

Abstract
The c-Jun N-terminal kinase (JNK) mitogen-activated protein kinase (MAPK) signaling pathway mediates stress responses in cells. JNK activity is regulated by a protein kinase cascade consisting of a MAPK kinase (MKK) and a MAPK kinase kinase (MAPKKK). β-Arrestin-2 acts as a scaffold by directly binding to the JNK3 isoform and also by recruiting MKK4 and the MAPKKK apoptosis-signaling kinase-1 (ASK1). In this study, we demonstrate by co-precipitation that the extended N-terminal region of JNK3 mediates binding to the C terminus of β-arrestin-2 and that the N terminus of JNK3 is required for its activation via β-arrestin-2. We have used site-specific mutagenesis to identify key residues within the N terminus of JNK3 that are essential for binding and demonstrate that this region represents an independent β-arrestin-2 binding motif that can be fused to other MAPKs and permit their recruitment to the scaffold complex. In addition, we demonstrate that JNK3 recruits MKK4 to the β-arrestin-2 scaffold complex by binding to the MAPK docking domain (D-domain) located within the N terminus of MKK4. These findings uncover molecular determinants of β-arrestin-2 scaffold complex assembly and assign a previously unrecognized role for the unique extended N terminus of JNK3.
The c-Jun N-terminal kinase (JNK)2 MAPK pathway is predominantly activated by stress stimuli and plays important roles in many cellular processes including development, apoptosis, cell growth, and immune responses (1, 2). There are ten JNK isoforms expressed in mammalian cells, and these are derived from the alternative splicing of three genes (3, 4). JNK1 and JNK2 isoforms are ubiquitous, while JNK3 isoforms are expressed in brain and testis and, to a lesser extent, heart (3, 5). There is a high level of sequence conservation between JNK isoforms, but JNK3 is unique in having an extended N terminus of 38 amino acids (3, 4). Gene-targeting studies in mice have revealed distinct and overlapping functions of JNK isoforms (1, 2). For example, JNK3-deficient mice are protected from neuronal death in response to excitotoxic stimuli, ischemia, and β-amyloid (6–8).
JNK, similar to other MAPKs, is activated by a signaling module that includes MKKs and MAPKKKs (1, 2). Components of JNK signaling modules include the MKKs MKK4 and MKK7 as well as multiple MAPKKKs including members of the MEK kinase (MEKK) and mixed-lineage kinase (MLK) families, apoptosis-signaling kinase-1 (ASK1), transforming growth factor-β-activated kinase-1 (TAK1), and tumor progression loci-2 kinase (Tpl2) (1, 2). The assembly, localization, and activity of JNK signaling modules can be coordinated by scaffold proteins (9). A number of scaffold proteins are implicated in regulating JNK signaling, including the JNK-interacting protein (JIP) family (9, 10), plenty of SH3 (POSH) (11), and β-arrestin-2 (12).
β-Arrestin-2 associates with many G protein-coupled receptors and members of the Frizzled and TGFβ receptor families and plays important roles in receptor desensitization, endocytosis, proteosomal degradation, and signaling (13). β-Arrestin-2 specifically binds to JNK3 but not to JNK1 or JNK2 (12). It also binds to the MAPKKK ASK1 and recruits MKK4, thereby nucleating a JNK signaling module (12). β-Arrestin-2 appears to facilitate ASK1-mediated signaling to JNK3 and angiotensin II-induced activation of JNK3 (12), although the precise functional role of the scaffold complex is not clear. Under certain conditions, β-arrestin-2 may also regulate JNK3 inactivation via the recruitment of MAPK phosphatases (MKPs) (14).
As well as scaffolding a JNK3 signaling module, there is evidence that β-arrestin-2 scaffolds an extracellular signal-regulated kinase (ERK) MAPK signaling module (15) and also integrates signals regulating other intracellular signaling pathways including the protein kinase B (PKB) and nuclear factor κB (NF-κB) pathways (15, 16). In addition to its roles in the cytoplasm, it is likely that β-arrestin-2 has a nuclear function as it is reported to shuttle between the cytoplasm and the nucleus and to control the subcellular distribution of JNK3 and other proteins that it binds (17, 18).
The molecular determinants underlying the assembly of the JNK3 signaling module with β-arrestin-2 remain to be fully elucidated. β-Arrestin-2 contains a sequence that resembles a D-domain docking motif that is present in many MAPK substrates, MKKs, MAPK phosphatases, and scaffold proteins (19, 20). Many studies have highlighted the importance of docking motifs for enzymatic specificity and efficient signaling of MAPK pathways (19, 20). The replacement of part of this sequence in β-arrestin-2 by the corresponding sequence in the closely related β-arrestin-1, which has a low binding affinity for JNK3, results in a loss of JNK3 binding (21). Similarly, insertion of this sequence into β-arrestin-1 results in a chimera with greatly improved binding to JNK3 (21).
Regions within JNK and other MAPKs that mediate docking interactions with D-domains have also been uncovered. These include the common docking (CD) domain and the ED site, which together have been proposed to form a docking groove for MAPK-interacting proteins (22, 23). Residues within or close to these domains in JNKs have been shown to be essential for binding to the D-domains of c-Jun, MKK4, MKP5, and JIP scaffold proteins (22, 24–27).
We set out to elucidate the molecular determinants of the interaction of the JNK3 signaling module with β-arrestin-2. Surprisingly, we found that JNK3 binding to β-arrestin-2 was mediated, not by the docking groove composed of the ED and CD domains, but by the unique extended N-terminal region that is absent from the other JNK isoforms. A deletion mutant consisting of the N-terminal region alone was capable of binding to β-arrestin-2, and the fusion of this region to either JNK1 or p38 promoted their binding to β-arrestin-2. We mapped residues essential for JNK3 binding and demonstrated that the mutation of these residues compromises β-arrestin-2-facilitated JNK3 activation. The JNK3 that bound to β-arrestin-2 was capable of recruiting MKK4 to the complex, and this was mediated by JNK3 interacting with the D-domain of MKK4.
Taken together, our results define novel molecular determinants of β-arrestin-2 scaffold complex assembly and increase our understanding of the mechanisms cells employ to ensure specific signaling.
EXPERIMENTAL PROCEDURES
Cell Culture and Transfections— COS-7 and 293T cells were maintained in Dulbecco's modified Eagle's medium supplemented with 5 mm glutamine, 100 units/ml penicillin/streptomycin, and 5% fetal bovine serum (Invitrogen). Cells were transfected using Lipofectamine (Invitrogen) according to the manufacturer's instructions. Where indicated, cells were incubated with 1 μm angiotensin II (Sigma) for 20 min.
