

Abstract
The two-pore domain K+ channel, TRESK (TWIK-related spinal cord K+ channel) is reversibly activated by the calcium/calmodulin-dependent protein phosphatase, calcineurin. In the present study, we report that 14-3-3 proteins directly bind to the intracellular loop of TRESK and control the kinetics of the calcium-dependent regulation of the channel. Coexpression of 14-3-3η with TRESK blocked, whereas the coexpression of a dominant negative form of 14-3-3η accelerated the return of the K+ current to the resting state after the activation mediated by calcineurin in Xenopus oocytes. The direct action of 14-3-3 was spatially restricted to TRESK, since 14-3-3η was also effective, when it was tethered to the channel by a flexible polyglutamine-containing chain. The effect of both the coexpressed and chained 14-3-3 was alleviated by the microinjection of Ser(P)-Raf259 phosphopeptide that competes with TRESK for binding to 14-3-3. The γ and η isoforms of 14-3-3 controlled TRESK regulation, whereas the β, ζ, ε, σ, and τ isoforms failed to influence the mechanism significantly. Phosphorylation of serine 264 in mouse TRESK was required for the binding of 14-3-3η. Because 14-3-3 proteins are ubiquitous, they are expected to control the duration of calcineurin-mediated TRESK activation in all the cell types that express the channel, depending on the phosphorylation state of serine 264. This kind of direct control of channel regulation by 14-3-3 is unique within the two-pore domain K+ channel family.
Members of the 14-3-3 family are dimeric proteins, and each subunit possesses a single polypeptide binding groove (1). Proteins that bind these grooves typically encode either the RSXpSXP (mode I) or RX(Y/F)XpSXP (mode II) consensus motif (where X may be any amino acid, and pS denotes phosphoserine (2, 3). Several different interacting partners of 14-3-3 have been described, and 14-3-3 proved to be an important constituent of large protein complexes implicated in such diverse processes as the initiation of DNA replication, transcription, control of cell cycle, intracellular trafficking, and the modulation of ion channel function (4, 5).
Two-pore domain potassium (2PK+) channels give rise to background (leak) K+ currents that are pivotal regulators of the excitability in neurons and other cell types (6). Members of this potassium channel family attracted particular attention as the stimulation of their currents essentially contributed to the therapeutically important action of volatile anesthetics (7–10). Among the 15 2PK+ channels, so far only TASK-1 and TASK-3 subunits have been shown to interact with 14-3-3β and -ζ through an unconventional (mode III) C-terminal motif. The binding of 14-3-3 overrides the endoplasmic reticulum retention signal and redirects these TASK channels to the cell surface (11–13).
TRESK,2 the 15th member of the 2PK+ channel family, was cloned from human spinal cord (14) and mouse cerebellum (15). Its mRNA is also expressed in the testis (15), spleen, thymus, placenta (16), and in the cerebrum (9). Recently, TRESK current was detected with painstaking work at the single channel level in dorsal root ganglion neurons (17), and others suggested that it was responsible for about 20% of the background K+ current in these cells (18). However, macroscopic TRESK current still could not be reliably separated from the current of other 2PK+ channels in the absence of specific inhibitors.
We have recently demonstrated that TRESK is regulated in a unique manner. The channel, expressed heterologously in Xenopus oocytes, is reversibly activated by calcium via the calcium/calmodulin-dependent protein phosphatase, calcineurin. Serine 276 was identified as the likely target of calcineurin-mediated dephosphorylation in the channel (15). In addition to its enzymatic action, calcineurin is also recruited to a nuclear factor of activated T cells (NFAT)-like binding motif (PQIVID) of TRESK (19). In the present study we demonstrate that 14-3-3 proteins also bind directly to the intracellular loop of TRESK and control the calcium-dependent regulation of the channel.
EXPERIMENTAL PROCEDURES
Plasmids—The sequences of pXEN and pXEN-pQ108 vectors were deposited to GenBank™ under the accession numbers EU267939 and EU267940, respectively. pXEN-pQ108, coding the flexible, artificial polypeptide chain (LEHQQQQQQQQQ)9, was obtained by inserting nine orientationally ligated pQ-s/pQ-a 5′-phosphorylated primer dimers into the Eco130I site of pXEN (see supplemental Table 1 for primer sequences). The (LEHQ9)9 polypeptide (approximating a random coil of 108 amino acids with a predicted contour length of about 41 nm) was designed on the basis of the known structure of polyglutamine peptides (20, 21) and the N-terminal inactivation chain of Drosophila Shaker K+ channel (22). The coding regions of mouse TRESK and human 14-3-3η (the latter differs only in two amino acids from the mouse ortholog) were amplified by PCR from our pEXO-mTRESK clone (15) and pcDNA3.1-h14-3-3η (received from Andrey S. Shaw) with the mTRESK-s/mTRESK-a and h14-3-3η-s/h14-3-3η-a primer pairs, respectively. The 14-3-3 and TRESK products were double-digested with PaeI/XhoI and EcoRI/Kpn2I, respectively, and cloned between the corresponding sites of pXEN-pQ108, resulting in the pXEN-mTRESK-pQ108-h14-3-3η (“chained”) construct.
Wild type and R57A,R61A mutant human 14-3-3η were subcloned from the mammalian expression vectors to pXEN with EcoRI/XhoI. The other six 14-3-3 isoforms were amplified with Pfu polymerase (Fermentas, Vilnius, Lithuania) from mouse brain RNA after reverse transcription, applying the m14-3-3x-s/m14-3-3x-a primer pairs (where x stands for β, γ, ε, ζ, σ and τ, respectively). The PCR protocol was 2 min initial denaturation at 98 °C, 34 cycles of 30-s denaturation at 98 °C, 1 min annealing, 80–90-s extension at 72 °C, and 5 min final extension at 72 °C; the annealing temperature was 64, 61, 59, and 56 °C in the first 3, second 3, third 3, and final 25 cycles, respectively. The products were digested at the respective restriction enzyme sites (see supplemental Table 1) and ligated to EcoRI/XhoI double-digested pXEN. The dominant negative versions of these isoforms (R58A,R62A β; R57A,R61A γ; R57A,R61A ε; R56A,R60A ζ; R56A,R60A σ, and RA56,R60A τ) were produced with QuikChange site-directed mutagenesis (see the sequences of the sense primers in supplemental Table 1). The resulting clones were verified by automatic sequencing.
The construction of the GST-TRESKloop-TAPtag fusion protein was described previously (19). The GST-h14-3-3η constructs were obtained by subcloning the EcoRI/XhoI inserts of wild type and dominant negative h14-3-3η (see above) to pGEX-6P-3 (Amersham Biosciences). The thioredoxin-hexahistidine-h14-3-3η (Trx-His6-h14-3-3η) plasmids were produced by ligating the above inserts to the EcoRI/XhoI double-digested pET32-ΔKpn vector (pET32-ΔKpn derived from pET32a+ (Novagen, Madison, WI) by digesting the plasmid with KpnI, polishing its sticky ends with Klenow polymerase, and religating). To produce the TRESKloop-H8 construct, the DNA coding for amino acids 185–292 of mouse TRESK was amplified by two consecutive rounds of PCR with the mTRloop-s/mTRloop-H8-a1 and mTRloop-s/mTRloop-H8-a2 primer combinations. The PCR product (coding also the C-terminal eight histidines) was double-digested with NcoI/XhoI and ligated to pET15b (Novagen). The 10 serines and 1 threonine of TRESKloop-H8 were sequentially mutated to alanine in different combinations by QuikChange site-directed mutagenesis (Stratagene, La Jolla, CA) to obtain the proteins containing only 1, 2, or 3 serines in the desired patterns (see the sense primers used for the mutations in supplemental Table 1).
