

Abstract
When the catalytic A subunits of the castor bean toxins ricin and Ricinus communis agglutinin (denoted as RTA and RCA A, respectively) are delivered into the endoplasmic reticulum (ER) of tobacco protoplasts, they become substrates for ER-associated protein degradation (ERAD). As such, these orphan polypeptides are retro-translocated to the cytosol, where a significant proportion of each protein is degraded by proteasomes. Here we begin to characterize the ERAD pathway in plant cells, showing that retro-translocation of these lysine-deficient glycoproteins requires the ATPase activity of cytosolic CDC48. Lysine polyubiquitination is not obligatory for this step. We also show that although RCA A is found in a mannose-untrimmed form prior to its retro-translocation, a significant proportion of newly synthesized RTA cycles via the Golgi and becomes modified by downstream glycosylation enzymes. Despite these differences, both proteins are similarly retro-translocated.
As in mammalian and yeast cells, the plant cell endoplasmic reticulum (ER)3 is a major protein folding compartment. Newly synthesized soluble and membrane proteins destined to remain in the ER, or to be transported to the Golgi complex, vacuoles, or apoplast, are scrutinized by a stringent quality control system (1). Such surveillance ensures that newly synthesized proteins assume their native conformation and, where appropriate, assemble into oligomers. In mammalian and yeast cells, proteins that fail to fold and/or assemble correctly are usually retro-translocated to the cytosol and degraded by cytosolic proteasomes, in a tightly coupled pathway referred to as ER-associated protein degradation (ERAD) (2, 3). For the majority of misfolded or unassembled substrates, extraction from the ER membrane requires a cytosolic ubiquitin-interacting AAA-ATPase complex designated CDC48 in yeast (p97, also known as valosin-containing protein, in mammalian cells), in association with the adaptor proteins Ufd1p and Npl4p (4–8). This complex is believed to use its ATPase activity to segregate ubiquitinated ERAD substrates (7, 9), and/or mechanically extract them from the ER membrane in readiness for delivery to proteasomes (5, 8). However, not all ERAD substrates are modified by ubiquitin and extracted by this ATPase (8, 10–12), leading to the suggestion that an alternative driving force must exist. For prepro-α-factor, this has been shown to be the ATPases located at the base of the 19 S proteasome regulatory complex (10).
A pathway closely resembling ERAD also operates in plant cells but, by comparison, relatively little is known about substrate recognition, extraction from the ER membrane, or the pathway(s) and machineries for degradation (13). The Arabidopsis thaliana genome contains sequences for three CDC48-like proteins, one of which has been functionally shown to complement a yeast CDC48 mutant (14, 15). This AAA-ATPase has been found to play a role in the quality control of a mutant polytopic membrane protein, barley (Hordeum vulgare) powdery mildew resistance protein (MLO), but its involvement in ERAD is thus far limited to this one example (16). The process of retro-translocation, deglycosylation, and proteasomal degradation in plant cells was first characterized by studying the biosynthesis of the catalytic A subunit (RTA) of the A-B plant toxin ricin (17). This orphan polypeptide, once delivered into the ER by its signal peptide, was shown to retro-translocate in a manner unrelated to its propensity for ubiquitination (18). Although it was shown that ubiquitination of internal lysine residues is not a strict requirement for ER to cytosol transport of this protein, the involvement or otherwise of CDC48 was not investigated. Here we have studied the effects of a dominant negative form of Arabidopsis CDC48 on the export of the orphan catalytic polypeptides of ricin and its close relative, Ricinus communis agglutinin, in tobacco protoplasts. The data presented clearly demonstrate that the retro-translocation of these proteins requires the participation of CDC48, and is irrespective of their glycosylation or ubiquitination status.
EXPERIMENTAL PROCEDURES
Recombinant DNA—All DNA constructs were generated in the CaMV 35S-promoter-driven expression vectors pDHA (see Ref. 19, for toxin- and phaseolin-based constructs) and pamPAT-MCS (GenBank™ accession number AY436765 (16)) or pGreenII-0029 (see Ref. 20, for CDC48- or fluorescent protein-based constructs). Expression constructs encoding RTA, phaseolin (pDHE-T343F), CDC48, and cytosolic YFP have been described previously (16, 21–23). The ricin active site substitution E177D, lysine substitutions K4R and K239R, and the removal of the proricin signal peptide and N-terminal propeptide have also been previously documented (18, 24, 25). All derivative constructs used in this work were generated using the QuikChange™ in vitro mutagenesis system (Stratagene, La Jolla, CA) using the following mutagenic primers (and their reverse complements, not shown): a stop codon was introduced into the prepro-RCA sequence (26) immediately after the open reading frame of the A chain to generate pRCA A using 5′-CCTCCACCGTCGTCAGAGTTTTAGTTGCTTATAAGGCCAGTGGTGCC-3′, and the equivalent active site substitution E176D was introduced into pRCA A using 5′-GGTTTGCATCCAAATGATTTCAGACGCAGCAAGATTCCAGTACATTG-3′. Sites of amino acid mutations are underlined. wtCDC48 and CDC48QQ were cloned into the HindIII-SmaI sites of the CaMV 35S-cassette using primers 5′-ATATATATAAGCTTATGTCTACCCCAGCTG-3′ and 5′-AACGAAGCCCGGGCTAATTGTAGAGATC-3′, for subsequent insertion into EcoRV-cut pGreenII-0029, used for tobacco leaf infiltration. Restriction enzyme sites are underlined. To generate cytosolic RFP, the monomeric RFP1 coding region was amplified by PCR from pcDNA1-mRFP (27) and cloned into the XbaI-SacI sites of the CaMV 35S-cassette using primers 5′-GCGCGCGTCTAGAATGGCCTCCTCCGAGGAC-3′ and 5′-TAATGATGGAGCTCTTAGGCGCCGGTGGAGTGGC-3′, again for subsequent insertion into EcoRV-cut pGreenII-0029. Restriction enzyme sites are underlined.
Transient Transfection of Tobacco Leaf Protoplasts and Pulse-Chase Experiments—Protoplasts were prepared from axenic leaves (4 to 7 cm long) of Nicotiana tabacum cv. Petit Havana SR1 (28), and subjected to polyethylene glycol-mediated transfection with one or more plasmids as previously described (22). Cells were radiolabeled with Pro-Mix (a mixture of [35S]cysteine and [35S]methionine (GE Healthcare)), and chased for the times indicated in the figures, as previously described (21). In some experiments, before radioactive labeling, protoplasts were incubated for 1 h at 25 °C in K3 medium (3.78 g/liter Gamborgs B5 basal medium with minimal organics, 750 mg/liter CaCl2·2H2O, 250 mg/liter NH4NO3, 136.2 g/liter sucrose, 250 mg/liter xylose, 1 mg/liter 6-benzylaminopurine, 1 mg/liter α-naphthalenacetic acid) supplemented with either 50 μg/ml tunicamycin (Sigma; 5 mg/ml stock in 10 mm NaOH) or 5 mm 1-deoxymannojirimycin (Sigma; 0.2 m stock in sterile H2O). When indicated, clasto-lactacystin β-lactone (Calbiochem, San Diego, CA; 20 mm stock in dimethyl sulfoxide) was added to a concentration of 80 μm at the beginning of the labeling period. At the desired time points, 3 volumes of cold W5 medium (9 g/liter NaCl, 0.37 g/liter KCl, 18.37 g/liter CaCl2·2H2O, 0.9 g/liter glucose) were added and protoplasts were pelleted by centrifugation at 60 × g for 10 min at 4 °C. Separated cell and media samples were frozen on dry ice and stored at -80 °C, unless further manipulations were to be performed as below.
