

Abstract
Integrin cell-adhesion receptors transduce signals bidirectionally across the plasma membrane via the single-pass transmembrane segments of each α and β subunit. While the β3 transmembrane segment consists of a linear 29-residue α-helix, the structure of the αIIb transmembrane segment reveals a linear 24-residue α-helix (Ile-966 -Lys-989) followed by a backbone reversal that packs Phe-992-Phe-993 against the transmembrane helix. The length of the αIIb transmembrane helix implies the absence of a significant transmembrane helix tilt in contrast to its partnering β3 subunit. Sequence alignment shows Gly-991-Phe-993 to be fully conserved among all 18 human integrin α subunits, suggesting that their unusual structural motif is prototypical for integrin α subunits. The αIIb transmembrane structure demonstrates a level of complexity within the membrane that is beyond simple transmembrane helices and forms the structural basis for assessing the extent of structural and topological rearrangements upon αIIb-β3 association, i.e. integrin transmembrane signaling.
Integrins are a major class of adhesion receptors that mediate cell migration and extracellular matrix assembly, as well as inflammation, thrombosis, and tumor metastasis (1). In addition to responding to signals that originate inside the cell and regulate receptor affinity, integrins can respond to the binding of extracellular ligands by activating intracellular signaling pathways (1). Integrins are noncovalent heterodimers with large extracellular and small intracellular domains that are connected by single-pass transmembrane (TM)3 segments. An extensive set of experiments provides evidence that signals are transduced bidirectionally across the plasma membrane via the TM segments of the α and β subunits (2-4). Specifically, the inactive receptor state is stabilized by the hydrophobic packing of the TM helices and adjacent electrostatic αIIb-β3 interactions, whereas integrin activation ensues from the separation of the TM segments (2, 4-8). However, the precise structural and topological processes underlying integrin inside-out and outside-in signaling events are currently unknown.
Single-pass TM segments likely traverse the membrane as α-helices. The recently determined structure of the integrin β3 TM segment reconstituted in small bicelles fulfills this expectation, but the TM helix is not restricted to residues between the first charged residues, Asp-692 and Lys-716, on the extra- and intracellular sides, respectively (9). Rather, the five mostly hydrophobic residues following Lys-716 are part of the 29-residue TM helix, encoding a substantial helix tilt relative to the membrane. Apparently, this allows Lys-716 to snorkel its side chain out of the hydrophobic lipid core. Integrin α subunits exhibit a similar pattern of hydrophobicity; a lysine residue constitutes the first charged residue on the intracellular side and is followed by four mostly apolar residues (see Fig. 1). Nevertheless, the structure of the αIIb TM segment reported herein shows that the TM helix ends at Lys-989 and that the following four residues loop back to immerse Phe-992-Phe-993 in the hydrophobic lipid core. Moreover, the 24-residue TM helix formed can immerse in a typical bilayer without a significant helix tilt. The αIIb TM structure reveals an unexpected level of complexity within the integrin TM region that appears crucial for understanding the association with its partnering β3 TM segment. Furthermore, the structure highlights that single-pass TM segments are not restricted merely to helical conformations.
FIGURE 1.

Sequence alignment of selected human integrin α TM segments. Conserved amino acids are colored by the jalview multiple alignment editor using the Clustalx color scheme. Proposed minimal lipid tail-to-headgroup borders for monomeric α subunits are depicted. The alignment of all 18 α subunits is provided as supplemental Fig. 4.
EXPERIMENTAL PROCEDURES
Integrin αIIb Expression and Purification—Synthetic oligonucleotides coding for human integrin αIIb residues Ala-958 - Pro-998 were assembled by PCR and subcloned into the pET-44 expression vector (Novagen, Inc.) with the third IgG-binding domain of protein G (GB3) as N-terminal fusion protein and an intervening tobacco etch virus protease cleavage site. Expression and purification proceeded as described for the β3 TM peptide (9) except that the αIIb peptide was cleaved from the fusion protein using tobacco etch virus protease at a molar ratio of 1:50 overnight at room temperature in 50 mm Tris·HCl, pH 8.0, 0.5 mm EDTA, 1 mm dithiothreitol, and 0.5 m urea, leaving Gly as N-terminal residue. To introduce a high affinity Cu2+-binding site, the Gly-Gly-His ATCUN motif (10, 11) was introduced by PCR for residues corresponding to αIIb(957-959). GGH-αIIb peptide expression and purification proceeded in the same manner as with αIIb. Isotope labeling was achieved by supplying 15N-NH4 Cl and 13C-d-glucose and culturing in 99 and 50% D2O for highly and moderately deuterated peptide, respectively.