Animals, Tissue Preparation, Xenopus Oocyte Microinjection, and Two-electrode Voltage Clamp Measurements—The tissues for RNA preparation derived from NMRI mouse strain (Toxicop). The oocytes were prepared, the cRNA was synthesized and microinjected, and two-electrode voltage clamp measurements were performed as previously described (15, 23). Oocytes were injected 1 day after defolliculation. Fifty nanoliters of the appropriate RNA solution was delivered with the Nanoliter Injector (World Precision Instruments, Sarasota, FL). Electrophysiological experiments were performed 3 or 4 days after the injection. Low [K+] solution contained 95 mm NaCl, 2 mm KCl, 1.8 mm CaCl2, 5 mm HEPES (pH 7.5 adjusted with NaOH). High [K+] solution contained 80 mm K+ (78 mm Na+ of the low [K+] solution was replaced with K+). All treatments of the animals were conducted in accordance with state laws and institutional regulations. The experiments were approved by the Animal Care and Ethics Committee of Semmelweis University.
Production and Purification of Recombinant Proteins—The GST fusion constructs were expressed in the BL21 strain of Escherichia coli. Bacteria were sonicated in G-lysis solution containing 50 mm Tris-HCl (pH 7.6), 50 mm KCl, 50 mm NaCl, 1 mm EDTA, 1 mm dithiothreitol, 1 mm PMSF, 0.1 mm benzamidine. The lysate was centrifuged, and the GST fusion proteins were affinity-purified from the supernatant with glutathione-agarose (Sigma). In the GST pulldown experiments, these immobilized proteins were used. For other experiments, the proteins were eluted from the resin with G-lysis supplemented with 20 mm glutathione and dialyzed against a solution containing 10 mm HEPES, 100 mm NaCl, 50 mm KCl, 2 mm MgCl2, 1 mm β-mercaptoethanol (pH 7.4 with NaOH).
The bacteria expressing the thioredoxin-His-tag fusion proteins were lysed in N-lysis solution containing 30 mm phosphate, 200 mm NaCl, 2 mmMgCl2, 1 mm PMSF, 0.2 mm benzamidine, 5 mm imidazole, 5.6 mm β-mercaptoethanol (pH 7.8 with NaOH). The proteins were affinity-purified with Ni-NTA agarose (Qiagen, Chatsworth, CA). The resin was washed 6 times for 5 min with 12 ml of N-lysis solution (in the second and third pairs of washing steps, the imidazole concentration of N-lysis solution was increased to 20 and 50 mm, respectively). The proteins were eluted with N-lysis containing 256 mm imidazole and dialyzed against a solution containing 20 mm Tris-HCl (pH 7.3), 100 mm NaCl, 50 mm KCl, 2 mm MgCl2, 1 mm EGTA, and 0.1 mm dithiothreitol.
TRESKloop-H8 proteins (wild type and multiple mutants) accumulated in inclusion bodies; therefore, these proteins were purified under denaturing conditions. The inclusion bodies were dissolved at room temperature in IB-lysis solution containing 30 mm phosphate, 200 mm NaCl, 1 mm PMSF, 0.1 mm benzamidine, 5 mm β-mercaptoethanol, 5 mm imidazole, 7000 mm urea (pH 7.8 with NaOH). TRESKloop-H8 proteins were purified with Ni-NTA-agarose from this solution. The resin was washed (at room temperature) 3 times with IB-lysis solution of increased (50 mm) imidazole concentration, once with IB-lysis, twice with IB-lysis of pH 5.5, and once again with IB-lysis. TRESKloop-H8 proteins immobilized on Ni-NTA resin were stored as a 50% suspension in IB-lysis solution at 4 °C.
In Vitro Radioactive Phosphorylation—For the preparation of Xenopus oocyte cytosol, ≈1 g of ovarian lobes was homogenized in 1 ml of a solution containing 50 mm HEPES, 50 mm KCl, 10 mm MgCl2, 50 mm β-glycerol phosphate, 20 mm NaF, 20 mm para-nitrophenylphosphate, 0.2 mm sodium orthovanadate, 2 mm PMSF, 0.2 mm benzamidine, 2 mm β-mercaptoethanol, 10 mm imidazole supplemented with cyclosporine A (5 μm), FK506 (1 μm), leupeptin (0.5 mg/ml), aprotinin (0.5 mg/ml), and soybean trypsin inhibitor (1 mg/ml, Sigma, type IIS), pH adjusted to 7.2 with NaOH. The lysate was centrifuged 3 times at 16,000 × g for 10 min and always the middle phase (supernatant above the insoluble pellet and below the lipid layer at the surface) was taken to the next centrifugation. The cleared supernatant was incubated with Ni-NTA-agarose (200 μl) to deplete the proteins binding nonspecifically to this resin.
TRESKloop-H8 proteins immobilized on 12–25 μl of Ni-NTA resin were washed 3 times with EQ solution containing 50 mm HEPES, 50 mm KCl, 1 mm PMSF, 0.1 mm benzamidine, 2 mm β-mercaptoethanol (pH 7.2 with NaOH) 20 min before the phosphorylation reaction. The immobilized proteins were phosphorylated at room temperature for 40 min in the presence of 100 μl of Xenopus oocyte cytosol and 1 MBq [γ-32P]ATP with continuous shaking. The proteins were run on 15% SDS-PAGE gels, the gels were stained with Coomassie Brilliant Blue, and their radioactivity was detected with phosphorimaging (GS-525, Bio-Rad). The phosphorylation with protein kinase A holoenzyme (protein kinase A (PKA), 1 μg/reaction, Sigma P5511, 0.7 units/μg) was performed at 37 °C for 30 min in a solution containing 20 mm HEPES, 80 mm KCl, 10 mm MgCl2, 25 mm β-glycerol phosphate, 0.5 mm β-mercaptoethanol, 0.1 mm sodium orthovanadate, 1 mm cAMP (pH 7.5 with NaOH) supplemented with 50 μm Na2ATP and 50 kBq [γ-32P]ATP.
GST and His-tag Pulldown Assays—The immobilized GST fusion proteins were phosphorylated overnight with PKA at 37 °C in a solution containing 50 mm Tris-HCl (pH 7.5), 50 mm KCl, 10 mm MgCl2, 50 mm β-glycerol phosphate, 1.3 mm dithiothreitol, 0.2 mm sodium orthovanadate, 1 mm cAMP, and 5 mm Na2ATP supplemented with 1% Triton X-100. The resins were washed twice with 0.2 ml G-binding solution containing 50 mm Tris-HCl (pH 7.5), 50 mm NaCl, 50 mm KCl, 1 mm PMSF, 0.1 mm benzamidine, 1 mm EDTA, 1 mm dithiothreitol. In the GST pulldown assay (with the Trx-His6-h14-3-3η proteins) the 1-h binding reaction was performed at 4 °C by rotating the beads in G binding solution containing 1% Triton X-100. The unbound proteins were removed by two washing steps (each 1 min) with 1 ml of G binding solution (+1% Triton X-100 in the first step).
