

Abstract
Ferrochelatase catalyzes the insertion of ferrous iron into protoporphyrin IX to form heme. Robust kinetic analyses of the reaction mechanism are complicated by the instability of ferrous iron in aqueous solution, particularly at alkaline pH values. At pH 7.00 the half-life for spontaneous oxidation of ferrous ion is approximately 2 min in the absence of metal complexing additives, which is sufficient for direct comparisons of alternative metal ion substrates with iron. These analyses reveal that purified recombinant ferrochelatase from both murine and yeast sources inserts not only ferrous iron but also divalent cobalt, zinc, nickel, and copper into protoporphyrin IX to form the corresponding metalloporphyrins but with considerable mechanistic variability. Ferrous iron is the preferred metal ion substrate in terms of apparent kcat and is also the only metal ion substrate not subject to severe substrate inhibition. Substrate inhibition occurs in the order Cu2+ > Zn2+ > Co2+ > Ni2+ and can be alleviated by the addition of metal complexing agents such as β-mercaptoethanol or imidazole to the reaction buffer. These data indicate the presence of two catalytically significant metal ion binding sites that may coordinately regulate a selective processivity for the various potential metal ion substrates.
Ferrochelatase (EC 4.99.1.1) catalyzes the insertion of ferrous iron into protoporphyrin IX to form heme (1, 2). This enzymatic reaction represents the intersection of the coordinately regulated porphyrin biosynthesis and iron transport pathways. Mutations in the human ferrochelatase gene have been linked to erythropoietic protoporphyria, a disease characterized by protoporphyrin IX accumulation and acute photosensitivity (3).
Ferrochelatases from a variety of sources, including mammal, plant, yeast, and bacterial, have been purified and at least partially characterized (4–7). Crystal structures are available for the human (8), (PDB3 number 1HRK), Saccharomyces cerevisiae (9), (PDB number 1LBQ) and Bacillus subtilis (10) (PDB number 1AK1) enzymes, and cDNA sequences for well more than 100 species-specific ferrochelatases can be found in the public data base. Remarkably, only six residues are currently known to be strictly conserved (see supplemental data Fig. 1.). The low sequence similarity of ferrochelatases may be reflective of the relatively facile nature of the porphyrin metallation reaction, which readily occurs in solution or by catalysis with abzymes (11), ribozymes (12), or DNAzymes (13). The catalytic efficiencies of these artificial enzymes with metal ion substrates are dramatically lower than those reported for ferrochelatase, however (13), and a key biological function of the enzyme may be to bind ferrous iron tightly and thereby minimize any toxicity associated with its release (14). The spectroscopic characterization and biological significance of a [2Fe-2S] cluster present in many ferrochelatases, including those from mammals, have received intense scrutiny, but assignment of a precise biochemical function remains elusive (2, 15–24).
The process whereby selective insertion of ferrous iron into protoporphyrin IX is achieved in vivo is unresolved. Ferrochelatase is known to catalyze insertion of divalent transition metal ions other than iron in vitro, most notably zinc, but cobalt, nickel, and copper have also each been reported to act as substrates, although species-specific differences have been noted (6, 25–27). Plausible mechanisms leading to ferrous iron specificity in vivo include the degree of protoporphyrin IX distortion after binding (28, 29), channeling of ferrous iron to the enzyme (30–33), and differential metal ion binding affinities and catalytic rates. An improved understanding of the mechanisms underlying metal ion selectivity in vivo could lead to novel and more effective approaches to treatment of diseases associated with heme and iron metabolism.
The intractable chemistries of the natural substrates have hindered the development of methods for routine acquisition of robust and unambiguous kinetic data for ferrochelatases. Protoporphyrin IX is highly hydrophobic and aggregates or “stacks” in aqueous solution (34, 35). This can be overcome by the addition of detergents to the assay buffer, but the use of more water soluble porphyrin substrates, such as mesoporphyrin and deuteroporphyrin IX, is common (36, 37). Furthermore, the severe instability of ferrous ion in aqueous solution (38, 39), particularly at neutral to basic pH, may be ameliorated by the inclusion of complexing agents such as β-mercaptoethanol and Tris buffer (40), but the use of complexing agents casts doubt on the kinetic data collected under these conditions due to the possibility that ferrochelatase reacts differentially with the complexed and uncomplexed metal ion pools. Consequently, the more stable metal ion substrate zinc is often utilized to assess ferrochelatase activity (6, 41). The routine use of nonnatural substrates to determine activity is a source of confusion that has made it difficult to achieve a holistic understanding of the reaction mechanism due to the distinct possibility that results obtained with alternate substrates are not good indicators of the results that would be obtained with the natural substrates under similar conditions (2).
To begin to address these problems we have defined conditions allowing direct kinetic comparisons of the naturally occurring substrates to alternative metal ion substrates, in aqueous solution devoid of strong metal ion complexing agents. The results indicate that multiple divalent transitions metal ions are good substrates for ferrochelatases from both mouse and yeast, but only iron is not subject to substrate inhibition. The presence of two catalytically significant metal ion binding sites suggests several future experiments, as discussed below.