NMR Sample Preparation—αIIb peptide concentration was measured in acetonitrile-water solution by UV spectroscopy (ε280 nm = 16,500 m-1 cm-1), and defined amounts of peptide were freeze-dried. Peptide was taken up in 320 μl of bicelle solution to give an αIIb peptide concentration of ∼0.6 mm. The bicelle solution was comprised of 105 mm long-chain lipids and 350 mm 1,2-dihexanoyl-sn-glycero-3-phosphocholine (DHPC), 25 mm HEPES·NaOH, pH 7.4, 6% D2O, 0.02% w/v NaN3.As long-chain lipids, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and 1,2-dimyristoyl-sn-glycero-3-phosphocholine) were used, respectively. No significant H-N αIIb chemical shift changes were detected between the two lipid environments (supplemental Fig. 1), showing that equivalent structures were adopted. Supplemental Fig. 2 illustrates the spectral quality obtained.
To align the αIIb-bicelle complex relative to the magnetic field stretched, negatively charged polyacrylamide gels were used (12, 13). A 320-μl gel was polymerized in 150 mm Tris·HCl, pH 8.0, from a 4.2% w/v solution of acrylamide, 2-acrylamido-2-methyl-1-propanesulfonate (AMPS), and bisacrylamide with a monomer-to-cross-linker ratio of 49:1 (w/w) and a molar ratio of 94:6 of acrylamide to AMPS. For easier comparisons between gels, AMPS was counted as acrylamide in the given weight ratios (14). The gel was dialyzed overnight in 50 ml of 100 mm NaH2PO4/Na2HPO4, pH 6.8, followed by H2O for 10 h and another H2O dialysis overnight. The gel was dried completely and soaked in 320 μl of αIIb-bicelle (POPC:DHPC) solution for 24 h and transferred into an open-ended NMR tube, as described (15). A 2H splitting of 1.5 Hz was observed.
NMR Spectroscopy—NMR experiments were carried out on a cryoprobe-equipped Bruker Avance 700 spectrometer at 35 °C except for the gel-aligned sample, which was measured at 40 °C. Data were processed and analyzed with the nmrPipe package (16) and CARA. Throughout all experiments, the solvent was kept at +Iz whenever possible (17, 18), and TROSY-type H-N detection was used (19).
Highly deuterated αIIb peptide (99% D2O cultures) was used for the following procedures: HN, N, Cα, Cβ, and C′ assignments from HNCA, HNCACB, HNCO, and HN(CA)CO experiments. 3JC′Cγ and 3JNCγ couplings for aromatic and aliphatic residues were obtained from quantitative J-correlation spectroscopy (20, 21) with dephasing times of 50 and 100 ms, respectively. 1JNH, 1JCαC′,1JC′N and 1JNH+1DNH,1JCαC′+1DCαC′, and 1JC′N+1DC′N couplings were determined from 1JNH-scaled HNCO experiments (22) and from quantitative J-correlation HNCO experiments (23, 24) on isotropic and aligned samples, respectively. [1H]-15N NOE measurements were carried out using 5 s of presaturation preceded by a recycling delay of 4 s for the NOE experiment and a 9-s recycle delay for the reference experiment. To measure HN-HN and HN-HO NOEs, a 15N-edited NOESY spectrum (150-ms mixing time) was recorded.
Moderately deuterated αIIb peptide (50% D2O cultures) was used for the detection of side-chain NOEs. Moreover, deuterated lipids, 1,2-dihexanoyl(d22)-sn-glycero-3-phosphocholine-1,1,2,2-d4-N,N,N-trimethyl-d9 and 1,2-dimyristoyl-d54-sn-glycerol-3-phosphocholine-1,1,2,2-d4-N,N,N-trimethyl-d9 (Avanti Polar Lipids, Inc.) were employed. 13C-edited NOESY spectra, optimized for the detection of aromatic and aliphatic nuclei, respectively, and a 15N-edited NOESY spectrum were recorded with mixing times of 130 ms. Aliphatic side-chain assignments were achieved using an (H)CCH-COSY experiment (25). Full side-chain assignments of the two Phe and partial assignments of the three Trp side chains resulted directly from CT-HSQC spectra and their NOE patterns (26).
