hydrophobic surfaces interacting with the adenine ring in MTA. The two surfaces stacking with the adenine ring are in magenta and yellow. MTA is shown in stick-and-ball representation, with carbon atoms in gray.
SPD in the SpmSyn·MTA·SPD ternary complex adopts a liner conformation (Fig. 5A). The aminopropyl moiety of SPD lies on the hydrophobic bed formed by the side chain of Tyr351 and is flanked on both sides by Trp355 and Val168. SPD N-1 is anchored through hydrogen bonds with the backbone carbonyl of Tyr351 and the side chain of Glu353. N-5 of SPD forms a hydrogen bond with the side chain of Asp167. The -(CH2)4 portion of SPD formed by C-6 -C-9 makes van der Waals contacts with the side chain of Asn169. The SPD N-10 atom participates in a network of hydrogen bonds involving the side chains of Asp276 and Tyr177 and the backbone carbonyl of Leu277 (Fig. 5, A and C). These extensive interactions position the SPD molecule for aminopropyl transfer.
Strikingly, the SPM molecule in the SpmSyn·MTA·SPM structure is found in a bent conformation (Fig. 5, B and D). It bends at the -(CH2)4 moiety even though it has almost the same interaction network as SPD in the SpmSyn·MTA·SPD structure with the exception that Asp276 and Asp201 make hydrogen bonds with N-14 of SPM, and the added aminopropyl moiety makes van der Waals contacts with Leu170 and the amide side chain of Gln315.
The SPD-binding pocket revealed by the structure of the human SpmSyn·MTA·SPD complex is a narrow channel that leads to the MTA-binding pocket with the amino group of SPD containing N-10 pointing to the sulfur atom of the MTA molecule (Fig. 7A). The N-10 atom of SPD is located in a highly negatively charged environment. This provides an ideal environment for deprotonation of the reactive amino group of SPD.
FIGURE 7.

dcAdoMet-binding site of SpmSyn and illustration of the general catalytic mechanism for aminopropyl transfer. A, surface of the substrate-binding pocket of human SpmSyn with SPD and MTA shown in stick-and-ball representation. The carbon atoms of MTA are gray, whereas the carbon atoms of SPD are yellow. Negatively charged surfaces are red. The residues involved in the catalytic reaction are shown in stick-and-ball representation, with carbon atoms in gray. B, superimposition of SpdSyn and SpmSyn active sites. The structures of SpmSyn (greencyan) and SpdSyn (gray) are shown in schematic representation and superimposed. SPM and MTA bound to SpmSyn and dcAdoMet bound to SpdSyn are shown in stick-and-ball representation. The key residues lined in the pocket for aminopropyl group transfer are also shown in stick-and-ball representation. C, reaction mechanism. The spermidine and dcAdoMet substrates are shown in black, and the key protein residues are shown in green. The red arrows indicate the proposed attack by the spermidine amino group on the methylene carbon of the aminopropyl group. This attack is facilitated by interactions with the carbonyl group of Asp276 (which is shown in the charged state), the hydroxyl of Tyr177, and the carbonyl group of Leu277, which deprotonate the attacking N-10 atom.
The SPM-binding pocket is next to the sulfur group of MTA and supports the scenario that the aminopropyl moiety in dcAdoMet occupies the same pocket. Because of the structural similarity between the SpmSyn and SpdSyn catalytic domains, we compared these two structures to gain information on the binding pocket of dcAdoMet, even though we have so far been unable to obtain a structure of SpmSyn in complex with dcAdoMet. Overlaid structures of SpmSyn·MTA·SPM and SpdSyn·dcAdoMet (Protein Data Bank code 2O0L) are shown in Fig. 7B. The adenine and ribose rings of MTA superimpose very well with those in dcAdoMet. All of the residues lined in the pocket for the aminopropyl group are highly conserved in the sequences and in the structures. In the SpdSyn structure, the pocket for the aminopropyl group of dcAdoMet consists of Tyr79, Asp104, and Asp173. In the SpmSyn·MTA·SPM structure, the corresponding pocket for the relevant aminopropyl group of SPM consists of Tyr177, Asp201, and Asp276.