For His-tag pulldown assays, the different immobilized TRESKloop-H8 proteins were washed 3 times with H-phos solution containing 50 mm HEPES, 50 mm KCl, 10 mm MgCl2, 50 mm β-glycerol phosphate, 2 β-mercaptoethanol, and 1% Triton X-100 (pH 7.5 with NaOH). The proteins were phosphorylated overnight with PKA at 37 °C in H-phos solution supplemented with 20 mm imidazole, 5 mm Na2ATP, 1 mm cAMP, and 200 μm sodium orthovanadate (pH 7.5 with NaOH). The His-tag pulldown assays (of the GST-h14-3-3η proteins) were performed in H-binding solution containing 50 mm HEPES, 50 mm NaCl, 50 mm KCl, 1 mm PMSF, 0.1 mm benzamidine, 2 mm β-mercaptoethanol, 30 mm imidazole (pH 7.4 with NaOH).
RESULTS
14-3-3 Proteins Determine the Recovery of TRESK Current from the Calcium-dependent Activation—The intracellular loop of mouse TRESK contains two putative 14-3-3 binding sites (KWRSLP194 and RSNSCP266, identified with Motif Scan). Assuming that 14-3-3 is really anchored to either of these sites in TRESK, it is reasonable to expect that 14-3-3 is also involved in a function related to the background K+ channel. Therefore, we examined whether the experimental manipulation of the cellular level of 14-3-3 proteins modified the functional properties of TRESK. Because the coexpression of 14-3-3η with mouse or human TRESK did not influence the resting TRESK current (data not shown), we tested whether 14-3-3 modulated the calcium-dependent regulation of the channel.
We have previously reported (15) that the calcium ionophore, ionomycin, induced a large (5–15-fold) activation of TRESK current expressed in Xenopus oocytes. The calcium-dependent activation was reversible after the cessation of the calcium signal, but the return of the background K+ current to its resting state (decrease in the current, hereafter called “recovery,” Fig. 1A) varied widely among the different oocyte preparations (e.g. the recovery was 20 ± 3% (n = 6) after 5 min in one oocyte preparation, whereas it was 73 ± 6% (n = 8) in another). These highly different kinetics of recovery may have reflected different concentrations of endogenous 14-3-3. To detect the effect of 14-3-3 irrespective of the endogenous expression, we experimentally manipulated the functional 14-3-3 levels in both direction for each oocyte preparation. In one of the compared groups of oocytes, the concentration of 14-3-3 was increased by the coexpression of the human 14-3-3η with TRESK. In the other group, the functional 14-3-3 level was reduced by the coexpression of the dominant negative R57A,R61A mutant form of human 14-3-3η with the K+ channel (the literature frequently refers to the dominant negative mutant as R56A,R60A 14-3-3η, although this numbering does not apply to the η isoform). To illustrate the effect of endogenous 14-3-3, we also show the results from oocytes expressing only TRESK (gray curves in Fig. 1).
FIGURE 1.
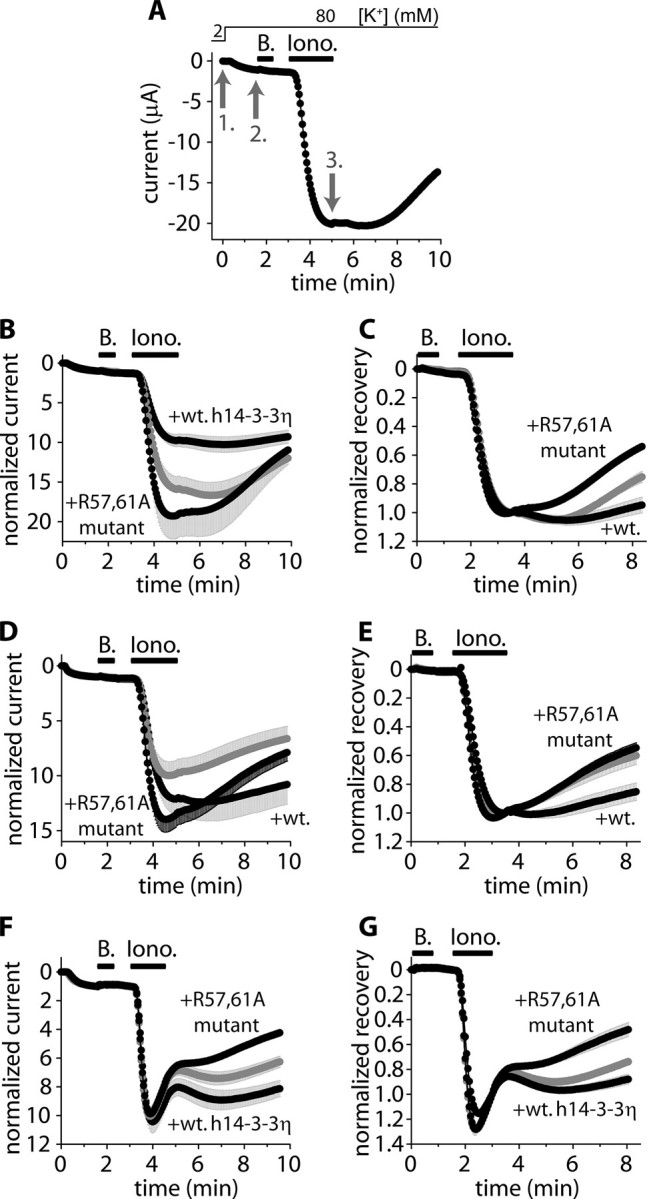
The coexpression of wild type 14-3-3η with TRESK inhibits, whereas the dominant negative 14-3-3η accelerates, the recovery of TRESK current from the calcium-dependent activation. A, currents of a representative Xenopus oocyte expressing mouse TRESK were measured at the ends of 300-ms voltage steps to –100 mV applied every 3 s from a holding potential of 0 mV. Extracellular [K+] was increased from 2 to 80 mm, and the oocyte was then challenged with benzocaine (B, 1 mm) and ionomycin (Iono., 0.5 μm) as indicated by the horizontal black bars (the initial resting state of TRESK was verified by the application of benzocaine, which inhibits the resting and calcineurin-activated channel by 10–15 and 50–60%, respectively (19). For normalized current, the current measured in 2 mm EC [K+](1. gray arrow) was normalized to 0, and the current in 80 mm EC [K+](2. gray arrow) to 1. For normalized recovery, the current at the 2. gray arrow was normalized to 0, and the current at the end of ionomycin stimulation (3. gray arrow) to 1. B and C, normalized currents and recoveries of three groups of oocytes expressing mouse TRESK (gray curve) coexpressing wild type human 14-3-3η or the dominant negative R57A,R61A 14-3-3η with TRESK, respectively (n = 3 × 5). D and E, the same experiment as in B and C was performed with another oocyte preparation (n = 3 × 6). F and G, the same experiment as in B and C was performed with oocytes expressing (or coexpressing) human TRESK (n = 3 × (5–7)) (ionomycin also caused a slow, calcium-independent inhibition of the human channel in addition to the calcineurin-dependent activation). The error bars represent S.E.
In addition to the conventional normalization of the data to the resting current (“normalized current,” see the legend to Fig. 1A), the same recordings were also presented in another form; they were normalized to the value measured at the end of ionomycin stimulation (normalized recovery, also defined in the legend to Fig. 1A). The latter normalization shows to what degree the activated current returned to the resting level in a given time after the withdrawal of ionomycin.