Tobacco Leaf Infiltration—The lower epidermis of 3 to 5-cm long tobacco leaves from 3 to 4-week-old plants were pressure infiltrated with a culture of Agrobacterium tumefaciens (transformed with empty vector, pGreenII-0029, or with either wild type or mutant CDC48) diluted to an A600 of 0.1 in infiltration media (50 mm MES, pH 5.6, 0.5% (w/v) glucose, 2 mm Na3PO4, 100 μm acetosyringone (10 mm stock in EtOH)). The plant was then incubated in greenhouse conditions for a further 3 days before preparation of protoplasts.
Protoplast Fractionation—Protoplast pellets (from 500,000 cells) were resuspended in 140 μl of 12% sucrose buffer (100 mm Tris-HCl, pH 7.6, 10 mm KCl, 1 mm EDTA, 12% (w/w) sucrose, supplemented immediately before use with Complete™ protease inhibitor mixture (Roche Applied Science)) and homogenized by pipetting 50 times with a Gilson-type micropipette through a 200-μl tip. Intact cells and debris were removed by centrifugation at 500 × g for 5 min at 4 °C. 130 μl was loaded onto a 17% (w/w) sucrose pad and centrifuged at 100,000 × g for 30 min at 4 °C. Pellets (microsomes) and supernatants (soluble proteins) were frozen on dry ice and stored at -80 °C.
Protease Protection Assay—Protoplast pellets (from 500,000 cells) were homogenized in 12% sucrose buffer as described above, this time omitting protease inhibitors, and cell debris was removed by centrifugation at 500 × g for 5 min at 4 °C. Supernatants were divided into three aliquots and incubated for 30 min at 25 °C with either buffer (negative control) or proteinase K (5 mg/ml stock in 50 mm Tris-HCl, pH 8.0, 1 mm CaCl2) at a final concentration of 75 μg/ml in the presence or absence of 1% Triton X-100. Phenylmethylsulfonyl fluoride was added to a final concentration of 20 mm to inhibit proteinase K before immunoprecipitation. Samples were frozen on dry ice and stored at -80 °C.
Preparation of Protein Extracts and Immunoprecipitation— Frozen samples were homogenized by adding 2 volumes of cold protoplast homogenization buffer (150 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1.5 mm EDTA, 1.5% (w/v) Triton X-100, supplemented immediately before use with Complete™ protease inhibitor mixture). Homogenates were used for immunoprecipitation with polyclonal rabbit anti-RTA, anti-BiP (22), anti-phaseolin (22), anti-calreticulin (29), or anti-GRP94 (30) antisera. Immunoselected polypeptides were analyzed by 15% SDS-PAGE. Gels were fixed, treated with Amplify™ (GE Healthcare), and radioactive polypeptides revealed by fluorography. Band intensity was determined using TotalLab 2003 software (Non-linear Dynamics, Newcastle-upon-Tyne, UK).
Toxicity Measurements—Triplicate aliquots of 330,000 protoplasts were co-transfected with a toxin-encoding plasmid or empty vector (pDHA), a CDC48-encoding plasmid or empty vector (pamPAT-MCS), and the phaseolin-encoding plasmid (pDHE-T343F). After 16 h of recovery, protoplasts were pulse-labeled for 1 h before being pelleted as described above. Polypeptides immunoselected from homogenates using anti-phaseolin antiserum were separated by SDS-PAGE, before fluorography and densitometry as before. Toxicity of the various constructs was expressed as a percentage of phaseolin synthesis with respect to protoplasts co-transfected with empty vector instead of toxin.
Endoglycosidase H Treatment—Protein A-Sepharose beads carrying immunoprecipitated protein were resuspended in 20 μl of sodium citrate buffer (0.25 m sodium citrate, pH 5.5, 0.2% (w/v) SDS) and boiled for 5 min. Supernatants were treated with 10 milliunits of endoglycosidase H (Roche Applied Science; 5 milliunits/μl of stock) at 37 °C for 16 h.
Confocal Microscopy—Transfected protoplasts were mounted in K3 and imaged with a Leica TCS SP5 confocal laser-scanning microscope, using a ×40 oil immersion objective lens (NA 1.25). YFP was excited at 514 nm and detected in the 525 to 583 nm range. RFP was excited at 561 nm and detected in the 592 to 635 nm range. Simultaneous detection of YFP or RFP was performed by combining the settings indicated above in the sequential scanning facility of the microscope, as instructed by the manufacturer.
Statistical Analysis—Each experiment was repeated 3 or 4 times (see individual figures) and data groups were analyzed using one-way analysis of variance. When a significant effect between sample groups was detected, the groups were compared using Tukey post hoc tests. Statistical analysis was performed using SPSS version 14.0.
RESULTS
Dominant Negative CDC48QQ Induces an Up-regulation of ER Chaperones—We rationalized that the presence of a dominant negative CDC48 would lead to the accumulation of aberrant proteins within the ER lumen as a prelude to an up-regulation of ER chaperones as part of the unfolded protein response (UPR). Such a phenotype would confirm the efficacy of the expressed CDC48 mutant. We therefore prepared protoplasts from sections of leaf tissue that had been subjected to Agrobacterium-mediated infiltration. The transformed protoplasts expressed empty vector, wild-type CDC48, or a mutant CDC48 in which the conserved glutamate residues of the Walker B motifs (Glu308 and Glu581) of the two ATPase domains had been replaced by glutamine (denoted CDC48QQ) (16). After 3 days, the levels of BiP, calreticulin, and GRP94 were increased in cells that were expressing CDC48QQ (Fig. 1A). The quantitation of these data is shown in Fig. 1B, where it can be seen that a statistically significant difference is observed in chaperone levels when CDC48QQ is expressed. It should be noted that the up-regulation observed is likely to be an underestimate because of the agrobacterial infiltration method used in these experiments, although more efficient that other methods of plant cell transformation, is unlikely to be 100% efficient. ER chaperones will therefore not be induced in a proportion of the cells taken for analysis.
FIGURE 1.

Expression of CDC48QQ promotes an up-regulation of ER chaperones. A, tobacco leaf cells were transfected, by Agrobacterium-mediated stomatal infiltration, with the pGreenII-0029 vector alone (vector) or with plasmids encoding wtCDC48 or CDC48QQ. After 3 days, protoplasts were recovered and pulsed with radioactive amino acids for 1 h. Proteins were immunoselected sequentially from cell homogenates using anti-calreticulin, anti-GRP94, and anti-BiP antisera and analyzed by reducing SDS-PAGE and fluorography. BiP immunoprecipitates were exposed to film for 2 days, whereas the GRP94 and calreticulin immunoprecipitates (IP) were exposed for 10 days. Numbers on the left indicate molecular mass markers in kilodaltons. B, quantitation of A, showing the average values from three independent experiments. The intensity of the immunoselected bands was measured by densitometry and expressed as a percentage of the control (vector). Bars indicate standard deviation and the presence of different letters above the bars indicate a significant difference (p < 0.05).