Site-directed Mutagenesis Reveals Key Residues for the Catalytic Mechanism of SpmSyn—To gain insight into the catalytic mechanism of human SpmSyn, we generated a series of mutants of SpmSyn and analyzed their kinetic properties. The crystal structure of human SpmSyn revealed that the active site with a bound SPD molecule contains an Asp residue (Asp276), a fully conserved residue (Fig. 3), which is in an ideal position to facilitate the deprotonation of the N-10 amino group of SPD that attacks the C-α atom of the aminopropyl group of dcAdoMet (Figs. 5C and 7C). Alteration of this residue to Asn reduced the kcat/Km for SPD by >200,000-fold (Table 2). Tyr351 (via backbone carbonyl and hydrophobic interactions) and Glu353 interact with the N-1 atom of the SPD substrate and the SPM product (Fig. 5D). Truncation of the protein by mutating the codon for Cys357 to a stop codon, which eliminates the 20 C-terminal residues including these 2 residues, led to a complete loss of activity (Fig. 4). Mutation of Glu353 to Gln reduced the kcat/Km by 800-fold (Table 2). Based on the model structure shown in Fig. 7B and the structure with the SPM product bound shown in Fig. 5D, the side chain of the fully conserved residue Asp201 interacts with the amino group of the aminopropyl group of the dcAdoMet substrate in the enzyme·substrate complex and with N-14 of the SPM product in the enzyme·product complex. Mutation of Asp201 to either Ala or Asn decreased the kcat/Km for dcAdoMet by >100,000-fold (Table 2). These results support the mechanism shown in Fig. 7C.
TABLE 2.
Kinetic properties of human SpmSyn mutants
| SpmSyn | kcat/Km for SPD | kcat/Km for dcAdoMet | kcat |
|---|---|---|---|
| s−1/M−1 | s−1/M−1 | s−1 | |
| Wild-type | 40,000 | 71 × 106 | 32 |
| D276N | 0.15 | 846 | 0.0022 |
| D201N | 1.0 | 124 | 0.0015 |
| D201A | 1.1 | 164 | 0.0015 |
| E353Q | 47 | 1.5 × 106 | 0.64 |
Phylogenetic Distribution of SpmSyn Orthologs—On the basis of our discovery that human SpmSyn has an N-terminal domain that adopts an AdoMetDC-like fold, we examined the amino acid sequences of other SpmSyns for a similar domain. Such AdoMetDC-like/aminopropyltransferase-like fusion proteins are found throughout the vertebrate species and in the Arthropoda, including fruit fly, mosquito, and bee (Figs. 3 and 8). They are also found in sea urchin, sea anemone, and the choanoflagellate Monosiga brevicollis. Noticeably, orthologs are absent in nematode worms, plants, fungi, and all sequenced single-cell eukaryotes with the exception of the ciliate Tetrahymena thermophila. Although structurally similar to AdoMetDC as shown in Fig. 2C, the N-terminal domain of human SpmSyn does not exhibit sequence similarity to any eukaryotic, eubacterial, or archaeal AdoMetDC sequence (supplemental Fig. S2). This is also the case for the N-terminal domains from other eukaryotic SpmSyn fusion proteins. However, the ciliate T. thermophila, a member of the Alveolata phylum, has three paralogous genes encoding aminopropyltransferases with an extended N-terminal domain. Two of the encoded proteins (EAS07189 and EAR85566) have significant amino acid sequence similarity in the N-terminal domain to bacterial AdoMetDC sequences (supplemental Fig. S2), whereas the third protein (XP_001013116) has similarity only to the N-terminal domains of the other two Tetrahymena proteins. In bacteria, there are several instances of fusions of N-terminal sequences exhibiting clear similarity to AdoMetDC with an aminopropyltransferase domain from α-, β-, and δ-Proteobacteria and Actinobacteria.
FIGURE 8.