Plasmids—The constructs pSRα-HA-JNK1α1 (28), pBJ5-HA-JNK3α1 (29), pcDNA3-HA-p38α (25), pEGFPC2-JNK3α1 (12), pcDNA3-FLAG-β-arrestin-2 (12), pCMV5-FLAG-JIP1 (30), pME-HA-ASK1 (12), pcDNA3-HA-MLK3 (30), pEBG-MKK4β (31), pEBG-MKK4β (L44A/L46A) (25), and pcDNA3-myc-MKK4β (25) were described previously. pEBG-β-arrestin-2 was generated by insertion of the rat β-arrestin-2 cDNA into the Spe1/Not1 sites of pEBG. The β-arrestin-1 expression vectors were generated by PCR amplification of human β-arrestin-1 cDNA (from IMAGE clone 3604829) and subcloning into pEBG or with an N-terminal FLAG tag into pCDNA3. pAMC-myc-CAMK1γ was generated PCR amplification of the human CAMK1γ cDNA (from IMAGE clone 5179957) and insertion into the EcoR1/Not1 sites of pAMC-myc. pEBG-JNK1α1, and pEBG-JNK3α1 were provided by C. Tournier (University of Manchester). pCDNA3-HA-AT1AR was provided by M. Caron (Duke University Medical Center).
Mutagenesis— JNK3 deletion mutants were generated by polymerase chain reaction (PCR) and subcloned into the BamH1/Not1 sites of pEBG. The deletion constructs were ΔN (aa 39–422), ΔN2 (aa 19–422), ΔN3 (aa 9–422), and JNK3-N (aa 1–38). JNK3/JNK1 chimeras and the JNK3/p38 chimera were generated by PCR mutagenesis and subcloning into BamH1/Not1 sites of pEBG or as HA tag fusions (Chim A: JNK3 aa 1–285/JNK1 aa 248–384, Chim B: JNK3 aa 1–38/JNK1 aa 1–384, JNK3/p38: JNK3 aa 1–38/p38α aa 1–360). The β-arrestin chimeras were subcloned into pEBG (Chim1: β-arrestin-1 aa 1–225/β-arrestin-2 aa 228–410, Chim 2: β-arrestin-2 aa 1–227/β-arrestin-1 aa 226–418). The β-arrestin-2 and β-arrestin-1 deletion constructs were generated by PCR and subcloned into pEBG. The deletion constructs used were β-arrestin-2 aa 1–229 and aa 228–410, and β-arrestin-1 aa 226–418. Site-directed mutagenesis was performed using the Quick-Change kit (Stratagene) according to the manufacturer's instructions and the introduced mutations were verified by DNA sequencing. The human JNK3 mutants used in this study are as follows: C9A, S10A, E11A/P12A, T13A, L14A, D15A, V16A, K17A, L14A/V16A, D344A/E347A/E349A (CDmut). The β-arrestin-2 mutants used were R196A/R197A, L199A/L201A, S198P, and the β-arrestin-1 mutation was P196S.
Immunoprecipitations and GST Pull-downs—COS-7 cells were washed once with ice-cold phosphate-buffered saline and then lysed in buffer containing 20 mm Tris, pH 7.4, 137 mm NaCl, 25 mm β-glycerophosphate, 2 mm sodium pyrophosphate, 2 mm EDTA, 1% Triton X-100, 10% glycerol, 1 mm phenylmethylsulfonyl fluoride, 1 mm sodium orthovanadate, and 5 μg/ml leupeptin. Lysates were centrifuged at 14,000 × g for 10 min to remove insoluble material. FLAG-tagged proteins were immunoprecipitated from lysates by incubation with M2 antibodies (Sigma) and protein G-Sepharose beads (Amersham Biosciences) for 3 h at 4 °C. GFP-tagged proteins were immunoprecipitated from lysates by incubation with anti-GFP antibodies (Santa Cruz Biotechnology) and protein A-Sepharose beads (Amersham Biosciences) for 3 h at 4 °C. GST proteins were isolated by incubating lysates with glutathione-Sepharose 4B (Amersham Biosciences). Beads were washed three times in lysis buffer, and bound proteins were eluted by the addition of an appropriate volume of 6× SDS loading buffer.
Immunoblotting—Samples were resolved by SDS-PAGE (10% gels) and transferred to Immobilon-P membranes (Millipore Inc.), which were immunoblotted with the following antibodies: anti-GST (Amersham Biosciences), M2 (Sigma), anti-HA (Cancer Research UK), anti-Myc, and anti-GFP (Santa Cruz Biotechnology), anti-phospho-JNK (Cell Signaling Technology), and horseradish peroxidase (HRP)-conjugated anti-HA (Santa Cruz Biotechnology). The β-arrestin antibody (A2CT) was provided by R. J. Lefkowitz (Duke University Medical Center). Immune complexes were detected using HRP-conjugated secondary antibodies followed by enhanced chemiluminescence (Pierce).
Immunofluorescence—293T cells were grown on glass coverslips coated in poly-l-lysine (Sigma). The cells were transfected with constructs expressing FLAG-β-arrestin-2 and either GFP, GFP-JNK3α1, or GFP-JNK3α1(L14A/V16A). Cells were washed in phosphate-buffered saline, fixed with 4% paraformaldehyde (Sigma) and lysed in 0.2% Triton X-100/phosphate-buffered saline. FLAG-β-arrestin-2 was detected using an Olympus BX51 microscope attached to a CCD Coolsnap-ES camera (Photometrics, UK) by indirect immunofluorescence using the M2 antibody and Alexor594-conjugated secondary antibody (Molecular Probes). GFP fluorescence was observed directly, and nuclei were detected with DAPI (Molecular Probes).
RESULTS
The N-terminal Extension of JNK3 Mediates Binding to β-Arrestin-2—JNK3 associates with β-arrestin-2 leading to enhanced activation by the ASK1-MKK4 signaling module (12). To determine the molecular determinants in JNK3 responsible for the binding to β-arrestin-2, we generated a chimera (J3/J1-A) between the N-terminal-half of JNK3 (amino acids 1–285) and the C-terminal-half of JNK1 (amino acids 248–384) (Fig. 1A). Co-precipitation experiments confirmed the binding of JNK3, but not JNK1, to β-arrestin-2 (Fig. 1B, lanes 2 and 3). The chimeric protein bound β-arrestin-2 similar to wild-type JNK3 (Fig. 1B, lane 4), indicating that the binding determinants are present in the N-terminal-half of JNK3. The primary sequences of JNK3 and JNK1 are highly conserved; however the major distinguishing feature is the extended N terminus of JNK3 that contains 38 amino acids that are not present in either JNK1 or JNK2 isoforms (3, 4). To find out if this region played a role in binding to β-arrestin-2, we generated a JNK3 deletion construct that lacked this region (JNK3ΔN; Fig. 1A). This JNK3 mutant did not bind to β-arrestin-2 (Fig. 1C), indicating that the extended N terminus of JNK3 is important for this interaction. Both the full-length and mutant JNK3 were capable of interacting with a different scaffold protein, JIP1 (Fig. 1D), demonstrating that the deletion is not overtly detrimental to the structure of JNK3. To ascertain whether the N terminus of JNK3 was sufficient for β-arrestin-2 binding, we performed co-precipitation experiments with a mutant JNK3 containing only the N-terminal 38 amino acids. This mutant bound to β-arrestin-2 similar to the full-length JNK3 (Fig. 1E), indicating that the major determinants of JNK3 binding to β-arrestin-2 reside in this N-terminal extension. Next we determined if the N-terminal of JNK3 could function as a portable β-arrestin-2 binding domain by generating fusion proteins with JNK1 and p38α (Fig. 1A). Unlike their wild-type counterparts these fusion proteins (J3/J1-B and J3/p38) bound to β-arrestin-2 to a similar degree as JNK3 (Fig. 1F, compare lanes 2, 4, and 6). Collectively these experiments demonstrate that the N-terminal 38 amino acids of JNK3 are sufficient to mediate binding to β-arrestin-2.