Expression of Dominant Negative CDC48 Increases the Stability of ER-sequestered RTA—We then followed the fate of ER-sequestered RTA in tobacco protoplasts 16 h after PEG-mediated co-transfection of plasmids encoding toxin together with either wild-type CDC48 or mutant CDC48, or in the presence of the glycosylation inhibitor (and UPR inducer) tunicamycin (31). To minimize inhibition of protein synthesis and thereby maximize the amount of newly made toxin that could be visualized, we used in these (and some of the subsequent) experiments a catalytic point mutant, RTAE177D, which has been shown to have virtually native structure (32) but a 70-fold reduced potency to ribosomes (24). As shown from the representative pulse-chase experiment in Fig. 2A, the glycosylated RTA made in a 1-h pulse with radiolabeled cysteine and methionine was degraded with a half-life of ∼3.5 h in cells expressing wild-type CDC48. In cells expressing CDC48QQ, the rate of RTA degradation was significantly and reproducibly reduced as shown by quantification and statistical analysis of bands taken from four independent repeats of this experiment (Fig. 2B). Although the expression of CDC48QQ was in itself toxic to cells, such that transfected cells synthesized only 40% as much RTA as cells expressing wild-type CDC48, the RTA that was made was more stable (Fig. 2), with a half-life of greater than 5 h (data not shown). The slightly increased gel mobility of the A chain bands during the chase (Fig. 2A, lanes 6–8 and 10–12) most likely represents the emergence of a mannose-trimmed species. Fig. 2B quantifies the kinetics of degradation averaged from four separate experiments. It is noticeable that, unlike CDC48QQ, tunicamycin treatment to block glycosylation and promote a generalized UPR does not impede the disappearance of RTA (Fig. 2A, lanes 13–16).
FIGURE 2.

Expression of mutant CDC48 increases the stability of ER-sequestered ricin A chain. A, protoplasts were transfected with pDHA vector alone (vector) or co-transfected with plasmids encoding ER-targeted ricin A chain with an active site mutation (RTAE177D), and either wtCDC48 or CDC48QQ. Where indicated, protoplasts were preincubated for 1 h with 50 μg/ml tunicamycin. Protoplasts were radiolabeled with 35S-labeled amino acids for 1 h and chased with unlabeled amino acids for the times indicated. RTA was immunoprecipitated from cell homogenates and analyzed by reducing SDS-PAGE and fluorography. The single asterisk indicates the position of glycosylated RTA, whereas the double asterisk marks the position of non-glycosylated RTA. The position of the 30-kDa molecular mass marker is indicated on the left. B, quantitation of the amount of immunoprecipitated RTA, made in the presence of wtCDC48, CDC48QQ, or tunicamycin, as measured by densitometry and expressed as a percentage of the total RTA present at the end of the pulse. The graph shows the average values from four independent experiments. Bars indicate standard deviation and different letters at the end of each series indicate a significant difference (p < 0.05) between treatments at all but the zero time point.
Expression of Mutant CDC48 Hampers the Retro-translocation of Toxin Subunits—We have previously shown that a proportion of retro-translocated RTA uncouples from the degradation pathway and refolds to inactivate ribosomes (18). A reduction in the protein biosynthetic capacity of these cells is therefore indicative of retro-translocation activity. To monitor protein synthesis in the transfected cell population alone we quantified, in triplicate, the levels of a reporter protein (phaseolin) encoded by a plasmid that was co-transfected (in a triple transfection) into protoplasts along with a plasmid encoding a cytosolic RTA, an ER-targeted RTA (RTA), or the nonrecombinant vector pDHA (no toxin) together with one of the CDC48 constructs (or vector alone (pamPAT-MCS)). Following overnight expression and a 1-h pulse with radiolabeled cysteine and methionine, the triplicate phaseolin immunoprecipitates were quantified from gels. As shown in Fig. 3A, protein synthesis in cells making ER-targeted RTA (with and without the wild-type CDC48 construct) is reduced by ∼60%. In contrast, protein synthesis in cells expressing RTA and the dominant negative CDC48QQ was reduced by only 30%, a statistically significant difference (p < 0.05). That such rescue was a general consequence of expressing CDC48QQ was ruled out from controls that show a consistent inhibition of protein synthesis (by more than 80%) when ricin A chain was deliberately expressed in the cytosol without a signal peptide (cytosolic RTA), either in the presence or absence of wild-type or mutant CDC48 (Fig. 3A). The significant rescue of protein synthesis shown in Fig. 3 therefore occurred only when RTA was initially targeted into the ER lumen in the presence of CDC48QQ. This would suggest that CDC48QQ mitigates the toxic effect of RTA by impeding its retro-translocation to the cytosol. Because this experiment relies on efficient co-transfection, we checked the ability of protoplasts to take up multiple plasmids. Fig. 3B is a representative set of images showing that protoplasts competent to take up a YFP expressing plasmid in all cases concomitantly co-express an RFP plasmid.
FIGURE 3.
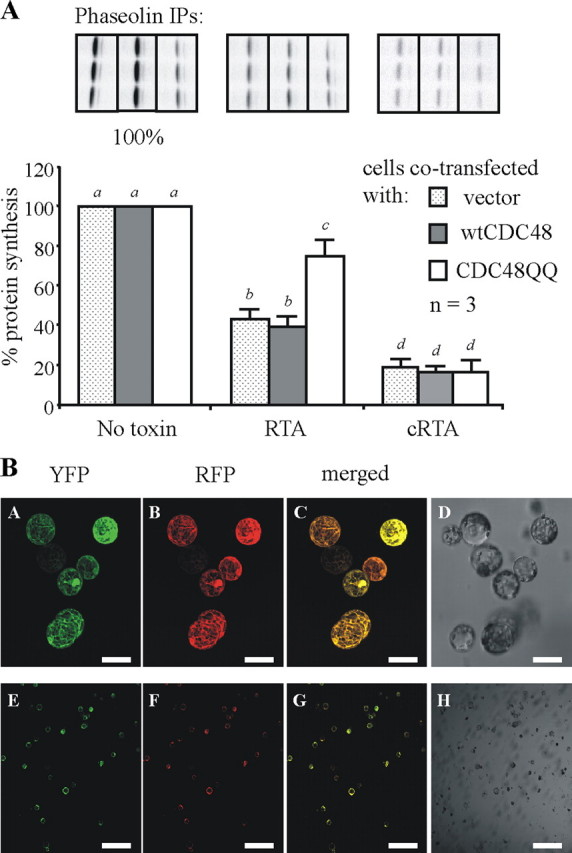
Expression of a mutant CDC48 decreases the toxicity of RTA to ribosomes. A, triplicate preparations of protoplasts were co-transfected with a plasmid encoding a phaseolin protein synthesis reporter along with either pDHA vector alone (no toxin) or a plasmid encoding ER-targeted RTA (RTA) or cytosolic RTA (cRTA). These co-transfections were carried out in combination with either the pamPAT-MCS vector alone (no CDC48), or this plasmid encoding either wtCDC48 or CDC48QQ. Protoplasts were pulsed for 1 h before cell lysis, and immunoprecipitates (IP) were subjected to SDS-PAGE, as described in the legend to Fig. 2. Fluorographs of a representative set of phaseolin immunoprecipitates taken from one of three experimental repeats are shown above the relevant densitometry quantifications. The levels of phaseolin made in the absence of any toxin expression are taken as 100%. The other data are expressed as a percentage of this and represent the average values from three independent experiments. Bars indicate standard deviation and different letters above bars indicate a significant difference (p < 0.05). B, protoplasts were co-transfected with plasmids encoding cytosolic YFP and cytosolic RFP. After overnight expression cells were analyzed by confocal microscopy. Panels A and E show YFP fluorescence (green), panels B and F show RFP fluorescence (red), and panels C and G show merged images. Panels D and H represent the total cell population. YFP was excited at 514 nm, and RFP was excited at 561 nm. All images shown were acquired using the same photo-multiplier gain and offset settings. For each sample, two magnifications are shown. Scale bars represent 50 μm (panels A–D) and 400 μm (panels E–H).