Neighbor joining phylogenetic tree of eukaryotic and bacterial aminopropyltransferase domains. Numbers on the branches refer to the bootstrap support for nodes based on 2,000 bootstrap replicates. The AdoMetDC-like N-terminal domains and part of the C-terminal domains have been removed to facilitate the alignment. Alignments were performed using ClustalW. APT, bacterial AdoMetDC/aminopropyltransferase fusion; SpdSyn*, confirmed SpdSyn; SpdSyn, putative SpdSyn; SpmSyn*, confirmed SpmSyn; SpmSyn, putative SpmSyn/AdoMetDC fusion (eukaryotic); TspmSynacl5*, thermospermine synthase. The GenBank™ accession numbers, bacterial phyla, and eukaryotic taxa are indicated in parentheses: T.th SpmSyn1-3, T. thermophila (EAS07189, EAR85566, and XP_001013116, respectively; Alveolata); D.ac APT, Delftia acidovorans (ZP_01582080; β-Proteobacteria); C.te APT, Comamonas testosteroni KF-1 (ZP_01519926; β-Proteobacteria); A.sp APT, Azoarcus sp. EbN1 (YP_158219; β-Proteobacteria); A.th TspmSynacl5*, A. thaliana (AAF01311; ACL5 (Acaulis 5); Viridiplantae); H.sa SpdSyn*, H. sapiens (NP_003123; Metazoa); D.re SpdSyn, D. rerio (NP_957328; zebrafish; Metazoa); S.pu SpdSyn, S. purpuratus (XP_796573; sea urchin; Metazoa); S.ce SpdSyn*, S. cerevisiae (AAC17191; fungi); C.el SpdSyn*, C. elegans (CAC37332; Metazoa); A.ga SpdSyn, A. gambiae strain PEST (XP_309520; mosquito; Metazoa); D.me SpdSyn, D. melanogaster (NP_731384; Metazoa); A.me SpdSyn, A. mellifera (XP_001120306; honey bee; Metazoa); S.ce SpmSyn*, S. cerevisiae (AAC19368; fungi); A.th SpmSyn*, A. thaliana (NP_568785; Viridiplantae); A.th SpdSyn1*, A. thaliana (CAB64644; Viridiplantae); P.fa SpdSyn*, P. falciparum 3D7 (CAB71155; Alveolata); L.do SpdSyn*, Leishmania donovani (AAG24612; Alveolata); B.ba APT, Bdellovibrio bacteriovorus HD100 (NP_970339; δ-Proteobacteria); E.co SpdSyn*, E. coli K12 (NP_414663; γ-Proteobacteria); T.ma SpdSyn*, T. maritima (Q9WZC2); H.sa SpmSyn*, H. sapiens (NP_004586; Metazoa); G.ga SpmSyn, G. gallus (NP_001025974; chicken; Metazoa); D.re SpmSyn, D. rerio (NP_571831; zebrafish; Metazoa); S.pu SpmSyn, S. purpuratus (XP_789223; sea urchin; Metazoa); D.me SpmSyn, D. melanogaster (NP_729798; Metazoa); A.ga SpmSyn, A. gambiae strain PEST (XP_315341; mosquito; Metazoa); A.me SpmSyn, A. mellifera (XP_393567; bee; Metazoa); S.ac APT, Syntrophus aciditrophicus SB (YP_460751; δ-Proteobacteria); CP.ub APT, Candidatus Pelagibacter ubique HTCC1002 (ZP_01264992; α-Proteobacteria); and N.fa APT, Nocardia farcinica IFM10152 (YP_119998; Actinobacteria).
Yeast and Arabidopsis thaliana SpmSyns, which lack the N-terminal domain of the metazoan enzymes (Fig. 3), cluster with the eukaryotic SpdSyns (Fig. 8), suggesting that the acquisition of the N-terminal domain has led to changes in the aminopropyltransferase domain of the metazoan fusion proteins. The A. thaliana thermospermine synthase (ACL5) does not cluster with known eukaryotic SpmSyns or SpdSyns.