Structure Calculations—Structures of the well folded Ile-966 -Lys-994 residues (see Fig. 4A) were calculated by simulated annealing, starting at 3000 K using the program XPLOR-NIH (27). The peptide termini were represented by random-coil conformations. Besides standard force field terms for covalent geometry (bonds, angles, and improper torsions) and nonbonded contacts (van der Waals repulsion), the following experimental and empirical potentials were included. The difference between predicted and experimental residual dipolar couplings was described by a quadratic harmonic potential. Dihedral angle restraints were implemented using a quadratic square-well potential and merely aided in the convergence of the residual dipolar coupling restraints. ϕ, θ backbone dihedral angle constraints were extracted from N, Cα, Cβ, and C′ chemical shifts using the program TALOS (28). χ1 side-chain angle restraints were derived from the 3JC′Cγ and 3JNCγ coupling constants (supplemental Table 2). NOE interproton distance restraints were referenced to the linear α-helical structure of Ile-966 -Lys-989 and were incorporated using a quadratic square-well potential. A backbone-backbone hydrogen-bonding potential (29) was used. A torsion angle potential of mean force (30), which was modified to implement the preferred rotamers in transmembrane helices (31, 32), was employed. Moreover, the higher side-chain rotamers (χ2-χ4) of Lys-989 were selected to extend toward the C terminus, i.e. snorkeling its side chain out of the membrane core. A total of 20 structures were calculated. The structural statistics are summarized in supplemental Table 1. The structures have been deposited in the Protein Data Bank (accession number 2k1a) together with the energy-minimized average structure.
FIGURE 4.
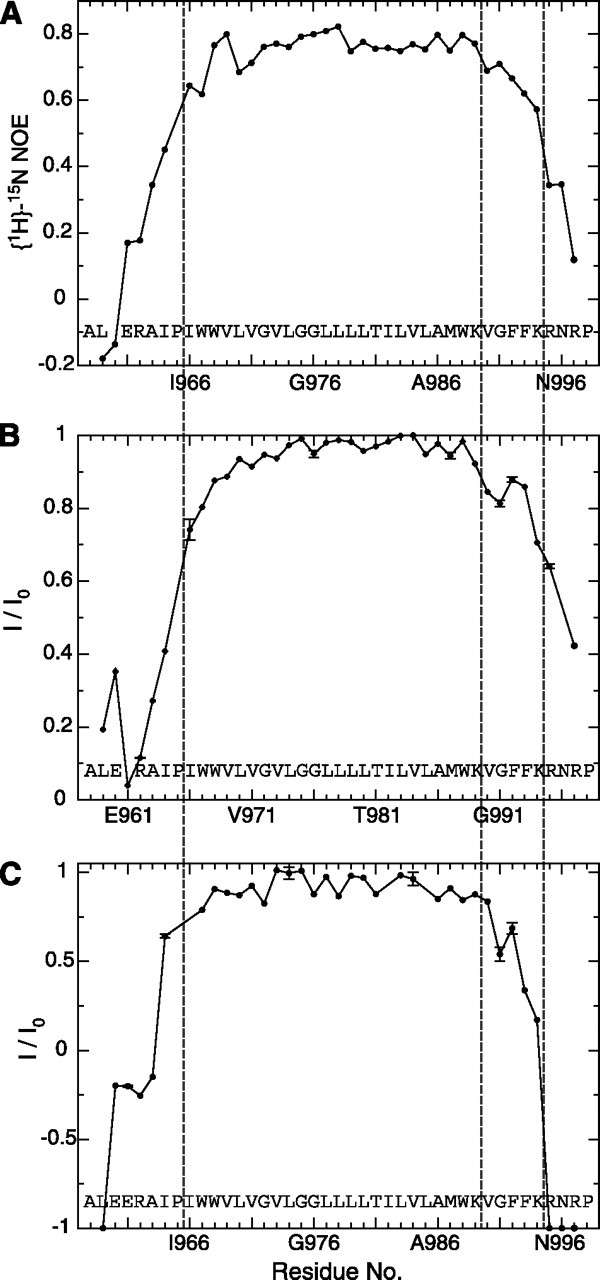
αIIb backbone dynamics, protection from paramagnetic Mn2+EDDA2- and HN-H2O chemical exchange. A, picoto nanosecond dynamics, correlative to backbone order (44), is evaluated in the form of heteronuclear [1H]-15N NOE values. Low values, as seen for the peptide termini, correspond to dynamically unstructured residues. B, signal broadening due to paramagnetic relaxation enhancement arising from the presence of net neutral Mn2+EDDA2- in the aqueous phase. The normalized ratio of H-N TROSY signal intensities in the presence and absence of 1 mm Mn2+EDDA2-, I/I0, is used to quantify signal broadening for each residue. C, exchange of the backbone amide protons (1HN) with water, depicted as the ratio of H-N TROSY signal intensities obtained with and without selective solvent inversion, I/I0, at pH 8.4 and 25 °C. The solvent was inverted 100 ms prior to the start of the pulse sequence, and radiation damping was suppressed by the application of a weak gradient pulse during this period. An I/I0 value of -1 corresponds to the exchange of the 1HN nucleus of a residue with water in all αIIb molecules present, whereas a value of +1 denotes the absence of any exchange. Resonances that were absent at pH 8.4, when compared with pH 7.4, were assigned I/I0 values of -1. The transmembrane helix (Ile-966 -Lys-989) borders and the Lys-994 -Arg-995 transition are marked by dashed lines in all panels.