To confirm toxin retro-translocation, we determined the location of the stabilized RTA. Cells expressing ER-targeted RTAE177D alone or together with either wild-type or mutant CDC48 were pulsed with 35S-labeled amino acids before being fractionated into membranes (M) and cytosol (C). As shown in Fig. 4A (lane 4), a significant amount of the RTAE177D synthesized was recovered in the cytosol fraction under these conditions. By contrast, virtually no toxin accumulated in the cytosol of cells expressing CDC48QQ (Fig. 4A, lane 8). BiP immunoprecipitates from the same samples indicate the integrity of the membrane fractions and show that the cytosolic RTA was unlikely to result from membrane leakage during cell lysis and membrane preparation. It is noticeable that the form accumulating in the cytosol was equivalent to the slowest migrating species observed in the membrane fractions. This higher RTA band is reminiscent of the mannose-untrimmed species seen in previous pulse-chase experiments (18). Regardless of the precise glycan forms, however, it is clear that co-expression of mutant CDC48QQ impaired the retro-translocation of RTA to the cytosol.
FIGURE 4.
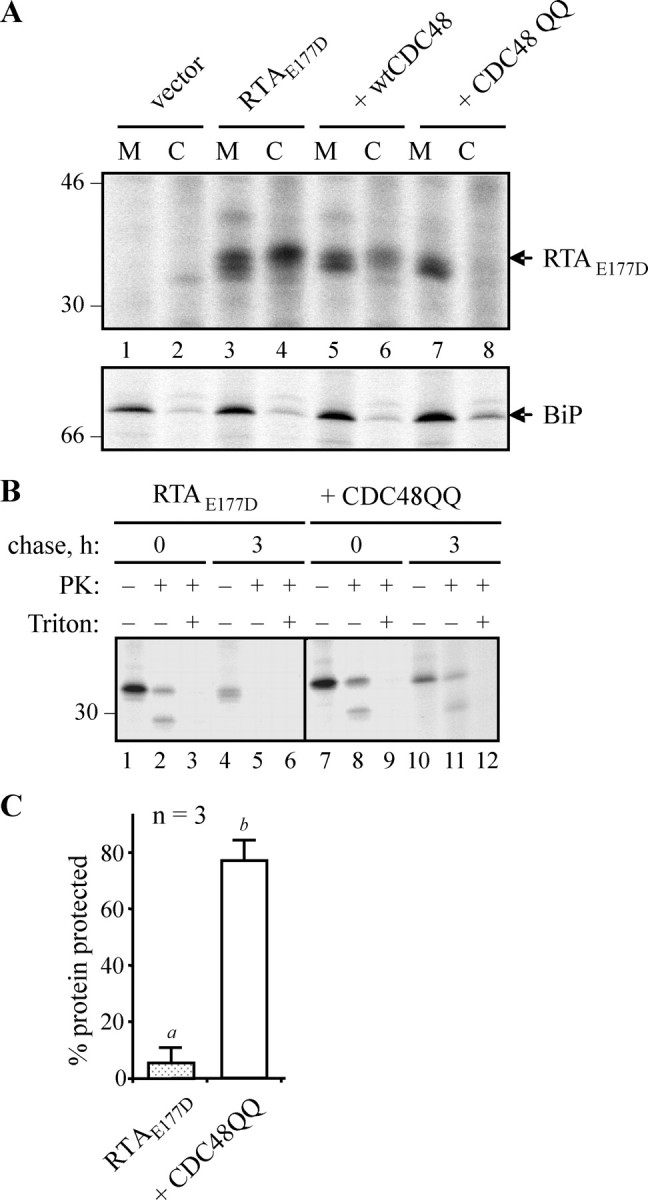
RTA is retained in the membrane fraction when CDC48-dependent retro-translocation is impaired. A, protoplasts were transfected with pDHA vector alone (vector) or the RTAE177D-encoding plasmid plus wtCDC48 or CDC48QQ plasmids. Protoplasts were radiolabeled with [35S]cysteine and [35S]methionine for 6 h, before being homogenized in the absence of detergent and fractionated to yield microsomes (M) and cytosol (C). Proteins were immunoselected sequentially using anti-RTA and anti-BiP antisera, and analyzed by reducing SDS-PAGE and fluorography. Numbers on the left indicate molecular mass markers in kilodaltons. B, protoplasts were transfected with the plasmids shown, before being radiolabeled for 1 h and chased as indicated. The cells were then homogenized as in A, before dividing three ways and incubating in the absence or presence of proteinase K (PK) and detergent (Triton X-100). RTA was immunoselected and resolved as in A. The position of the 30-kDa molecular mass marker is indicated on the left. C, quantitation, from three separate pulse-chase experiments, of the amount of RTA made in the absence or presence of CDC48QQ that was subsequently protected from proteinase K after 3 h. Bars indicate standard deviation and different letters above the bars indicate a significant difference (p < 0.05).
To investigate whether RTA was retained inside the membranes we carried out a protease protection experiment using cell extracts containing ER microsomes (25). Accordingly, at the 3-h chase point, when RTA was largely sensitive to proteinase K under normal conditions (Fig. 4B, lane 5), a proportion of this protein was clearly protected from protease when made in the presence of CDC48QQ (Fig. 4B, lane 11, and quantified from three independent repeats in Fig. 4C). Based on the statistical analysis, there is significantly more protease-protected RTA in the cells expressing CDC48QQ (Fig. 4C). The faster migrating species, visualized upon incubation of the samples with protease, most likely represents a membrane-protected remnant of the fraction of RTA that was in the process of being retro-translocated at the time of cell lysis and that may have been partially exposed to the cytosol. Consistent with this, we observed the complete disappearance of this fragment when the membranes were treated with protease concomitant with membrane solubilization (Fig. 4B, lanes 3, 6, 9, and 12).
Toxins Can Be Retro-translocated in Mannose-trimmed and Untrimmed Forms, but Only When CDC48 Is Active—We next used cells expressing either ER-targeted RTAE177D or a comparable inactive version of its relative, R. communis agglutinin (RCA AE176D (33)), and analyzed the presence or absence of these orphan proteins in the cytosol and membrane fractions over time. It was noticeable that RTA, but not RCA A, underwent a slight shift in size during pulse-chase experiments (Fig. 5, top panel). This slight downsizing of RTA can be blocked using the ER mannosidase inhibitor 1-deoxymannojirimycin (Fig. 5, second panel) to generate a species that is now comparable with the sharper bands observed for RCA A. Clearly, the appearance of RTA in the cytosol does not critically depend on prior mannose-trimming events. Taken together, these observations suggest that the core glycan of RCA A may not normally be a substrate for extensive mannosidase action, and that the retro-translocation machinery does not differentiate between the different glycosylated forms presented by RTA and RCA A.
FIGURE 5.