DISCUSSION
The structure of the active site of SpmSyn as revealed by the complexes with MTA and SPD or MTA and SPM and the results of the site-directed mutagenesis of the key acidic residues Asp201, Asp276, and Glu353 are in agreement with the general model for aminopropyl transfer that was proposed based on our studies of human SpdSyn (27). The amine substrate is positioned by interactions between acidic residues and the terminal nitrogen atoms and by hydrophobic interactions with the alkane portions. These acidic residues are Asp276 and Glu353 in SpmSyn, which correspond to Asp173 and Asp176, respectively, in SpdSyn. Interaction of the attacking amino group with Asp276 (SpmSyn) or Asp173 (SpdSyn), reinforced by interactions with the hydroxyl group of Tyr177 (SpmSyn) or Tyr79 (SpdSyn) and the backbone carbonyl of Leu277 (SpmSyn) or Ser174 (SpdSyn), facilitates deprotonation and the attack on C-α of the aminopropyl group of dcAdoMet. This attack is facilitated by the interaction of the 3-amino group of the dcAdoMet substrate with Asp201 (SpmSyn) or Asp104 (SpdSyn) (Fig. 7C). These residues are highly conserved in aminopropyltransferases (Fig. 3) (27), as are those involved in binding the adenosyl moiety of the dcAdoMet substrate (Asp255, Gln148, and Glu220 in SpmSyn and Asp155, Gln49, and Glu124 in SpdSyn). Thus, the general mechanism for aminopropyl transfer derived from SpdSyns also applies to SpmSyns.
Comparison of the structure of the active site in SpmSyn with that of SpdSyn provides a plausible explanation for the substrate specificity. In addition to the longer gatekeeping loop, SpmSyn lacks the steric block to substrate binding provided in human SpdSyn by the side chain of Trp28, which restricts the substrate specificity to putrescine (27). SpmSyn has Ala127 at the corresponding position and can easily accommodate longer substrates.
The absolute requirement for the N-terminal domain for SpmSyn activity is surprising because it appears that all of the residues making up the active site are in the C-terminal and central domains. Because the N-terminal domain provides the major factor in dimerization, it seems likely that dimerization is essential for activity. It is possible that the dimer is needed for correct formation of the two active sites, but because the truncated proteins did not form crystals under the conditions tested, this is currently only speculation. All characterized aminopropyltransferases are homodimers, except those from acute thermophiles, which are homotetramers formed by pairs of dimers (21, 27).
Despite a lack of obvious similarity in amino acid sequence, the N-terminal domain of human SpmSyn has a very similar overall structure to the AdoMetDC proenzyme from T. maritima (41). The domain structure is also similar to the human AdoMetDC proenzyme (r.m.s.d. of 2.99 Å) and potato AdoMetDC, which differ only slightly in structure from the T. maritima proenzyme with two additional β-strands (52, 53). SpmSyn has no AdoMetDC activity as expected because it does not contain the Ser residue necessary to generate, through an autoprocessing reaction, the essential pyruvoyl prosthetic group. Recent results have shown that trypanosomatids contain a paralog of AdoMetDC that has lost catalytic activity (54). This protein has a high affinity for AdoMetDC and forms a high affinity heterodimer with 1,200-fold higher activity, suggesting that it is an allosterically active subunit of AdoMetDC in these species. Although we could not detect any functional interactions upon mixing human recombinant AdoMetDC with SpmSyn and its N-terminal domain, it is possible that such interactions would occur upon co-translation of the two mRNAs but do not form after the production of the dimer, which is very stable in the case of both SpmSyn and the processed AdoMetDC αβ-unit. Such an interaction could regulate AdoMetDC activity. It is interesting in this context that transgenic overexpression of SpmSyn from a strong ubiquitous CAG promoter produces little or no increase in SPM and a decrease in SPD despite a very large increase in SpmSyn activity as measured in vitro using cell extracts (55).
Our structural studies may relate to the phenotype in Snyder-Robinson syndrome (19). This X-linked recessive mental retardation condition is associated with a mutation in the SpmSyn gene that leads to production of a truncated protein (S-R) of 110 amino acids. This S-R protein was expected to lack SpmSyn activity, and our present results confirm this (Fig. 4). However, the truncated S-R protein contains most of the N-terminal domain of SpmSyn, and the discrete and stable structure of this N-terminal fragment suggests that it may accumulate and form dimers with itself or with other proteins. It is noteworthy that there is little reduction in SPM in cell cultures from Snyder-Robinson patients, although SPD is increased (19). The maintenance of SPM is probably due to a small level of correct splicing of the SpmSyn gene product. It is certainly possible that the altered SPM/SPD ratio leads to the Snyder-Robinson phenotype, but the accumulation of S-R protein should also be considered.