EXPERIMENTAL PROCEDURES
Reagents—MOPS, Tween 80, sodium chloride, cobalt chloride hexahydrate, zinc chloride, nickel chloride hexahydrate, and cupric chloride dihydrate were from Sigma. Ferrous chloride tetrahydrate was obtained from Fisher. Porphyrins were from Frontier Scientific. Blue Sepharose was from GE Healthcare.
Overexpression, Purification, Storage, Handling, and Analysis of Ferrochelatase—The overexpression, purification, storage, and handling of mature murine ferrochelatase have been previously described (42). The purification of yeast ferrochelatase differed slightly from that for mouse. The lysis buffer contained 1.5% cholate and 1.5 m sodium chloride, and the ammonium sulfate cut used was 18–28% (w/v). The resulting pellet was dissolved in 0.1 m Tris-HCl, pH 8.0, containing 20% glycerol, and loaded onto the blue Sepharose column equilibrated in the same buffer. The resin column was washed with this buffer containing 1.5 m sodium chloride until the absorbance at 280 nm reached base line and then eluted by the further addition of 1.5% cholate to the buffer. Protein concentrations were determined by the bicinchoninic acid method using bovine serum albumin as the standard. Reported enzyme concentrations are based on monomeric molecular masses of 41,984 and 41,209 daltons for the murine and yeast enzymes, respectively, as calculated from the primary amino acid sequences encoded by the cDNAs for the two recombinant enzymes.
Preparation of Protoporphyrin IX and Metal Ion Solutions—Stock solutions of protoporphyrin IX were prepared from the commercially available hydrochloride salt by vortexing 1–3 mg in 100 μl of 2 m sodium hydroxide. This was followed by the sequential addition of 0.5 ml of 10% (w/v) Tween 80, 4 ml of deionized water, 90 μl of 2 m hydrochloric acid, and 0.5 ml of 1 m MOPS, pH 7.00, with thorough mixing after each addition. Stock solutions had a final pH of slightly greater than 7.00 and were stored at 4 °C wrapped in aluminum foil for no more than 1 week, whereupon a new stock solution was prepared. Stock concentrations were determined spectrophotometrically in 2.6 m hydrochloric acid using an extinction coefficient of 297,000 m–1·cm–1 for the Soret band at 408 nm (43). Metal ion solutions were prepared by completely dissolving 5 mmol of the solid (FeCl2·4H20, CoCl2·6H20, ZnCl2, NiCl2·6H20, or CuCl2·2H20) in 5 ml of concentrated HCl and diluting to 50 ml with deionized water to yield 0.1 m stock solutions in 1.2 m HCl. On the day of use aliquots of the stock solutions were serially diluted to 10, 1, and 0.1 mm with deionized water.
Determination of Ferrous Iron Stability in Ferrochelatase Reaction Buffer—Ferrous iron stability was assayed at 30 °C using a spectrophotometric assay wherein fluoride ion acts as a masking agent for ferric iron (38). Ferrochelatase reaction buffer is defined as 0.1 m MOPS, 0.4 m sodium chloride, and 0.2% (v/v) Tween 80, pH 7.00.
Progress Curve and Initial Rate Acquisition—Progress curves and initial rates were acquired using a Shimadzu UV2401PC spectrophotometer equipped with thermostatically controlled cuvette holders adjusted to 30 °C by monitoring the change in absorbance of the Soret band (44). Reaction mixtures contained ferrochelatase reaction buffer, 3 μm protoporphyrin IX, 0.2–100 μm divalent transition metal ion, and 0.1–0.5 μm ferrochelatase. Assays were conducted in a final volume of 2.00 ml in magnetically stirred quartz cuvettes and initiated by the addition of metal ion. Between reactions the cuvette was cleaned by sequentially rinsing with deionized water, 0.6 m HCl, and deionized water. Absorbance was continuously recorded at 407 nm in the case of iron and cobalt or at 417 nm for zinc, nickel, and copper. The time required to add the metal ion and close the observation chamber was consistently 3–4 s. Control reactions devoid of enzyme were also recorded and subtracted from those recorded in the presence of enzyme. The first 10–20 s of the reactions were analyzed using linear regression to obtain initial rates. Progress curves were exported as ASCII files for analysis using Dynafit software (45).
