

Abstract
Heparanase is an endoglycosidase that cleaves heparan sulfate (HS) side chains from heparan sulfate proteoglycans (HSPGs) present in extracellular matrix and cell membranes. Although HSPGs have many functions during development, little is known of the role of the enzyme that degrades HS, heparanase. We cloned and characterized the expression of two heparanase splicing variants from Xenopus laevis and studied their function in early embryonic development. The heparanase gene (termed xHpa) spans over 15 kb and consists of at least 12 exons. The long heparanase (XHpaL) cDNA encodes a 531-amino acid protein, whereas the short splicing variant (XHpaS) results in a protein with the same open reading frame but missing 58 amino acids as a consequence of a skipped exon 4. Comparative studies of both isoforms using heterologous expression systems showed: 1) XHpaL is enzymatically active, whereas XHpaS is not; 2) XHpaL and XHpaS interact with heparin and HS; 3) both proteins traffic through the endoplasmic reticulum and Golgi apparatus, but XHpaL is secreted into the medium, whereas XHpaS remains associated with the membrane as a consequence of the loss of three glycosylation sites; 4) overexpression of XHpaS but not XHpaL increases cell adhesion of glioma cells to HS-coated surfaces; 5) XHpaL and XHpaS mRNA and protein levels vary as development progresses; 6) specific antisense knock-down of both XHpaL and XHpaS, but not XHpaL alone, results in failure of embryogenesis to proceed. Interestingly, rescue experiments suggest that the two heparanases regulate the same developmental processes, but via different mechanisms.
Growth factors, cytokines, and extracellular matrix (ECM)2 molecules are important in controlling embryonic development. Their activity can be regulated by proteoglycans, including heparan sulfate proteoglycans (HSPGs) and chondroitin sulfate proteoglycans, which are families of molecules found in the ECM, and in basement and cell membranes (1). The basic molecular structure of HSPGs consists of a core protein to which are attached many heparan sulfate (HS) glycosaminoglycan chains (2).
HSPGs regulate development by controlling the migration, adhesion, and morphology of various cell types (3-5). For example, mutations in genes involved in HSPG synthesis have emerged from genetic screens that affect axon guidance and segment polarity in Drosophila (6-8). Similarly, defects in gas-trulation and aberrant axonal pathfinding were observed when HSPGs were either knocked down or degraded enzymatically during Xenopus laevis development (9-12). HSPGs affect biological events by several different means. For example, the structural integrity and selective permeability of components of the ECM and basement membrane, such as laminin, fibronectin, or collagen IV, depend on interactions with HSPGs. HSPGs also interact with enzymes, growth factors, cytokines, and chemokines, sequestering them in the ECM as an inactive reservoir (5, 13, 14). These molecules are then released and become bioactive upon enzymatic degradation of HSPG (13-15). Although HS and enzymes involved in HSPG biosynthesis are recognized as being important for normal development, little is known about the contribution that enzymes that cleave HS, like heparanase, may have.
Heparanase is an endoglucuronidas that degrades HS side chains of the HSPG. It is synthesized as a latent pre-proheparanase of ∼65 kDa that contains a signal peptide. A post-translational maturation process of the polypeptide generates amino (8 kDa) and carboxyl (50 kDa) termini products, which associate noncovalently to generate the active enzyme (16, 17). Heparanase genes from several species have been identified (18-23). Heparanase activity is associated with migration and invasion processes during pathological events such as tumor invasion and tumor-mediated neovascularization (24-26). As well, heparanase has normal physiological functions in the migration and extravasation of a number of immune cells (27-29). Interestingly, heparanase can, in a manner not associated with its enzymatic activity, increase the adhesion of immune and cancer cells (29-31). During development, heparanase expression has been detected in the early mouse embryo, the chick vascular and nervous systems, as well as the human fetal colon and liver (21, 32, 33).
Although each species has a single gene coding for heparanase, some species-specific properties have been reported. For example, the secretion and cellular localization of human and chicken heparanase differ as a consequence of their specific signal peptides (21). Moreover, recent reports in human and rat indicate the presence of two different heparanase splice variants, only one of which can be secreted (34, 35), suggesting that secreted and non-secreted heparanase forms are evolutionarily conserved. The biological significance, however, of two heparanase isoforms that are differentially secreted is unknown.
Here we report the cloning and characterization of two splice variants of Xenopus heparanase. The proteins encoded by these two splice variants differ by 58 amino acids due to a skipped exon 4, but conserve the same open reading frame. We found that the two variants are differentially expressed during development, and differ functionally in several important ways, including their enzymatic activity, secretion, and adhesive properties. Despite these differences, both variants when overexpressed are able to rescue early embryonic death that results from knockdown of endogenous heparanase levels with an antisense oligonucleotide. These data suggest the intriguing possibility that the two isoforms use distinct means to regulate the same molecular events in the developing embryo.
EXPERIMENTAL PROCEDURES
Cloning of Xenopus Heparanase cDNA—We used the chicken and human heparanase cDNA sequences to screen the EST data base for a homologous Xenopus candidate mRNA sequence (Sanger Institute Xenopus EST data base). Several Xenopus tropicalis and X. laevis ESTs showed homology with both cDNAs (gi:28279837; DN075994; CX971553, CX934009), but they all corresponded to partial sequences. Overlapping and alignment of these partial ESTs let us identify a putative X. tropicalis heparanase sequence. Specific primers designed based on this sequence were used to amplify two products by RT-PCR from total cDNA from stage 37/38 X. laevis embryos. The amplification products, named xHpaL (Xenopus heparanase long) and xHpaS (Xenopus heparanase short), were cloned and sequenced.
RT-PCR—Total RNA was obtained from whole embryos using TRIzol (Invitrogen) according to the manufacturer's protocol. Single-stranded cDNA was produced from DNase I-treated RNA samples (5 μg) by priming with oligo(dT) primers using SuperScript™ II RNase H reverse transcriptase (Invitrogen) according to the manufacturer's instructions. All PCR amplifications were carried out in a total volume of 25 μl with 200 ng of cDNA, 400 nm primers, 50 mm KCl, 10 mm Tris-HCl, pH 8.0, 1.5 mm MgCl2, 200 μm dNTP, and 1.25 units of recombinant PWO polymerase (Roche). To compare xHpaL and xHpaS mRNA expression, the number of PCR cycles was determined to be within the linear range (heparanase: 35 cycles and EF1α: 25 cycles). The annealing temperature for each gene analyzed was 55 °C. Primers used to amplify heparanase were forward, 5′-aaacccacgattcctctctgt-3′ and reverse 5′-gcttcctgtcatctctgcttg-3′, whereas primers for EF1α were reported previously (36).
Generation of Expression Constructs—PCR products obtained from cDNA were cloned into either TOPO-pCRII (Invitrogen) or p-JET (Fermentas, Canada) vectors and sequenced. The heparanase cDNA expression constructs (XHpaL and XHpaS) were generated by subcloning into the pCS2 eukaryotic vector or pCS2-MT that inserts 6 myc tags in-frame at the carboxyl terminus of the protein. Xenopus heparanase chimeras with the human immunoglobulin κ leader sequence (Igκ-SP) were generated by subcloning the signal sequence from pSecTagB vector (Invitrogen) into the pCS2-MT expression vector using BamHI and ClaI. Subsequently, Xenopus heparanases without the first 63 nucleotides (21 aa) were amplified by PCR using primers containing the corresponding restriction enzyme sites and further subcloned into the Igκ leader sequence expression vector. These constructs were named pCS2-IgK-XHpaL-MT and pCS2-IgK-XHpaS-MT. Expression vectors of XHpaL and XHpaS without signal peptides were also generated using the same strategy and subcloned into pCS2-MT expression vectors. The Stratagene QuikChange® Site-directed Mutagenesis Kit (Stratagene) was used, according to the manufacturer's protocol, to substitute asparagines, within potential glycosylation consensus sequences of XHpaL and XHpaS, for glutamines, as well as to generate mutant isoforms immune to antisense oligonucleotide knock down. Nucleotide changes and sequence integrity of all the expression vectors were confirmed by automated sequencing at the DNA services facility, University of Calgary.
Sense and antisense (AS) oligonucleotides were synthesized based on the xHpa sequence targeting exon 3 (nucleotides 483 to 505), or exon 4 (AS4) (nucleotides 553 to 575), to knock down both XHpaL and XHpaS or only XHpaL, respectively (37). Briefly, oligonucleotides contained a 6′-carboxyfluorescein aminohexylphosphate (6-FAM) at the 5′ end, plus five nucleotides on either end with 2′-O-methyl (2′-OMe) sugar modifications and an internucleoside linkage backbone with phosphorothioates to resist nuclease degradation (6-FAM-5 (2′O-Me)-13 phosphorothioates-5 (2′-OMe)).
Cells Lines and Transient Transfections—COS-7 and C6 glioma cell lines were maintained in growth medium (Dulbecco's modified Eagle's medium plus 0.02% glutamine supplemented with 10% fetal bovine serum (Invitrogen)) without antibiotics, and grown in a humidified 5% CO2, 95% air atmosphere at 37 °C. Transient transfection was carried out on 70-80% confluent monolayers in 6- or 24-well dishes with Lipofectamine 2000 (Invitrogen). Cells were keep in Dulbecco's modified Eagle's medium without fetal bovine serum for 6-8 h followed by additional recovery and expression time of 24-28 h in Dulbecco's modified Eagle's medium with 10% fetal calf serum. Conditioned medium were obtained 24 h after transfection, centrifuged, and filtered through a 22-μm pore filter (Pall Corporation, MI).