Because the AdoMetDC-like N-terminal domain is not found in plant and fungal SpmSyns, it is interesting to consider at which evolutionary stage SpmSyn acquired this domain. Multicellular animals (Metazoa) consist of two branches: sponges and Eumetazoa. The Eumetazoa consists of comb jellies (ctenophores), cnidarians (anemones, jelly fish, and hydras), and bilaterians (all other multicellular animals). Within the Bilateria are two branches: Protostomia (including mollusks, nematodes, and insects) and Deuterostomia (including non-chordates such as sea urchins and chordates, which include the Vertebrata). The SpmSyn fusion protein is found in vertebrates, elsewhere in the Deuterostomia (the sea urchin Strongylocentrotus purpuratus), and in some of the Protostomia (fruit fly, mosquito, and bee). Within the Protostomia, nematode worms lack a SpmSyn fusion protein. However, this is not due to loss of the N-terminal domain but to loss of SpmSyn, as only one aminopropyltransferase gene is present in nematodes, encoding SpdSyn (24). The last common ancestor of cnidarians and bilaterians also possessed the fusion protein, as the gene is present in the sea anemone Nematostella vectensis. Acquisition of the N-terminal domain of the SpmSyn fusion protein must have predated the origin of the Metazoa, as the fusion protein is present in the Choanoflagellata, a close, single-cell sister group of the Metazoa (56).
The presence of three genes encoding SpmSyn fusion-like proteins in the genome of Tetrahymena could suggest that the fusion protein arose very early in eukaryotic evolution and has been subsequently lost in sister ciliate groups such as Paramecium and in major divisions such as algae, plants, and fungi. However, although one of the Tetrahymena genes does not possess sequence similarity to bacterial AdoMetDC in the N-terminal domain, the other two genes have N-terminal domains with strong similarity to bacterial sequences, especially from the Firmicutes phylum. This suggests that the Tetrahymena fusion protein genes have been recently acquired from bacteria by horizontal gene transfer, a relatively common occurrence in ciliates (57). The most parsimonious explanation for the origin of the SpmSyn N-terminal domain is that it was acquired after the divergence of the fungal and metazoan ancestors but before the origin of multicellularity in animals, suggesting that the N-terminal domain was eukaryotic in origin. AdoMetDC/aminopropyltransferase fusion proteins found in bacteria are likely to have arisen by convergent evolution.
Supplementary Material
The atomic coordinates and structure factors (codes 3C6K and 3C6M) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
This work was supported by the Structural Genomics Consortium and by United States Public Health Service Grant R01 GM26290 (to A. P.). The Structural Genomics Consortium is a registered charity (Number 1097737) supported by the Canadian Institutes of Health Research, the Canadian Foundation for Innovation, and Genome Canada through the Ontario Genomics Institute, GlaxoSmithKline, the Karolinska Institute, the Knut and Alice Wallenberg Foundation, the Ontario Innovation Trust, the Ontario Ministry for Research and Innovation, Merck & Co., Inc., the Novartis Research Foundation, the Swedish Agency for Innovation Systems, the Swedish Foundation for Strategic Research, and the Wellcome Trust. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
This article was selected as a Paper of the Week.
Footnotes
The abbreviations used are: SPD, spermidine; SPM, spermine; dcAdoMet, decarboxylated S-adenosylmethionine; SpdSyn, spermidine synthase; SpmSyn, spermine synthase; AdoMetDC, S-adenosylmethionine decarboxylase; MTA, 5′-methylthioadenosine; MES, 4-morpholineethanesulfonic acid; BisTris, 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol; r.m.s.d., root mean square deviation.
The nomenclature used in this study for the atoms in the polyamines is as follows: spermidine, H2N1C2H2C3H2C4H2N5HC6H2C7H2C8H2C9H2N10H2; and spermine, H2N1C2H2C3H2C4H2N5HC6H2C7H2C8H2C9H2N10HC11H2C12H2C13H2N14H2.