The coexpression of the wild type or the dominant negative 14-3-3 with mouse TRESK had significantly different effects on the activation of the K+ current in one oocyte preparation. The activation was 9.4 ± 0.7-fold (n = 5) at the end of the application of ionomycin in the cells coexpressing the wild type 14-3-3, whereas it was 19.1 ± 2.8-fold (n = 5) in the cells coexpressing the dominant negative 14-3-3 with mouse TRESK (p < 0.02 at 276s(t test), Fig. 1B), indicating that 14-3-3 inhibited the activation of mouse TRESK. However, in another oocyte preparation no significant change in mouse TRESK activation was observed (Fig. 1D). As was also verified by the microinjection of 14-3-3 protein or the immobilization of 14-3-3 to the channel (see below), the activation of mouse TRESK was undoubtedly suppressed by high concentration of 14-3-3. Perhaps this inhibitory effect was not apparent in every coexpression experiment, because it also depended on the level of overexpression of 14-3-3. The activation of human TRESK was not modified by the coexpression of the wild type or the dominant negative 14-3-3η with the channel (Fig. 1F).
The recovery of mouse TRESK (see the end of the recordings in Fig. 1, C and E) was reduced to 5 ± 6 and 15 ± 6% in the first and second oocyte preparation (n = 5 and 6), respectively, by the coexpression of wild type 14-3-3η with the channel. In the cells coexpressing the dominant negative 14-3-3 with mouse TRESK, the recovery was 46 ± 1 and 45 ± 4% (n = 5, p < 10–4 and n = 6, p < 0.003) in the two preparations, respectively (Fig. 1, C and E). In general, care must be taken when evaluating the recovery if the activation is also different in the compared groups (as in the first oocyte preparation, see Fig. 1, B and C). However, in this case the rate of recovery in the cells coexpressing the dominant negative 14-3-3 was substantially higher than that in the cells coexpressing the wild type 14-3-3. At the end of the measurement, neither the currents (11.6 ± 1.9 μAinthe wild type and 10.0 ± 1.6 μA in the dominant negative group) nor the normalized currents (9.3 ± 0.8- and 11.0 ± 1.8-fold, respectively, at 591 s in Fig. 1B) were significantly different. Despite this, the rate of recovery (estimated by the slope of the normalized currents at the end of the measurement in Fig. 1B) was about 5-fold higher in the dominant negative than in the wild type group (3.9 ± 1.1 versus 0.8 ± 0.2% of the resting current per second, p < 0.03). Because TRESK expression was nearly equal in the different oocytes in this experiment, the rates calculated even from the original current recordings were significantly different at the end of the measurement (26.3 ± 9.0 and 5.7 ± 3.1 nA/s, p < 0.05, respectively). This clearly indicates that 14-3-3 inhibits the recovery of mouse TRESK in addition to its effect on the activation of the channel. This result was also confirmed by the data obtained in the second oocyte preparation, where the activation was not significantly influenced (Fig. 1D), but the recovery was robustly inhibited by 14-3-3 (Fig. 1E).
Although the degree of activation of human TRESK was not affected by 14-3-3, its recovery was inhibited. The recovery was 12 ± 4% in the cells coexpressing the wild type (n = 7), whereas it was 52 ± 6% in the group coexpressing the dominant negative 14-3-3η with the channel (n = 7, p < 0.002 at 483 s, Fig. 1G).
In summary, 14-3-3η suppressed the calcium-dependent activation of mouse TRESK (this effect was not significant in all of the examined oocyte preparations). However, the major effect of 14-3-3 was statistically significant in all of our experiments; 14-3-3η inhibited the recovery of both human and mouse TRESK currents.
The Effect of 14-3-3η Is Acute and Spatially Restricted to TRESK in the Cell—14-3-3 proteins are known to affect a plethora of cellular functions including the regulation of the cell cycle and gene expression. In the above experiments, a high amount of coexpressed 14-3-3η was present in the oocytes for days (see supplemental Fig. S1). To exclude that gene expression changes or other long term alterations were responsible for the effects of 14-3-3η on TRESK regulation, we examined whether the microinjection of a recombinant 14-3-3 protein also inhibited TRESK recovery similarly to the coexpression of 14-3-3.
In this experiment R57A,R61A 14-3-3η was coexpressed with TRESK to reduce the effect of endogenous 14-3-3 proteins. Before the ionomycin application, the cells were microinjected either with wild type GST-14-3-3η or (as a control) with R57A,R61A mutant GST-14-3-3η protein. The activation of mouse TRESK was smaller in the wild type (10.1 ± 1.3-fold, n = 6) than in the R57A,R61A group (19.5 ± 1.2-fold activation (n = 5), p < 0.001 at 213 s, Fig. 2A). The microinjection of GST-14-3-3η also inhibited the return of TRESK current to the resting state (1 ± 10% recovery in the GST-14-3-3η-injected and 41 ± 2% in the GST-14-3-3η-R57A,R61A-injected cells, p < 0.01 at 429s, Fig. 2B). The rate of recovery was also significantly different at the end of the measurement in the two groups (2.8 ± 0.3 and 5.1 ± 0.4% of the resting current per second, respectively, p < 0.01, Fig. 2A; at this time the currents and the normalized currents were not different in the two groups (data not shown)). The same microinjection experiment was also performed with oocytes expressing only mouse TRESK, and the activation and the recovery were also significantly attenuated by the microinjection of wild type GST-14-3-3η in these cells (results not shown).
FIGURE 2.

The microinjection of recombinant GST-h14-3-3η protein inhibits the activation of mouse TRESK and delays the recovery from activation of both human and mouse TRESK channels. A, normalized currents of two groups of oocytes microinjected with 50 nl of GST-h14-3-3η (wild type, wt., black curve) or R57A,R61A mutant GST-h14-3-3η fusion protein (gray curve). The cells were coexpressing mouse TRESK with R57A,R61A 14-3-3η (in both groups). Extracellular [K+] was increased from 2 to 80 mm, and the oocytes were challenged with ionomycin (Iono., 0.5 μm, as indicated by the horizontal black bar) 108–248 min after the microinjection of the proteins. B, normalized recovery was calculated from the same recordings as represented in panel A. C and D, a similar microinjection experiment as in A and B was performed with human TRESK in another oocyte preparation. The proteins were microinjected 188–236 min before the application of ionomycin.
The same proteins were tested on human TRESK in another oocyte preparation (Fig. 2, C and D) (the dominant negative 14-3-3η was coexpressed with TRESK also in these oocytes to suppress the effect of endogenous 14-3-3). The activation of the K+ current was not different after the microinjection of the wild type or the R57A,R61A mutant GST-14-3-3η (Fig. 2C). In contrast, the recovery was clearly different. The K+ current did not recover in the cells injected with the wild type, whereas it rapidly recovered in the cells injected with the R57A,R61A mutant protein (–20 ± 12 and 48 ± 7% recovery (n = 11 and 12), respectively, at 644 s in Fig. 2D, p < 10–4; the negative recovery in the wild type group indicated that TRESK current was even larger at the end of the washout period than at the end of the ionomycin stimulation). Thus, the microinjection of GST-14-3-3η protein inhibited the recovery of both human and mouse TRESK but suppressed the activation of only the mouse channel. These results verified the identical conclusion derived from the coexpression experiments.