FIGURE 1.

The extended N terminus of JNK3 binds to β-arrestin-2. A, schematic of the JNK and p38 constructs used in the experiments and a summary of their binding to β-arrestin-2 (- represents no binding; + represents binding). B, constructs expressing GST, GST-JNK3, GST-JNK1, or the chimera GST-J3/J1-A were introduced into COS-7 cells together with an expression vector for FLAG-β-arrestin-2. GST-containing complexes were isolated from cell lysates with glutathione-Sepharose beads and FLAG-β-arrestin-2 (β-Arr2) present in the pull-down (PD) was examined by immunoblot analysis using the M2 antibody. The expression of the GST-JNK constructs and FLAG-β-arrestin-2 in the lysates was examined by immunoblotting. C and D, constructs expressing GST, GST-JNK3, or GST-JNK3ΔN were introduced into COS-7 cells together with an expression vector for FLAG-β-arrestin-2 (C) or FLAG-JIP1 (D). GST-containing complexes were isolated from cell lysates with glutathione-Sepharose beads and the β-arrestin-2 or JIP1 present in the pull-down (PD) was examined by immunoblot using M2 antibody. The expression of the JNK mutants, β-arrestin-2 and JIP1 was examined by immunoblotting the lysates with anti-GST and M2 antibodies, respectively. E and F, constructs expressing GST or the indicated GST-JNK3 mutants were introduced into COS-7 cells together with an expression vector for FLAG-β-arrestin-2. GST-containing complexes were isolated from cell lysates with glutathione-Sepharose beads and the β-arrestin-2 present in the pull-downs (PD) was examined by immunoblot using M2 antibody. The expression of the JNK mutants and β-arrestin-2 was examined by immunoblotting the lysates with anti-GST and M2 antibodies, respectively. Experiments were performed either two or three times and representative immunoblots are shown.
Mapping Residues Essential for JNK3 Binding to β-Arrestin-2—To further delineate the region of JNK3 responsible for binding to β-arrestin-2, we made a series of progressive deletions into the N terminus of JNK3 (Fig. 2A) and performed co-precipitation analysis. A deletion mutant lacking the first 8 residues of JNK3 (JNK3ΔN3) bound to β-arrestin-2, while a deletion mutant lacking the first 18 residues (JNK3ΔN2) did not (Fig. 2C), suggesting that key binding determinants were located between amino acids 9 and 18. To identify specific residues required for binding we performed alanine-scanning mutagenesis of this region within JNK3ΔN3 and probed the ability of the mutants to bind to β-arrestin-2. We uncovered several point mutants that did not bind (L14A, D15A, V16A, and K17A) and two mutants with barely detectable binding (E11A/P12A and T13A) (Fig. 2D). We then generated a full-length JNK3 mutant (JNK3 L14A/V16A) that failed to bind to either exogenously expressed β-arrestin-2 (Fig. 2E) or to endogenous β-arrrestin-2 (Fig. 2F). To determine if the binding region in JNK3 represented a more general β-arrestin-2 binding module we performed a homology search. The protein we identified that had the most significant homology to this region was another protein kinase, calcium/calmodulin-dependent protein kinase-1γ (CAMK1γ) (Fig. 2B). Co-precipitation experiments demonstrated that CAMK1γ also bound to β-arrestin-2 (Fig. 2G) and that JNK3 and CAMK1γ can compete for binding.3 This suggests that both kinases may use similar determinants to bind to β-arrestin-2.
FIGURE 2.

Mapping residues critical for JNK3 binding to β-arrestin-2. A, 38-amino acid N-terminal extension of JNK3. The positions of the N-terminal deletion mutants are indicated. The residues mutated to Ala are in bold italics. B, alignment of the N-terminal sequences of human JNK3 and CAMK1γ. Conserved residues are boxed. Numbers refer to amino acids. C–E, constructs expressing GST or GST-tagged JNK3 mutants were introduced into COS-7 cells together with an expression vector for FLAG-β-arrestin-2. GST-containing complexes were isolated from cell lysates with glutathione-Sepharose beads, and the β-arrestin-2 present in the pull-down (PD) was examined by immunoblot using M2 antibody. The expression of the JNK3 deletions and β-arrestin-2 was examined by immunoblotting the lysates with anti-GST and M2 antibodies, respectively. F, constructs expressing GFP, GFP-JNK3, and GFP-JNK3(L14A/V16A) were introduced into 293T cells and JNK3-containing complexes isolated using anti-GFP antibodies. The presence of endogenous β-arrestin-2 in the precipitates (IP) was detected using an anti-β-arrestin antibody. The expression of the JNK3 proteins and β-arrestin-2 was examined by immunoblotting the lysates with anti-GFP and anti-β-arrestin antibodies, respectively. G, constructs expressing GST or GST-tagged β-arrestin-2 were introduced into COS-7 cells together with an expression vector for Myc-tagged CAMK1γ. GST-containing complexes were isolated from cell lysates with glutathione-Sepharose beads, and the CAMK1γ present in the pull-down (PD) was examined by immunoblot using anti-Myc antibody. Experiments were performed either two or three times and representative immunoblots are shown.
Binding of JNK3 to β-Arrestin-2 Can Regulate Its Subcellular Localization—It was previously reported that β-arrestin-2 can regulate the subcellular localization of JNK3 (12, 17, 18). To determine whether the binding of JNK3 to β-arrestion-2 was required for this regulation we utilized the binding defective JNK3 mutant (JNK3 L14A/V16A). We co-expressed GFP-tagged wild-type or mutant JNK3 in cells together with β-arrestin-2 and analyzed their localization. As expected GFP-JNK3 is found in the cytoplasm and nucleus, but when co-expressed with β-arrestin-2, it is located exclusively in the cytoplasm (Fig. 3, A and B). The localization of the mutant JNK3 does not change in the presence of β-arrestin-2, suggesting that JNK3 binding to β-arrestin-2 is essential for β-arrestin-2 regulation of JNK3 localization (Fig. 3B).
FIGURE 3.