References
- 1.Gerner, E. W., and Meyskens, F. L., Jr. (2004) Nat. Rev. Cancer 4 781-792 [DOI] [PubMed] [Google Scholar]
- 2.Casero, R. A., Jr., and Marton, L. J. (2007) Nat. Rev. Drug Discov. 6 373-390 [DOI] [PubMed] [Google Scholar]
- 3.Pendeville, H., Carpino, N., Marine, J. C., Takahashi, Y., Muller, M., Martial, J. A., and Cleveland, J. L. (2001) Mol. Cell. Biol. 21 6459-6558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nishimura, K., Nakatsu, F., Kashiwagi, K., Ohno, H., Saito, H., Saito, T., and Igarashi, K. (2002) Genes Cells 7 41-47 [DOI] [PubMed] [Google Scholar]
- 5.Park, M. H. (2006) J. Biochem. (Tokyo) 139 161-169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wallace, H. M., Fraser, A. V., and Hughes, A. (2003) Biochem. J. 376 1-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jänne, J., Alhonen, L., Pietilä, M., Keinanen, T. A., Uimari, A., Hyvonen, M. T., Pirinen, E., and Jarvinen, A. (2006) J. Biochem. (Tokyo) 139 155-160 [DOI] [PubMed] [Google Scholar]
- 8.Pegg, A. E. (1986) Biochem. J. 234 249-262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hamasaki-Katagiri, N., Katagiri, Y., Tabor, C. W., and Tabor, H. (1998) Gene (Amst.) 210 195-210 [DOI] [PubMed] [Google Scholar]
- 10.Chattopadhyay, M. K., Tabor, C. W., and Tabor, H. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 13869-13874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mackintosh, C. A., and Pegg, A. E. (2000) Biochem. J. 351 439-447 [PMC free article] [PubMed] [Google Scholar]
- 12.Nilsson, J., Gritli-Linde, A., and Heby, O. (2000) Biochem. J. 352 381-387 [PMC free article] [PubMed] [Google Scholar]
- 13.Korhonen, V.-P., Niranen, K., Halmekyto, M., Pietilä, M., Diegelman, P., Parkkinen, J. J., Eloranta, T., Porter, C. W., Alhonen, L., and Jänne, J. (2001) Mol. Pharmacol. 59 231-238 [DOI] [PubMed] [Google Scholar]
- 14.Meyer, R. A., Jr., Henley, C. M., Meyer, M. H., Morgan, P. L., McDonald, A. G., Mills, C., and Price, D. K. (1998) Genomics 48 289-295 [DOI] [PubMed] [Google Scholar]
- 15.Imai, A., Akiyama, T., Kato, T., Sato, S., Tabata, S., Yamamoto, K. T., and Takahashi, T. (2004) FEBS Lett. 556 148-152 [DOI] [PubMed] [Google Scholar]
- 16.Clay, N. K., and Nelson, T. (2005) Plant Physiol. 138 767-777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamaguchi, K., Takahashi, Y., Berberich, T., Imai, A., Takahashi, T., Michael, A. J., and Kusano, T. (2007) Biochem. Biophys. Res. Commun. 352 486-490 [DOI] [PubMed] [Google Scholar]
- 18.Knott, J. M., Romer, P., and Sumper, M. (2007) FEBS Lett. 581 3081-3086 [DOI] [PubMed] [Google Scholar]
- 19.Cason, A. L., Ikeguchi, Y., Skinner, C., Wood, T. C., Lubs, H. A., Martinez, F., Simensen, R. J., Stevenson, R. E., Pegg, A. E., and Schwartz, C. E. (2003) Eur. J. Human Genet. 11 937-944 [DOI] [PubMed] [Google Scholar]
- 20.Pegg, A. E., Poulin, R., and Coward, J. K. (1995) Int. J. Biochem. 27 425-442 [DOI] [PubMed] [Google Scholar]
- 21.Ikeguchi, Y., Bewley, M., and Pegg, A. E. (2006) J. Biochem. (Tokyo) 139 1-9 [DOI] [PubMed] [Google Scholar]