Stable cell lines expressing myc-tagged heparanases were generated after transfection of C6 cells with pcDNA3.1 vector (control) or pcDNA3.1-XHpaL-MT or pcDNA3.1-XHpaS-MT vectors and selected with Genticin® (750 μg/ml) (Invitrogen). After 3 weeks of selection, specific clones were isolated. Expression of heparanase in each clone was determined by Western blot.
Measurement of Heparanase Activity—To detect HS degradation we developed a sensitive technique, which combined two previously published methods (22, 38). Fluorescein isothiocyanate (FITC)-labeled HS (a gift from Syntex SA, Buenos Aires, Argentina) was used as substrate for heparanase as reported by Toyoshima and Nakajima (22). Instead of detecting degradation products by chromatography, a shift of mobility using native non-denaturing gel electrophoresis was employed as reported by Rozenberg and co-workers (38). The advantages of this method are the direct detection of the fluorescently labeled FITC-HS, and the speed and simplicity of electrophoresis. Heparanase activity was determined for extracts from COS-7 cells transfected with control vector, pCS2-XHpaL, or pCS2-XHpaS. After 24 h of transfection, cell extracts (100 μl; 5 mg/ml) in phosphate-buffered saline (PBS, pH 6) were sonicated and incubated with FITC-HS (final concentration: 10 μg/ml) at 37 °C for 24 h. Aliquots of the reaction were boiled 10 min with nondenaturing loading buffer and run on a 20-cm long, 15% polyacrylamide gel. FITC-HS was detected with a DyNA light Dual Intensity UV transilluminator (Labnet, NJ).
Interaction of Heparanases with Heparin—The interaction of XHpaL and XHpaS with heparin was determined as described previously (39), but with a few modifications. Briefly, transfected COS-7 cells were lysed by five rounds of freezing (-80 °C) and thawing (4 °C) in PBS, pH 6, and 0.1% Nonidet P-40 (Igepal, Sigma). Cell debris was pelleted by centrifugation and supernatants (1 mg/ml) were added to 30 μl of heparinacrylic beads (Sigma) and incubated for 3 h at 4 °C. Beads were then washed five times in cold PBS, centrifuged, and boiled in reducing SDS sample buffer for 5 min. Supernatants were analyzed by Western blot.
Western Blot Analysis—Protein was measured with bicinchoninic acid using a BCA™ protein assay kit (Thermo Scientific, IL). Forty μg of protein/lane were separated on 10% polyacrylamide gels (unless specifically described), transferred to polyvinylidene difluoride membranes (Bio-Rad), and immunoblotted with anti c-myc (rabbit A-14, 1/1000 dilution; Santa Cruz Biotechnology Inc., or mouse 9E10 monoclonal, 1/2000 dilution, Covance, CA), goat anti-actin (C-11, 1/1000 dilution; Santa Cruz Biotechnology Inc.), anti-human heparanase (HPA1 monoclonal clone HP3/17, 1/500 dilution; Cedarlane Lab, Canada; and goat polyclonal C20, 1/500 dilution, Santa Cruz Biotechnology Inc.) antibodies. Specific peroxidase-conjugated secondary antibodies were used to detect protein expression by enhanced chemiluminescence (PerkinElmer Life Sciences). Densitometry analysis, of either digital images of agarose gels taken on a UV transilluminator or scanned films from Western blots, was performed by using the public domain ImageJ software version 1.37.
Immunocytochemistry—COS-7 and C6 glioma cells were transiently transfected in an 8-well chamber slide. After 24 h, the cells were fixed with 4% paraformaldehyde, blocked in 5% goat serum, and incubated overnight at 4 °C in primary antibody diluted in PBT (PBS, 0.1% bovine serum albumin (Sigma), 0.5% Triton (BDH)) with 5% goat serum (Invitrogen). Rabbit anti-myc (1/500 dilution), anti-calnexin (1/200 dilution; BD Transduction Laboratories), and anti-golgin 97 from conditioned medium of mouse CDF4 hybridoma (dilution 1/1000; gift from Dr M. Fritzler, University of Calgary) were used to determine the subcellular localization of specific proteins. Fluorescent-conjugated Alexa Fluor 488 and Alexa Fluor 546 (1/2000 dilution; Invitrogen) secondary antibodies were used. Controls without primary or secondary antibodies were run to verify the specificity of the staining. Interaction of HS with cells overexpressing heparanase was determined by incubation of transfected cells with FITC-HS (50 μg/ml) for 2 h. Digital images of samples were obtained by using a Spot II camera and Spot Advanced software (Diagnostics Instruments) and processed for brightness and contrast using Adobe Photoshop (7.0) software.
Heparan Sulfate Adhesion Assays—Confluent monolayers of C6-vector, C6-XHpaL, and C6-XHpaS stable cell lines were deattached by gentle pipetting and passed through a 25-guage needle several times to obtain a monodispersed cell suspension. Cells were left to recover in suspension for 30 min and then plated on 24-well FITC-HS-coated plates at a concentration of 1 × 105 cell/ml (0.5 ml/well) for different times (0 to 90 min). At the end of the assay, cells were carefully washed with ice-cold PBS and the number of attached cells was evaluated by protein determination with bicinchoninic acid using the BCA protein assay kit (Thermo Scientific, IL) according to the manufacturer's instructions. The rate of adhesion was expressed as a percentage of the total cells seeded into the well. HS-coated plates were obtained by incubation with FITC-HS (50 μg/100 μl water/well) and overnight drying.
Embryo and Blastomere Injections—X. laevis embryos were obtained from chorionic gonadotrophin (Intervet Canada Ltd.) induced egg production and in vitro fertilization according to standard procedures (40). For overexpression and knock-down experiments, both blastomeres of two-cell stage embryos were injected with ∼10 nl of solution containing pCS2-XHpaL or pCS2-XHpaS constructs (40 ng/μl), or 30 μm of the sense, AS, and AS4 oligonucleotides (final concentrations of oligonucleotides within the embryos were ∼260-577 nm). Procedures involving frogs and embryos were approved by the Animal Care and Use Committee, University of Calgary.
RESULTS
Cloning of the Xenopus Heparanases cDNA—A search of the Xenopus EST data base (Sanger Institute Xenopus EST data base) identified several X. tropicalis and X. laevis partial ESTs with homology for chicken and human heparanase mRNA sequences. Overlapping and alignment of these partial ESTs let us identify a putative X. tropicalis heparanase sequence. We performed RT-PCR, using specific primers designed based on this sequence, on total cDNA from stage 37/38 X. laevis embryos. Two products were amplified, cloned, and sequenced, and found to encode proteins of 531 and 473 aa that share the same open reading frame, other than 58 aa missing in the short variant (Fig. 1, A and B). These two variants were called XHpaL (Xenopus heparanase long) and XHpaS (Xenopus heparanase short). The predicted amino acid sequence encoded by XHpaL showed 57 and 54% identity, and 78 and 74% similarity, with human and chicken heparanase, respectively. The presence of two heparanase cDNAs with the same open reading frame suggests the presence of a splicing variant rearrangement with a putative skipped exon. This was confirmed by identification of the exon-intron boundaries through comparison of the X. tropicalis genomic sequence with the XHpaL cDNA sequence. The Xenopus heparanase gene spans over 15 kb, and consists of at least 12 exons. The main difference between xHpaL and xHpaS is the elimination of the 174 nucleotides of exon 4 in xHpaS (Fig. 1A). Comparative analysis between the X. laevis cDNA and the putative X. tropicalis heparanase predicted from the genomic DNA sequence showed 87% identity and 92% similarity at the protein level.
FIGURE 1.
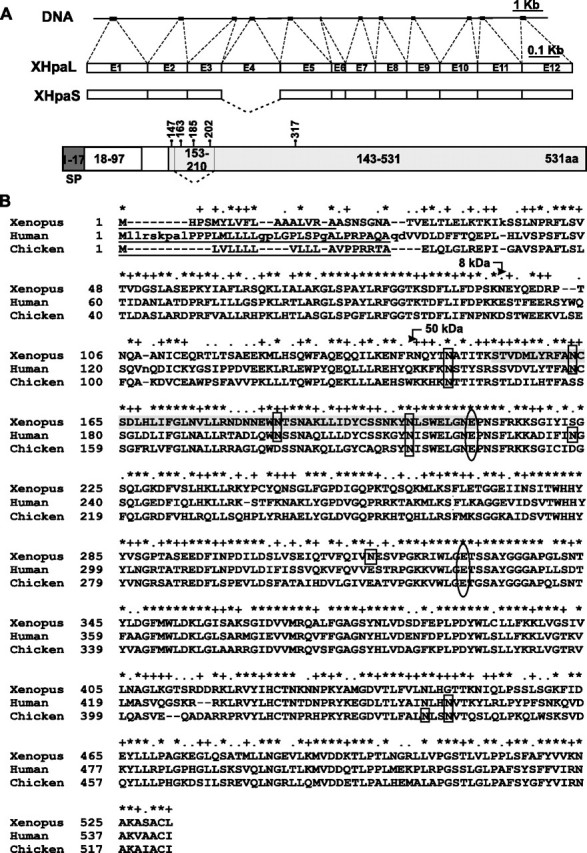
Cloning of Xenopus heparanase. A, schematic representation of the X. tropicalis DNA showing the exons (boxes) and introns (lines) of the heparanase gene. DNA is aligned against the mRNA of long (XHpaL) and short (XHpaS) heparanase splicing variants cloned from X. laevis (drawn to scale). Exon 4 is eliminated in XHpaS. A schematic diagram of the Xenopus heparanase protein, indicating the position in the amino acid sequence of the five putative asparagines susceptible to N-glycosylation, as well as the putative peptides (the signal peptide (SP), the 8-kDa peptide (white box), and the 50-kDa peptide (gray box)) generated after processing, based on comparison to mammalian heparanases. The sequence missing in the XHpaS, corresponding to amino acids 153-210 and containing 3 glycosylation sites, is also indicated. B, predicted amino acid sequence of the Xenopus heparanase aligned with human and chicken heparanases. The asterisk and plus symbols indicate conserved and similar residues, respectively. Dashes indicate spaces introduced for alignment. The putative glycosylation sites are indicated by rectangles. The putative proton donor at Glu210 (missing in XHpaS) and a nucleophile residue at Glu329 are indicated by ovals. The 58-amino acid sequence spliced out of the XHpaS variant is shadowed in gray. Numbers on the left indicate the amino acid residue. The predicted signal peptides of the three sequences are underlined. Predicted sites of cleavage in the proheparanase to generate the 8- and 50-kDa proteins that form the active enzyme are shown with arrows.
