

Abstract
Firefly luciferase utilizes the chemical energy of ATP and oxygen to convert its substrate, D-luciferin, into an excited state oxyluciferin molecule. Relaxation of this molecule to the ground state is responsible for the yellow-green light emission. Synthetic cyclic alkylaminoluciferins are described that allow robust red-shifted light emission with the modified luciferase Ultra-Glo. Overall light emission is higher than that of acyclic alkylaminoluciferins, aminoluciferin, and the native substrate, D-luciferin.
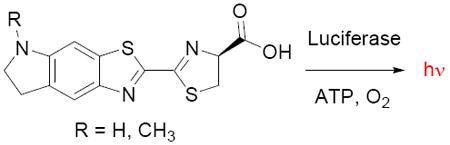
Beetle luciferases chemically generate light by converting D-luciferin into an excited-state molecule of oxyluciferin (Figure 1).1,2 The energy difference between the excited state and ground state determines the wavelength of the bioluminescence.3 Despite the fact that the light emission properties of beetle luciferases are inherently limited by the luciferin structure, there has been little work on the synthesis of modified luciferin substrates since the late sixties.4,5,6 Here we describe conformationally-restricted cyclic alkylaminoluciferin substrates that result in red-shifted and more intense light emission with Ultra-Glo luciferase.7
Figure 1.

Beetle luciferases catalyze the conversion of D-luciferin (R = OH) or 6’-aminoluciferin (R = NH2) into the corresponding oxyluciferin. Light emission occurs from the oxyluciferin excited state.
White and McElroy established that the 6’-hydroxyl of D-luciferin (LH2) can be replaced with an amino group;8 6’-aminoluciferin (6’-NH2LH2) emits light at a longer wavelength and has ~10-fold higher affinity for luciferase than LH2.9 On the other hand, no light is emitted when the 6’-hydroxyl is removed or replaced with 6’-methoxy, ethoxy, or N-acetylamino groups. This is not due to a lack of binding – conversely, these and other bulky modifications are well-tolerated.10 Rather, these modifications prevent light emission.
We hypothesized that replacement of the phenolic hydroxyl of luciferin with more strongly electron-donating alkylamino groups would red-shift the spectral properties of the luciferin chromophore while maintaining high affinity for the luciferase. Furthermore, we hypothesized that restriction of the conformational flexibility of the alkylaminoluciferin by cyclization of the aryl amine would maintain a high quantum yield of bioluminescence. Conversely, free rotation around the aryl-nitrogen bond in acyclic alkylaminoluciferins was predicted to result in a reduced quantum yield due to poor overlap of the nitrogen lone pair with the conjugated system of the luciferin, particularly in the context of the luciferin binding pocket.
We first synthesized acyclic alkylaminoluciferins from 1 (Scheme 1a). Reductive alkylation of 3a followed by nucleophilic substitution with cyanide and condensation with D-cysteine afforded 6’-MeNHLH2 (5a) and 6’-Me2NLH2 (5b). However, attempts to construct cyclic alkylamino modifications from 3a were stymied by the exclusive formation of the undesired 6,7 isomer rather than the 5,6 ring fusion (Scheme 1b).
Scheme 1.

Synthesis of alkylaminoluciferins. a) Reductive alkylation of intermediate 3a afforded acyclic alkylaminoluciferins; b) Attempts to form cyclic alkylaminoluciferins from 3a gave exclusive formation of the undesired 6’,7’-fused isomer (e.g., 8); c) Synthesis of 5’,6’-fused cyclic alkylaminoluciferins 16 required reversal of the order of ring formation. i) HNO3, H2SO4; ii) SnCl2-2H2O, EtOH, reflux; iii) NaBH(OAc)3, 37% HCHO, DCE, HOAc; iv) KCN, DMSO, 130°C; v) D-Cys, aq. MeOH, pH 8; vi) 5 mol% In(OTf)3, acetone, rt; vii) TFAA, TEA, rt; viii) KSCN, Br2, AcOH, rt; ix) t-BuONO2, CuCl, CH3CN; x) NaBH4, EtOH.
Thwarted by the positional reactivity preferences of 3a, we examined approaches to construct the benzothiazole ring as a final step (Scheme 1c). Protection of 9 as a trifluoroacetamide followed by reduction of the nitro group and oxidative ring formation yielded the desired 2-aminobenzothiazole. Diazotization of the 2-amino group and concomitant displacement by chlorine, followed by reductive removal of the trifluoroacetamide protecting group afforded 14a. Introduction of the 2-cyano group and subsequent treatment with D-cysteine afforded the respective luciferin analog 16a (CycLuc1). The N-methylated derivative 16b (CycLuc2) was similarly constructed from 14b.