- 22.Ohnuma, M., Terui, Y., Tamakoshi, M., Mitome, H., Niitsu, M., Samejima, K., Kawashima, E., and Oshima, T. (2005) J. Biol. Chem. 280 30073-30082 [DOI] [PubMed] [Google Scholar]
- 23.Korolev, S., Ikeguchi, Y., Skarina, T., Beasley, S., Arrowsmith, C., Edwards, A., Joachimiak, A., Pegg, A. E., and Savchenko, A. (2002) Nat. Struct. Biol. 9 27-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dufe, V. T., Luersen, K., Eschbach, M. L., Haider, N., Karlberg, T., Walter, R. D., and Al-Karadaghi, S. (2005) FEBS Lett. 579 6037-6043 [DOI] [PubMed] [Google Scholar]
- 25.Dufe, V. T., Qiu, W., Muller, I. B., Hui, R., Walter, R. D., and Al-Karadaghi, S. (2007) J. Mol. Biol. 373 167-177 [DOI] [PubMed] [Google Scholar]
- 26.Lu, P. K., Tsai, J. Y., Chien, H. Y., Huang, H., Chu, C. H., and Sun, Y. J. (2007) Proteins 67 743-754 [DOI] [PubMed] [Google Scholar]
- 27.Wu, H., Min, J., Ikeguchi, Y., Zeng, H., Dong, A., Loppnau, P., Pegg, A. E., and Plotnikov, A. N. (2007) Biochemistry 46 8331-8339 [DOI] [PubMed] [Google Scholar]
- 28.Cacciapuoti, G., Porcelli, M., Moretti, M. A., Sorrentino, F., Concilio, L., Zappia, V., Liu, Z. J., Tempel, W., Schubot, F., Rose, J. P., Wang, B. C., Brereton, P. S., Jenney, F. E., and Adams, M. W. (2007) J. Bacteriol. 189 6057-6067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Almrud, J. J., Oliveira, M. A., Kern, A. D., Grishin, N. V., Phillips, M. A., and Hackert, M. L. (2000) J. Mol. Biol. 295 7-16 [DOI] [PubMed] [Google Scholar]
- 30.Tolbert, D. W., Ekstrom, J. L., Mathews, I. I., Secrist, J. A. I., Kapoor, P., Pegg, A. E., and Ealick, S. E. (2001) Biochemistry 40 9484-9494 [DOI] [PubMed] [Google Scholar]
- 31.Wiest, L., and Pegg, A. E. (1998) in Methods in Molecular Biology: Polyamine Protocols (Morgan, D. M. L., ed) Volume 79, pp. 51-58, Humana Press, Totowa, NJ [Google Scholar]
- 32.Otwinowski, Z., and Minor, W. (1997) Methods Enzymol. 276 307-326 [DOI] [PubMed] [Google Scholar]
- 33.Vagin, A., and Teplyakov, A. (1997) J. Appl. Crystallogr. 30 1022-1025 [Google Scholar]
- 34.Perrakis, A., Harkiolaki, M., Wilson, K. S., and Lamzin, V. S. (2001) Acta Crystallogr. Sect. D Biol. Crystallogr. 57 1445-1450 [DOI] [PubMed] [Google Scholar]
- 35.Emsley, P., and Cowtan, K. (2004) Acta Crystallogr. Sect. D Biol. Crystallogr. 60 2126-2132 [DOI] [PubMed] [Google Scholar]
- 36.Ikeguchi, Y., Mackintosh, C. A., McCloskey, D. E., and Pegg, A. E. (2003) Biochem. J. 373 885-892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shantz, L. S., and Pegg, A. E. (1998) in Methods in Molecular Biology: Polyamine Protocols (Morgan, D. M. L., ed) Volume 79, pp. 45-50, Humana Press, Totowa, NJ [Google Scholar]
- 38.Brooks, W. H., McCloskey, D. E., Daniel, K. G., Ealick, S. E., Secrist, J. A., III, Waud, W. R., Pegg, A. E., and Guida, W. C. (2007) J. Chem. Inf. Model. 47 1897-1905 [DOI] [PubMed] [Google Scholar]
- 39.Martin, J. L., and McMillan, F. M. (2002) Curr. Opin. Struct. Biol. 12 783-793 [DOI] [PubMed] [Google Scholar]
- 40.Holm, L., and Sander, C. (1995) Trends Biochem. Sci. 20 478-480 [DOI] [PubMed] [Google Scholar]