Computational and comparative analyses of the XHpaL amino acid sequence against those of heparanases from other species (human, chicken, mouse, and rat) suggest that Xenopus heparanase contains a signal peptide (Signal-P program) that spans the NH2-terminal 17 aa, an 8-kDa peptide from Ala18 to Lys97, and a 50-kDa peptide corresponding to Asp143 to Leu531 (Fig. 1, A and B).
Expression and Enzymatic Activity of XHpaL and XHpaS—To analyze possible biochemical differences between XHpaL and XHpaS, constructs with a COOH-terminal myc tag were generated. These constructs were used to transfect two mammalian expression systems: COS-7 cells, which show high rates of expression of transfected plasmids, and glioma C6 cells, which lack endogenous heparanase activity (21, 41). To ensure that the entire heparanase protein was expressed, and at sufficient levels, extracts from the cells were analyzed by Western blot by using two commercially available anti-human heparanase antibodies. A monoclonal antihuman antibody recognized both Xenopus heparanase variants, generating protein bands of ∼67 and 57 kDa, corresponding to the XHpaL and XHpaS pro-heparanases, respectively (Fig. 2A). This antibody also recognized the myc-tagged heparanases (74- and 63-kDa band for XHpaL-MT and XHpaS-MT, respectively), although its immunoreactivity was substantially lower than that of the anti-myc antibody (Fig. 2A). A goat polyclonal antibody raised against the human heparanase COOH terminus region did not recognize either Xenopus heparanase variants (data not shown).
FIGURE 2.

XHpaL and XHpaS differ in their secretion and endoglucuronidase properties. A, expression of XHpaL and XHpaS. COS-7 cells were transfected with heparanase XHpaL and XHpaS constructs with or without the myc tag. After 48 h, 40 μg of cell extract were analyzed by Western blot using anti-myc or anti-human heparanase (Clone HP3/17) antibodies.β-Actin detection was used as equal loading control. The same Western blot was exposed 20 s (top) to detect the myc tags and 10 min (bottom) for detection with the anti-heparanase antibody. B, heparanase activity of XHpaL. COS-7 cells were transfected with XHpaL, XHpaS, or mock control (vector). One day after transfection, cell extracts were incubated with FITC-HS for an additional 24 h and degradation products of FITC-HS were detected on a native, 15% polyacrylamide gel. C, XHpaL and XHpaS interact with heparin. Cellular extracts from COS-7 cells transiently transfected with myc-tagged XHpaL, XHpaS, and LIMK1 were incubated with heparin-acrylic beads. Bound proteins were evaluated by Western blotting with anti-myc antibodies. A Western analysis of myc-tagged proteins from input is also shown. D, secretion of XHpaL but not XHpaS. COS-7 cells were transfected with the empty vector, constructs expressing myc-tagged XHpaL and XHpaS, or constructs expressing Xenopus heparanases with a deleted signal peptide (XHpaL and XHpaS no SP) or with the Xenopus signal peptide replaced with the human immunoglobulin κ leader sequence (IgKHpaL and IgKHpaS). After 24 h, cell extracts and conditioned medium were analyzed by Western blotting utilizing anti-myc antibodies. β-Actin in the cell extract and the presence of a nonspecific band (NS) in conditioned medium were used as equal loading controls.
The heparanase activity of XHpaL and XHpaS was evaluated by determining the capacity of the two isoforms to degrade HS. For this purpose, FITC-labeled HS was used as substrate and incubated with cell lysates obtained from COS-7 cells transfected with pCS2-XHpaL or pCS2-XHpaS heparanase expression vectors, or pCS2 vector alone as control. After 24 h, the degradation products were separated by gel electrophoresis and labeled FITC-HS detected with a transilluminator. Lanes with XHpaL-transfected cell extracts showed a faster migrating smear of FITC-HS degradation products as compared with the samples from XHpaS and control transfected cells, which suggest that XHpaL extracts contain heparanase activity (Fig. 2B). No difference in FITC-HS electrophoresis mobility was detected between lysates from cells transfected with vector or XHpaS, suggesting that the short heparanase variant lacks enzymatic activity. A small but noticeable downshift of mobility of FITC-HS smear products was detected with samples from mock and XHpaS-transfected cells as compared with the heated-treated (20 min at 80 °C) control lysate, which may reflect loss of endogenous COS-7 cell heparanase activity upon heating.
XHpaL and XHpaS Both Interact with Heparin—Because XHpaS has no apparent enzymatic activity, we next determined if it could still interact with heparanase substrates. Heparin and HS are two closely related molecules that are known to interact with human heparanase (39). Heparin-acrylic beads were incubated with cell extracts of COS-7 cells transfected with a myc-tagged XHpaL and XHpaS construct, and any bound heparanase was identified by Western blot with α-myc antibodies. A protein not known to interact with heparin, the cytoplasmic serine/threonine kinase LIM kinase 1 (LIMK1), was used as a negative control. Both splicing variants, but not LIMK1, associated with the heparin beads (Fig. 2C). This result shows that both splicing variants interact with heparin and presumably with HS substrata.
XHpaL but Not XHpaS Is Secreted—The literature indicates that distinct heparanases are either secreted or non-secreted (21, 34, 35), and that differences in their signal peptides explain the differential secretion properties of human and chicken heparanases (21). Because the Xenopus heparanase signal peptide shows no homology with those of the human and chicken heparanases (Fig. 1B), the capacity of the XHpaL and XHpaS isoforms to be secreted was investigated. COS-7 cells were transiently transfected with myc-tagged XHpaL and XHpaS constructs and protein levels in cell extracts and conditioned media were analyzed by Western blot. Similar protein levels of XHpaL and XHpaS were detected in COS-7 cell extracts; however, only XHpaL was detected in the conditioned medium (Fig. 2D), suggesting that the short variant is not secreted. Moreover, this result indicates that differences in the secretion properties of XHpaL and XHpaS are not related to the signal peptide, which they have in common. This was confirmed by assaying for secretion of engineered long and short heparanases, where the signal peptide on each was replaced with the signal peptide from the human immunogloblin κ. Similar to the results obtained with the Xenopus heparanase signal peptide, only the long isoform was secreted from the cells and appeared in the conditioned medium (Fig. 2D). Deletion of 21 amino acids, containing the signal peptide, completely abrogated secretion of XHpaL, suggesting that the signal peptide is necessary but not sufficient to direct heparanase secretion. Similar results were obtained using glioma C6 cells (data not shown). These data support the idea that the pronounced difference in secretion between XHpaL and XHpaS is not due to the signal peptide, which leaves the amino acid sequence encoded by exon 4 as the most obvious molecular explanation.
Effect of Glycosylation on XHpaL and XHpaS Secretion—Glycosylation of heparanase is necessary for secretion (42), raising the possibility that inclusion and exclusion of exon 4, respectively, may be a biological mechanism to generate a secreted, glycosylated long isoform, and a nonsecreted, minimally glycosylated short heparanase isoform. Indeed, computational analysis searching for potential post-translational modifications of the amino acid sequence showed that 3 of the 5 potential N-glycosylation sites present in XHpaL are in the sequence encoded by exon 4 (Fig. 1). To test this, we first investigated if Xenopus heparanase can be glycosylated. Glycosylation of both heparanase isoforms was demonstrated in COS-7 cells by transiently transfecting with XHpaL and XhpaS and treating with tunicamycin A (TM), an inhibitor of glycosylation. A dose-dependent electrophoretic mobility gel shift was detected with TM treatment for both heparanases, suggesting that both forms are glycosylated (Fig. 3A). Although 0.1 μg of TM partially inhibited N-glycosylation and generated a protein with faster mobility, concentrations of 1 and 10 μg/ml completely inhibited glycosylation.
FIGURE 3.