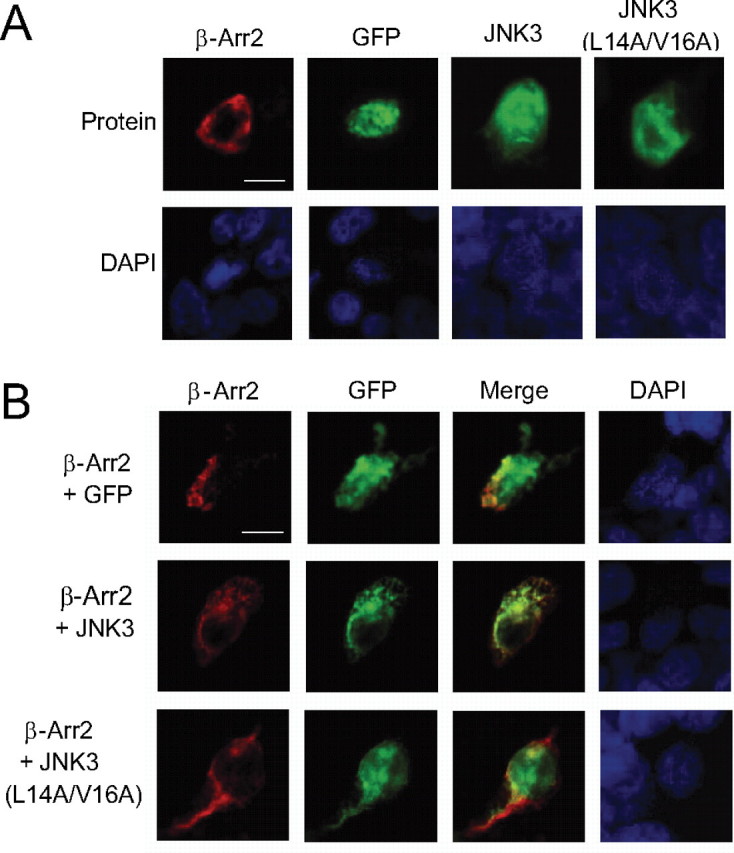
Co-localization of JNK3 and β-arrestin-2. A and B, constructs expressing the indicated combinations of GFP, GFP-JNK3α1, GFP-JNK3α1(L14A/V16A) with FLAG-β-arrestin-2 were introduced into 293T cells. GFP fluorescence was observed directly (green), and the FLAG-β-arrestin-2 was detected by indirect immunofluorescence using the M2 antibody and an Alexor594-conjugated secondary antibody (red). Nuclei were detected with DAPI (blue). Scale bars, 10 μm.
The Binding-defective Mutant of JNK3 Is Not Activated by β-Arrestin-2 and ASK1—We next sought to determine the importance of JNK3 binding for β-arrestion-2-mediated JNK3 activation. β-Arrestin-2 enhances ASK1 activation of JNK3 (Ref. 12 and Fig. 4A, lane 5) but not of the binding-defective JNK3 mutant (Fig. 4A, lane 9) demonstrating that the interaction of JNK3 with β-arrestin-2 is essential for optimal activation of JNK3 by ASK1. Both wild-type and mutant JNK3 were capable of being activated by co-expression with a different MAPKKK, MLK3 (Fig. 4B). This suggests that the N terminus of JNK3 is an important specificity determinant of signaling by the β-arrestin-2 scaffold complex but that it is not a general requirement for JNK3 activation. To further elucidate the importance of the JNK3 N terminus for β-arrestin-2-mediated signaling, we utilized the chimeric protein featuring the N terminus of JNK3 fused to JNK1 (J3/J1-B). β-Arrestin-2 enhanced ASK1-mediated phosphorylation of this chimera similar to wild-type JNK3 (Fig. 4C, compare lanes 2 and 3 with lanes 5 and 6). However, β-arrestin-2 did not influence ASK1-mediated phosphorylation of JNK1 (Fig. 4C, lanes 8 and 9). Previously, β-arrestin-2 has been implicated in mediating angiotensin II-induced activation of JNK3 (12). We confirmed this (Fig. 4D, compare lanes 2 and 5) and demonstrated that the binding-defective JNK3 mutant was not activated by angiotensin II (Fig. 4D, compare lanes 5 and 6). These data are supportive of the N terminus of JNK3 being the key determinant that permits regulation by β-arrestin-2.
FIGURE 4.

JNK3 binding to β-arrestin-2 is required to mediate ASK1 activation of JNK3. A, constructs expressing HA-JNK3 or HA-JNK3 (L14A/V16A) were introduced into COS-7 cells together with expression vectors for HA-ASK1 and FLAG-β-arrestin-2 as indicated. Lysates were immunoblotted with phospho-JNK, HA, and M2 antibodies. B, constructs expressing HA-JNK3 or HA-JNK3 (L14A/V16A) were introduced into COS-7 cells together with an expression vector for HA-MLK3 as indicated. Lysates were immunoblotted with phospho-JNK and HA antibodies. * indicates a nonspecific band detected by the HA antibody. C, constructs expressing HA-JNK3 or the JNK3/JNK1 chimera (HA-J3/J1-B) or HA-JNK1 were introduced into COS-7 cells together with expression vectors for HA-ASK1 and FLAG-β-arrestin-2 as indicated. Lysates were immunoblotted with phospho-JNK, HA, and M2 antibodies. * indicates nonspecific bands detected by the HA antibody. D, constructs expressing GFP, GFP-JNK3, or GFP-JNK3(L14A/V16A) were introduced into 293T cells together with expression vectors for FLAG-β-arrestin-2 and the angiotensin II receptor HA-AT1AR. Cells were treated with or without angiotensin II (Ang II; 1 μm, 20 min) as indicated. Immunoblotting was performed with phospho-JNK, GFP, HA, and M2 antibodies. Experiments were performed three times, and representative immunoblots are shown.
JNK3 Recruits MKK4 to the β-Arrestin-2 Scaffold Complex— It is proposed that MKK4 is recruited to β-arrestin-2 complexes by binding to ASK1 and JNK3 (12). Consistent with these data we have shown that ASK1 is present in β-arrestin-2 immunoprecipitates from transfected COS-7 cells (Fig. 5A) and that MKK4 does not directly bind to β-arrestin-2 (Fig. 5E, lane 1). We also detect binding of MKK4 to ASK1 (Fig. 5B) and JNK3 (Fig. 5C). To determine the relative importance of ASK1 or JNK3 for recruiting MKK4 to β-arrestin-2 we co-expressed MKK4 and β-arrestin-2 in COS-7 cells along with either ASK1 or JNK3 and looked for β-arrestin-2 in MKK4 precipitates. We detected strong association of MKK4 with β-arrestin-2 in the presence of JNK3, but not in the presence of ASK1 (Fig. 5D), indicating that JNK3 plays a major role in recruiting MKK4 to β-arrestin-2 complexes. To support this conclusion we performed a similar experiment using the JNK3 mutant (JNK3 L14A/V16A) that does not bind to β-arrestin-2 and also the chimeric construct featuring the N terminus of JNK3 fused to JNK1. As predicted neither the JNK3 mutant or JNK1 (which binds to MKK4 but not β-arrestin-2) were able to mediate MKK4 binding, while the chimera did recruit MKK4 to β-arrestin-2 (Fig. 5E). These experiments demonstrate that JNK3 recruits MKK4 to the β-arrestin-2 scaffold complex.
FIGURE 5.