CDC48-dependent retro-translocation does not distinguish between different glycosylated forms. Protoplasts were transfected with pDHA vector alone (control), or constructs encoding ER-targeted RTAE177D or R. communis agglutinin A chain with the equivalent active site point mutation (RCA AE176D). Where indicated, protoplasts were co-transfected with a plasmid expressing CDC48QQ, preincubated for 1 h with 5 mm 1-deoxymannojirimycin (DMM) before radiolabeling, and/or incubated with 80 μm clasto-lactacystin β-lactone during the pulse. Protoplasts were subjected to pulse-chase before being homogenized in the absence of detergent. Homogenates were centrifuged to yield microsomal (M) and cytosol (C) fractions and proteins immunoprecipitated sequentially using anti-RTA and anti-BiP antisera before analysis by reducing SDS-PAGE and fluorography. A representative gel showing the distribution of BiP in the membrane and cytosol fractions is shown in the bottom panel. The asterisk at the right-hand side of the β-lactone panel indicates the likely deglycosylated RTA species that is more clearly observed when proteasomes are inhibited (17). Numbers on the left indicate molecular mass markers in kilodaltons.
As observed before for RTA (17, 18), there is a qualitative loss of both toxin chains from membrane fractions with time (Fig. 5, top two panels). That this is due to retro-translocation and cytosolic degradation is indicated from experiments in which proteasome activity was compromised using clasto-lactacystin β-lactone (Fig. 5, third panel). We have previously observed that this conventional inhibitor of mammalian proteasomes does not completely block RTA degradation. Nevertheless, under these conditions, the cytosolic turnover of both toxin A chains was sufficiently hindered to permit the more rapid appearance of a faster migrating intermediate (Fig. 5, third panel, asterisk) that was absent in nontransfected controls. This species of RTA was previously shown to be a deglycosylation product of cytosolic peptide:N-glycanase that becomes more noticeable when proteasomes are partially inhibited (17). Strikingly, when 1-deoxymannojirimycin was used (to maintain both toxins as tight homogenous bands) together with the proteasome inhibitor and CDC48QQ, neither A chain was observed in cytosolic fractions (Fig. 5, compare cytosol fractions in the top three panels with the fourth panel). The bottom panel reveals the distribution of ER BiP to indicate the integrity of the membrane fractions. Clearly, the effect of CDC48QQ was to severely compromise emergence of both Ricinus toxins into the cytosol.
Cycling via the Golgi Can Preface Retro-translocation—RTA and RCA A, although very closely related, appear to be handled differently upon their synthesis in the plant cell. Both polypeptides become core-glycosylated at a single site (N10) upon entering the ER lumen, and such glycans can be removed by treatment with endoglycosidase H. Although we know that RTA, in the absence of its normal partner B chain, is neither secreted nor delivered to vacuoles in tobacco cells (21), a significant proportion of this newly made protein was found to be Endo H-resistant (Fig. 6A, top panel, lanes 4–6). This supports a cycling of RTA from ER to Golgi to ER, and an encounter with Golgi glycan modification enzymes. By contrast, RCA A does not acquire Endo H resistance under identical conditions (Fig. 6A, lower panel, lanes 4–6) suggesting that cycling of these soluble proteins in the early secretory pathway is not obligatory for their retro-translocation. In the presence of CDC48QQ, a greater proportion of the total population of newly made RTA became Endo H resistant (Fig. 6A, top panel, compare lanes 4–6 with lanes 10–12), presumably because cycling continues even when retro-translocation is perturbed. Indeed, when retro-translocation is inhibited, a proportion of RCA A also reveals Endo H resistance (Fig. 6A, lower panel, lanes 11 and 12). The quantification of the proportion of Endo H-resistant species at the 5-h chase points from three independent experiments are shown in Fig. 6B. They reveal a statistically significant difference between the fraction of Endo H-resistant RTA and RCA A chains, and also between RCA A made in the absence or presence of CDC48QQ. A possible rationalization for the latter is that when retrotranslocation is compromised, RCA A has greater opportunity to be cycled via the Golgi and to acquire Endo H resistance. Note that both RTA and RCA A appear to be degraded at comparable rates during the chase when wild-type CDC48 is present, indicating that retro-translocation is independent of glycan structure. This was confirmed for RTA where both the Endo H-sensitive and Endo H-resistant forms were observed in the cytosolic fraction (Fig. 6C).
FIGURE 6.
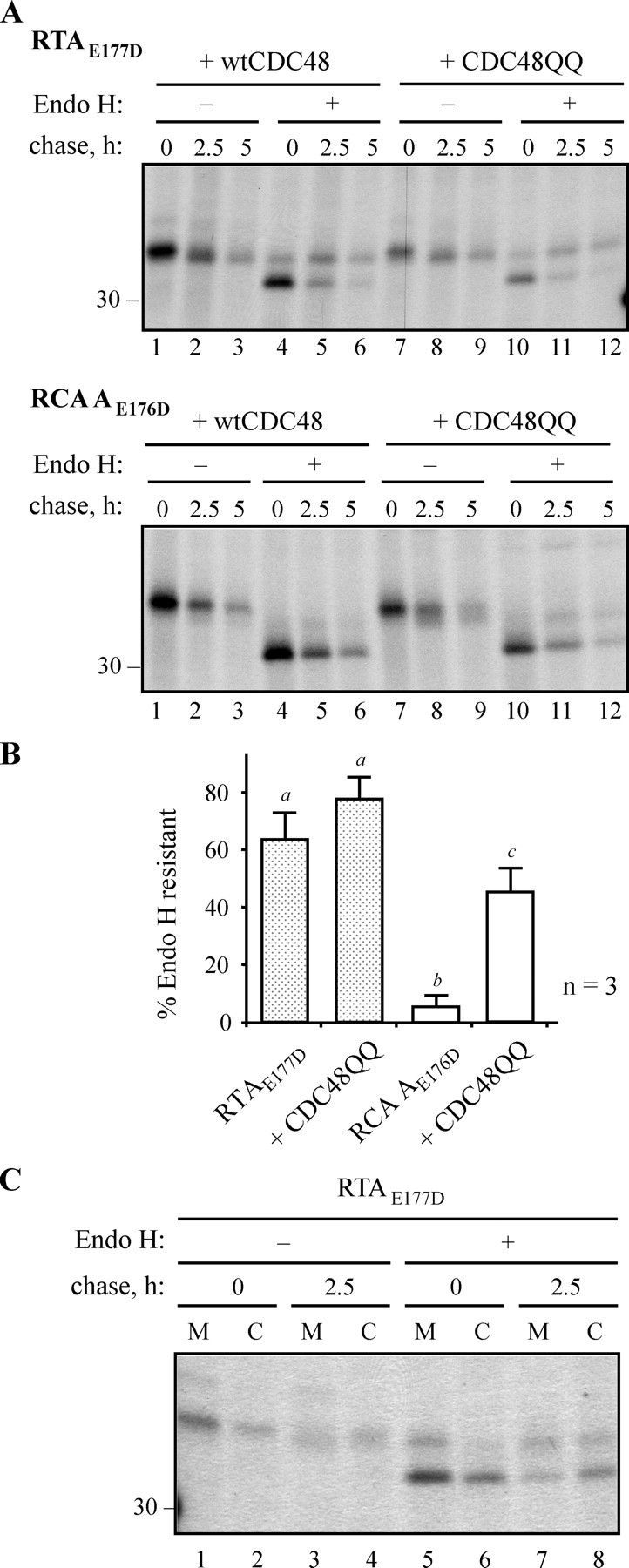
A plant ERAD substrate may cycle through the Golgi prior to its CDC48-dependent retro-translocation. A, protoplasts were co-transfected with plasmids encoding RTAE177D or RCA AE176D, and wtCDC48 or CDC48QQ, before pulse-chase analysis. RTA or RCA A were immunoprecipitated, and the immunoprecipitates treated for 16 h in the absence or presence of Endo H before analysis by reducing SDS-PAGE and fluorography. The position of the 30-kDa molecular mass marker is indicated on the left. B, quantitation, from three separate pulse-chase experiments, of the amounts of RTA or RCA A, made in the absence or presence of CDC48QQ, that were resistant to Endo H treatment after 5 h. Bars indicate standard deviation and different letters above the bars indicate a significant difference (p < 0.05). C, protoplasts were transfected to express RTAE177D, radiolabeled as in A, and chased with unlabeled amino acids for 0 and 2.5 h, before being homogenized in the absence of detergent. Homogenates were fractionated into microsomes (M) and cytosol (C) and RTA immunoselected, treated with Endo H, and analyzed as in A. The position of the 30-kDa molecular mass marker is indicated on the left.