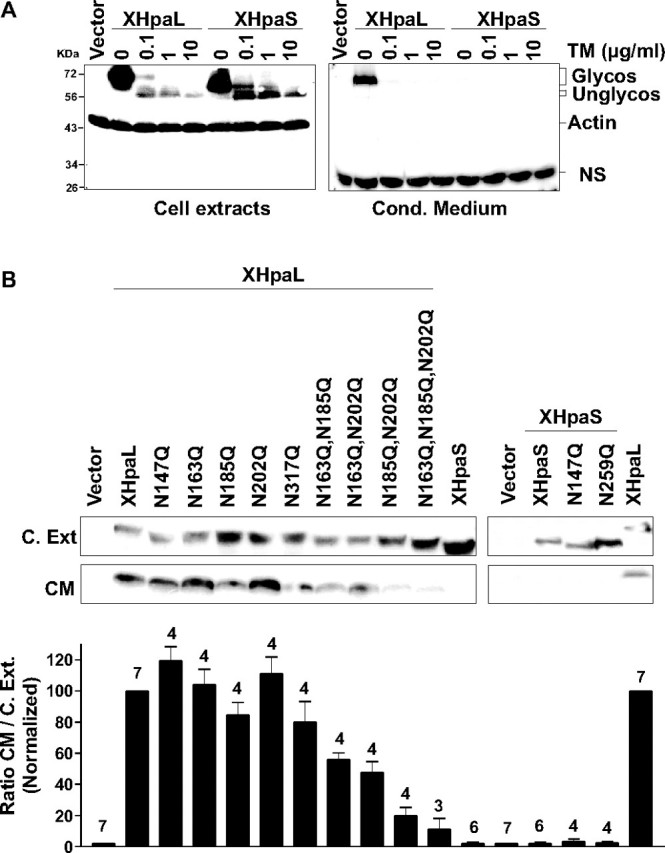
Effect of glycosylation on heparanase secretion. A, inhibition of glycosylation and secretion by TM treatment. COS-7 cells transiently transfected with myc-tagged XHpaL or XHpaS were treated with various concentrations of TM for 18 h. Cell extracts and conditioned medium were immunoblotted with anti-myc and anti-β-actin antibodies. A nonspecific band (NS) detected in the conditioned medium with anti-myc antibodies is also shown. B, glycosylation and secretion of specific mutated forms (asparagines (N) to glutamine (Q)) of heparanase. COS-7 cells were transiently transfected with myc-tagged heparanase constructs that encode wild type XHpaL or XHpaS, or with one (147, 163, 185, 202, and 317; N249Q on XHpaS is equivalent to N317Q on XHpaL), two, or three asparagines substituted. Cell extract lysates and conditioned medium were analyzed as in A. Densitometric quantification expressed as the ratio between protein levels in conditioned medium versus cell extract normalized to XHpaL (100%) is shown at the bottom. Error bars represent the S.D. and the number of determinations (n) is indicated above each bar.
A small difference in the molecular weight (electrophoresis mobility) of XHpaL and XHpaS in their unglycosylated forms (10 μg of TM) was observed, consistent with the differences detected between the two heparanases lacking the signal peptide (Fig. 2D, lanes 6 and 7) and the molecular mass deduced from their amino acid sequence (unglycosylated forms: 59 and 52.5 kDa for XHpaL and XHpaS, respectively). The greater difference in the molecular weights of the glycosylated forms, however, further indicated that post-translational modifications, such as glycosylation, may occur in the region removed by splicing in XHpaS.
To analyze the contribution of each potential glycosylation site to heparanase secretion, we generated heparanase constructs in which each putative glycosylated asparagine (consensus sequence Asn-Xaa-Ser/Thr, where Xaa represents any amino acid except Pro) was substituted by glutamine. The amount of heparanase expression in cell extracts was compared with the level of heparanase in the conditioned medium by Western blot (Fig. 3B). Electrophoresis mobilities of the five single mutant forms, XHpaL-N147Q, XHpaL-N163Q, XHpaL-N185Q, XHpaL-N202Q, and XHpaL-N317Q, were faster than the wild type XHpaL. Also, the short splicing variants in which the two remaining potential N-glycosylated asparagines were replaced by glutamine (XHpaS-N149Q and XHpaS-N259Q) showed a downshift in their electrophoretic mobility, indicating a reduction in their molecular weight. These results, together with the tunicamycin data, suggest that in both variants all the asparagines are N-glycosylated. Densitometry analysis showed that the ratios of secretion (conditioned medium)/expression (cell extract) for all single mutant forms were not different from that of wild type XHpaL, indicating that modification of only one asparagine does not affect secretion. In contrast, mutation of any two asparagines in this region (XHpaL: N147W,N163Q; N163Q,N185Q, and N185Q,N202Q) reduced secretion by more than 50% (Fig. 3B). The maximum effect was detected in the XHpaL-N185Q,N202Q double mutant, which was secreted almost 80% less than the wild type. Finally, the secretion of the XHpaL was almost completely abrogated when the third asparagine present in the sequence encoded by exon 4 was substituted (N163Q,N185Q,N202Q), which strongly argues that glycosylation of these sites is required for normal secretion of XHpaL. All together, these results indicate that both splice variant products are glycosylated, but that exon 4, and its three glycosylation sites, accounts largely for the differential secretion of XHpaL and XHpaS.
Cellular Localization of XHpaL and XHpaS—Differential secretion of the two splicing variants could be associated with differences in intracellular trafficking. To investigate this possibility, we carried out co-localization studies in transiently transfected COS-7 and C6 cells. Specific markers for the endoplasmic reticulum (ER) (calnexin) and Golgi apparatus (golgin 97) were used concomitant with anti-myc antibodies to detect heparanase. XHpaL and XHpaS proteins showed a reticular staining outside of the nucleus, whereas constructs lacking the first 21 amino acids containing the signal peptide, XHpaL no SP (Fig. 4A) and XHpaS no SP (data not shown), exhibited diffuse staining, characteristic of cytosolic proteins. Moreover, both heparanase isoforms co-localize with calnexin (Fig. 4B) and golgin 97 (data not shown). All together, these results suggest that both heparanases are transported through the ER and Golgi apparatus, ruling out the possibility that lack of secretion of XHpaS results from its failure to be trafficked through the ER-Golgi.
FIGURE 4.
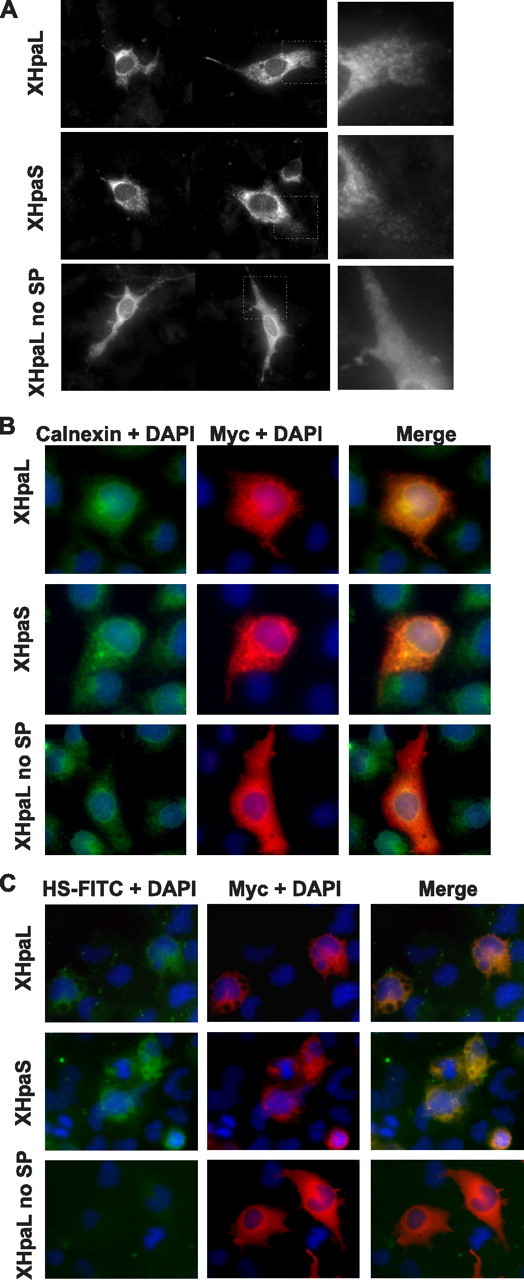
Cellular localization of XHpaL and XHpaS. A, glioma C6 cells were transiently transfected with myc-tagged XHpaL, XHpaS, or XHpaL without signal peptide (XHpaL no SP). After 24 h, cells were fixed and stained with anti-myc antibodies. Note the reticular and pycnotic staining in XHpaL- and XHpaS-transfected cells (higher magnification on the right panel), whereas cell staining is diffuse for XHpaL no SP. B, COS-7 cells transiently transfected were stained with antibody against the myc epitope (red) and calnexin (green). 4′,6-Diamidino-2-phenylindole (DAPI) (blue) was used to detect the nucleus. C, COS-7 cells were transfected as in A. After 24 h, cells were incubated with FITC-HS for 2 h. HS is detected as green fluorescence, and heparanase expression was detected using anti-myc antibodies (red). Co-localization of heparanase and calnexin (B) and heparanase and HS (C) are detected in the merged images as yellow.
Finally, we examined cell surface expression of the two heparanase isoforms by analyzing the interaction of FITC-HS with COS-7 cells transiently transfected with constructs containing myc-tagged XHpaL, XHpaS, or XHpaL no SP. After 24 h the cells were incubated with FITC-HS (50 μg/ml) for 2 h, the cultures fixed, and anti-myc used to detect heparanase expression. Cells overexpressing XHpaL and XHpaS showed strong labeling with FITC-HS, whereas staining of untransfected cells or cells overexpressing heparanase without a signal peptide was considerably less intense (Fig. 4C). Similar results were obtained with cells incubated at 4 °C, to prevent internalization, and exposed to FITC-HS (data not shown). These data argue that whereas XHpaS is not secreted into the media, it is present on the outer cell surface and can interact with HS.