RESULTS AND DISCUSSION
Structure Determination and Description—The structure of the αIIb TM segment reconstituted in small bicelles was determined by multidimensional heteronuclear NMR spectroscopy, making use of 2H/13C/15N-labeled peptide encompassing residues Ala-958 -Pro-998 (Fig. 1). Bicelles provide a small, symmetric lipid bilayer whose rim is stabilized by short-chain lipids (33, 34), and they appear well suited for the study of single-pass TM segments (9). Under the experimental conditions employed, the presence of a paramagnetic tag on αIIb was not sensed by untagged αIIb peptides, and no significant concentration-dependent chemical shift changes were observed (supplemental Fig. 3), clearly showing that the αIIb structure is not affected by any homo-oligomerization tendency. To validate the choice of bicelle lipids, the effects of lipid headgroup and tail length on αIIb spectral parameters were examined. Between glycerophosphatidylcholine-based lipids with 16/18 and 14/14 hydrocarbon tails, no significant 1HN and 15N chemical shift changes were detected (supplemental Fig. 1). Moreover, the introduction of lipids carrying phosphatidylserine headgroups had no effects on the αIIb backbone structure, as evidenced by the invariance of 13Cα secondary shifts (Fig. 2A), which correlate with backbone torsion angles (35). The newly introduced lipids were sensed predominantly by αIIb residues on the extracellular face (Fig. 2, B and C). Thus, the detected αIIb structure is stable in lipid environments that resemble the composition of its native platelet membrane (36).
FIGURE 2.
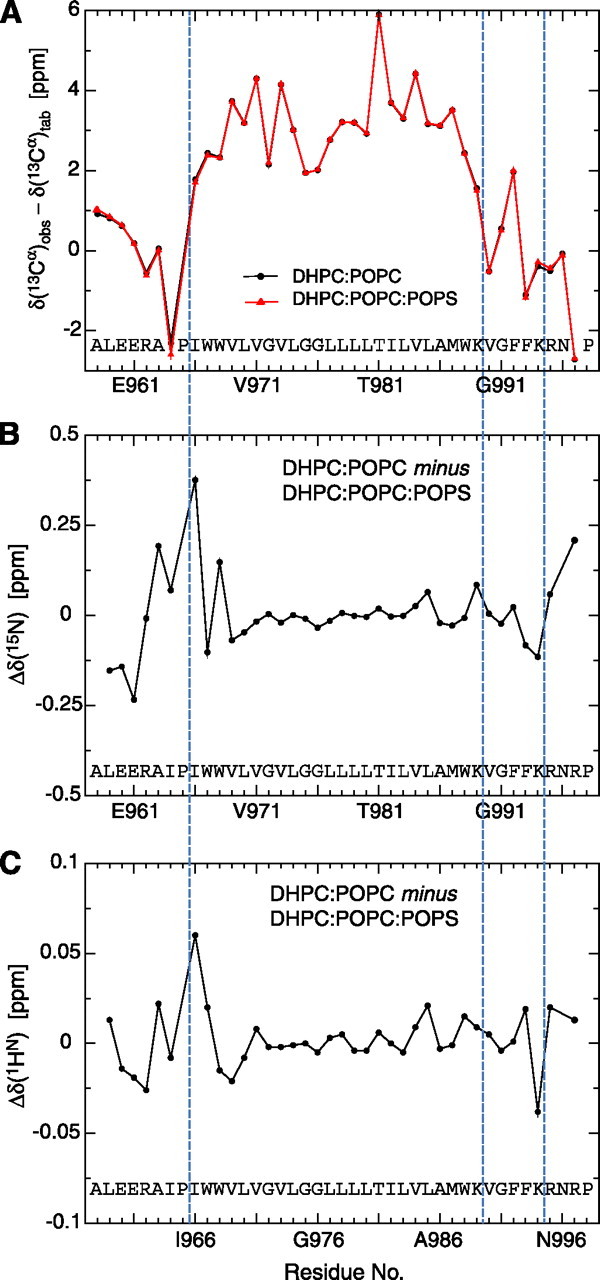
Effect of bicelle lipid composition on αIIb chemical shifts. The substitution of one-third of bicellar POPC lipids with 1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-l-serine] (POPS) lipids has no effects on the 13Cα secondary shifts (A) and, thus, αIIb backbone structure. Secondary 13Cα chemical shifts, defined as the difference between the observed and tabulated random-coil 13Cα shift of a residue, correlate with the underlying backbone conformation (35). Pro-965 and Pro-998 limit the backbone conformation of their preceding residues to extended conformations (42), as evidenced by their large negative 13Cα secondary shifts. 13Cα secondary shifts are reported relative to the random-coil values of Ref. (35) but have not been corrected for the ∼+0.5-ppm isotope shift resulting from perdeuteration of the carbon-attached hydrogens. B and C, the introduction of POPS is sensed predominantly by the αIIb backbone 1HN and 15N nuclei on the extracellular aIIb TM helix side. Changes in chemical shifts for these nuclei types are sensitive to changes in the chemical environment as well as to structural changes (43). However, overall 1HN and 15N chemical shift changes are relatively small. The transmembrane helix (Ile-966 -Lys-989) borders and the Lys-994 -Arg-995 transition are marked by blue dashed lines in all panels.