Estimation of Extinction Coefficients—Extinction coefficients for protoporphyrin IX in ferrochelatase reaction buffer at 30 °C were determined by observation of the signal amplitudes produced from a stock solution prepared as described above (also see supplemental Fig. 2). The observed extinction coefficients were 1.07 × 105 m–1·cm–1 at 407 nm and 8.05 × 104 m–1·cm–1 at 417 nm. Apparent extinction coefficients for conversion of protoporphyrin IX to metallated protoporphyrins in this buffer were determined in a similar fashion using 0.2 μm yeast ferrochelatase and excess metal ion to convert known concentrations of protoporphyrin IX into the corresponding metalloporphyrins. The apparent extinction coefficients were: iron protoporphyrin IX, 4.15 × 104 m–1·cm–1 (407 nm); cobalt protoporphyrin IX, 7.54 × 104 m–1·cm–1 (407 nm); zinc protoporphyrin IX, 1.49 × 105 m–1·cm–1 (417 nm); nickel protoporphyrin IX, 4.19 × 104 m–1·cm–1 (417 nm); copper protoporphyrin IX, 6.26 × 104 m–1·cm–1 (417 nm). Spectral changes associated with the metallation reactions are provided as supplemental Fig. 3.
RESULTS
Iron Stability in Ferrochelatase Reaction Buffers—The rate of ferrous iron oxidation in aqueous solutions is dependent not only on the concentration of dissolved oxygen but also on the square of the hydroxide ion concentration (38). The effect of pH on ferrous ion oxidation in ferrochelatase reaction buffer at 30 °C was investigated near neutral pH in the experiments recorded in Fig. 1. Strong pH dependence was observed with half-lives at pH 6.75–7.25 and 30 °C on the scale of minutes, as recorded in Table 1. The addition of 5 mm β-mercaptoethanol approximately doubled the stability of ferrous iron, whereas 5 mm imidazole increased stability by about 20%.
FIGURE 1.

Stability of ferrous iron in ferrochelatase assay buffer. Ferrous chloride (100 μm) in 100 mm MOPS, 0.4 m NaCl, and 0.2% Tween 80 was assayed at 30 °C as described (38). The pH values were 6.75 (circles), 7.00 (triangles), and 7.25 (squares). The half-lives at each pH are recorded in Table 1.
TABLE 1.
Effect of pH and buffer additives on the half-life of ferrous iron oxidation in 100 mm MOPS, 400 mm NaCl, and 0.2% Tween 80 at 30 °C
| pH | Buffer additive | Half-life |
|---|---|---|
| min | ||
| 6.75 | None | 6.2 |
| 7.00 | None | 1.8 |
| 7.00 | 5 mm β-mercaptoethanol | 3.8 |
| 7.00 | 5 mm imidazole | 2.2 |
| 7.25 | None | 1.2 |
Progress Curves for Reaction of Murine and Yeast Ferrochelatase with Protoporphyrin IX and Metal Ions—Progress curves for the reaction of yeast ferrochelatase with 3 μm protoporphyrin IX and 1 μm divalent iron, zinc, or copper indicated widely divergent kinetic behavior for insertion of these metal ions (Fig. 2). The progress curve for iron is hyperbolic, approximating a first-order decay. This simplest numerical model adequate to describe this progress curve is the Michaelis-Menten equation, as described by Equation 1, where it is assumed that the enzyme is saturated with protoporphyrin IX during the time course. In Equation 1 EP is the enzyme-protoporphyrin IX complex, M is metal ion, EPM is the Michaelis complex, and E is the free enzyme. The hyperbolic shape of the curve occurs due to depletion of EPM as the reaction proceeds and the requirement that kcat < k–1.
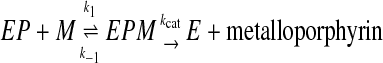 |
(Eq. 1) |
FIGURE 2.

Non-Michaelis-Menten kinetics of ferrochelatase from progress curves with transition metal ion substrates in the absence of chelating agents. Shown are progress curves of 0.1 μm yeast ferrochelatase with 3 μm protoporphyrin IX and 1 μm ferrous iron (○), zinc (•), and copper (□). The inset includes the progress curves for ferrous iron, cobalt, and nickel, from fastest to slowest. The progress curve for ferrous iron was corrected to account for the rate of spontaneous oxidation.
The progress curve for zinc is more consistent with zero-order kinetics, wherein the metal is not in rapid equilibrium with the enzyme, and the concentration of EPM remains relatively constant for the duration of the reaction. Equation 1 is again adequate to describe this progress curve, but here, unlike the case for ferrous iron described above, the rate that zinc dissociates from the Michaelis complex must be comparable with or slower than kcat (i.e. k–1 is comparable to or slower than kcat). A close inspection of the data for zinc reveals a structured deviation in the residual error that arises because the reaction rate increases slightly as the substrates are consumed, suggesting substrate inhibition may be a more appropriate model for this progress curve.
The progress curve for copper (Fig. 2) is sigmoidal rather than hyperbolic. The increase in reaction rate as copper is depleted suggests substrate inhibition that is progressively relieved as the instantaneous concentration of copper decreases, and the simplest kinetic model providing a numerical fit for this progress curve while remaining consistent with the data for iron and zinc requires the addition of the possibility of binding a second, inhibitory, metal ion to the enzyme as depicted in Equation 2.