XHpaS but Not XHpaL Increases Cell Adhesion to HS—Because XHpaL and XHpaS exhibit differential secretion and enzymatic properties, we were interested in determining if they have distinct biological functions. Previously, it was shown that an engineered chimeric heparanase, which lacked enzymatic activity, induced cell adhesion of glioma cells to the ECM (30, 43). This led us to ask if the presence of a naturally occurring enzymatically inactive heparanase, which localizes to the cell membrane and retains HS/heparin binding capacity, regulates cell adhesion. To test this idea, we generated stable glioma C6 cell lines expressing comparable levels of myc-tagged forms of XHpaL and XHpaS (Fig. 5A).
FIGURE 5.

Effect of XHpaL and XHpaS on adhesion of glioma C6 cells to a HS substrate. A, Western blot of C6-stable cell lines. Cell extract obtained from C6 cells stably expressing the mock control vector (C6-vector), the long (C6-XHpaL), or the short (C6-XHpaS) myc-tagged heparanase were blotted using anti-myc antibodies. B, phase-contrast photomicrography of stable cell lines. C, cell adhesion kinetics. Cell suspensions of stable cell lines were seeded on dishes previously coated with (filled symbols) or without (open symbols) HS(50 μg/ml). At various times, the extent of adhesion was determined and expressed as percentage of the total cells seeded. Curves were fit to a one-phase exponential association (zero to top). *, p < 0.05; analysis of variance. D, stable cell lines were transfected with sense (S) or antisense oligonucleotides against exon 3 (AS) or exon 4 (AS4). After 24 h, cell extracts were analyzed by Western blot using anti-β-actin and anti-myc antibodies to detect heparanase expression. Densitometry analysis between heparanase and β-actin expression normalized against sense-transfected cells is shown at the bottom. A representative example of two independent experiments is shown. E, C6-XHpaS cells were transfected with sense and antisense oligonucleotides as explained for B. After 48 h, cell suspensions were obtained and the extent of adhesion was determined following 20 min incubation. For C and E, one each of three independent experiments are shown (*, p < 0.05; analysis of variance).
The parental C6 cell line, the mock cell line transfected with the vector (C6-vector), as well the heparanase-expressing C6-XHpaL and C6-XHpaS cell lines, all exhibited similar cellular morphologies (Fig. 5B). Interestingly, however, we noticed that in general the C6-XHpaS clones required longer exposures to trypsin during routine subculture than did the C6-XHpaL and C6-vector clones, suggesting that they were more firmly attached to the tissue culture plastic or the ECM generated by the cells. As such, we analyzed the time course of adhesion of stable heparanase-expressing clones to the culture dish immediately after plating. Overexpression of neither heparanase substantially modified the adhesion kinetics, because C6-XHpaL and C6-HpaS cell lines showed similar adhesion slopes as the C6-vector cell line. About 50% of cells attached to the dishes at 14.4 ± 3.4, 24.6 ± 8.4, and 25.8 ± 4.8 min for C6-vector, C6-XHpaL, and C6-XHpaS, respectively (n = 3 independent experiments; p > 0.05) (Fig. 5C). We also assessed cell adhesion to HS. Although C6-vector and C6-XHpaL cell lines showed similar adhesion kinetics to wells with or without HS (C6-vector, 16.8 ± 3.4 versus 14.4 ± 3.4 min; C6-XHpaL, 21.7 ± 6.6 versus 24.7 ± 8.4 min; n = 3), clones expressing the short splicing variant adhered more rapidly to HS-coated dishes. Adhesion of 50% of the C6-XHpaS cells was obtained in ∼14 min less time on dishes with HS than those without (11.7 ± 1.9 versus 25.8 ± 4.8 min, respectively; n = 3; p < 0.05, analysis of variance) (Fig. 5C).
To confirm that XHpaS plays a causal role in HS-mediated cell adhesion, we specifically knocked down XHpaL and XHpaS isoforms with AS oligonucleotides in the stable heparanase-expressing cell lines, and measured the rate of adhesion of the cells to HS. Transfection of stables cell lines with an AS against the region corresponding to exon 3 reduced by about 70% the expression of XHpaS and XHpaL after 24 h, whereas a specific AS oligonucleotide against exon 4 (AS4), significantly reduced XHpaL expression (80% less expression of XHpaL than in sense-transfected cells) without affecting XHpaS expression (Fig. 5D). The accelerated adhesion seen with C6-XHpaS cells on HS was abolished when the cells were transfected with AS oligonucleotides against exon 3, but not with either sense or AS4 oligonucleotides (Fig. 5E).
Taken together, these results demonstrate that the short splice form of heparanase, even though enzymatically inactive, is expressed on the cell surface and increases cell adhesion to HS. In contrast, the long variant is secreted and has no effect on adhesion to HS.
Heparanase Is Required for Normal Xenopus Embryonic Development—An important question that arises is whether the two heparanase variants, with distinct functional properties, control the same or distinct biological processes. To answer this question, we differentially modified the expression levels of the isoforms in the early Xenopus embryo. In support of a role for heparanases in early embryonic development, expression of the only heparanase cloned in mouse is detected in morula and blastula embryos (32), and chicken heparanase is found in cells migrating from the epiblast layer and later on in tissues involved in vascular and nervous system organogenesis (21). To guide these experiments, we first determined whether XHpaL and XHpaS are expressed in early Xenopus embryos by RT-PCR. Because XHpaL and XHpaS differ in one exon, forward and reverse primers, upstream and downstream of exon 4, respectively, were used to recognize both splice variants in the same PCR. The elongation factor EF1α was used to ensure that similar amounts of cDNA were added to each reaction: EF1α levels initially increase but remain relatively stable after stage 12 (36). At stages 4 and 12, xHpaL cDNA is 3-5 times more abundant than xHpaS cDNA, which is only weakly expressed (Fig. 6A). Interestingly, the ratio of XHpaL to XHpaS cDNA is dynamic through to at least tadpole stages. For example, whereas XHpaL cDNA is more abundant than XHpaS cDNA at early stages (4/12), the ratio of XHpaL/XHpaS approaches one at mid-neurula and tailbud stages (18/23), due both to a reduction in XHpaL levels and an increase in the levels of the short splice variant. At tadpole stages (35/36 to 37/38), a strong up-regulation in XHpaL and XHpaS cDNA expression was observed but the ratio XHpaL/XHpaS remains stable (Fig. 6A).
FIGURE 6.
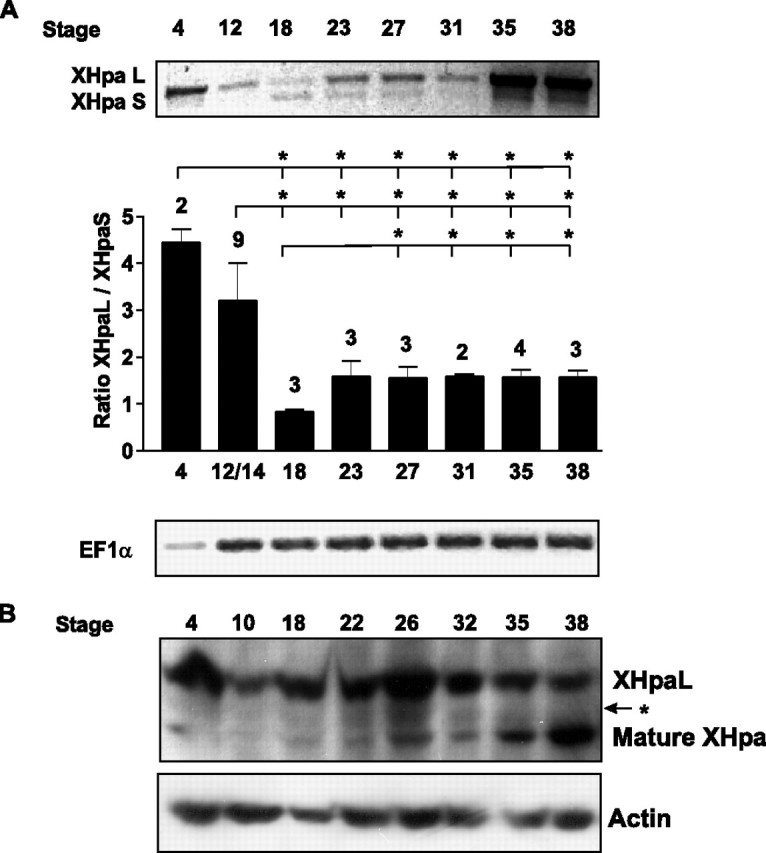
Differential expression of XHpaL and XHpaS during development. A, RT-PCR products for XHpaL, XHpaS, and EF1α generated from whole embryos at different stages of development. RT-PCR of one representative experiment is shown. Densitometric quantification expressed as the ratio between xHpaL and xHpaS and the number of determinations (n) indicated above each bar is also shown. Error bars represent the S.D. *, p < 0.05; analysis of variance. B, Western blot analysis of extracts from whole embryos (150 μg) at different stages of development using the anti-human heparanase antibody (clone HP3/17).β-Actin detection was used as the equal loading control.
To verify that the changes observed in mRNA expression reflect changes in XHpaL and XHpaS protein expression, protein levels were assessed by Western blot with the anti-human heparanase monoclonal antibody (clone HP3/17), that recognizes the Xenopus heparanases with low affinity (Fig. 2A). Two main bands were detected in extracts from embryos of different development stages. The upper band, which corresponds to the proheparanase form of the long heparanase splice variant, showed high expression levels at early stages (stage 4), diminished at stage 10 and rose at later stages (stages 18 to 26) (Fig. 6B). This expression pattern is in agreement with the RT-PCR results obtained for XHpaL. A second band of 48 kDa was also detected (lower band), whose intensity was low at early stages (stages 4 to 10) and became more evident after stage 18. An up-regulation of the 48-kDa band at late stages (32 to 37/38), was concomitant with a reduction of XHpaL proheparanase levels, suggesting that it corresponds to the cleaved and mature heparanase form (Fig. 6B). An intermediate band, with a molecular weight similar to the XHpaS variant, was also slightly detected (Fig. 6B, arrow).