We next measured the emission wavelength of bioluminescence for our synthetic aminoluciferin substrates. Relative to LH2, 6’-NH2LH2 exhibits red-shifted light emission from wild-type firefly luciferase (593 nm vs. 557 nm). We found that 6’-MeNHLH2 extended this emission shift to 609 nm, while 6’-Me2NLH2 was even more red-shifted to 623 nm. This emission wavelength is identical to the railroad worm beetle luciferase PxhRE, the most red-shifted luciferase yet reported.11 Surprisingly, luciferase emission with the cyclic aminoluciferins was less red-shifted than with their acyclic counterparts (599 nm for CycLuc1 and 607 nm for CycLuc2; SI Table S1).
Measurement of the emission intensity as a function of wavelength revealed a significant reduction in light output for all aminoluciferins relative to D-luciferin. The lower intensity of light emission could arise from either kinetic factors (lower rate of enzymatic turnover or product inhibition), or an inherently lower efficiency of light production (lower quantum yield or failure to produce oxyluciferin), or both. We therefore performed a rapid injection experiment in which the light output was measured during and immediately after mixing of the luciferin and luciferase.12 All of the substrates gave a robust initial burst of light and are thus capable of rapid conversion to a light-emitting oxyluciferin (Figure 2). However, sustained light output is low after the initial burst, consistent with product inhibition.13 Addition of coenzyme A, which has been shown to reduce product inhibition by L-AMP,1 failed to prevent this effect (SI Figure S1). Encouragingly, the initial flash of light from cyclic substrate CycLuc1 substantially exceeded that of all other aminoluciferins (Figure 2).
Figure 2.
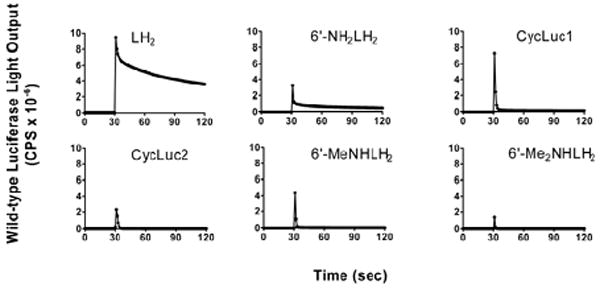
Light emission from 10 μM luciferin substrate after rapid injection of wild-type firefly luciferase at the 30 second time point.
The burst kinetics of firefly luciferase, even with its native substrate D-luciferin, is well-known (Figure 2).1 To provide a steady level of light emission, Promega has developed Ultra-Glo, a highly mutated form of luciferase designed to be more stable and resistant to inhibition during high-throughput screening assays.7,14 When used with P450-Glo buffer, this luciferase has also been shown to allow sustained light emission from substrates that led to product inhibition of wild-type firefly luciferase.5,15
When tested with Ultra-Glo luciferase in P450-Glo buffer at a concentration of 1 μM, CycLuc1 and CycLuc2 exhibited dramatically higher light output than any other substrate (Figure 3). CycLuc1 emitted 5.7-fold more light than aminoluciferin, and 3.2-fold more light than D-luciferin. Similarly, CycLuc2 emitted 4.7-fold more light than aminoluciferin, and 2.6-fold more light than D-luciferin. The superior light output of CycLuc1 held true across a wide concentration range (1 – 100 μM; SI Figure S2), indicating that this behavior is not simply due to higher affinity for the luciferase. Even when tested at exorbitantly high substrate concentrations (750 μM), the light emission from CycLuc1 greatly exceeds all other aminoluciferins and is equivalent to that of D-luciferin (SI Figure S2). In contrast, the acyclic substrate 6’-MeNHLH2 emitted ~50% more light than aminoluciferin at 1 μM but was no better at concentrations over 100 μM, and 6’-Me2NLH2 had uniformly lower light emission at all tested concentrations (SI Figure S2).
Figure 3.
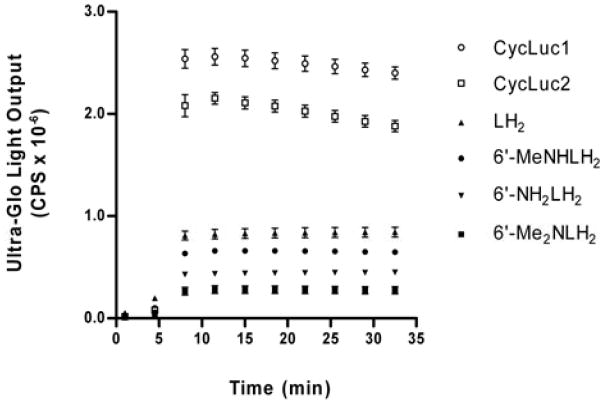
Ultra-Glo light output with 1 μM luciferin substrate.
The superior light output of CycLuc1 and CycLuc2 demonstrates that they are more efficient light emitters than their respective acyclic counterparts and that both monoalkyl and dialkylaminoluciferins can be efficient substrates (Figure 3). We hypothesize that this is due to the increased rigidity and restricted bond rotation of the cyclic alkylaminoluciferins, leading to an increase in the relative quantum yield.16 In the context of wild-type firefly luciferase, product inhibition overshadowed this effect and limited light output (Figure 2).