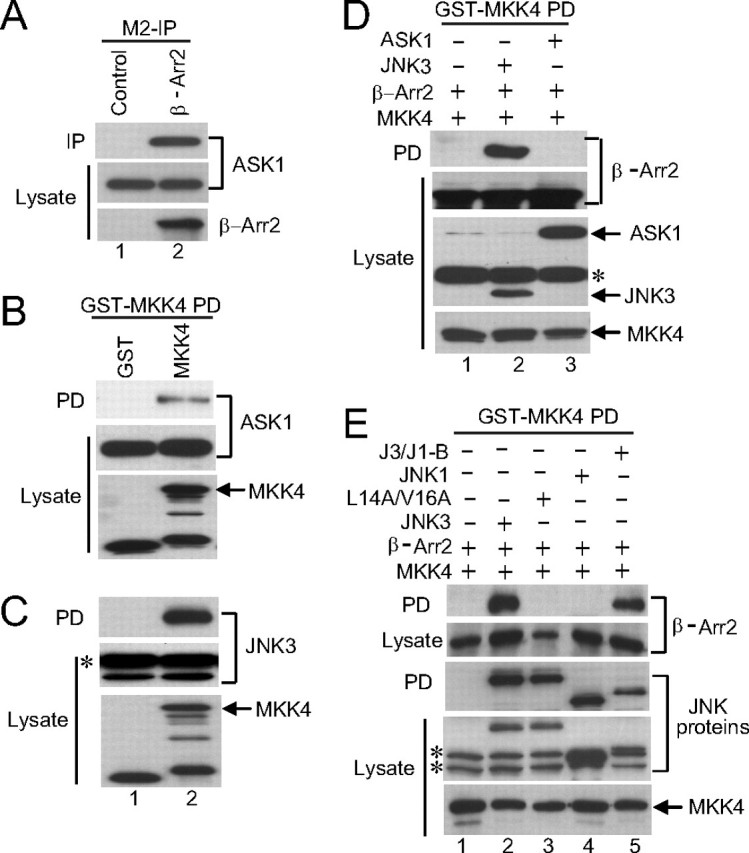
JNK3 recruits MKK4 to β-arrestin-2. A, constructs expressing HA-ASK1 and FLAG-β-arrestin-2 were introduced into COS-7 cells. In the control an empty vector was transfected in place of the β-arrestin-2 expression vector. β-Arrestin-2 was immunoprecipitated with the M2 antibody (M2-IP) and ASK1 present in the precipitates detected with anti-HA antibody. The expression of ASK1 and β-arrestin-2 was examined by immunoblotting the lysates with anti-HA and M2 antibodies, respectively. B and C, constructs expressing GST or GST-MKK4 were introduced into COS-7 cells together with an expression vector for HA-ASK1 (B) or HA-JNK3 (C). GST-containing complexes were isolated from cell lysates with glutathione-Sepharose beads, and the ASK1 or JNK3 present in the pull-down (PD) was examined by immunoblot using anti-HA antibody. The expression of the MKK4, ASK1, and JNK3 was examined by immunoblotting the lysates with anti-GST and anti-HA antibodies, respectively. D, constructs expressing GST-tagged MKK4 were introduced into COS-7 cells together with an expression vector for FLAG-β-arrestin-2 and either HA-JNK3 or HA-ASK1. GST-containing complexes were isolated from cell lysates with glutathione-Sepharose beads and the β-arrestin-2 present in the pull-down (PD) was examined by immunoblot using M2 antibody. The expression of the MKK4, β-arrestin-2, ASK1, and JNK3 was examined by immunoblotting the lysates with anti-GST, M2, and anti-HA antibodies, respectively. E, construct expressing GST-MKK4 was introduced into COS-7 cells together with an expression vector for FLAG-β-arrestin-2 and the indicated JNK mutants. GST-containing complexes were isolated from cell lysates with glutathione-Sepharose beads and the β-arrestin-2 present in the pull-down (PD) was examined by immunoblot using M2 antibody. The expression of the MKK4, β-arrestin-2, and the JNK mutants was examined by immunoblotting the lysates with anti-GST, M2, and anti-HA antibodies, respectively. * indicates nonspecific bands detected by the HA antibody. Experiments were performed either two or three times and representative immunoblots are shown.
The D-domain of MKK4 Is Required for Its Recruitment to β-Arrestin-2 via JNK3—Previous studies have demonstrated that a D-domain sequence in the N terminus of MKK4 mediates its binding to JNK1 and JNK2 (24, 25). It is possible therefore that JNK3 recruits MKK4 to the β-arrestin-2 scaffold by binding to the D-domain of MKK4. Co-precipitation analysis demonstrated that, like JNK1 and JNK2, the D-domain of MKK4 was required for binding to JNK3 as the mutation of the D-domain (L44A/L46A) blocked the interaction (Fig. 6A). To confirm the importance of the MKK4 D-domain for the recruitment of MKK4 to the β-arrestin-2 scaffold by JNK3 we demonstrated that the D-domain mutant of MKK4 was not recruited to β-arrestin-2 by JNK3 (Fig. 6B). JNK isoforms interact with MKK4 via a docking groove comprising the ED and CD domains (24, 25). Therefore, we mutated critical acidic residues in the CD domain of JNK3. This JNK3 mutant could bind to β-arrestin-2 (Fig. 6C) but failed to bind to MKK4 (Fig. 6D) or to recruit MKK4 to the β-arrestin-2 complex (Fig. 6E). Our results support the idea that MKK4 is recruited to β-arrestin-2 by JNK3 via an interaction between the JNK3 docking groove and the D-domain of MKK4.
FIGURE 6.