The differential expression of XHpaL and XHpaS during development, coupled to their disparate functional properties, suggests a model whereby the variants control distinct events in the embryo. To test this possibility, we overexpressed and knocked down the levels of heparanase variants in the early Xenopus embryo. First, two cell-stage embryos were injected with full-length XHpaL (n = 3 independent experiments; 64 total embryos) and XHpaS (n = 3; 67 total embryos). Overexpression of neither heparanase splicing variant (confirmed by Western blot) had an obvious effect, and embryos developed apparently normally over the stages analyzed (until stage 37/38; Fig. 7A). We next analyzed the consequences of knocking down XHpaL or XHpaS on early embryonic development by injecting embryos at the two-cell stage with the sense, AS, or AS4 oligonucleotides. The functionality of the oligonucleotides in knocking down endogenous heparanase was confirmed by RT-PCR of mRNA collected from stage 12-14 embryos (Fig. 7C, left panel). Injection of sense oligonucleotides (n = 4; 84 embryos), as well as the AS4 (n = 4; 89 embryos), designed to knock down only XHpaL, had no effect on embryo survival (Fig. 7B). In contrast, injection of AS, designed to interfere with both XHpaL and XHpaS, resulted in a significant increase in the numbers of embryos that failed to complete gastrulation, with subsequent embryo death. Approximately 40% of the embryos died shortly after gastrulation (stage 17) and death continued over the next day with almost 75% of the embryos dying by stage 28 (n = 4, 95 total embryos; p < 0.05, log rank test).
FIGURE 7.

Effect of altering expression of heparanase on embryo survival. A, overexpression of XHpaL and XHpaS. X. laevis embryos were injected in each blastomere after the first cleavage with pCS2-XHpaL or pCS2-XHpaS expression constructs (400 pg/10 nl), or were untreated (C; control). Embryo survival was monitored over time. B and E, knock down of endogenous heparanase and rescue experiments. Embryos were injected as indicated in A with sense (S) or antisense oligonucleotides directed against exon 3 (AS) or exon 4 (AS4) (10 nl/embryo/30 μm solution), either alone (B) or in combination with constructs encoding mutated XHpaL and XHpaS that escape from AS down-regulation (E). C, RT-PCR from embryos collected at stage 12-14. cDNA was generated from normal embryos (C) or embryos injected at the two-cell stage with oligonucleotides (S, AS, and AS4), alone or in combination with mutated heparanase constructs. D, nucleotide sequence targeted by the AS oligonucleotides. Show are the six mutations generated in the XHpaL and XHpaS constructs to prevent knock down by the AS oligonucleotides, whereas conserving the amino acid sequences. A, B, and E, combined data of three (A) or four (B and E) independent experiments normalized against survival of untreated embryos (100%). *, p < 0.05; log rank test.
The embryonic death observed with the AS can be explained if both XHpaL and XHpaS are required in early embryonic development, or alternatively by the lone involvement of XHpaS. To address which of these possibilities is occurring, and to verify that embryo death resulted from the specific knock down of heparanase, we carried out rescue experiments. We first engineered full-length XHpaL and XHpaS rescue mutants that would be immune to the knockdown effects of the AS oligonucleotide, by introducing six point mutations in the region that should anneal with the AS (Fig. 7D). Escape of these mutated heparanases from the knocking down properties of the AS oligonucleotides was confirmed by RT-PCR (Fig. 7C, right panel). To test for rescue of the AS-induced phenotype, embryos were co-injected at the two-cell stage with the AS plus either the XHpaL or XHpaS rescue construct. Surprisingly, embryo survival was restored by either construct (Fig. 7E), with percentages of embryos surviving at stage 35 reaching those seen with injection of the sense oligonucleotide (XHpaL, 77%, n = 4, 86 embryos; XHpaS, 70%, n = 4, 92 embryos; sense, 83%, n = 4; 65 embryos; nonsignificant differences). In contrast, almost 70% of embryos injected with only the AS oligonucleotide eventually died (n = 4, 79 embryos; p < 0.05 versus AS plus XHpaL and AS plus XHpaS; log rank test) (Fig. 7E).
We therefore conclude that although XHpaL and XHpaS are proteins with distinct properties, cellular localization, and temporal patterns of expression during development, either of them is sufficient to rescue embryonic lethality, suggesting that they regulate the same developmental processes through different mechanisms.
DISCUSSION
In this paper we identify the gene encoding Xenopus heparanase and report the cloning and expression of two splice variants, a long variant (XHpaL; exon 1-12) and a short splice variant (XHpaS), where 174 nucleotides (58 aa) from exon 4 are skipped. The two isoforms that result from the XHpaL and XHpaS splice variants show specific biochemical and cellular properties. Notably, XHpaL can degrade HS and is secreted into the conditioned medium, whereas XHpaS lacks enzymatic activity and is retained on the external cell surface rather than being secreted by the cell. However, both splice variant products interact with heparin and HS. The presence of two heparanase splice variants, one secreted and the other not, is evolutionary conserved, being present in Xenopus, humans, and the blind subterranean mole rat (20, 23, 34, 35), which argues that both isoforms are important functionally. In this regard, we show for the first time that a native inactive splice variant increases cell adhesion, and that both variants are necessary in early embryonic development, but likely by different means.
Xenopus Heparanase Gene and Biochemical Differences between XHpaL and XHpaS—The 12 exons of Xenopus heparanase span over 15 kb, whereas the human heparanase gene consists of 14 exons that span 50 kb and include the 5′- and 3′-untranslated regions, corresponding to exons 1 and 14, respectively (44). The Xenopus heparanase sequence that was amplified by PCR would not have contained the 5′- and 3′-untranslated regions, so the Xenopus heparanase gene may also consist of 14 exons. In support, the Xenopus cDNA sequence (xHpa, exons 1 to 12) aligns with exons 2 to 13 of human heparanase, and the exon-intron organization predicted from the X. tropicalis genome indicates the presence of two additional putative exons that correspond to the 5′- and 3′-untranslated regions of xHpa mRNA. XHpaL resembles the only heparanase to be described in chicken, in that it is enzymatically active and readily secreted (21). In contrast, the amino acid sequence and biochemical characteristics of XHpaS indicate that it is the Xenopus homologue of a recently reported human splicing variant, which results from a skipped exon 5 and similar to the Xenopus form is inactive and not secreted (35).
The latent 65-kDa form of heparanase, which does not show significant enzymatic activity, is cleaved to generate an active 50-kDa enzyme (45). By Western blot, we observed no cleavage of either short or long heparanase when overexpressed in a mammalian system. However, a 48-kDa band that likely corresponds to the mature heparanase form was detected in Xenopus embryos. The lack of cleavage of myc-tagged heparanase constructs is not surprising as COOH terminus myc and GFP-tagged human heparanase are also uncleaved (42, 46). The fact that processing of the non-myc-tagged constructs was not detected, however, is unexpected and is either due to a low sensitivity of the human anti-heparanase antibody for the Xenopus protein, or to a species-specific processing requirement. Interestingly, despite no detectable processing of the heparanase variants, heparanase activity was detected in cell extracts from XHpaL but not XHpaS-overexpressing cells, which is in agreement with the recently reported human heparanase variants (35), and suggests the pro-heparanase retains some limited ability to process HS substrates. The lack of glycosidase activity of the Xenopus and human short heparanases may be related to the absence of a proton donor at Glu210, which is critical for heparanase enzymatic properties (47). Despite differences in their ability to be secreted, both Xenopus isoforms were associated with the reticular staining often indicative of the ER-Golgi apparatus. Intriguingly, we found that whereas XHpaS is not secreted it is found on the cell surface and is able to interact with both heparin and HS, properties that are yet to be ascribed to the human splicing variant. Indeed, the full-length and short human heparanase variants appear to be differentially localized within the cell. The full-length human heparanase showed a reticular staining that was predominantly perinuclear and associated with lysosomes and the Golgi apparatus (46), whereas the short variant was described as having a more diffuse cytosolic staining (35). These differences in subcellular localization might suggest different functions of the short isoforms in Xenopus and human.
Mechanisms to Regulate Heparanase Secretion—Although unique signal peptides appear to explain the differential secretion of chicken and the long human heparanase proteins (21), an alternate mechanism likely accounts for the differential secretion of the two Xenopus splice variants. XHpaL and XHpaS share the same signal peptide, which shows no sequence homology with either the chicken or human peptides. Moreover, changing the Xenopus heparanase signal peptide for the human immunoglobulin κ signal peptide did not modify the cellular localization and secretion properties of either splice variant. Instead, glycosylation appears to be the main mechanism regulating differential secretion between the Xenopus splice variants. Indeed, XHpaL secretion was almost completely inhibited by either tunicamycin A treatment or mutation of the three glycosylation sites present in the sequence encoded by exon 4. These results argue that glycosylation of residues encoded by exon 4 is required for secretion, and the absence of this exon in XHpaS results in a non-secreted heparanase isoform. Glycosylation sites outside of exon 4 and/or alternative regulatory mechanisms are also likely involved, however, because substitution of all the asparagines in the sequence encoded by exon 4 still resulted in a protein that could be secreted, although at 10% of wild type levels. Of note, although the role of glycosylation in differential secretion of the human heparanase splice variants remains to be determined, the human short heparanase also loses three glycosylation sites by skipping exon 5. Interestingly, whereas the single asparagine mutations only slightly affected the secretion of XHpaL, one asparagine substitution in exon 5 dramatically inhibited the secretion of the human long isoform (e.g. Asp178 replacement -163 in Xenopus-inhibited secretion by more than 90%) (42). Finally, mole rats appear to have evolved a third mechanism by which heparanase secretion can be regulated. Here, a splicing variant that is missing 16 aa (skipping of exon 7), possesses the same signal peptide and glycosylation sites as the longer heparanase splice isoform, but is not secreted (34).