CDC48-mediated Retro-translocation of RTA Does Not Require Polyubiquitination of Lysine Residues—The CDC48-p97 complex is believed to function in retro-translocation by interacting with polyubiquitin chains on exposed client proteins and mechanically extracting them from the ER membrane. Studies in mammalian cells with the non-ubiquitinated but, nevertheless, still retro-translocated substrate cholera toxin A1 chain (12) have shown that cytosolic p97 was not required. A similar situation may exist for RTA, because only the degradation of this protein rather than its retrotranslocation was affected by a modulation of lysine content (18). It is evident that a lysyl-free toxin variant (RTAE177D0K) is able to retro-translocate when endogenous or endogenous plus overexpressed wild-type CDC48 are present (Fig. 7, lanes 7–12 and 13–18, respectively). However, when the mutant ATPase is present, the dislocation of the lysine-free toxin to the cytosol is completely prevented (Fig. 7, lanes 20, 22, and 24).
FIGURE 7.

CDC48-dependent retro-translocation can be independent of lysyl ubiquitination. Protoplasts were transfected with pDHA vector alone (vector) or with a plasmid encoding RTAE177D lacking its two endogenous lysine residues (RTAE177D 0K), plus a wtCDC48 or CDC48QQ construct where indicated. Protoplasts were subjected to a 1-h pulse and chased as shown. Cell homogenates were centrifuged to yield microsomal (M) and cytosol (C) fractions, and proteins immunoselected sequentially using anti-RTA and anti-BiP antisera before analysis by reducing SDS-PAGE and fluorography. Numbers on the left indicate molecular mass markers in kilodaltons.
DISCUSSION
To test for a dominant negative effect of CDC48, we monitored the up-regulation of ER chaperones that follows the inhibition of retro-translocation and that forms part of the unfolded protein response. Fig. 1 reveals a significant induction of chaperones in cells expressing CDC48QQ, but not in cells that make the wild-type protein, and confirms, in plant cells, an effect of the mutant ATPase that is well known in other systems (12, 34, 35).
The process of ERAD typically begins with the recognition of a substrate as being misfolded or orphan. The structural features recognized are not known in most cases, but for newly made RTA (and its close relative RCA A) it is likely that, in the absence of cognate B chain(s), exposed hydrophobic stretches of amino acids normally masked in the holotoxin (36) may trigger chaperone or lipid interactions (37) to prevent aggregation. Persistent absence of the partner B chain appears, however, to lead to a redirection of the catalytic A subunit to the retro-translocation machinery (17), which is likely to be a protein-conducting channel in the ER membrane (38). Once exposed on the cytosolic face of the ER membrane, most ERAD substrates become oligoubiquitinated on lysyl residues in a process requiring specific membrane-associated ubiquitin-conjugating enzymes and ligases (e.g. the Doa10p and Hrd1p complexes in yeast (39–43)). This is usually followed by an ATP-dependent extraction process, during which the ubiquitin-binding CDC48 (p97 in mammals) and its cofactors Ufd1p and Npl4p are recruited to the membrane by Ubx2 (44). The substrate is usually then polyubiquitinated by E3 enzymes such as Ufd2 (45) or the N-glycan-specific SCFFbs1 and SCFFbs2 complexes (46, 47), followed by deglycosylation where appropriate (48), and passage on to proteasomes for destruction.
In comparison to the extensive characterization of mammalian and yeast ERAD, the molecular and mechanistic details of this process in plant cells remains very poorly understood. Earlier identification of plant ERAD substrates, either misfolded (16, 49) or orphan as here (17, 18), now opens the way to identifying the key players and requirements in this system. It should be noted that the behavior of newly synthesized ricin A chain in the ER of plant cells reflects, in outline at least, the situation observed when disulfide-bonded A-B ricin holotoxin is endocytosed into the mammalian ER, whereupon reduction sparks the retro-translocation of the catalytic subunit. In both scenarios (biosynthesis of RTA in the plant cell or its liberation in the ER of mammalian cells following holotoxin endocytosis and reduction), most retro-translocated RTA appears to be locked in a tightly coupled pathway leading to proteasomal degradation (50). However, a fraction of RTA is known to uncouple from this pathway, and to refold and intoxicate either the ultra-sensitive mammalian ribosomes or the more recalcitrant plant ribosomes (51). Such degradation avoidance is known to be linked to the scarcity of lysyl residues in the toxin moiety that prevents this protein becoming efficiently ubiquitinated (52).
The observation that most retro-translocated substrates are ubiquitinated and pulled from the ER membrane by virtue of this modification begs the question, what happens to proteins that are not ubiquitinated (10–12)? It has been shown for non-ubiquitinated prepro-α-factor that ATPases in the 19 S proteasome regulatory complex can provide an alternative driving force (10, 53) for membrane extraction. It is also evident from functional analyses that p97 is not required for the retro-translocation of ubiquitin-free cholera toxin A1 chain in mammalian cells (12). The driving force in this latter case is not known with certainty. Such findings have led to the suggestion that the p97 (CDC48)-Ufd1-Npl4 complex may operate only for substrates that are modified by ubiquitin (12). However, it is clear that there are other instances where CDC48 is known to be dispensable (54) or to play a more ancillary than obligatory role (55). Here, we have shown that CDC48 does participate in the retro-translocation of the orphan toxin subunits (RTA and RCA A; Fig. 5), even when completely lacking in lysine residues (as with RTA 0K; Fig. 7). Assuming that atypical N-terminal ubiquitination is not occurring in this instance, this finding would suggest that the ATPase can facilitate the extraction of a non-ubiquitinated protein. It remains to be seen whether or not the requirement for CDC48 involves direct interaction with the toxin polypeptides.
In the presence of CDC48QQ, glycosylated RTA is partially stabilized (Fig. 2), is considerably less toxic to ribosomes (Fig. 3), and is localized to the membrane fraction (Fig. 4). Likewise, RCA A is similarly stabilized and retained in a membrane fraction when CDC48 function is impaired (Fig. 5). That CDC48QQ was responsible for these effects rather than the UPR itself is shown in Fig. 2. From this, it is clear that induction of the UPR by a different means (blocking glycosylation by treatment of cells with tunicamycin, thus increasing the levels of misfolded proteins) does not result in stabilization of RTA.