As an alternative approach to demonstrate the acutely reversible nature and specificity of the 14-3-3 action, a phosphopeptide ligand, Ser(P)-Raf259 (LSQRQRSTpSTPNVHA (2, 24)), competing with TRESK for binding to 14-3-3η (see the supplemental material), was microinjected into the oocytes. In the oocytes coexpressing 14-3-3η and TRESK, the inhibition of the recovery was promptly eliminated by the microinjection of Ser(P)-Raf259. In these peptide-injected cells, the recovery of the current after ionomycin stimulation accelerated dramatically (88 ± 14% compared with –1 ± 11% in the water-injected control oocytes (n = 2 × 6, p < 0.001), Fig. 3A). Thus, the elimination of the functional (endogenous and overexpressed) 14-3-3 proteins by saturating their peptide binding grooves with Ser(P)-Raf259 rapidly alleviated the inhibition of TRESK recovery. This indicates that the 14-3-3 action is immediate and specific and not attributable to chronic alterations in the molecular environment of TRESK (e.g. induced by secondarily modified protein expression).
FIGURE 3.

The 14-3-3 inhibitor Ser(P)-Raf259 accelerates the recovery of TRESK and the chained construct. A, normalized currents of oocytes microinjected with the phosphopeptide (pS-Raf259, 50 nl, 10 mm, 37–112 min before the application of ionomycin) or with water (control). The cells were coexpressing mouse TRESK with 14-3-3η (experimental protocol and current normalization were described in the legend to Fig. 1). B, schematic topology of the chained construct. TRESK was connected to 14-3-3η via the custom-designed, flexible polypeptide chain (gray). The NFAT-like calcineurin binding motif (PQIVID), serine 264, and 276 were also depicted. The chained 14-3-3η (black) is shown to be dimerized with another 14-3-3 subunit (gray) deriving from the other chained construct (of the functional TRESK dimer; not shown) or from the endogenous pool of the oocyte. C and D, normalized currents and recoveries of the oocytes expressing wild type TRESK (wt. TRESK, gray) or the chained construct. The cells expressing the chained construct were microinjected with the phosphopeptide (+pS-Raf259, 50 nl, 10 mm, 37–156 min before the application of ionomycin) or with water (chained).
Although the kinetics of TRESK recovery was determined by the functional 14-3-3 levels in a short timescale, the coexpressed (or microinjected) 14-3-3η may have affected different regulatory proteins acutely. This raised the question of whether the action of 14-3-3 was direct or was based on a distant signaling component. Therefore, we examined how TRESK recovery was regulated if the K+ channel and 14-3-3 protein were integrated into a single polypeptide chain. The N terminus of 14-3-3η was tethered to the C terminus of mouse TRESK via a flexible polypeptide linker (LEHQ9)9 (chained construct, Fig. 3B, see “Experimental Procedures”for details). Both the calcium-dependent activation (4.3 ± 0.5-fold) and the recovery (–47 ± 22%, n = 6) of this construct were reduced compared with wild type TRESK (7.4 ± 0.7-fold activation and 23 ± 3% recovery (n = 5); p < 0.01 for the activation at 213 s (Fig. 3.C); p < 0.02 for the recovery at 429 s (Fig. 3D)). Thus, the effects of the coexpressed 14-3-3η were completely reproduced by the chained protein (compare the gray control and +wt. h14-3-3η curves in Fig. 1B to the gray control and chained curves in Fig. 3C). To verify that the altered kinetics of regulation was the consequence of the protein-protein interaction between TRESK and the tethered 14-3-3, the competing 14-3-3-binding ligand, Ser(P)-Raf259 phosphopeptide, was also tested on the chained construct. In the oocytes microinjected with Ser(P)-Raf259, the recovery of the chained construct completed within 5 min (99 ± 9%, n = 6). This was in sharp contrast to the near absence of recovery in the control (water-injected) cells over the same time period (p < 10–22 at 429 s, Fig. 3D), confirming that the reduced recovery of the chained construct was induced by the specific binding of tethered 14-3-3 to TRESK. The efficiency of chained 14-3-3 indicates that 14-3-3 modulation of recovery kinetics is mediated by a direct interaction with TRESK or through a signaling complex which contains TRESK rather than through a distant regulatory element.
The Binding of 14-3-3η Depends on the Phosphorylation of Serine 264 of Mouse TRESK—Phosphorylation of mode I motifs is generally required for 14-3-3 binding; therefore, we examined whether the potential phosphorylation sites in the intracellular loop sequence of TRESK could be phosphorylated. The loop region of TRESK was produced as TRESKloop-H8 recombinant protein containing amino acids 185–292 of mouse TRESK extended with eight C-terminal histidines. This wild type construct had 10 serines and 1 threonine. Several mutant versions of TRESKloop-H8 were also produced by repeated site-directed mutagenesis, each retaining only one, two, or three serines in different combinations (the eliminated serines/threonine were replaced by alanine). TRESKloop-H8, attached to Ni-NTA-agarose, was phosphorylated by Xenopus oocyte cytosol in the presence of [γ-32P]ATP. All the TRESKloop-H8 mutants containing serine 264 were also phosphorylated (Fig. 4A), indicating that this residue was the target of an active oocyte kinase. PKA phosphorylation of the TRESKloop-H8 substrates exhibited the same phosphorylation pattern as did the cytosol (Fig. 4B). Therefore, PKA was used to phosphorylate serine 264 in our further in vitro experiments.
FIGURE 4.

Serine 264 of mouse TRESK is phosphorylated in vitro by Xenopus oocyte cytosol and protein kinase A. A, TRESKloop-H8 proteins were phosphorylated with Xenopus oocyte cytosol in the presence of [γ-32P]ATP. The mutant proteins contained only the serines indicated by the numbers above the lanes. The upper panel shows the SDS-PAGE gel stained with Coomassie Blue, whereas the autoradiogram of the same gel is on the lower panel. wt, wild type. B, a similar experiment as in A was performed with PKA (note that the protein containing only serine 264 was phosphorylated by both the cytosol and PKA).
GST pulldown assays were performed to demonstrate that serine 264 was required for the phosphorylation-dependent binding of 14-3-3. In these experiments the interaction of GST-TRESKloop-TAPtag, containing the entire intracellular loop of mouse TRESK (amino acids 164–292), and Trx-His6-h14-3-3η (thioredoxin-His-tag fusion protein of human 14-3-3η) was tested. GST-TRESKloop-TAPtag bound Trx-His6-h14-3-3η only if the GST fusion protein was previously phosphorylated with PKA (compare lanes 3 and 4 in Fig. 5A). 14-3-3 attached specifically by its phosphopeptide binding groove, since the binding of the R57A,R61A mutant Trx-His6-h14-3-3η was strongly reduced (compare lanes 4 and 6 in Fig. 5A) (in this mutant two phosphoserine-interacting, positively charged amino acids of the peptide binding groove are neutralized (25). The specificity of the interaction was also confirmed by the application of Ser(P)-Raf259. Increasing concentrations of the phosphopeptide displaced the PKA-phosphorylated GST-TRESKloop-TAPtag from the peptide binding groove of 14-3-3, preventing the pull down of Trx-His6-h14-3-3η (Fig. 5B).
FIGURE 5.