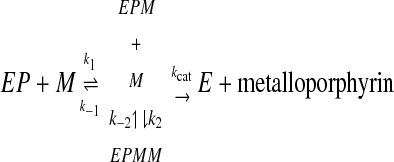 |
(Eq. 2) |
The raw data for copper protoporphyrin IX production were fit to both Equations 1 and 2. Equation 1 was inadequate to describe the progress curve, whereas Equation 2 yielded a good fit to the experimental data.
The progress curves with cobalt and nickel as substrates were hyperbolic, in analogy to iron (Fig. 2, inset), and those obtained with murine ferrochelatase under identical conditions were similar to those obtained with the yeast enzyme for each of the five metal ions (not shown). Analysis of progress curves over an extended range of zinc (Fig. 3A) or copper (Fig. 3B) concentrations clearly indicated the inhibitory effects of these metal ions on ferrochelatase.
FIGURE 3.

Concentration-dependence of progress curves for Zn2+ and Cu2+. In A, 0.1 μm yeast ferrochelatase was reacted with 3 μm protoporphyrin IX and 0.2–50 μm ZnCl2. The initial rate decreased with increasing zinc concentration, such that the curves represent, from right to left, 50, 20, 10, 5, 2, 1, 0.5, and 0.2 μm zinc. The 0.2, 0.5, and 1 μm curves are also plotted in the inset. In B, 0.2 μm yeast ferrochelatase was reacted with 3 μm protoporphyrin IX, 0.5–10 μm CuCl2. From right to left the curves are 10, 5, 2, 1, and 0.5 μm copper.
Initial Rates as a Function of Metal Ion Concentration—The possibility of substrate inhibition was investigated for Fe2+, Co2+, Ni2+, Zn2+, and Cu2+ ions for both the murine and mouse ferrochelatases by varying the metal ion concentration while keeping all other potential variables fixed. Assays were conducted in ferrochelatase reaction buffer at 30 °C in triplicate with the results in Fig. 4 and Table 2. In each case an attempt was made to fit the data to Equation 3 for a substrate-inhibited reaction using SigmaPlot graphing software.
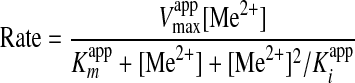 |
(Eq. 3) |
Good fits were observed for the cobalt, nickel, and zinc data, whereas the data for copper could not be fit due to the severity of the inhibition. An apparent inhibitory constant could not be estimated for ferrous iron over the concentration range tested, although the data for yeast ferrochelatase with ferrous iron led us to suggest that inhibition might be observed at concentrations higher than 100 μm (Fig. 4F). The data for iron were instead fit to the Michaelis-Menten equation. The specific activity of the yeast enzyme was consistently 6–10-fold higher than the murine, but the utilization of each metal ion was otherwise very similar for the two enzymes.
FIGURE 4.

Ferrochelatase initial rates indicate selective substrate inhibition. In A–E the initial rates are for murine ferrochelatase, whereas in F–J initial rates are for yeast ferrochelatase. Initial rates are in units of μm product/min/μm enzyme.
TABLE 2.
Apparent kinetic constants for insertion of divalent transition metals into 3 μm protoporphyrin IX in the presence of ferrochelatase
NA, not applicable.
|
Murine FC |
Yeast FC |
|||||
|---|---|---|---|---|---|---|
| Km | Ki | kcat | Km | Ki | kcat | |
| μm | μm | min-1 | μm | μm | min-1 | |
| Iron | 0.79 ± 0.19 | n/a | 6.6 ± 0.3 | 0.5 ± 0.1 | n/a | 45 ± 1 |
| Cobalt | 0.12 ± 0.04 | 22 ± 3 | 1.3 ± 0.1 | 0.2 ± 0.1 | 20 ± 10 | 11 ± 4 |
| Nickel | 1.1 ± 0.2 | 70 ± 10 | 1.3 ± 0.1 | 5.0 ± 0.5 | 74 ± 9 | 18 ± 1 |
| Zinc | NA | 12 ± 2 | 1.4 ± 0.1 | 0.19 ± 0.06 | 1.8 ± 0.4 | 23 ± 3 |
Effect of β-Mercaptoethanol and Imidazole on Ferrochelatase Progress Curves and Initial Rates—The substrate inhibition of ferrochelatase observed in the absence of strong metal ion complexing agents indicated the presence of at least two distinct metal binding sites on the enzyme. The possibility that metal ion-coordinating buffer additives would compete with the inhibitory site and thereby alleviate or mask substrate inhibition was investigated using murine ferrochelatase (Fig. 5). The effect of β-mercaptoethanol on incorporation of divalent zinc ion into protoporphyrin IX in ferrochelatase reaction buffer at pH 8.0 can be seen in Fig. 5, panel A. The initial rates increased at low concentrations of β-mercaptoethanol, consistent with alleviation of substrate inhibition via metal ion complexation, and then decreased at higher concentrations, suggesting metal ion depletion. The shape of the progress curves went from nearzero order to first order as the concentration of β-mercaptoethanol increased.
FIGURE 5.