The αIIb TM structure was calculated from an extensive set of bond vector orientation, internuclear distance, and torsion angle constraints (supplemental Tables 1 and 2). For the ensemble of 20 calculated simulated annealing structures (Fig. 3A), coordinate precisions of 0.22 Å were obtained for the backbone-heavy atoms of the structured αIIb residues (Ile-966 - Lys-994). The structure is characterized by a linear α-helix for Ile-966 -Lys-989 followed by a reversal of backbone direction that conversely immerses Phe-992-Phe-993 toward the lipid hydrocarbon core of the intracellular membrane face (Fig. 3, D and E). By exhibiting increased protection from the paramagnetic Mn2+EDDA2- agent present in the aqueous phase as well as from HN-H2O exchange, the increased Phe-992-Phe-993 immersion was directly detectable within the bicelle membrane model (Fig. 4, B and C). Phe-993 packs against the TM helix, as evidenced by close interproton distances between its aromatic ring and nuclei from Trp-988, Leu-985, and Val-984 and its well defined χ1 rotameric state (supplemental Table 2). The packing of Phe-992 is characterized by hydrophobic contacts to Val-990 and Met-987; however, overall packing appears less defined, leading to an averaged χ1 rotameric state (supplemental Table 2). C-terminal to Lys-994, fast time scale backbone dynamics increase significantly (Fig. 4A), leading to dynamically unstructured conformations, with Arg-997 forced into extended backbone conformations by Pro-998 (Fig. 2A). Incidentally, the extracellular side of the TM segment is also strongly affected by a proline residue (Fig. 3, B and C); Pro-965 restricts Ile-964 to extended conformations, as evidenced by its large negative 13Cα secondary shift (Fig. 2A), which may benefit dynamic interactions with the hydrophobic lipid tails (Fig. 3, B and C). Backbone dynamics increase steadily from the start of the TM helix at Ile-966 to the N terminus (Fig. 4A).
FIGURE 3.

Structure of the αIIb integrin transmembrane segment. A, superposition of the ensemble of 20 calculated simulated annealing structures. B and C, views of the extracellular face of the energy-minimized, average structure. Residues N-terminal to Ile-966 become dynamically unstructured (Fig.4A), and their depicted conformation is but one of many possible conformations. D and E, views of the intracellular face of the energy-minimized, average structure. Residues C-terminal to Lys-994 become dynamically unstructured. To clearly depict the side-chain orientations, helix periodicity is not maintained between the extra- and intracellular views.
Structural similarities to the integrin αIIb TM segment were found in the Protein Data Bank for a β peptide (1 nkz, chain F) of the light-harvesting complex II from Rhodopseudomonas acidophila (37) (supplemental Fig. 5A). Somewhat more remotely related is a TM helix of cytochrome c oxidase from Thermus thermophilus (1xme, chain A) (38) (supplemental Fig. 5B).
Membrane Embedding and Relationship to Other Integrin α Subunits—Sequence alignment of all 18 human integrin α subunits (Fig. 1 and supplemental Fig. 4) suggests that the αIIb extra- and intracellular transitions to the TM helix are characteristic of α subunits. At the extracellular helix border, half of all α subunits exhibit a proline residue preceded by one that is hydrophobic. The Gly-Phe-Phe motif on the intracellular side is the only fully conserved tri-residue sequence element among integrin α TM segments, suggesting that the observed backbone reversal and Phe-Phe membrane immersion are general structural features of integrin α subunits. These transitions are fundamentally different from the β3 TM segment, where irregular structure and helix fraying characterized the transitions to the extra- and intracellular domains, respectively (9). Moreover, in contrast to the 29-residue β3 TM helix, the 5-residue-shorter αIIb TM helix does not require any tilt within a typical lipid bilayer (Fig. 5). The structural complexity of the Gly-991-Phe-993 motif and ensuing complicated Mn2+EDDA2- protection make it difficult to determine the TM helix center within the bicelle (Fig. 4B); in the absence of this information, the helix is assumed to be centered in the membrane (Fig. 5).
FIGURE 5.