The RSNSCP266 motif of mouse TRESK binds 14-3-3η. A, GST pulldown assays with GST-TRESKloop-TAPtag or GST, phosphorylated with PKA or not, in the presence or absence of wild type (wt.) or R57A,R61A mutant (m.) Trx-His6-h14-3-3η, as indicated at the bottom of the panel. In lanes 1 and 2, only Trx-His6-h14-3-3η and GST-TRESKloop-TAPtag were loaded, respectively. Incompletely translated/degraded products were also present in lane 2, one of them being especially abundant at the size of GST. This was also marked as GST in B and C. B, the binding of Trx-His6-h14-3-3η to PKA-phosphorylated GST-TRESKloop-TAPtag was tested in the presence of different concentrations of Ser(P)-Raf259 phosphopeptide. In lane 6 no Trx-His6-h14-3-3η was added. C, The pulldown of Trx-His6-h14-3-3η was tested with wild type, S192A, or S264A mutant GST-TRESKloop-TAPtag, phosphorylated with PKA or not, as shown at the bottom of the panel. D, His-tag pulldown assays with TRESKloop-H8 phosphorylated with PKA or not in the presence or absence of wild type (wt.) or R57A,R61A mutant (m.) GST-h14-3-3η. In lanes 6 and 7, only wild type and R57A,R61A GST-h14-3-3η were loaded, respectively. The barely visible TRESKloop-H8 bands are marked with an asterisk (as in E and F). E, the binding of GST-h14-3-3η to PKA-phosphorylated (wild type) TRESKloop-H8 was tested in the presence of different concentrations of Ser(P)-Raf259 phosphopeptide. In lane 7 only GST-h14-3-3η was loaded (as in F). F, His-tag pulldown assays with the same (wild type and mutant) TRESKloop-H8 proteins as in Fig. 4A (all phosphorylated with PKA) in the presence of GST-h14-3-3η. SDS-PAGE gels were stained with Coomassie Blue.
Next we examined which predicted mode I motif in GST-TRESKloop-TAPtag was required for the interaction. The S192A mutant of GST-TRESKloop-TAPtag pulled down Trx-His6-h14-3-3η like the wild type, but the S264A mutation almost completely eliminated the binding (compare lanes 2, 4, and 6 in Fig. 5C). The diminished binding of the S264A mutant unequivocally indicated that serine 264 (in contrast to Ser-192) was indispensable for the interaction, and the other regions of the entire intracellular loop could not compensate for its loss regardless of the phosphorylation state.
In the tag-reversed case, His-tag pulldown experiments were performed with the TRESKloop-H8 constructs and GST-h14-3-3η fusion protein. GST-h14-3-3η bound to wild type TRESK-loop-H8 specifically, since the binding was phosphorylation-dependent, and it was diminished by the R57A,R61A mutation of GST-h14-3-3η (Fig. 5D). The addition of Ser(P)-Raf259 phosphopeptide also inhibited this binding in a concentration-dependent manner (Fig. 5E). All the TRESKloop-H8 mutants containing serine 264 bound GST-h14-3-3η and all the others which did not contain this residue failed to do so (Fig. 5F). Most importantly, the PKA-phosphorylated mutant, in which Ser-264 was the only serine residue, also pulled down GST-h14-3-3η, indicating that serine 264 was not only necessary but also sufficient for the binding of 14-3-3.
The Interaction of TRESK with 14-3-3 Is Isoform-specific—We have cloned the cDNAs of the six other 14-3-3 isoforms (β, γ, ε, ζ, σ, and τ) from mouse brain and also prepared their double-mutant, dominant negative versions (analogous to R57A,R61A of 14-3-3η). These twelve 14-3-3 proteins were coexpressed with mouse TRESK, and the activations and recoveries were measured (see supplemental Fig. S2). The coexpression of wild type γ isoform with TRESK diminished the activation (4.5 ± 0.4-fold, n = 5) and eliminated the recovery (–8 ± 14%, both p < 0.001, compared with the corresponding dominant negative form (16.1 ± 2.7-fold activation and 45 ± 3% recovery, n = 5), analysis of variance, and Tukey honestly significant difference post hoc test). The effects of the β, ε, ζ, σ, and τ isoforms were not significantly different (wild type versus dominant negative). In the wild type β, ε, and ζ groups, the averages of both the activation and the recovery were smaller than those in the oocytes coexpressing the respective dominant negative form with TRESK. However, even if the differences of these averages reflected an existing interaction, their small extent suggested that the overexpressed wild type β, ε, and ζ isoforms influenced TRESK weakly or these isoforms were completely ineffective, and only their dominant negative forms attenuated the action of the endogenous Xenopus 14-3-3s by heteromerization. Therefore, theσ andτ isoforms are ineffective; theβ, ε, and ζ proteins may evoke only a mild effect, whereas the γ and η isoforms are the major controllers of TRESK regulation.
14-3-3 Inhibits the Recovery of TRESK Current after the Stimulation of M1 Muscarinic Receptor—The stimulation of Gq protein-coupled receptors activates a complex machinery of signal transduction, and the calcium signal is only one important consequence of this activation. Because the receptor-mediated regulation of TRESK depended on the calcium signal (15), ionomycin was applied in all of the above experiments to elevate the cytoplasmic calcium concentration independently of the other signaling mechanisms, which would have been triggered by receptor stimulation. Thus, the question was raised of whether the inhibitory effect of 14-3-3 on the recovery of TRESK current could be reproduced after the more physiological (but at the same time more complex) receptor stimulation. Therefore, we coexpressed M1 muscarinic receptor with human TRESK and dominant negative R57A,R61A 14-3-3η. We used human TRESK because the activation of the human channel was not influenced by 14-3-3 in the above experiments, and thus, it was anticipated that the recovery of the current could be more easily evaluated. R57A,R61A 14-3-3η was coexpressed to reduce the effect of Xenopus 14-3-3. These (triple coexpressing) oocytes were microinjected with GST-14-3-3η or R57A,R61A mutant GST-14-3-3η protein and subsequently stimulated with carbachol (1 μm, Fig. 6). The receptor-mediated activation was identical in the two groups (10.9 ± 1.2-fold (n = 7) in the GST-14-3-3η-injected and 10.5 ± 1.1-fold (n = 8) in the R57A,R61A GST-14-3-3η-injected group at the end of carbachol-stimulation (336 s); see Fig. 6A). However, the K+ current recovered more slowly in the GST-14-3-3η-injected cells (41 ± 4% recovery at 644 s in Fig. 6B) than in the R57A,R61A GST-14-3-3η-injected oocytes (77 ± 4% recovery, p < 10–4). This indicates that the return of human TRESK current to the resting state was also blocked by 14-3-3 after the stimulation of M1 muscarinic receptor.
FIGURE 6.

14-3-3 inhibits the recovery of TRESK current after the stimulation of M1 muscarinic receptor. A, normalized currents of two groups of oocytes microinjected with 50 nl of GST-h14-3-3η (wild type, wt., black curve) or R57A,R61A mutant GST-h14-3-3η fusion protein (gray curve). The cells were coexpressing M1 muscarinic receptor with human TRESK and R57A,R61A 14-3-3η (triple coexpression in both groups). Extracellular [K+] was increased from 2 to 80 mm, and the oocytes were stimulated with carbachol (Carb., 1 μm, as indicated by the horizontal black bar) 200–233 min after the microinjection of the proteins. B, normalized recovery was calculated from the same recordings as represented in panel A.