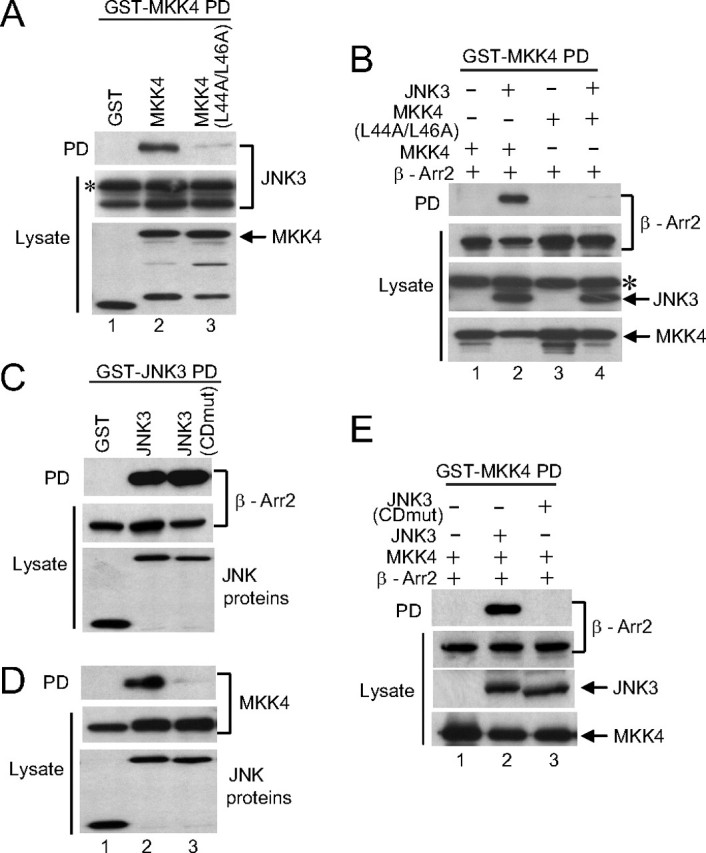
MKK4 is recruited to the β-arrestin-2 scaffold complex via its N-terminal D-domain. A, constructs expressing GST, GST-MKK4, or GST-MKK4 (L44A/L46A) were introduced into COS-7 cells together with an expression vector for HA-JNK3. GST-containing complexes were isolated from cell lysates with glutathione-Sepharose beads and JNK3 present in the pull-down (PD) was examined by immunoblot using anti-HA antibody. The expression of the MKK4 and JNK3 was examined by immunoblotting the lysates with anti-GST and anti-HA antibodies, respectively. B, constructs expressing GST-MKK4 or GST-MKK4 (L44A/L46A) were introduced into COS-7 cells together with expression vectors for HA-JNK3 and FLAG-β-arrestin-2. GST-containing complexes were isolated from cell lysates with glutathione-Sepharose beads, and the β-arrestin-2 present in the pull-down (PD) was examined by immunoblot using M2 antibody. The expression of the MKK4, JNK3, and β-arrestin-2 was examined by immunoblotting the lysates with anti-GST, anti-HA, and M2 antibodies, respectively. C and D, constructs expressing GST, GST-JNK3, or GST-JNK3(CDmut) were introduced into COS-7 cells together with expression vectors for either FLAG-β-arrestin-2 (C) or Myc-tagged MKK4 (D). GST-containing complexes were isolated from cell lysates with glutathione-Sepharose beads, and β-arrestin-2 and MKK4 present in the pull-down (PD) were examined by immunoblot using M2 and anti-Myc antibodies, respectively. The expression of the JNK3, β-arrestin-2, and MKK4 was examined by immunoblotting the lysates with anti-GST, M2, and anti-Myc antibodies, respectively. E, constructs expressing GST-tagged MKK4 were introduced into COS-7 cells together with expression vectors for FLAG-β-arrestin-2 and either HA-JNK3 or HA-JNK3 (CDmut). GST-containing complexes were isolated from cell lysates with glutathione-Sepharose beads, and the β-arrestin-2 present in the pull-down (PD) was examined by immunoblot using M2 antibody. The expression of the MKK4, JNK3, and β-arrestin-2 was examined by immunoblotting the lysates with anti-GST, anti-HA, and M2 antibodies, respectively. * indicates nonspecific bands detected by the HA antibody. Experiments were performed either two or three times, and representative immunoblots are shown.
The C Terminus of β-Arrestin-2 Binds to JNK3—A previous study identified a D-domain-like sequence in β-arrestin-2 that was important for determining the specificity of binding to JNK3 (21). However, our results suggest that JNK3 is unlikely to be binding directly to the D-domain on β-arrestin-2 as it binds to the D-domain on MKK4 to recruit it to the scaffold complex (Fig. 6, A and B). Secondly, the mutation of the CD domain of JNK3, which has previously been demonstrated to be important for binding to D-domains (20, 22, 25), does not block JNK3 binding to β-arrrestin-2 (Fig. 6C). To address this issue, we generated deletion mutants of β-arrestin-2 and demonstrated by co-precipitation analysis that JNK3 binds to the C-terminal-half of β-arrestin-2 and not to an N-terminal fragment that contains the putative D-domain (Fig. 7, A and B). ASK1, on the other hand, bound to the N-terminal β-arrestin-2 fragment (Fig. 7C).
FIGURE 7.

Specificity determinants within β-arrestin-2 for binding to JNK3. A, schematic of the β-arrestin constructs used in the experiments and a summary of their binding to JNK3 (- represents weak or no binding; + represents strong binding). The position of the D-domain like sequence is indicated. B and C, constructs expressing GST-tagged β-arrestin-2 deletion mutants were introduced into COS-7 cells together with expression vectors for HA-JNK3 (B) and HA-ASK1 (C). GST-containing complexes were isolated from cell lysates with glutathione-Sepharose beads, and the JNK3 or ASK1 present in the pull-down (PD) was examined by immunoblot using anti-HA antibody. The expression of the ASK1, JNK3, and β-arrestin-2 was examined by immunoblotting the lysates with anti-HA and anti-GST antibodies, respectively. D and E, constructs expressing the indicated GST-tagged β-arrestin proteins were introduced into COS-7 cells together with an expression vector for HA-JNK3. GST-containing complexes were isolated from cell lysates with glutathione-Sepharose beads and the JNK3 present in the pull-down (PD) was examined by immunoblot using anti-HA antibody. The expression of the JNK3 and the β-arrestin proteins was examined by immunoblotting the lysates with anti-HA and anti-GST antibodies, respectively. F, sequence alignment of the putative D-domains in β-arrestin-1 and β-arrestin-2. The D-domains are boxed, and the key Ser and Pro residues highlighted. Numbers refer to the amino acid position. G, constructs expressing the indicated FLAG-tagged β-arrestin-2 mutants were introduced into COS-7 cells together with an expression vector for either GST or GST-JNK3. GST-containing complexes were isolated from cell lysates with glutathione-Sepharose beads, and the β-arrestin proteins present in the pull-down (PD) was examined by immunoblot using the M2 antibody. The expression of the JNK3 and the β-arrestin proteins was examined by immunoblotting the lysates with anti-GST and M2 antibodies, respectively. H, constructs expressing the indicated FLAG-tagged β-arrestin proteins were introduced into COS-7 cells together with an expression vector for GST-JNK3. FLAG-β-arrestin-2 complexes were isolated from cell lysates by immunoprecipitation with the M2 antibody (M2-IP), and JNK3 present in the precipitates was examined by immunoblot using an anti-GST antibody. The expression of the JNK3 and the β-arrestin proteins was examined by immunoblotting the lysates with anti-GST and M2 antibodies, respectively. I, scheme of β-arrestin-2 scaffold assembly. The C terminus of β-arrestin-2 (denoted C) binds to the extended N terminus of JNK3 (denoted N), and this binding is controlled by residues within the D-domain of β-arrestin-2 (denoted D). MKK4 is recruited to the complex via its D-domain (denoted D) binding to JNK3. ASK1 binds to the N terminus of β-arrestin-2 (denoted N). Experiments were performed either two or three times, and representative immunoblots are shown.