Substrate inhibition of ferrochelatase can be masked by β-mercaptoethanol or imidazole. In A, 0.2 μm murine ferrochelatase was reacted with 3 μm protoporphyrin IX and 2 μm ZnCl2 at the following concentrations of β-mercaptoethanol: none (solid line), 0.5 mm (short dashes), 1 mm (long dashes), 2 mm (dash-dot line), and 3 mm (dash-dot-dot-dash line). In B, 0.5 μm murine ferrochelatase was reacted with 3 μm protoporphyrin IX and 2 μm CuCl2 at the following concentrations of imidazole: none (solid line), 0.2 mm (short dashes), 0.5 mm (long dashes), 1 mm (dotted line), 2 mm (dash-dot line), and 3 mm (dash-dot-dot-dash line).
The severe substrate inhibition observed for incorporation of copper into protoporphyrin IX was completely eliminated by inclusion of 3 mm imidazole into ferrochelatase reaction buffer, pH 7.00 (Fig. 5, panel B). The observed initial rate was increased by more than 2 orders of magnitude.
DISCUSSION
The d orbitals of transition metal ions form stable complexes with a wide variety of common buffer components, including carboxylates, phosphates, amines, sulfhydryls, sulfates, halides, dissolved oxygen, hydroxide ion, and alcohols (including water) (46). The formation of these coordination complexes is so energetically favorable that virtually none of the dissolved metal ions exist in an unliganded or “free” state in aqueous solution, and the term “free metal ion” is generally understood to refer to a complexed form of the transition metal ion in which the ligands are weakly bound and readily exchangeable, such as with water or chloride ions. In this study we eliminated the complexing agents Tris and β-mercaptoethanol from our ferrochelatase reaction buffer. Tris forms coordination complexes with transition metal ions with equilibrium constants in the range of 100–10,000 (40), and sulfhydryls are well known to coordinate metal ions strongly (47). In contrast, MOPS buffer does not form stable metal ion complexes (48, 49).
The kinetic behavior of ferrochelatase in the presence of complexing agents such as β-mercaptoethanol and imidazole is entirely consistent with sequestration of metal ion in forms that are not substrates for the enzyme. This exogenous equilibrium depletes the free metal ion pool and can have dramatic effects on the measured ferrochelatase activity. Two examples of this are shown in Fig. 5 for the most inhibitory metal ion substrates, zinc and copper, where lower concentrations of β-mercaptoethanol or imidazole accelerate activity, presumably by effectively competing with the inhibitory metal binding site on the enzyme for free metal ion. At higher concentrations of complexing agents, depletion of the free metal ion pool results in conversion of the non-Michaelis-Menten progress curves observed with these substrates into first-order decays that could be, albeit erroneously, assumed to represent typical Michaelis-Menten enzyme kinetics rather than complex equilibria. With ferrous iron, which is not substrate inhibited, the effect of complexing agents is to slow the reaction rate with no effect on the shape of progress curves (not shown). The activity modifying effects of complexing agents likely extends to all metal ion substrates. Elimination of metal ion complexing agents allows more unambiguous studies of the ferrochelatase mechanism to be conducted by maintaining the substrate metal ion pool in a relatively homogenous form utilizable by the enzyme, and in this study allowed us to demonstrate that the enzyme is substrate inhibited by nonferrous iron metal ion substrates.
The stability of ferrous iron in ferrochelatase reaction buffer was highly pH-dependent, as expected (38, 39), and at pH 7.00 was reasonably stable and assays could be conducted aerobically. Determination of ferrous iron stability is indispensable to assessing the validity of ferrochelatase assay conditions and should be a routine control for ferrochelatase assays when iron is used as the metal ion substrate.
The inhibition and progress curve data indicate that ferrochelatase contains at least two catalytically important metal binding sites. The biological significance of an inhibitory binding site remains to be elucidated, and multiple possibilities can be envisioned. The inhibitory metal ion binding site may represent an evolutionary adaptation toward metal ion specificity. In this scenario the inhibitory binding site would preferentially bind transition metal ions other than iron, resulting in inhibition of activity and thereby preventing biosynthesis of metalloporphyrins other than heme. This would be of heightened importance in diseases involving imbalances in metal metabolism. For instance, the accumulation of zinc protoporphyrin observed during iron insufficiency might be limited due to substrate inhibition of ferrochelatase (50), and the accumulation of porphyrins observed in a mouse model of Wilson disease, a copper transport disorder characterized by toxic copper accrual, may be directly related to ferrochelatase inhibition (51). It is interesting to note that the ratio of the apparent kcat for iron to that for any other metal ion is higher for murine ferrochelatase than for yeast ferrochelatase (Table 2), suggesting the mouse enzyme has evolved to be more iron-specific. Future studies with ferrochelatases from other diverse evolutionary sources or with the structurally homologous cobalt chelatases (52–54) should further clarify the likelihood of evolution of metal ion specificity in free metal ion pools and indirectly shed light on the probability of direct metal ion channeling to chelatases in vivo.