Model of integrin αIIb and β3 membrane embedding as well as initial association. This figure depicts likely orientations of the αIIb and β3 TM segments, shown in green and cyan, respectively, relative to the indicated functional groups of a lipid bilayer composed of 1,2-dioleoyl-sn-glycero-3-phosphocholine (45), which is closely related to the lipids employed herein. The side chains of Val-696, Leu-697, Val-700, Gly-708, Trp-715, and Lys-716 are shown for the β3 subunit; for αIIb, the side chains of Trp-968, Gly-672, Gly-976, Thr-981, Trp-988, Lys-989, Phe-992, and Phe-993 are depicted (2, 8). Initial αIIb-β3 association is compatible with the individual αIIb and β3 structures; the final dimer structure will involve additional structural rearrangements.
The lipid tail-to-headgroup transition on the extracellular side is estimated to take place before the TM helix commences, i.e. between Pro-965-Ile-966 (Fig. 1). In this region, protection from paramagnetic Mn2+EDDA2- is significantly reduced (Fig. 4B). This transition is also in agreement with the Pro-998 -Leu-999 transition detected for the α5 subunit (39), which is similar to αIIb (Fig. 1). It is also noted that, at the extracellular helix border, no sharp hydrophobic-hydrophilic transition takes place (Fig. 1), which may result in a border variation between α subunits (39). The membrane borders depicted in Fig. 1 should therefore be considered as minimal for monomeric α subunits. On the intracellular side, the αIIb TM segment is expected to leave the lipid core after Phe-993 at the earliest. Lys-994 dynamics and protection are still somewhat high (Fig. 4), but, in conjunction with glycosylation mapping studies of integrin α2 and α5 subunits (40), the membrane border is estimated to lie between Phe-993-Lys-994. By virtue of a higher sequence homology on the intracellular TM face (Fig. 1 and supplemental Fig. 4), this transition appears to be more uniform among integrin α subunits. Finally, it should be noted that, on the basis of the Mn2+EDDA2- protection data of the αIIb and β3TMsegments (9) (Fig. 4B), immersion of the αIIb TM helix in the membrane is nevertheless slightly deeper than is the case for the β3 TM helix, as depicted in Fig. 5.
A stretch of continuous α-helix for residues Val-990 -Phe-993 appears less preferable than the observed conformation for three reasons. First, the two Phe-992-Phe-993 aromatic rings prefer to immerse in the hydrophobic lipid tail environment instead of in the electrostatically complex headgroup interface (41). Second, any TM helix extension would necessitate a helix tilt, which will require Phe-992-Phe-993 to reposition further outward into the headgroup region because they would come to lie on the same helix side as snorkeling Lys-989. In contrast, the β3 subunit has Ile-719 and Thr-720 following Lys-716 as the third and fourth residues (supplemental Fig. 4). Third, Gly-991 may destabilize helical conformation. Thus, as a paradigm for integrin α and β subunits, the four residues following αIIb(Lys-989) dispose its TM helix toward a short helix and the absence of a tilt, whereas the five residues following β3(Lys-716) create a bias for a long TM helix and a substantial tilt (Fig. 5).
The use of the Gly-Phe-Phe sequence motif is remarkable since, for example, the introduction of a second charged residue after αIIb(Lys-989) would equally well cause a short αIIb TM helix (39, 40). It appears likely that the highly conserved Gly-Phe-Phe motif has additional functional meaning.
Implications for Integrin αIIb β3 Association—Integrin bidirectional signaling proceeds with changes in α-β TM helix association (2, 4-8). The individual αIIb and β3 TM structures, which correspond to activated integrin conformations (2-4), therefore lay the foundation for evaluating the extent of structural and topological rearrangements upon αIIb-β3 association, i.e. integrin TM signaling. Moreover, the structures show that, in the activated receptor, the α-β extracellular domains connect to their respective transmembrane segments at different membrane crossing angles (Fig. 5).
Alanine substitution of Phe-992 or Phe-993 leads to receptor activation (5), showing that interactions of these residues with the β3 subunit are required for maintaining the inactive state. By either interacting with the β3 subunit in the conformation of the monomeric αIIb TM segment or shifting Val-990 -Phe-993 to another, for example helical, conformation, the Gly-991-Phe-993 structural motif is central to determining the αIIb-β3 relative orientation and topology not only in the activated but also in the resting receptor states (Fig. 5). Interestingly, in the reported cytoplasmic αIIb-β3 complex structure, which extends up to αIIb(Lys-989), Phe-992-Arg-997 are in helical conformation (7). Cysteine scanning mutagenesis of resting integrin αIIb-β3 has identified close interhelical contacts (2), whereas leucine scanning has revealed additional dimerization-sensitive residues (8). These interhelical contacts are mainly clustered near the extracellular helix sides, and the association of the individual αIIb and β3 structures appears able to fulfill these contacts (Fig. 5). Likewise, the salt bridge detected between αIIb(Arg-995) and β3(Asp-723) (5) is compatible with such a structure, although this is less significant since αIIb(Arg-995) is highly dynamic in the monomeric αIIb TM structure (Fig. 3). Clearly, additional structural information is required to define the associated αIIb-β3 structure. In conclusion, the unusual Gly-991-Phe-993 structural motif of αIIb, which appears prototypical for integrin α subunits, reveals a high degree of structural complexity at the TM level and lays the foundation for understanding the transition from activated to resting receptor states.