It is also noticeable that the two toxin subunits are processed differently in preparation for dislocation. In the case of RTA, its emergence into the cytosol appears to involve two species: an initial mannose-untrimmed version and, at later chase points, a mannose-trimmed version (Fig. 5, top panel). The mannose-trimmed version appearing at later chase times may well represent the fraction of RTA that has undergone cycling via the Golgi (Fig. 6A), because only at such a time point is an Endo H-resistant form present in the cytosol fraction (Fig. 6C). This species would need to have undergone mannose trimming and anterograde export from the ER to reach the enzymes that render the toxin glycan resistant to Endo H. By contrast, RCA A is not mannose-trimmed or cycled via the Golgi (Figs. 5 and 6). Despite these differences, both polypeptides are retro-translocated with similar kinetics in a CDC48-dependent manner. The reason for such a striking difference between two proteins that are 94% identical at the primary sequence level and that possess an identical glycosylation site is not clear. It may be connected with the fact that RCA A, unlike RTA, is normally required to assemble into an A2-B2 tetramer in which disulfide bonds exist between each A and B chain and between the two A chains. This additional burden may lead to prolonged interactions and scrutiny by ER resident oxidoreductases that prevent the entry of this protein into an ER to Golgi cycling pathway. The cycling observed with RTA is reminiscent of some soluble substrates in yeast that rely on transport to the ER from the Golgi (56–58).
It is evident from the limited number of studies that have been carried out that an ERAD-like protein disposal pathway does indeed operate in plant cells. However, the present dearth of known substrates means that many details and variations are yet to emerge, not least the range of cytosolic proteins dedicated to the extraction, modification, and targeting of ER-sequestered substrates to the plant proteasome, and possibly to other cytosolic proteases. The finding that a CDC48 complex is required for the natural plant substrates described to date (16) suggests it is likely that the plant homologues of other mammalian and yeast ERAD components, found in Arabidopsis (59) and in other plants (60), may well have a functional role in this disposal pathway. In Arabidopsis, over 1300 genes (5% of the proteome) are devoted to the selective breakdown of proteins by the extensive ubiquitin/26 S proteasome pathway (61), although virtually nothing at present is known about these components in relation to ERAD. It should also be noted that because plant cells have other proteolytic repositories, in the form of lytic vacuoles (62), where proteins can be degraded (63, 64), the impact of this ERAD pathway to overall protein degradation, including the turnover of conformationally regulated proteins that may have critical roles in photomorphogenesis, pathogen defense, and hormonal regulation in the plant cell, remains to be determined.
Acknowledgments
We thank Paul Hunter for assistance with confocal microscopy, Ralph Panstruga (Max Planck Institute for Plant Breeding Research, Köln, Germany) for provision of the CDC48 plasmids, and Alessandro Vitale (IBBA-CNR, Milano, Italy) for provision of antisera against plant endoplasmin and calreticulin.
This work was supported in part by grants from The Leverhulme Trust (to L. M. R. and L. F.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: ER, endoplasmic reticulum; ERAD, endoplasmic reticulum-associated degradation; RTA, ricin toxin A chain; RCA A, Ricinus communis agglutinin A chain; Endo H, endoglycosidase H; RFP, red fluorescent protein; UPR, unfolded protein response; YFP, yellow fluorescent protein; MES, 2-N-morpholinoethanesulfonic acid.
References
- 1.Vitale, A. (2001) Plant Cell 131260 -1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Romisch, K. (2005) Annu. Rev. Cell Dev. Biol. 21435 -456 [DOI] [PubMed] [Google Scholar]
- 3.Meusser, B., Hirsch, C., Jarosch, E., and Sommer, T. (2005) Nat. Cell Biol. 7 766-772 [DOI] [PubMed] [Google Scholar]
- 4.Braun, S., Matuschewski, K., Rape, M., Thoms, S., and Jentsch, S. (2002) EMBO J. 21 615-621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ye, Y., Meyer, H. H., and Rapoport, T. A. (2001) Nature 414652 -656 [DOI] [PubMed] [Google Scholar]
- 6.Rabinovich, E., Kerem, A., Frohlich, K.-U., Diamant, N., and Bar-Nun, S. (2002) Mol. Cell. Biol. 22 626-634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jarosch, E., Taxis, C., Volkwein, C., Bordallo, J., Finley, D., Wolf, D. H., and Sommer, T. (2002) Nat. Cell Biol. 4134 -139 [DOI] [PubMed] [Google Scholar]
- 8.Ye, Y., Meyer, H. H., and Rapoport, T. A. (2003) J. Cell Biol. 16271 -84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rape, M., Hoppe, T., Gorr, I., Kalocay, M., Richly, H., and Jentsch, S. (2001) Cell 107667 -677 [DOI] [PubMed] [Google Scholar]
- 10.Lee, R. J., Liu, C. W., Harty, C., McCracken, A. A., Latterich, M., Romisch, K., DeMartino, G. N., Thomas, P. J., and Brodsky, J. L. (2004) EMBO J. 232206 -2215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Worthington, Z. E., and Carbonetti, N. H. (2007) Infect. Immun. 752946 -2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kothe, M., Ye, Y., Wagner, J. S., De Luca, H. E., Kern, E., Rapoport, T. A., and Lencer, W. I. (2005) J. Biol. Chem. 28028127 -28132 [DOI] [PubMed] [Google Scholar]
- 13.Ceriotti, A., and Roberts, L. M. (2006) in Plant Cell Monographs (Robinson, D. G., ed) Vol.4 , pp. 75-98, Springer Verlag, Berlin/Heidelberg [Google Scholar]
- 14.Feiler, H. S., Desprez, T., Santoni, V., Kronenberger, J., Caboche, M., and Traas, J. (1995) EMBO J. 145626 -5637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rancour, D. M., Dickey, C. E., Park, S., and Bednarek, S. Y. (2002) Plant Physiol. 1301241 -1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Muller, J., Piffanelli, P., Devoto, A., Miklis, M., Elliott, C., Ortmann, B., Schulze-Lefert, P., and Panstruga, R. (2005) Plant Cell 17149 -163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Di Cola, A., Frigerio, L., Lord, J. M., Ceriotti, A., and Roberts, L. M. (2001) Proc. Natl. Acad. Sci. U. S. A. 9814726 -14731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Di Cola, A., Frigerio, L., Lord, J. M., Roberts, L. M., and Ceriotti, A. (2005) Plant Physiol. 137287 -296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tabe, L. M., Wardley-Richardson, T., Ceriotti, A., Aryan, A., McNabb, W., Moore, A., and Higgins, T. J. (1995) J. Anim. Sci. 732752 -2759 [DOI] [PubMed] [Google Scholar]