To try and reconcile our results with the previously reported role of the putative D-domain of β-arrestin-2 in regulating JNK3 binding, we generated chimeric proteins between β-arrestin-2 and β-arrestin-1, which does not bind to JNK3 (Fig. 7A). Consistent with a role for the D-domain in determining specificity, the chimera including the D-domain of β-arrestin-2 and the C terminus of β-arrestin-1 bound to JNK3 similar to wild-type β-arrestin-2, while the chimera that included the D-domain of β-arrestin-1 and the C terminus of β-arrestin-2 bound very weakly (Fig. 7D). This suggested that, although the C terminus of β-arrestin-2 contained the JNK3 binding site, there were residues within the D-domain that could influence the binding. In addition, it suggested that the C terminus of β-arrestin-1 may be capable of binding to JNK3 as the chimera with the C terminus of β-arrestin-1 bound JNK3 (Fig. 7D). To confirm this, we generated a construct expressing the C terminus of β-arrestin-1 and we found that it bound to JNK3 similar to the β-arrestin-2 C terminus (Fig. 7E). To further characterize the role of the D-domain in modulating binding to the β-arrestin-2 C terminus we mutated the basic and hydrophobic residues that are known to be required for D-domain docking functions (20, 22). These mutations did not affect JNK3 binding to β-arrestin-2 (Fig. 7, F and G). The lack of importance of the basic residues for determining binding specificity is supported by the fact that the N terminus of JNK3 binds to both the rat β-arrestin-2 used in this study and human β-arrestin-2,3 even though the D-domain is not entirely conserved with the initial Arg missing in the human protein (Fig. 7F). Next we mutated Ser-198 within the β-arrestin-2 D-domain to Pro, which is found at the corresponding position in β-arrestin-1, and performed the reverse mutation in β-arrestin-1 (Fig. 7F). The β-arrestin-2 S198P mutant displayed greatly reduced binding to JNK3, while the β-arrestin-1 P196S mutant displayed enhanced binding (Fig. 7H). Taken, together, these results indicate that Ser-198 is a key specificity determining residue in β-arrestin-2 that permits the binding of JNK3 to the C terminus.
DISCUSSION
The JNK MAPK pathway plays pivotal roles in regulating many cellular functions (1, 2). The elucidation of the molecular mechanisms by which specific components within the JNK pathway interact to transmit a signal is important for our understanding of these functions. In this study, we investigated the docking interactions between components of the β-arrestin-2 scaffold complex, which contains JNK3, MKK4, and ASK1. In particular we focused on the role of JNK3 binding to β-arrestin-2 and the recruitment of MKK4.
JNK MAPK isoforms demonstrate high sequence similarity. However JNK3 is unique in binding to β-arrestin-2. Previous studies suggested that JNK3 binding to β-arrestin-2 was mediated by a MAP kinase docking motif on β-arrestin-2 that resembles a D-domain (21). D-domains are found in many MAPK binding partners and are proposed to bind to a docking groove in the MAPK that is formed in part by the N-terminal ED domain and the C-terminal CD domain (22, 23). Surprisingly our findings demonstrate that β-arrestin-2 does not bind to this docking groove in JNK3 but that the binding is mediated via the extended N terminus of JNK3 (Figs. 1 and 2) and that this is required for β-arrestin-2-mediated activation of JNK3 (Fig. 4). Indeed, the JNK3 N terminus can serve as an independent β-arrestin-2 targeting motif as when fused to JNK1 it permits the recruitment of JNK1 to the β-arrestin-2 scaffold and confers β-arrestin-2 control over JNK1 phosphorylation and activation (Figs. 1F and 4C). Our study is the first demonstration of a specific role for the N-terminal extension of JNK3.
It is currently not clear whether the interaction between JNK3 and β-arrestin-2 is constitutive or if it is regulated by signaling events. We have not observed changes in binding in response to ASK1 activation of JNK3 or in response to angiotensin II.3 However, it is possible that the interaction is controlled by other stimuli or that there are other modes of regulation.
Interestingly, homology searches identified CAMK1γ as having a region with significant similarity to the N terminus of JNK3 (Fig. 2B) and we demonstrated that it can also bind to β-arrestin-2 (Fig. 2G). CAMK1γ is a recently characterized membrane-associated neuronal isoform of the CAMK1 family (32) reported to play a role in dendritogenesis (33, 34). Previously, a connection between arrestins and CAMKs has been reported in Drosophila photoreceptors where phosrestin I, the Drosophila homologue of photoreceptor arrestin, is phosphorylated by CAMK (35). However, the functional significance of the interplay between arrestins and CAMKs remains to be determined. Our results suggest that β-arrestin-2 may use similar binding determinants to recruit functionally distinct protein kinases and raise the possibility that these kinases may compete for binding to β-arrestin-2.
In addition to identifying key binding determinants on JNK3 that are required for its interaction with β-arrestin-2, we also demonstrated that JNK3 does not directly bind to the putative D-domain in β-arrestin-2 but instead binds to a C-terminal region (Fig. 7). However, residues within the D-domain do determine the specificity of β-arrestin-2 binding to JNK3 (Fig. 7). It is plausible that substituting Ser-198 in β-arrestin-2 for Pro, as found in this position in β-arrestin-1, alters the conformation of the β-arrestin-2 C terminus to reduce its affinity for JNK3.
The binding of β-arrestin-2 to the N terminus of JNK3 means that the docking groove in JNK3 that contains the ED and CD domains may be available for interacting with other D-domain-containing proteins. Indeed, we find that MKK4 is recruited to the β-arrestin-2 scaffold complex via its N-terminal D-domain interacting with JNK3 (Fig. 5). Our results suggest a model whereby the N terminus of JNK3 is recruited to the C terminus of β-arrestin-2 and can then bind to the N-terminal D-domain of MKK4 thus nucleating a MAPK signaling module (Fig. 7I). In addition, they also raise the possibility that JNK3 could recruit other D-domain-containing proteins to the β-arrestin-2 scaffold complex including substrates and phosphatases.
In conclusion, our study provides an important insight into how signaling specificity is achieved by the JNK pathway. The identification of a specific docking determinant within the N terminus of JNK3 explains why this JNK isoform is specifically recruited to and activated by the β-arrrestin-2 scaffold complex.
Acknowledgments
We thank C. Tournier, R. J. Davis, R. J. Lefkowitz, and M. Caron for reagents. We thank A. Sharrocks, S.-H. Yang, and C. Tournier for critical reading of the manuscript.
This work was supported in part by the Medical Research Council (MRC), the Wellcome Trust, BBSRC, and a Lister Institute-Jenner Research Fellowship (to A. J. W.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: JNK, c-Jun N-terminal kinase; ASK1, apoptosis-signaling kinase-1; CAMK1γ, calcium/calmodulin-dependent kinase-1γ; CD, common docking domain; DAPI, 4′,6-diamidino-2-phenylindole; D-domain, docking domain; GFP, green fluorescent protein; GST, glutathione S-transferase; HA, hemagglutinin; JIP, JNK-interacting protein; MAPK, mitogen-activated protein kinase; MKK, mitogen-activated protein kinase kinase; MAPKKK, mitogen-activated protein kinase kinase kinase; MEKK, MEK kinase; MKP, MAPK phosphatase; MLK, mixed lineage kinase; aa, amino acids.