Supplementary Material
Acknowledgments
We thank Mark Ginsberg and Diana Gegala for critically reading the manuscript.
The atomic coordinates and structure factors (code 2k1a) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
This work was supported by an award from the American Heart Association (to T. S. U.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains two supplemental tables and five supplemental figures.
Footnotes
The abbreviations used are: TM, transmembrane; EDDA2-, ethylenediamine-N,N′-diacetate; NOE, nuclear Overhauser effect; NOESY, NOE spectroscopy; TROSY, transverse relaxation optimized spectroscopy; DHPC, 1,2-dihexanoyl-sn-glycero-3-phosphocholine; POPC, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine; POPS, 1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-l-serine]; AMPS, 2-acrylamido-2-methyl-1-propanesulfonate.
References
- 1.Hynes, R. O. (2002) Cell 110 673-687 [DOI] [PubMed] [Google Scholar]
- 2.Luo, B. H., Springer, T. A., and Takagi, J. (2004) PLoS Biol. 2 776-786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu, J. Q., Carman, C. V., Kim, M., Shimaoka, M., Springer, T. A., and Luo, B. H. (2007) Blood 110 2475-2483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Partridge, A. W., Liu, S. C., Kim, S., Bowie, J. U., and Ginsberg, M. H. (2005) J. Biol. Chem. 280 7294-7300 [DOI] [PubMed] [Google Scholar]
- 5.Hughes, P. E., DiazGonzalez, F., Leong, L., Wu, C. Y., McDonald, J. A., Shattil, S. J., and Ginsberg, M. H. (1996) J. Biol. Chem. 271 6571-6574 [DOI] [PubMed] [Google Scholar]
- 6.Li, W., Metcalf, D. G., Gorelik, R., Li, R. H., Mitra, N., Nanda, V., Law, P. B., Lear, J. D., DeGrado, W. F., and Bennett, J. S. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 1424-1429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vinogradova, O., Velyvis, A., Velyviene, A., Hu, B., Haas, T. A., Plow, E. F., and Qin, J. (2002) Cell 110 587-597 [DOI] [PubMed] [Google Scholar]
- 8.Luo, B. H., Carman, C. V., Takagi, J., and Springer, T. A. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 3679-3684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lau, T.-L., Partridge, A. P., Ginsberg, M. H., and Ulmer, T. S. (2008) Biochemistry 47 4008-4016 [DOI] [PubMed] [Google Scholar]
- 10.Donaldson, L. W., Skrynnikov, N. R., Choy, W. Y., Muhandiram, D. R., Sarkar, B., Forman-Kay, J. D., and Kay, L. E. (2001) J. Am. Chem. Soc. 123 9843-9847 [DOI] [PubMed] [Google Scholar]
- 11.Harford, C., and Sarkar, B. (1997) Acc. Chem. Res. 30 123-130 [Google Scholar]
- 12.Tycko, R., Blanco, F. J., and Ishii, Y. (2000) J. Am. Chem. Soc. 122 9340-9341 [Google Scholar]
- 13.Ulmer, T. S., Ramirez, B. E., Delaglio, F., and Bax, A. (2003) J. Am. Chem. Soc. 125 9179-9191 [DOI] [PubMed] [Google Scholar]
- 14.Ulmer, T. S., Bax, A., Cole, N. B., and Nussbaum, R. L. (2005) J. Biol. Chem. 280 9595-9603 [DOI] [PubMed] [Google Scholar]
- 15.Chou, J. J., Gaemers, S., Howder, B., Louis, J. M., and Bax, A. (2001) J. Biomol. NMR 21 377-382 [DOI] [PubMed] [Google Scholar]
- 16.Delaglio, F., Grzesiek, S., Vuister, G. W., Zhu, G., Pfeifer, J., and Bax, A. (1995) J. Biomol. NMR 6 277-293 [DOI] [PubMed] [Google Scholar]
- 17.Grzesiek, S., and Bax, A. (1993) J. Am. Chem. Soc. 115 12593-12594 [Google Scholar]