The use of cyclic aminoluciferin substrates or pro-substrates for in vitro assays with Ultra-Glo is expected to improve the sensitivity of detection.17 However, despite its utility for high-throughput screening applications, Ultra-Glo is not available as a genetic construct that can be introduced into living organisms for bioluminescence imaging.9,18 Future work will be directed toward mutation of luciferase in order to relieve product inhibition and allow sustained light emission from cyclic aminoluciferins under physiological conditions, using a luciferase that can be readily expressed within cells and whole organisms.
Supplementary Material
Acknowledgments
This work was supported by a grant from the NCI (NIH R21CA127196). We thank Bill Kobertz for critical reading of the manuscript.
Footnotes
SUPPORTING INFORMATION AVAILABLE: Experimental procedures and full spectroscopic data for all new compounds. This material is available free of charge at http://pubs.acs.org
References
- 1.Fraga H. Photochem Photobiol Sci. 2008;7:146–158. doi: 10.1039/b719181b. [DOI] [PubMed] [Google Scholar]
- 2.Nakatsu T, Ichiyama S, Hiratake J, Saldanha A, Kobashi N, Sakata K, Kato H. Nature. 2006;440:372–376. doi: 10.1038/nature04542. [DOI] [PubMed] [Google Scholar]
- 3.Naumov P, Ozawa Y, Ohkubo K, Fukuzumi S. J Am Chem Soc. 2009;131:11590–11605. doi: 10.1021/ja904309q. [DOI] [PubMed] [Google Scholar]
- 4.Branchini BR. Meth Enzymol. 2000;305:188–195. doi: 10.1016/s0076-6879(00)05488-4. [DOI] [PubMed] [Google Scholar]
- 5.Woodroofe CC, Shultz JW, Wood MG, Osterman J, Cali JJ, Daily WJ, Meisenheimer PL, Klaubert DH. Biochemistry. 2008;47:10383–10393. doi: 10.1021/bi800505u. [DOI] [PubMed] [Google Scholar]
- 6.Branchini BR, Murtiashaw MH, Magyar RA, Portier NC, Ruggiero MC, Stroh JG. J Am Chem Soc. 2002;124:2112–2113. doi: 10.1021/ja017400m. [DOI] [PubMed] [Google Scholar]
- 7.Hall MP, Gruber MG, Hannah RR, Jennens-Clough ML, Wood KV. Bioluminescence and Chemiluminescence Perspectives for the 21st Century. John Wiley & Sons; Chichester, U.K: 1998. pp. 392–395. [Google Scholar]
- 8.White EH, Worther H, Seliger HH, McElroy WD. J Am Chem Soc. 1966;88:2015–2019. [Google Scholar]
- 9.Shinde R, Perkins J, Contag CH. Biochemistry. 2006;45:11103–12. doi: 10.1021/bi060475o. [DOI] [PubMed] [Google Scholar]
- 10.Denburg JL, Lee RT, McElroy WD. Arch Biochem Biophys. 1969;134:381–394. doi: 10.1016/0003-9861(69)90297-5. [DOI] [PubMed] [Google Scholar]
- 11.Viviani VR, Bechara EJ, Ohmiya Y. Biochemistry. 1999;38:8271–8279. doi: 10.1021/bi9900830. [DOI] [PubMed] [Google Scholar]
- 12.Branchini BR, Magyar RA, Murtiashaw MH, Anderson SM, Zimmer M. Biochemistry. 1998;37:15311–15319. doi: 10.1021/bi981150d. [DOI] [PubMed] [Google Scholar]
- 13.While it was qualitatively apparent that these substrates had higher affinity for luciferase than LH2 or 6’-NH2LH2, their kinetic behavior prevented accurate determination of their true Km values.
- 14.Auld DS, Zhang Y, Southall NT, Rai G, Landsman M, Maclure J, Langevin D, Thomas CJ, Austin CP, Inglese J. J Med Chem. 2009;52:1450–1458. doi: 10.1021/jm8014525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.It is likely that the presence of components such as detergent and/or competitive inhibitors in the assay buffer assist steady light emission.
- 16.Ando Y, Niwa K, Yamada N, Enomoto T, Irie T, Kubota H, Ohmiya Y, Akiyama H. Nat Photon. 2008;2:44–47. [Google Scholar]
- 17.Fan F, Wood KV. Assay Drug Dev Technol. 2007;5:127–136. doi: 10.1089/adt.2006.053. [DOI] [PubMed] [Google Scholar]
- 18.Prescher JA, Contag CH. Curr Op Chem Biol. 2010;14:80–89. doi: 10.1016/j.cbpa.2009.11.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.