A more speculative alternative is that both metal ion binding sites are part of a general processing pathway that magnifies the “stickiness” of ferrous iron, minimizing the cellular toxicity that would be associated with release of ferrous iron into the cellular milieu. The requirement for such a mechanism is seldom discussed. Ferrochelatase is typically treated as a classical Michaelis-Menten-type enzyme, wherein the free and bound substrates are in rapid equilibrium with the enzyme (37, 41). This common kinetic situation would be quite deleterious to the cell, however, because any ferrous iron released by ferrochelatase into the mitochondrial matrix, which is approximately pH 8.0, would be rapidly oxidized via Fenton chemistry, wasting the considerable energy investment involved in delivering iron to ferrochelatase in a non-toxic form and causing severe oxidative stress to the cell. The data reported here indicate that ferrochelatase has the capacity to bind and processes divalent metal ion into product faster than it is released back into solution. If this were the case with ferrous iron in vivo, it would represent a tremendous cellular advantage. The rapid and toxic decomposition of ferrous iron in aqueous solution makes it unlikely that ferrochelatase binds iron weakly in vivo.
The residues comprising the inhibitory metal ion binding site have not been identified. Indeed, the identity of the primary, catalytic metal ion binding site is not generally agreed upon. One model proposes that the metal is bound and inserted at residues arginine 164 and tyrosine 165 (55), whereas crystal structures for ferrochelatase from S. cerevisiae with bound cobalt (9) (PDB 1LX8) and for the enzyme from B. subtilis with bound iron (PDB 2HK6), zinc (PDB 1LD3), or cadmium (56, 57) (PDB 1NOI) all identify histidine 263 and glutamate 343 at the opposite face of the porphyrin substrate as the catalytic metal binding site.4 Several other metal ion binding sites have also been observed in ferrochelatase crystal structures, and each of these are good candidates for inhibitory binding sites (Refs. 9 (PDB 1L8X; multiple cobalt binding sites), 56 (PDB 2HK6; total of four iron binding sites), and 57 (PBD 1LD3; zinc in catalytic site and magnesium in π -helix, and PDB 1N0I, cadmium in catalytic site and at His-287)). Many of these sites cluster around a π -helix predicted to undergo substantial conformational change during the course of the catalytic cycle (9, 58). This helix is rich in acidic residues, and metal binding in this region could impact activity by modulating the enzyme dynamics presumably required for catalytic turnover. Alternatively, the well conserved histidine 341 residue at the base of the active site cleft appears conspicuously placed for metal binding, and crystals of B. subtilis ferrochelatase soaked in the inhibitory metal ion cadmium reveal metal ion bound to both this histidine and the histidine 263 glutamate 343 pair, with a distance of 9 Å between the two metal atoms (57). Detailed analyses of point mutants in these regions should help delineate their contribution to substrate inhibition. It is possible that mutation to an inhibitory site might result in substantially enhanced activity with non-ferrous iron substrates, in analogy to what is seen when a competitor is introduced into the buffer with the wild-type enzyme (Fig. 5B).
Considerable evidence for channeling of ferrous iron to ferrochelatase exists (30, 59, 60). Direct channeling, wherein the metal atom is delivered as part of a chaperone-ferrochelatase complex without ever being released into the surrounding aqueous environment has not been definitively proven, but it has been shown that the mitochondrial iron chaperone frataxin can provide a source of ferrous iron for ferrochelatase in vitro (30). The experimental approaches reported here most closely approximate a free metal ion system and may help to provide an unambiguous kinetic benchmark against which to judge whether frataxin, mitoferrin (31), or some other iron chaperone might directly channel ferrous iron to ferrochelatase.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grant GM080270. This work was also supported by American Heart Association Florida Affiliate Grant 0655091B (to G. C. F.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–3.
Footnotes
The abbreviations used are: PDB, Protein Data Bank; MOPS, 3-[N-morpholino]propanesulfonic acid.
Amino acid position numbering throughout the text is for the human enzyme for convenience only.