DISCUSSION
Several members of the two-pore domain potassium (2PK+) channel family are regulated by calcium-mobilizing hormones and neurotransmitters (for review, see Ref. 26). It is well documented that the most extensively examined channels of the TASK and TREK subfamilies (TASK-1, TASK-3, TREK-1, and TREK-2) are inhibited in response to the activation of the G protein subunit, Gαq. Although the signaling pathway is still debated (the breakdown of membrane phosphoinositides (27, 28), the direct binding of Gαq to the channel (29), the production of diacylglycerol (28), and the phosphorylation by protein kinase C (30, 31) were all suggested as possible mechanisms for the different channels), there is a general consensus that the formation of inositol-1,4,5-trisphosphate and the following calcium signal are not involved in the inhibition of TASK and TREK subfamily members.
In contrast to TASK and TREK channels, TRESK is activated by the Gq protein-coupled receptor pathway. We have previously demonstrated that both human and mouse TRESK channels, expressed in Xenopus oocytes, were activated by the stimulation of the endogenous lysophosphatidic acid and the heterologously expressed M1 muscarinic or AT1a angiotensin receptors (15). It has been recently reported that TRESK, expressed in dorsal root ganglion neurons, is activated by G protein-coupled receptor agonists (32). In cell-attached membrane patches, the lamotrigine-sensitive component of the K+ current (attributed to TRESK) was activated by 30–100% in response to the application of acetylcholine, glutamate, or histamine. In the same study acetylcholine activated TRESK by 80% when the channel was coexpressed with M3 muscarinic receptors in COS-7 cells. These reported levels of activation in the mammalian cells were robust enough to be biologically important although smaller than the severalfold activation in the Xenopus expression system. The difference could be due to variations in the phosphorylation state of TRESK, the expression of calcium-mobilizing receptors, and the properties of the calcium signal.
In two previous papers (15, 19), we elucidated the mechanism of TRESK activation; TRESK was shown to be activated by calcineurin. Moreover, we demonstrated that the susceptibility to calcineurin-mediated activation is hardwired to TRESK; it is specifically defined at the protein sequence level. Whereas scaf-folding proteins may be required for the localization of the phosphatase to other substrates (33), the intracellular loop of TRESK contains an NFAT-like docking motif for calcineurin (PQIVID in mouse and PQIIIS in human TRESK). To our knowledge this kind of motif (34–36) has not been reported in any other ion channels so far. We have shown that the anchoring of calcineurin to this motif is absolutely required for TRESK activation (19). Because the ability to bind calcineurin is an inherent property of TRESK protein, this background K+ channel is expected to be generally activated by the calcium signal if calcineurin expression in the cell and the basal inhibitory phosphorylation of the channel are sufficient.
In the present study we report a novel and direct protein-protein interaction of TRESK subunit. Our biochemical data indicate unequivocally that 14-3-3 adaptor protein directly binds to the RSNSCP motif, conserved in the intracellular loop region of both human and mouse TRESK. The phosphorylation of the second serine residue in the motif (Ser-264 in mouse TRESK) was required for the interaction. The RSNSCP motif is located between the PQIVID calcineurin docking site (amino acids 210–215) and the putative regulatory serine of channel activation (Ser-276 in mouse TRESK). The close proximity of the 14-3-3 binding motif to these functionally important regions suggested that the binding of 14-3-3 may be implicated in the calcineurin-dependent regulation of the channel.
Indeed, experimental manipulation of the functional 14-3-3 levels (by the coexpression or microinjection of wild type or dominant negative 14-3-3 or the application of the competing phosphopeptide) profoundly influenced TRESK regulation in the Xenopus oocyte expression system. The most substantial effect of 14-3-3 was the inhibition of the return of TRESK current to the resting state after the calcineurin-mediated activation. In addition to this, 14-3-3 also evoked a mild auxiliary effect on mouse TRESK; the adaptor protein also inhibited the calcineurin-dependent activation of the mouse channel. 14-3-3 controlled TRESK regulation identically, even if it was tethered to the channel with a flexible polypeptide chain, and thus, the chained 14-3-3 could not reach the multitude of its other distant binding sites in the cell. This result indicates that 14-3-3 functions within the signaling complex of TRESK (of which the only other known element is calcineurin at present). Although it cannot be excluded that the dimeric 14-3-3 also binds to another component of this signaling complex (to an unknown component as calcineurin does not bind 14-3-3, to our knowledge), it is highly unlikely that the canonical 14-3-3 binding motif is present in TRESK only by chance and at the same time 14-3-3 acts exclusively somewhere else within the signaling complex. Instead of this, the presented data strongly support the conclusion that 14-3-3 also binds directly to TRESK in the living cell (as in vitro), and this is the reason that the recovery of the background K+ current becomes decelerated.
What is the mechanism by which the binding of 14-3-3 decelerate TRESK recovery? We have previously demonstrated that both the docking of calcineurin to the NFAT-like motif and the enzymatic activity of the phosphatase are required for the calcium-dependent activation (19). It logically follows that TRESK is inhibited by a kinase acting at the same residue that is dephosphorylated by calcineurin. Although the identification of this “TRESK kinase” is beyond the scope of the present study (the region of Ser-276 does not match the consensus sequence of any known kinase, and under our basic experimental conditions it was not phosphorylated by the oocyte cytosol), it is feasible to assume that the recovery depends on this kinase.
We postulate that the binding of 14-3-3γ and -η interferes with the interaction of TRESK kinase and TRESK (perhaps by steric hindrance). This would explain why the overexpression of 14-3-3 substantially attenuated the recovery, whereas the suppression of 14-3-3 function (by the coexpression of a dominant negative form or the application of the competing phosphopeptide) accelerated the return of the K+ current to the resting state. (As an additional minor effect, 14-3-3 also reduced the activation of mouse TRESK, suggesting that (when overexpressed) 14-3-3 could also interfere with the action of calcineurin). The postulated inhibition of TRESK kinase (by the binding of 14-3-3 to the phosphorylated Ser-264) would hinder the re-phosphorylation of the regulatory residue of TRESK (probably Ser-276) and, thus, decelerate the recovery of the channel from the calcium-dependent activation.
The binding of 14-3-3 extends the period of enhanced TRESK activity after the calcium signal. The activation of the background K+ current stabilizes the resting membrane potential at negative values and reduces the excitability in the cells that express TRESK abundantly. Therefore, the phosphorylation-dependent binding of 14-3-3γ and -η isoforms is expected to adjust the period of time after which the hyperpolarization and the reduced excitability cease. Because 14-3-3 proteins are ubiquitous and their interaction with TRESK is determined by a well defined binding motif, 14-3-3 is envisioned to be a standard constituent in TRESK signaling complexes and to regulate the duration of reduced excitability after the calcium signal.
Supplementary Material
Acknowledgments
We thank Prof. Andrey S. Shaw for the wild type and R57A,R61A mutant human 14-3-3η clones. The skillful technical assistance of Irén Veres and Beáta Busi is acknowledged. Prof. David Lotshaw's useful comments to the manuscript are highly appreciated.
This work was supported by the Hungarian National Research Fund (OTKA F-67743), by the Hungarian Medical Research Council (ETT-417/2006), and by a Semmelweis University research grant. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Table 1 and Figs. S1 and S2. The nucleotide sequence(s) reported in this paper has been submitted to the GenBank™/EBI Data Bank with accession number(s) EU267939 and EU267940.
Footnotes
The abbreviations used are: TRESK, TWIK-related spinal cord K+ channel; PKA, protein kinase A; NFAT, nuclear factor of activated T cells; GST, glutathione S-transferase; PMSF, phenylmethylsulfonyl fluoride; Ni-NTA, nickelnitrilotriacetic acid.