Differential Functions of Heparanase Splice Isoforms—Although XHpaS is not secreted, it is still trafficked through the ER and Golgi, and makes it to the cell surface where it can interact with externally applied HS. Presumably, this interaction affects the adhesive properties of cells expressing XHpaS to substrates containing HS. Yet, whereas cells overexpressing the secreted XHpaL were also labeled by FITC-HS, they showed comparable adhesion kinetics to control cells. Cellular labeling with FITC-HS could occur if FITC-HS interacted either with XHpaL at the cell membrane during the process of the long form being secreted, or during XHpaL internalization. Indeed, addition of exogenous recombinant pro-heparanase to human fibroblasts results in the cell surface binding of this precursor, followed by its internalization and proteolytic activation (48). Because after 24 h all COS-7 cells would be exposed to high levels of secreted XHpaL in the media, one would have expected to see all cells labeled with FITC-HS if an interaction during internalization was the explanation. Yet, we found only cells overexpressing XHpaL were labeled, which suggests that in the process of XHpaL secretion the enzyme interacts with the bath-applied FITC-HS. Whether this occurs biologically, or is a consequence of overexpression of the protein is unclear.
The fact that XHpaS has no heparanase activity, and yet can interact both with heparin and HS, argues that it has a different function than XHpaL, and one that is independent of enzymatic activity. Such roles for heparanase and descriptions of inactive splice variants have recently come to light (29-31, 34, 35, 43), although their importance in normal physiological events in the organism is unknown. Our data in Xenopus start to address this issue. We found that enzymatic and non-enzymatic, possibly adhesive, functions of heparanase are important in embryonic development in that either heparanase isoform was able to rescue the embryonic lethality of knockdown of both heparanase splicing variants. Furthermore, our results suggest that the distinct functions are separated into two independent molecules, with XHpaL retaining enzyme activity and XHpaS capable of promoting adhesion to HS: cells expressing the short, cell-associated variant increased adhesion to HS-coated dishes, whereas cells stably expressing the long splicing variant failed to do so. As far as we know, this is the first demonstration that a native, nonenzymatic, heparanase controls cell adhesion, and confirms previous reports of the adhesion promoting capacity of an artificially engineered human inactive enzyme (30) and a recombinant human proheparanase (31). In agreement with our data, cells expressing the surface-associated heparanases showed a higher degree of adhesion than those expressing noncell surface-associated human heparanase (30, 45). Moreover, highly invasive and migratory cells expressed the secreted heparanase (26). The exact mechanism by which XHpaS induces rapid cell adhesion to HS or ECM is still unclear. The fact that specific signal transduction pathways are activated during heparanase-mediated cell adhesion, including phosphorylation of Akt and ERK and Pyk-2 activation (31, 43, 49), argues against the explanation that heparanase simply forms a bridge between the HS cell surface substratum and the cells. Future studies will be required to understand how XHpaS mediates HS-cell adhesion, and its biological significance.
Overexpression of either the short or long heparanase resulted in embryos with no gross defects. These results are in agreement with those obtained with homozygous transgenic mice that overexpress the long human heparanase (32, 50-52). These animals appeared normal and exhibited a normal life span, despite decreased food consumption and body weight. Interestingly, the pathologies associated with heparanase overexpression were only detected in adults and included kidney malfunction and an accelerated rate of wound healing and hair growth, suggesting a tissue-specific role of heparanase in adults.
Our data indicate that XHpaL and XHpaS are proteins with distinct properties, cellular localization, and temporal patterns of expression during development. As such, one might expect that the proteins encoded by the two splice variants would have unique, non-overlapping roles in the organism. Instead, our data in the developing Xenopus embryo support a model whereby the two isoforms regulate the same developmental processes, but do so by different functional mechanisms. Failure of embryos to proceed through development, starting at gastrulation, occurred with knock down of both XHpaL and XHpaS isoforms, but not when only the XHpaL variant was down-regulated (AS4). The rescue experiments are particularly informative in differentiating between whether XHpaS alone or XHpaS and XHpaL is required for normal embryonic development. The capacity of either XHpaL or XHpaS rescue constructs to abrogate the embryonic death resulting from knockdown of both variants with AS indicates at first glance that the two isoforms can replace each other's function in early embryonic development. Given the fact that the two heparanases showed different characteristics and properties, however, this seems unlikely. Indeed, overexpression of XHpaL and XHpaS in C6 cells resulted in different effects on cell adhesion. Rather, we propose that the two isoforms regulate the same process, which is critical for embryonic development. Cleavage of HSPGs could regulate the availability of a factor required for early events in the embryo, as is recognized for the developmentally important fibroblast growth factors (13, 15, 53, 54). The levels of the factor could be in part determined by the enzyme activity of XHpaL, which would aid its release from HSPGs, and alternatively by non-enzymatic mechanisms controlled by XHpaS. For instance, XHpaS could bind and retain the factor near its site of cleavage and/or bring XHpaL-secreting cells into close proximity to HSPGs in the ECM or cell membranes. Presumably, if XHpaL is knocked down, but not eliminated, enough of the factor is available to allow embryonic development to proceed normally. However, in the absence of both heparanase functions, levels of the factor fall below a critical threshold and embryonic development arrests. Providing back excess of either heparanase function, proximity to HSPGs and/or retention of the factor by XHpaS overexpression, or enzymatic activity with XHpaL overexpression, would make available sufficient levels of the factor to permit normal development. If true, temporal alterations in the relative levels of XHpaL and XHpaS may translate into careful control of HSPG cleavage and the levels of developmentally important regulators. Future studies will need to unravel which developmental events the two splice variants regulate and how.
Acknowledgments
We thank Drs. V. Zaremberg and J. Hocking for comments on the manuscript. We thank C. L. Hehr and J. Johnston for excellent technical assistance.
This work was supported in part by an operating grant from the Natural Sciences and Engineering Research Council of Canada and a University of Calgary Research grant. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: ECM, extracellular matrix; ER, endoplasmic reticulum; HS, heparan sulfate; HSPGs, heparan sulfate proteoglycans; XHpaL, Xenopus heparanase long; XHpaS, Xenopus heparanase short; TM, tunicamycin A; RT, reverse transcriptase; aa, amino acid(s); Me, methyl; FITC, fluorescein isothiocyanate; PBS, phosphate-buffered saline; EST, expressed tag sequence; AS, antisense.