References
- 1.Ferreira, G. C. (1999) Int. J. Biochem. Cell Biol. 31 995–1000 [DOI] [PubMed] [Google Scholar]
- 2.Dailey, H. A., Dailey, T. A., Wu, C. K., Medlock, A. E., Wang, K. F., Rose, J. P., and Wang, B. C. (2000) Cell. Mol. Life Sci. 57 1909–1926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bottomley, S. S. (2004) Porphyria, pp. 1057–1087, Lippincott Williams & Wilkins, Baltimore. MD
- 4.Okuda, M., Kohno, H., Furukawa, T., Tokunaga, R., and Taketani, S. (1994) Biochim. Biophys. Acta 1200 123–128 [DOI] [PubMed] [Google Scholar]
- 5.Suzuki, T., Masuda, T., Inokuchi, H., Shimada, H., Ohta, H., and Takamiya, K. (2000) Plant Cell Physiol. 41 192–199 [DOI] [PubMed] [Google Scholar]
- 6.Gora, M., Grzybowska, E., Rytka, J., and Labbe-Bois, R. (1996) J. Biol. Chem. 271 11810–11816 [DOI] [PubMed] [Google Scholar]
- 7.Hansson, M., and Hederstedt, L. (1994) Eur. J. Biochem. 220 201–208 [DOI] [PubMed] [Google Scholar]
- 8.Wu, C. K., Dailey, H. A., Rose, J. P., Burden, A., Sellers, V. M., and Wang, B. C. (2001) Nat. Struct. Biol. 8 156–160 [DOI] [PubMed] [Google Scholar]
- 9.Karlberg, T., Lecerof, D., Gora, M., Silvegren, G., Labbe-Bois, R., Hansson, M., and Al-Karadaghi, S. (2002) Biochemistry 41 13499–13506 [DOI] [PubMed] [Google Scholar]
- 10.Al-Karadaghi, S., Hansson, M., Nikonov, S., Jonsson, B., and Hederstedt, L. (1997) Structure 5 1501–1510 [DOI] [PubMed] [Google Scholar]
- 11.Cochran, A. G., and Schultz, P. G. (1990) Science 249 781–783 [DOI] [PubMed] [Google Scholar]
- 12.Conn, M. M., Prudent, J. R., and Schultz, P. G. (1996) J. Am. Chem. Soc. 118 7012–7013 [Google Scholar]
- 13.Li, Y., and Sen, D. (1997) Biochemistry 36 5589–5599 [DOI] [PubMed] [Google Scholar]
- 14.Crichton, R. R., Wilmet, S., Legssyer, R., and Ward, R. J. (2002) J. Inorg. Biochem. 91 9–18 [DOI] [PubMed] [Google Scholar]
- 15.Shepherd, M., Dailey, T. A., and Dailey, H. A. (2006) Biochem. J. 397 47–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dailey, T. A., and Dailey, H. A. (2002) J. Bacteriol. 184 2460–2464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schneider-Yin, X., Gouya, L., Dorsey, M., Rufenacht, U., Deybach, J. C., and Ferreira, G. C. (2000) Blood 96 1545–1549 [PubMed] [Google Scholar]
- 18.Crouse, B. R., Sellers, V. M., Finnegan, M. G., Dailey, H. A., and Johnson, M. K. (1996) Biochemistry 35 16222–16229 [DOI] [PubMed] [Google Scholar]
- 19.Sellers, V. M., Johnson, M. K., and Dailey, H. A. (1996) Biochemistry 35 2699–2704 [DOI] [PubMed] [Google Scholar]
- 20.Franco, R., Moura, J. J., Moura, I., Lloyd, S. G., Huynh, B. H., Forbes, W. S., and Ferreira, G. C. (1995) J. Biol. Chem. 270 26352–26357 [DOI] [PubMed] [Google Scholar]
- 21.Furukawa, T., Kohno, H., Tokunaga, R., and Taketani, S. (1995) Biochem. J. 310 533–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferreira, G. C., Franco, R., Lloyd, S. G., Pereira, A. S., Moura, I., Moura, J. J., and Huynh, B. H. (1994) J. Biol. Chem. 269 7062–7065 [PubMed] [Google Scholar]
- 23.Dailey, H. A., Finnegan, M. G., and Johnson, M. K. (1994) Biochemistry 33 403–407 [DOI] [PubMed] [Google Scholar]
- 24.Lloyd, S. G., Franco, R., Moura, J. J. G., Moura, I., Ferreira, G. C., and Huynh, B. H. (1996) J. Am. Chem. Soc. 118 9892–9900 [Google Scholar]
- 25.Hanson, J. W., and Dailey, H. A. (1984) Biochem. J. 222 695–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taketani, S., and Tokunaga, R. (1982) Eur. J. Biochem. 127 443–447 [DOI] [PubMed] [Google Scholar]
- 27.Jones, M. S., and Jones, O. T. (1970) Biochem. J. 119 453–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karlberg, T., Hansson, M. D., Yengo, R. K., Johansson, R., Thorvaldsen, H. O., Ferreira, G. C., Hansson, M., and Al-Karadaghi, S. (2008) J. Mol. Biol. 378 1074–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Al-Karadaghi, S., Franco, R., Hansson, M., Shelnutt, J. A., Isaya, G., and Ferreira, G. C. (2006) Trends Biochem. Sci. 31 135–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yoon, T., and Cowan, J. A. (2004) J. Biol. Chem. 279 25943–25946 [DOI] [PubMed] [Google Scholar]