- 41.Toms, A. V., Kinsland, C., McCloskey, D. E., Pegg, A. E., and Ealick, S. E. (2004) J. Biol. Chem. 279 33837-33846 [DOI] [PubMed] [Google Scholar]
- 42.Ekstrom, J. E., Matthews, I. I., Stanley, B. A., Pegg, A. E., and Ealick, S. E. (1999) Structure 7 583-595 [DOI] [PubMed] [Google Scholar]
- 43.Plotnikov, A. N., Vasilenko, K. S., Kirkitadze, M. D., Kotova, N. V., Motuz, L. P., Korotkov, K. V., Semisotnov, G. V., and Alakhov Iu, B. (1996) Bioorg. Khim. 22 489-502 [PubMed] [Google Scholar]
- 44.Ptitsyn, O. B., and Semisotnov, G. V. (1991) in Conformations and Forces in Protein Folding (Nall, B. T., and Dill, K. A., eds) pp. 155-168, AAAS Publishing, Washington, DC
- 45.Hackert, M. L., and Pegg, A. E. (1997) in Comprehensive Biological Catalysis (Sinnott, M. L., ed) pp. 201-216, Academic Press, London
- 46.Pajula, R.-L., Raina, A., and Eloranta, T. (1979) Eur. J. Biochem. 101 619-626 [DOI] [PubMed] [Google Scholar]
- 47.Pajula, R.-L., and Raina, A. (1979) FEBS Lett. 99 343-345 [DOI] [PubMed] [Google Scholar]
- 48.Hibasami, H., Borchardt, R. T., Chen, S. Y., Coward, J. K., and Pegg, A. E. (1980) Biochem. J. 187 419-428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pegg, A. E., and Williams-Ashman, H. G. (1969) Biochem. J. 115 241-247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Basu, I., Cordovano, G., Das, I., Belbin, T. J., Guha, C., and Schramm, V. L. (2007) J. Biol. Chem. 282 21477-21486 [DOI] [PubMed] [Google Scholar]
- 51.Raina, A., Hyvonen, T., Eloranta, T., Voutilainen, M., Samejima, K., and Yamanoha, B. (1984) Biochem. J. 219 991-1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tolbert, W. D., Zhang, Y., Bennett, E. M., Cotter, S. E., Ekstrom, J. L., Pegg, A. E., and Ealick, S. E. (2003) Biochemistry 42 2386-2395 [DOI] [PubMed] [Google Scholar]
- 53.Bennett, E. M., Ekstrom, J. L., Pegg, A. E., and Ealick, S. E. (2002) Biochemistry 41 14509-14517 [DOI] [PubMed] [Google Scholar]
- 54.Willert, E. K., Fitzpatrick, R., and Phillips, M. A. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 8275-8280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ikeguchi, Y., Wang, X., McCloskey, D. E., Coleman, C. S., Nelson, P., Hu, G., Shantz, L. M., and Pegg, A. E. (2004) Biochem. J. 381 701-707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lang, B. F., O'Kelly, C., Nerad, T., Gray, M. W., and Burger, G. (2002) Curr. Biol. 12 1773-1778 [DOI] [PubMed] [Google Scholar]
- 57.Ricard, G., McEwan, N. R., Dutilh, B. E., Jouany, J. P., Macheboeuf, D., Mitsumori, M., McIntosh, F. M., Michalowski, T., Nagamine, T., Nelson, N., Newbold, C. J., Nsabimana, E., Takenaka, A., Thomas, N. A., Ushida, K., Hackstein, J. H., and Huynen, M. A. (2006) BMC Genomics 7 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Laskowski, R. A., MacArthur, M. W., Moss, D. S., and Thornton, J. M. (1993) J. Appl. Crystallogr. 26 283-291 [Google Scholar]
- 59.Thompson, J. D., Higgins, D. G., and Gibson, T. J. (1994) Nucleic Acids Res. 22 4673-4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.