- 20.Hellens, R. P., Edwards, E. A., Leyland, N. R., Bean, S., and Mullineaux, P. M. (2000) Plant Mol. Biol. 42 819-832 [DOI] [PubMed] [Google Scholar]
- 21.Frigerio, L., Vitale, A., Lord, J. M., Ceriotti, A., and Roberts, L. M. (1998) J. Biol. Chem. 27314194 -14199 [DOI] [PubMed] [Google Scholar]
- 22.Pedrazzini, E., Giovinazzo, G., Bielli, A., de Virgilio, M., Frigerio, L., Pesca, M., Faoro, F., Bollini, R., Ceriotti, A., and Vitale, A. (1997) Plant Cell 91869 -1880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hunter, P. R., Craddock, C. P., Di Benedetto, S., Roberts, L. M., and Frigerio, L. (2007) Plant Physiol. 1451371 -1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chaddock, J. A., and Roberts, L. M. (1993) Protein Eng. 6425 -431 [DOI] [PubMed] [Google Scholar]
- 25.Jolliffe, N. A., Di Cola, A., Marsden, C. J., Lord, J. M., Ceriotti, A., Frigerio, L., and Roberts, L. M. (2006) J. Biol. Chem. 28123377 -23385 [DOI] [PubMed] [Google Scholar]
- 26.Roberts, L. M., Lamb, F. I., Pappin, D. J., and Lord, J. M. (1985) J. Biol. Chem. 26015682 -15686 [PubMed] [Google Scholar]
- 27.Campbell, R. E., Tour, O., Palmer, A. E., Steinbach, P. A., Baird, G. S., Zacharias, D. A., and Tsien, R. Y. (2002) Proc. Natl. Acad. Sci. U. S. A. 997877 -7882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maliga, P., Sz-Breznovits, A., and Marton, L. (1973) Nat. New Biol. 24429 -30 [DOI] [PubMed] [Google Scholar]
- 29.Denecke, J., Carlsson, L. E., Vidal, S., Hoglund, A. S., Ek, B., van Zeijl, M. J., Sinjorgo, K. M., and Palva, E. T. (1995) Plant Cell 7391 -406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klein, E. M., Mascheroni, L., Pompa, A., Ragni, L., Weimar, T., Lilley, K. S., Dupree, P., and Vitale, A. (2006) Plant J. 48657 -673 [DOI] [PubMed] [Google Scholar]
- 31.Martinez, I. M., and Chrispeels, M. J. (2003) Plant Cell 15561 -576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Allen, S. C. H., Moore, K. A. H., Marsden, C. J., Fulop, V., Moffat, K. G., Lord, J. M., Ladds, G., and Roberts, L. M. (2007) FEBS J. 2745586 -5599 [DOI] [PubMed] [Google Scholar]
- 33.Roberts, L. M., and Lord, J. M. (1981) Eur. J. Biochem. 11931 -41 [DOI] [PubMed] [Google Scholar]
- 34.Wojcik, C., Yano, M., and DeMartino, G. N. (2004) J. Cell Sci. 117281 -292 [DOI] [PubMed] [Google Scholar]
- 35.Dalal, S., Rosser, M. F., Cyr, D. M., and Hanson, P. I. (2004) Mol. Biol. Cell 15 637-648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Simpson, J. C., Lord, J. M., and Roberts, L. M. (1995) Eur. J. Biochem. 232458 -463 [DOI] [PubMed] [Google Scholar]
- 37.Day, P. J., Pinheiro, T. J., Roberts, L. M., and Lord, J. M. (2002) Biochemistry 412836 -2843 [DOI] [PubMed] [Google Scholar]
- 38.Tsai, B., Ye, Y., and Rapoport, T. A. (2002) Nat. Rev. Mol. Cell Biol. 3 246-255 [DOI] [PubMed] [Google Scholar]
- 39.Bays, N., Wilhovsky, S., Goradia, A., Hodgkiss-Harlow, K., and Hampton, R. (2001) Mol. Biol. Cell 124114 -4128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Denic, V., Quan, E. M., and Weissman, J. S. (2006) Cell 126349 -359 [DOI] [PubMed] [Google Scholar]
- 41.Carvalho, P., Goder, V., and Rapoport, T. A. (2006) Cell 126361 -373 [DOI] [PubMed] [Google Scholar]
- 42.Swanson, R., Locher, M., and Hochstrasser, M. (2001) Genes Dev. 152660 -2674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deak, P. M., and Wolf, D. H. (2001) J. Biol. Chem. 27610663 -10669 [DOI] [PubMed] [Google Scholar]
- 44.Schuberth, C., and Buchberger, A. (2005) Nat. Cell Biol. 7999 -1006 [DOI] [PubMed] [Google Scholar]
- 45.Richly, H., Rape, M., Braun, S., Rumpf, S., Hoege, C., and Jentsch, S. (2005) Cell 120 73-84 [DOI] [PubMed] [Google Scholar]
- 46.Yoshida, Y., Chiba, T., Tokunaga, F., Kawasaki, H., Iwai, K., Suzuki, T., Ito, Y., Matsuoka, K., Yoshida, M., Tanaka, K., and Tai, T. (2002) Nature 418438 -442 [DOI] [PubMed] [Google Scholar]
- 47.Yoshida, Y., Tokunaga, F., Chiba, T., Iwai, K., Tanaka, K., and Tai, T. (2003) J. Biol. Chem. 27843877 -43884 [DOI] [PubMed] [Google Scholar]
- 48.Wiertz, E., Jones, T., Sun, I., Bogyo, M., Geuze, H., and Ploegh, H. (1996) Cell 84 769-779 [DOI] [PubMed] [Google Scholar]
- 49.Brandizzi, F., Hanton, S., DaSilva, L. L., Boevink, P., Evans, D., Oparka, K., Denecke, J., and Hawes, C. (2003) Plant J. 34269 -281 [DOI] [PubMed] [Google Scholar]
- 50.Roberts, L. M., and Smith, D. C. (2004) Toxicon 44469 -472 [DOI] [PubMed] [Google Scholar]
- 51.Lord, M. J., Jolliffe, N. A., Marsden, C. J., Pateman, C. S., Smith, D. C., Spooner, R. A., Watson, P. D., and Roberts, L. M. (2003) Toxicol. Rev. 22 53-64 [DOI] [PubMed] [Google Scholar]
- 52.Deeks, E. D., Cook, J. P., Day, P. J., Smith, D. C., Roberts, L. M., and Lord, J. M. (2002) Biochemistry 413405 -3413 [DOI] [PubMed] [Google Scholar]
- 53.Wahlman, J., DeMartino, G. N., Skach, W. R., Bulleid, N. J., Brodsky, J. L., and Johnson, A. E. (2007) Cell 129943 -955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wojcik, C., Rowicka, M., Kudlicki, A., Nowis, D., McConnell, E., Kujawa, M., and DeMartino, G. N. (2006) Mol. Biol. Cell 174606 -4618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carlson, E. J., Pitonzo, D., and Skach, W. R. (2006) EMBO J. 254557 -4566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vashist, S., Kim, W., Belden, W. J., Spear, E. D., Barlowe, C., and Ng, D. T. (2001) J. Cell Biol. 155355 -368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vashist, S., and Ng, D. T. (2004) J. Cell Biol. 16541 -52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Caldwell, S. R., Hill, K. J., and Cooper, A. A. (2001) J. Biol. Chem. 27623296 -23303 [DOI] [PubMed] [Google Scholar]
- 59.Schmid, M., Davison, T. S., Henz, S. R., Pape, U. J., Demar, M., Vingron, M., Scholkopf, B., Weigel, D., and Lohmann, J. U. (2005) Nat. Genet. 37501 -506 [DOI] [PubMed] [Google Scholar]
- 60.Kirst, M. E., Meyer, D. J., Gibbon, B. C., Jung, R., and Boston, R. S. (2005) Plant Physiol. 138218 -231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Smalle, J., and Vierstra, R. D. (2004) Annu. Rev. Plant Biol. 55 555-590 [DOI] [PubMed] [Google Scholar]
- 62.Carter, C., Pan, S., Zouhar, J., Avila, E. L., Girke, T., and Raikhel, N. V. (2004) Plant Cell 163285 -3303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Contento, A. L., Kim, S. J., and Bassham, D. C. (2004) Plant Physiol. 1352330 -2347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pimpl, P., Taylor, J. P., Snowden, C., Hillmer, S., Robinson, D. G., and Denecke, J. (2006) Plant Cell 18 198-211 [DOI] [PMC free article] [PubMed] [Google Scholar]