References
- 1.Xiao, B., Smerdon, S. J., Jones, D. H., Dodson, G. G., Soneji, Y., Aitken, A., and Gamblin, S. J. (1995) Nature 376 188–191 [DOI] [PubMed] [Google Scholar]
- 2.Muslin, A. J., Tanner, J. W., Allen, P. M., and Shaw, A. S. (1996) Cell 84 889–897 [DOI] [PubMed] [Google Scholar]
- 3.Yaffe, M. B., Rittinger, K., Volinia, S., Caron, P. R., Aitken, A., Leffers, H., Gamblin, S. J., Smerdon, S. J., and Cantley, L. C. (1997) Cell 91 961–971 [DOI] [PubMed] [Google Scholar]
- 4.Aitken, A. (2006) Semin. Cancer Biol. 16 162–172 [DOI] [PubMed] [Google Scholar]
- 5.Gardino, A. K., Smerdon, S. J., and Yaffe, M. B. (2006) Semin. Cancer Biol. 16 173–182 [DOI] [PubMed] [Google Scholar]
- 6.Lotshaw, D. P. (2007) Cell Biochem. Biophys. 47 209–256 [DOI] [PubMed] [Google Scholar]
- 7.Patel, A. J., Honore, E., Lesage, F., Fink, M., Romey, G., and Lazdunski, M. (1999) Nat. Neurosci. 2 422–426 [DOI] [PubMed] [Google Scholar]
- 8.Heurteaux, C., Guy, N., Laigle, C., Blondeau, N., Duprat, F., Mazzuca, M., Lang-Lazdunski, L., Widmann, C., Zanzouri, M., Romey, G., and Lazdunski, M. (2004) EMBO J. 23 2684–2695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu, C. H., Au, J. D., Zou, H. L., Cotten, J. F., and Yost, C. S. (2004) Anesth. Analg. 99 1715–1722 [DOI] [PubMed] [Google Scholar]
- 10.Keshavaprasad, B., Liu, C., Au, J. D., Kindler, C. H., Cotten, J. F., and Yost, C. S. (2005) Anesth. Analg. 101 1042–1049 [DOI] [PubMed] [Google Scholar]
- 11.O'Kelly, I., Butler, M. H., Zilberberg, N., and Goldstein, S. A. N. (2002) Cell 111 577–588 [DOI] [PubMed] [Google Scholar]
- 12.Rajan, S., Preisig-Muller, R., Wischmeyer, E., Nehring, R., Hanley, P. J., Renigunta, V., Musset, B., Schlichthorl, G., Derst, C., Karschin, A., and Daut, J. (2002) J. Physiol. (Lond.) 545 13–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Renigunta, V., Yuan, H., Zuzarte, M., Rinne, S., Koch, A., Wischmeyer, E., Schlichthorl, G., Gao, Y., Karschin, A., Jacob, R., Schwappach, B., Daut, J., and Preisig-Muller, R. (2006) Traffic 7 168–181 [DOI] [PubMed] [Google Scholar]
- 14.Sano, Y., Inamura, K., Miyake, A., Mochizuki, S., Kitada, C., Yokoi, H., Nozawa, K., Okada, H., Matsushime, H., and Furuichi, K. (2003) J. Biol. Chem. 278 27406–27412 [DOI] [PubMed] [Google Scholar]
- 15.Czirják, G., Tóth, Z. E., and Enyedi, P. (2004) J. Biol. Chem. 279 18550–18558 [DOI] [PubMed] [Google Scholar]
- 16.Kang, D., Mariash, E., and Kim, D. (2004) J. Biol. Chem. 279 28063–28070 [DOI] [PubMed] [Google Scholar]
- 17.Kang, D., and Kim, D. (2006) Am. J. Physiol. Cell Physiol. 291 138–146 [Google Scholar]
- 18.Dobler, T. M., Springauf, A., Tovornik, S., Weber, M., Schmitt, A., Sedlmeier, R., Wischmeyer, E., and Döring, F. (2007) J. Physiol. (Lond.) 585 867–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Czirják, G., and Enyedi, P. (2006) J. Biol. Chem. 281 14677–14682 [DOI] [PubMed] [Google Scholar]
- 20.Masino, L., Kelly, G., Leonard, K., Trottier, Y., and Pastore, A. (2002) FEBS Lett. 513 267–272 [DOI] [PubMed] [Google Scholar]
- 21.Sen, S., Dash, D., Pasha, S., and Brahmachari, S. K. (2003) Protein Sci. 12 953–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoshi, T., Zagotta, W. N., and Aldrich, R. W. (1990) Science 250 533–538 [DOI] [PubMed] [Google Scholar]
- 23.Czirják, G., and Enyedi, P. (2002) J. Biol. Chem. 277 5426–5432 [DOI] [PubMed] [Google Scholar]
- 24.Thorson, J. A., Yu, L. W., Hsu, A. L., Shih, N. Y., Graves, P. R., Tanner, J. W., Allen, P. M., Piwnica-Worms, H., and Shaw, A. S. (1998) Mol. Cell. Biol. 18 5229–5238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang, L., Wang, H., Liu, D., Liddington, R., and Fu, H. (1997) J. Biol. Chem. 272 13717–13724 [DOI] [PubMed] [Google Scholar]
- 26.Mathie, A. (2007) J. Physiol. (Lond.) 578 377–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Czirják, G., Petheoő, G. L., Spät, A., and Enyedi, P. (2001) Am. J. Physiol. Cell Physiol. 281 700–708 [DOI] [PubMed] [Google Scholar]
- 28.Chemin, J., Girard, C., Duprat, F., Lesage, F., Romey, G., and Lazdunski, M. (2003) EMBO J. 22 5403–5411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen, X., Talley, E. M., Patel, N., Gomis, A., McIntire, W. E., Dong, B., Viana, F., Garrison, J. C., and Bayliss, D. A. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 3422–3427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kang, D., Han, J., and Kim, D. (2006) Am. J. Physiol. Cell Physiol. 291 649–656 [DOI] [PubMed] [Google Scholar]
- 31.Veale, E. L., Kennard, L. E., Sutton, G. L., MacKenzie, G., Sandu, C., and Mathie, A. (2007) Mol. Pharmacol. 71 1666–1675 [DOI] [PubMed] [Google Scholar]
- 32.Kang, D., Kim, G. T., Kim, E. J., La, J. H., Lee, J. S., Lee, E. S., Park, J. Y., Hong, S. G., and Han, J. (2008) Biochem. Biophys. Res. Commun. 367 609–615 [DOI] [PubMed] [Google Scholar]
- 33.Dodge, K. L., and Scott, J. D. (2003) Biochem. Biophys. Res. Commun. 311 1111–1115 [DOI] [PubMed] [Google Scholar]
- 34.Li, H., Rao, A., and Hogan, P. G. (2004) J. Mol. Biol. 342 1659–1674 [DOI] [PubMed] [Google Scholar]
- 35.Li, H., Zhang, L., Rao, A., Harrison, S. C., and Hogan, P. G. (2007) J. Mol. Biol. 369 1296–1306 [DOI] [PubMed] [Google Scholar]
- 36.Roy, J., Li, H., Hogan, P. G., and Cyert, M. S. (2007) Mol. Cell 25 889–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.