References
- 1.Lindahl, U., Kusche-Gullberg, M., and Kjellen, L. (1998) J. Biol. Chem. 273 24979-24982 [DOI] [PubMed] [Google Scholar]
- 2.Casu, B., and Lindahl, U. (2001) Adv. Carbohydr. Chem. Biochem. 57 159-206 [DOI] [PubMed] [Google Scholar]
- 3.Kreuger, J., Spillmann, D., Li, J. P., and Lindahl, U. (2006) J. Cell Biol. 174 323-327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rapraeger, A. C. (2001) Semin. Cell Dev. Biol. 12 107-116 [DOI] [PubMed] [Google Scholar]
- 5.Allen, B. L., and Rapraeger, A. C. (2003) J. Cell Biol. 163 637-648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hacker, U., Nybakken, K., and Perrimon, N. (2005) Nat. Rev. Mol. Cell Biol. 6 530-541 [DOI] [PubMed] [Google Scholar]
- 7.Bellaiche, Y., The, I., and Perrimon, N. (1998) Nature 394 85-88 [DOI] [PubMed] [Google Scholar]
- 8.Binari, R. C., Staveley, B. E., Johnson, W. A., Godavarti, R., Sasisekharan, R., and Manoukian, A. S. (1997) Development 124 2623-2632 [DOI] [PubMed] [Google Scholar]
- 9.Walz, A., McFarlane, S., Brickman, Y. G., Nurcombe, V., Bartlett, P. F., and Holt, C. E. (1997) Development 124 2421-2430 [DOI] [PubMed] [Google Scholar]
- 10.Yamane, Y., Tohno-oka, R., Yamada, S., Furuya, S., Shiokawa, K., Hirabayashi, Y., Sugino, H., and Sugahara, K. (1998) J. Biol. Chem. 273 7375-7381 [DOI] [PubMed] [Google Scholar]
- 11.Galli, A., Roure, A., Zeller, R., and Dono, R. (2003) Development 130 4919-4929 [DOI] [PubMed] [Google Scholar]
- 12.Ohkawara, B., Yamamoto, T. S., Tada, M., and Ueno, N. (2003) Development 130 2129-2138 [DOI] [PubMed] [Google Scholar]
- 13.Vlodavsky, I., Bar-Shavit, R., Ishai-Michaeli, R., Bashkin, P., and Fuks, Z. (1991) Trends Biochem. Sci. 16 268-271 [DOI] [PubMed] [Google Scholar]
- 14.Johnson, K. G., Ghose, A., Epstein, E., Lincecum, J., O'Connor, M. B., and Van Vactor, D. (2004) Curr. Biol. 14 499-504 [DOI] [PubMed] [Google Scholar]
- 15.Aviezer, D., Levy, E., Safran, M., Svahn, C., Buddecke, E., Schmidt, A., David, G., Vlodavsky, I., and Yayon, A. (1994) J. Biol. Chem. 269 114-121 [PubMed] [Google Scholar]
- 16.Levy-Adam, F., Miao, H. Q., Heinrikson, R. L., Vlodavsky, I., and Ilan, N. (2003) Biochem. Biophys. Res. Commun. 308 885-891 [DOI] [PubMed] [Google Scholar]
- 17.McKenzie, E., Young, K., Hircock, M., Bennett, J., Bhaman, M., Felix, R., Turner, P., Stamps, A., McMillan, D., Saville, G., Ng, S., Mason, S., Snell, D., Schofield, D., Gong, H., Townsend, R., Gallagher, J., Page, M., Parekh, R., and Stubberfield, C. (2003) Biochem. J. 373 423-435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kizaki, K., Yamada, O., Nakano, H., Takahashi, T., Yamauchi, N., Imai, K., and Hashizume, K. (2003) Placenta 24 424-430 [DOI] [PubMed] [Google Scholar]
- 19.Miao, H. Q., Navarro, E., Patel, S., Sargent, D., Koo, H., Wan, H., Plata, A., Zhou, Q., Ludwig, D., Bohlen, P., and Kussie, P. (2002) Protein Expression Purif. 26 425-431 [DOI] [PubMed] [Google Scholar]
- 20.Hulett, M. D., Freeman, C., Hamdorf, B. J., Baker, R. T., Harris, M. J., and Parish, C. R. (1999) Nat. Med. 5 803-809 [DOI] [PubMed] [Google Scholar]
- 21.Goldshmidt, O., Zcharia, E., Aingorn, H., Guatta-Rangini, Z., Atzmon, R., Michal, I., Pecker, I., Mitrani, E., and Vlodavsky, I. (2001) J. Biol. Chem. 276 29178-29187 [DOI] [PubMed] [Google Scholar]
- 22.Toyoshima, M., and Nakajima, M. (1999) J. Biol. Chem. 274 24153-24160 [DOI] [PubMed] [Google Scholar]
- 23.Vlodavsky, I., Friedmann, Y., Elkin, M., Aingorn, H., Atzmon, R., Ishai-Michaeli, R., Bitan, M., Pappo, O., Peretz, T., Michal, I., Spector, L., and Pecker, I. (1999) Nat. Med. 5 793-802 [DOI] [PubMed] [Google Scholar]
- 24.Vreys, V., and David, G. (2007) J. Cell Mol. Med. 11 427-452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vlodavsky, I., Goldshmidt, O., Zcharia, E., Atzmon, R., Rangini-Guatta, Z., Elkin, M., Peretz, T., and Friedmann, Y. (2002) Semin. Cancer Biol. 12 121-129 [DOI] [PubMed] [Google Scholar]
- 26.Goldshmidt, O., Zcharia, E., Abramovitch, R., Metzger, S., Aingorn, H., Friedmann, Y., Schirrmacher, V., Mitrani, E., and Vlodavsky, I. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 10031-10036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parish, C. R., Hindmarsh, E. J., Bartlett, M. R., Staykova, M. A., Cowden, W. B., and Willenborg, D. O. (1998) Immunol. Cell Biol. 76 104-113 [DOI] [PubMed] [Google Scholar]
- 28.Bartlett, M. R., Underwood, P. A., and Parish, C. R. (1995) Immunol. Cell Biol. 73 113-124 [DOI] [PubMed] [Google Scholar]
- 29.Gilat, D., Hershkoviz, R., Goldkorn, I., Cahalon, L., Korner, G., Vlodavsky, I., and Lider, O. (1995) J. Exp. Med. 181 1929-1934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goldshmidt, O., Zcharia, E., Cohen, M., Aingorn, H., Cohen, I., Nadav, L., Katz, B. Z., Geiger, B., and Vlodavsky, I. (2003) FASEB J. 17 1015-1025 [DOI] [PubMed] [Google Scholar]
- 31.Sotnikov, I., Hershkoviz, R., Grabovsky, V., Ilan, N., Cahalon, L., Vlodavsky, I., Alon, R., and Lider, O. (2004) J. Immunol. 172 5185-5193 [DOI] [PubMed] [Google Scholar]
- 32.Revel, A., Helman, A., Koler, M., Shushan, A., Goldshmidt, O., Zcharia, E., Aingorn, H., and Vlodavsky, I. (2005) Fertil. Steril. 83 580-586 [DOI] [PubMed] [Google Scholar]
- 33.Goldshmidt, O., Yeikilis, R., Mawasi, N., Paizi, M., Gan, N., Ilan, N., Pappo, O., Vlodavsky, I., and Spira, G. (2004) J. Pathol. 203 594-602 [DOI] [PubMed] [Google Scholar]
- 34.Nasser, N. J., Nevo, E., Shafat, I., Ilan, N., Vlodavsky, I., and Avivi, A. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 15161-15166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nasser, N. J., Avivi, A., Shushy, M., Vlodavsky, I., and Nevo, E. (2007) Biochem. Biophys. Res. Commun. 354 33-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sindelka, R., Ferjentsik, Z., and Jonak, J. (2006) Dev. Dyn. 235 754-758 [DOI] [PubMed] [Google Scholar]
- 37.Lennox, K. A., Sabel, J. L., Johnson, M. J., Moreira, B. G., Fletcher, C. A., Rose, S. D., Behlke, M. A., Laikhter, A. L., Walder, J. A., and Dagle, J. M. (2006) Oligonucleotides 16 26-42 [DOI] [PubMed] [Google Scholar]
- 38.Rozenberg, G. I., Espada, J., de Cidre, L. L., Eijan, A. M., Calvo, J. C., and Bertolesi, G. E. (2001) Electrophoresis 22 3-11 [DOI] [PubMed] [Google Scholar]
- 39.Levy-Adam, F., Abboud-Jarrous, G., Guerrini, M., Beccati, D., Vlodavsky, I., and Ilan, N. (2005) J. Biol. Chem. 280 20457-20466 [DOI] [PubMed] [Google Scholar]
- 40.Gimlich, R. L., and Gerhart, J. C. (1984) Dev. Biol. 104 117-130 [DOI] [PubMed] [Google Scholar]
- 41.Shteper, P. J., Zcharia, E., Ashhab, Y., Peretz, T., Vlodavsky, I., and Ben-Yehuda, D. (2003) Oncogene 22 7737-7749 [DOI] [PubMed] [Google Scholar]
- 42.Simizu, S., Ishida, K., Wierzba, M. K., and Osada, H. (2004) J. Biol. Chem. 279 2697-2703 [DOI] [PubMed] [Google Scholar]
- 43.Zetser, A., Bashenko, Y., Miao, H. Q., Vlodavsky, I., and Ilan, N. (2003) Cancer Res. 63 7733-7741 [PubMed] [Google Scholar]
- 44.Dong, J., Kukula, A. K., Toyoshima, M., and Nakajima, M. (2000) Gene (Amst.) 253 171-178 [DOI] [PubMed] [Google Scholar]
- 45.Abboud-Jarrous, G., Rangini-Guetta, Z., Aingorn, H., Atzmon, R., Elgavish, S., Peretz, T., and Vlodavsky, I. (2005) J. Biol. Chem. 280 13568-13575 [DOI] [PubMed] [Google Scholar]
- 46.Goldshmidt, O., Nadav, L., Aingorn, H., Irit, C., Feinstein, N., Ilan, N., Zamir, E., Geiger, B., Vlodavsky, I., and Katz, B. Z. (2002) Exp. Cell Res. 281 50-62 [DOI] [PubMed] [Google Scholar]
- 47.Hulett, M. D., Hornby, J. R., Ohms, S. J., Zuegg, J., Freeman, C., Gready, J. E., and Parish, C. R. (2000) Biochemistry 39 15659-15667 [DOI] [PubMed] [Google Scholar]
- 48.Nadav, L., Eldor, A., Yacoby-Zeevi, O., Zamir, E., Pecker, I., Ilan, N., Geiger, B., Vlodavsky, I., and Katz, B. Z. (2002) J. Cell Sci. 115 2179-2187 [DOI] [PubMed] [Google Scholar]
- 49.Ben-Zaken, O., Gingis-Velitski, S., Vlodavsky, I., and Ilan, N. (2007) Biochem. Biophys. Res. Commun. 361 829-834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zcharia, E., Zilka, R., Yaar, A., Yacoby-Zeevi, O., Zetser, A., Metzger, S., Sarid, R., Naggi, A., Casu, B., Ilan, N., Vlodavsky, I., and Abramovitch, R. (2005) FASEB J. 19 211-221 [DOI] [PubMed] [Google Scholar]
- 51.Zcharia, E., Philp, D., Edovitsky, E., Aingorn, H., Metzger, S., Kleinman, H. K., Vlodavsky, I., and Elkin, M. (2005) Am. J. Pathol. 166 999-1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zcharia, E., Metzger, S., Chajek-Shaul, T., Aingorn, H., Elkin, M., Friedmann, Y., Weinstein, T., Li, J. P., Lindahl, U., and Vlodavsky, I. (2004) FASEB J. 18 252-263 [DOI] [PubMed] [Google Scholar]
- 53.Xu, X., Rao, G., Quiros, R. M., Kim, A. W., Miao, H. Q., Brunn, G. J., Platt, J. L., Gattuso, P., and Prinz, R. A. (2007) J. Biol. Chem. 282 2363-2373 [DOI] [PubMed] [Google Scholar]
- 54.Moscatelli, D. (1987) J. Cell Physiol. 131 123-130 [DOI] [PubMed] [Google Scholar]