- 18.Ulmer, T. S., Campbell, I. D., and Boyd, J. (2004) J. Magn. Reson. 166 190-201 [DOI] [PubMed] [Google Scholar]
- 19.Pervushin, K., Riek, R., Wider, G., and Wuthrich, K. (1997) Proc. Natl. Acad. Sci. U. S. A. 94 12366-12371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu, J. S., and Bax, A. (1997) J. Biomol. NMR 9 323-328 [DOI] [PubMed] [Google Scholar]
- 21.Hu, J. S., Grzesiek, S., and Bax, A. (1997) J. Am. Chem. Soc. 119 1803-1804 [Google Scholar]
- 22.Kontaxis, G., Clore, G. M., and Bax, A. (2000) J. Magn. Reson. 143 184-196 [DOI] [PubMed] [Google Scholar]
- 23.Jaroniec, C. P., Ulmer, T. S., and Bax, A. (2004) J. Biomol. NMR 30 181-194 [DOI] [PubMed] [Google Scholar]
- 24.Chou, J. J., Delaglio, F., and Bax, A. (2000) J. Biomol. NMR 18 101-105 [DOI] [PubMed] [Google Scholar]
- 25.Gehring, K., and Ekiel, I. (1998) J. Magn. Reson. 135 185-193 [DOI] [PubMed] [Google Scholar]
- 26.Lin, Z., Xu, Y. Q., Yang, S., and Yang, D. W. (2006) Angew. Chem. Int. Ed. Engl. 45 1960-1963 [DOI] [PubMed] [Google Scholar]
- 27.Schwieters, C. D., Kuszewski, J. J., Tjandra, N., and Clore, G. M. (2003) J. Magn. Reson. 160 65-73 [DOI] [PubMed] [Google Scholar]
- 28.Cornilescu, G., Delaglio, F., and Bax, A. (1999) J. Biomol. NMR 13 289-302 [DOI] [PubMed] [Google Scholar]
- 29.Grishaev, A., and Bax, A. (2004) J. Am. Chem. Soc. 126 7281-7292 [DOI] [PubMed] [Google Scholar]
- 30.Kuszewski, J. J., and Clore, G. M. (2000) J. Magn. Reson. 146 249-254 [DOI] [PubMed] [Google Scholar]
- 31.Gray, T. M., and Matthews, B. W. (1984) J. Mol. Biol. 175 75-81 [DOI] [PubMed] [Google Scholar]
- 32.Chamberlain, A. K., and Bowie, J. U. (2004) Biophys. J. 87 3460-3469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanders, C. R., II, and Landis, G. C. (1995) Biochemistry 34 4030-4040 [DOI] [PubMed] [Google Scholar]
- 34.Prosser, R. S., Evanics, F., Kitevski, J. L., and Al-Abdul-Wahid, M. S. (2006) Biochemistry 45 8453-8465 [DOI] [PubMed] [Google Scholar]
- 35.Spera, S., and Bax, A. (1991) J. Am. Chem. Soc. 113 5490-5492 [Google Scholar]
- 36.Garcia-Guerra, R., Garcia-Dominguez, J. A., and Gonzalez-Rodriguez, J. (1996) Platelets (Abingdon) 7 195-205 [DOI] [PubMed] [Google Scholar]
- 37.Papiz, M. Z., Prince, S. M., Howard, T., Cogdell, R. J., and Isaacs, N. W. (2003) J. Mol. Biol. 326 1523-1538 [DOI] [PubMed] [Google Scholar]
- 38.Hunsicker-Wang, L. M., Pacoma, R. L., Chen, Y., Fee, J. A., and Stout, C. D. (2005) Acta Crystallogr. Sect. D Biol. Crystallogr. 61 340-343 [DOI] [PubMed] [Google Scholar]
- 39.Stefansson, A., Armulik, A., Nilsson, I. M., von Heijne, G., and Johansson, S. (2004) J. Biol. Chem. 279 21200-21205 [DOI] [PubMed] [Google Scholar]
- 40.Armulik, A., Nilsson, I., von Heijne, G., and Johansson, S. (1999) J. Biol. Chem. 274 37030-37034 [DOI] [PubMed] [Google Scholar]
- 41.Wimley, W. C., and White, S. H. (1996) Nat. Struct. Biol. 3 842-848 [DOI] [PubMed] [Google Scholar]
- 42.Karplus, P. A. (1996) Protein Sci. 5 1406-1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang, Y. J., and Jardetzky, O. (2004) J. Biomol. NMR 28 327-340 [DOI] [PubMed] [Google Scholar]
- 44.Lipari, G., and Szabo, A. (1982) J. Am. Chem. Soc. 104 4546-4559 [Google Scholar]
- 45.Wiener, M. C., and White, S. H. (1992) Biophys. J. 61 434-447 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.