C. Guo and A. J. Whitmarsh, unpublished results.
References
- 1.Davis, R. J. (2000) Cell 103 239-252 [DOI] [PubMed] [Google Scholar]
- 2.Kyriakis, J. M., and Avruch, J. (2001) Phys. Revs. 81 807-869 [DOI] [PubMed] [Google Scholar]
- 3.Kyriakis, J. M., Banerjee, P., Nikolakaki, E., Dai, T., Rubie, E. A., Ahmad, M. F., Avruch, J., and Woodgett, J. R. (1994) Nature 369 156-160 [DOI] [PubMed] [Google Scholar]
- 4.Gupta, S., Barrett, T., Whitmarsh, A. J., Cavanagh, J., Sluss, H. K., Derijard, B., and Davis, R. J. (1996) EMBO J. 15 2760-2770 [PMC free article] [PubMed] [Google Scholar]
- 5.Mohit, A. A., Martin, J. H., and Miller, C. A. (1995) Neuron 14 67-78 [DOI] [PubMed] [Google Scholar]
- 6.Yang, D. D., Kuan, C.-Y., Whitmarsh, A. J., Rincon, M., Zheng, T. S., Davis, R. J., Rakic, P., and Flavell, R. A. (1997) Nature 389 865-870 [DOI] [PubMed] [Google Scholar]
- 7.Kuan, C.-Y., Whitmarsh, A. J., Yang, D. D., Liao, G., Schloemer, A. J., Dong, C., Bao, J., Banasiak, K. J., Haddad, G. G., Flavell, R. A., Davis, R. J., and Rakic, P. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 15184-15189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morishima, Y., Gotoh, Y., Zieg, J., Barrett, T., Takano, H., Flavell, R. A., Davis, R. J., Shirasaki, Y., and Greenberg, M. E. (2001) J. Neurosci. 21 7551-7560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morrison, D. K., and Davis, R. J. (2003) Annu. Rev. Cell Dev. Biol. 19 91-118 [DOI] [PubMed] [Google Scholar]
- 10.Whitmarsh, A. J. (2006) Biochem. Soc. Trans. 34 828-832 [DOI] [PubMed] [Google Scholar]
- 11.Xu, Z., Kukekov, N. V., and Greene, L. A. (2003) EMBO J. 22 252-261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McDonald, P. H., Chow, C.-W., Miller, W. E., Laporte, S. A., Field, M. E., Lin, F.-T., Davis, R. J., and Lefkowitz, R. J. (2000) Science 290 1574-1577 [DOI] [PubMed] [Google Scholar]
- 13.Lefkowitz, R. J., Rajagopal, K., and Whalen, E. J. (2006) Mol. Cell 24 643-652 [DOI] [PubMed] [Google Scholar]
- 14.Willoughby, E. A., and Collins, M. K. (2005) J. Biol. Chem. 280 25651-25658 [DOI] [PubMed] [Google Scholar]
- 15.DeWire, S. M., Ahn, S., Lefkowitz, R. J., and Shenoy, S. K. (2007) Annu. Rev. Physiol. 69 483-510 [DOI] [PubMed] [Google Scholar]
- 16.Ma, L., and Pei, G. (2006) J. Cell Sci. 120 213-218 [DOI] [PubMed] [Google Scholar]
- 17.Scott, M. G. H., Le Rouzic, E., Perianin, A., Pierotti, V., Enslen, H., Benichou, S., Marullo, S., and Benmerah, A. (2002) J. Biol. Chem. 277 37693-37701 [DOI] [PubMed] [Google Scholar]
- 18.Wang, P., Wu, Y., Ge, X., Ma, L., and Pei, G. (2003) J. Biol. Chem. 278 11648-11653 [DOI] [PubMed] [Google Scholar]
- 19.Sharrocks, A. D., Yang, S., and Galanis, A. (2000) Trends. Biochem. Sci. 25 448-453 [DOI] [PubMed] [Google Scholar]
- 20.Tanoue, T., and Nishida, E. (2003) Cell. Signal. 15 455-462 [DOI] [PubMed] [Google Scholar]
- 21.Miller, W. E., McDonald, P. H., Cai, S. F., Field, M. E., Davis, R. J., and Lefkowitz, R. J. (2001) J. Biol. Chem. 276 27770-27777 [DOI] [PubMed] [Google Scholar]
- 22.Tanoue, T., Adachi, M., Moriguchi, T., and Nishida, E. (2000) Nat. Cell Biol. 2 110-116 [DOI] [PubMed] [Google Scholar]
- 23.Tanoue, T., Maeda, R., Adachi, M., and Nishida, E. (2001) EMBO J. 20 466-479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ho, D. T., Bardwell, A. J., Abdollahi, M., and Bardwell, L. (2003) J. Biol. Chem. 278 32662-32672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mooney, L. M., and Whitmarsh, A. J. (2004) J. Biol. Chem. 279 11843-11852 [DOI] [PubMed] [Google Scholar]
- 26.Heo, Y. S., Kim, S. K., Seo, C. I., Kim, Y. K., Sung, B. J., Lee, H. S., Lee, J. I., Park, S. Y., Kim, J. H., Hwang, K. Y., Hyum, Y. L., Jeon, Y. H., Ro, S., Cho, J. M., Lee, T. G., and Yang, C. H. (2004) EMBO J. 23 2185-2195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barr, R. K., Hopkins, R. M., Watt, P. M., and Bogoyevitch, M. A. (2004) J. Biol. Chem. 279 43178-43189 [DOI] [PubMed] [Google Scholar]
- 28.Sluss, H. K., Barrett, T., Derijard, B., and Davis, R. J. (1994) Mol. Cell. Biol. 14 8376-8384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yasuda, J., Whitmarsh, A. J., Cavanagh, J., Sharma, M., and Davis, R. J. (1999) Mol. Cell. Biol. 19 7245-7254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Whitmarsh, A. J., Cavanagh, J., Tournier, C., Yasuda, J., and Davis, R. J. (1998) Science. 281 1671-1674 [DOI] [PubMed] [Google Scholar]
- 31.Tournier, C., Whitmarsh, A. J., Cavanagh, J., Barrett, T., and Davis, R. J. (1999) Mol. Cell. Biol. 19 1569-1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takemoto-Kimura, S., Terai, H., Takamoto, M., Ohmae, S., Kikumura, S., Segi, E., Arakawa, Y., Furuyashiki, T., Narumiya, S., and Bito, H. (2003) J. Biol. Chem. 278 18597-18605 [DOI] [PubMed] [Google Scholar]
- 33.Wayman, G. A., Impey, S., Marks, D., Saneyoshi, T., Grant, W. F., Derkach, V., and Soderling, T. R. (2006) Neuron 50 897-909 [DOI] [PubMed] [Google Scholar]
- 34.Takemoto-Kimura, S., Ageta-Ishihara, N., Nonaka, M., Adachi-Morishima, A., Mano, T., Okamura, M., Fujii, H., Fuse, T., Hoshino, M., Suzuki, S., Kojima, M., Mishina, M., Okuno, H., and Bito, H. (2007) Neuron 54 755-770 [DOI] [PubMed] [Google Scholar]
- 35.Matsumoto, H., Kurlen, B. J., Takagi, Y., Kahn, E. S., Kinumi, T., Komori, N., Yamada, T., Hayashi, F., Isono, K., Pak, W. L., Jackson, K. W., and Tobin, S. L. (1994) Neuron 12 997-1010 [DOI] [PubMed] [Google Scholar]