- 31.Shaw, G. C., Cope, J. J., Li, L., Corson, K., Hersey, C., Ackermann, G. E., Gwynn, B., Lambert, A. J., Wingert, R. A., Traver, D., Trede, N. S., Barut, B. A., Zhou, Y., Minet, E., Donovan, A., Brownlie, A., Balzan, R., Weiss, M. J., Peters, L. L., Kaplan, J., Zon, L. I., and Paw, B. H. (2006) Nature 440 96–100 [DOI] [PubMed] [Google Scholar]
- 32.Park, S., Gakh, O., O'Neill, H. A., Mangravita, A., Nichol, H., Ferreira, G. C., and Isaya, G. (2003) J. Biol. Chem. 278 31340–31351 [DOI] [PubMed] [Google Scholar]
- 33.O'Neill, H. A., Gakh, O., Park, S., Cui, J., Mooney, S. M., Sampson, M., Ferreira, G. C., and Isaya, G. (2005) Biochemistry 44 537–545 [DOI] [PubMed] [Google Scholar]
- 34.Brown, S. B., Shillcock, M., and Jones, P. (1976) Biochem. J. 153 279–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Margalit, R., and Rotenberg, M. (1984) Biochem. J. 219 445–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Najahi-Missaoui, W., and Dailey, H. A. (2005) Blood 106 1098–1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoggins, M., Dailey, H. A., Hunter, C. N., and Reid, J. D. (2007) Biochemistry 46 8121–8127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sung, W., and Morgan, J. (1980) Environ. Sci. Technol. 14 561–568 [Google Scholar]
- 39.Stumm, W., and Lee, G. F. (1961) Ind. Eng. Chem. 53 143–146 [Google Scholar]
- 40.Fischer, B. E., Haring, U. K., Tribolet, R., and Sigel, H. (1979) Eur. J. Biochem. 94 523–530 [DOI] [PubMed] [Google Scholar]
- 41.Shi, Z., and Ferreira, G. C. (2004) J. Biol. Chem. 279 19977–19986 [DOI] [PubMed] [Google Scholar]
- 42.Ferreira, G. C. (1994) J. Biol. Chem. 269 4396–4400 [PubMed] [Google Scholar]
- 43.Gunter, E. W., Turner, W. E., and Huff, D. L. (1989) Clin. Chem. 35 1601–1608 [PubMed] [Google Scholar]
- 44.Porra, R. J., Vitols, K. S., Labbe, R. F., and Newton, N. A. (1967) Biochem. J. 104 321–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuzmic, P. (1996) Anal. Biochem. 237 260–273 [DOI] [PubMed] [Google Scholar]
- 46.Zumdahl, S. S. (2004) Chemical Principles, Houghton Mifflin Co., pp. 891–936, Boston, MA
- 47.Kuchar, J., and Hausinger, R. P. (2004) Chem. Rev. 104 509–525 [DOI] [PubMed] [Google Scholar]
- 48.Mash, H. E., Chin, Y. P., Sigg, L., Hari, R., and Xue, H. (2003) Anal. Chem. 75 671–677 [DOI] [PubMed] [Google Scholar]
- 49.Yu, Q., Kandegedara, A., Xu, Y., and Rorabacher, D. B. (1997) Anal. Biochem. 253 50–56 [DOI] [PubMed] [Google Scholar]
- 50.Labbe, R. F., Vreman, H. J., and Stevenson, D. K. (1999) Clin. Chem. 45 2060–2072 [PubMed] [Google Scholar]
- 51.Nakayama, K., Takasawa, A., Terai, I., Okui, T., Ohyama, T., and Tamura, M. (2000) Arch. Biochem. Biophys. 375 240–250 [DOI] [PubMed] [Google Scholar]
- 52.Brindley, A. A., Raux, E., Leech, H. K., Schubert, H. L., and Warren, M. J. (2003) J. Biol. Chem. 278 22388–22395 [DOI] [PubMed] [Google Scholar]
- 53.Schubert, H. L., Raux, E., Wilson, K. S., and Warren, M. J. (1999) Biochemistry 38 10660–10669 [DOI] [PubMed] [Google Scholar]
- 54.Yin, J., Xu, L. X., Cherney, M. M., Raux-Deery, E., Bindley, A. A., Savchenko, A., Walker, J. R., Cuff, M. E., Warren, M. J., and James, M. N. (2006) J. Struct. Funct. Genomics 7 37–50 [DOI] [PubMed] [Google Scholar]
- 55.Sellers, V. M., Wu, C. K., Dailey, T. A., and Dailey, H. A. (2001) Biochemistry 40 9821–9827 [DOI] [PubMed] [Google Scholar]
- 56.Hansson, M. D., Karlberg, T., Rahardja, M. A., Al-Karadaghi, S., and Hansson, M. (2007) Biochemistry 46 87–94 [DOI] [PubMed] [Google Scholar]
- 57.Lecerof, D., Fodje, M. N., Alvarez Leon, R., Olsson, U., Hansson, A., Sigfridsson, E., Ryde, U., Hansson, M., and Al-Karadaghi, S. (2003) J. Biol. Inorg. Chem. 8 452–458 [DOI] [PubMed] [Google Scholar]
- 58.Medlock, A. E., Dailey, T. A., Ross, T. A., Dailey, H. A., and Lanzilotta, W. N. (2007) J. Mol. Biol. 373 1006–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bencze, K. Z., Yoon, T., Millan-Pacheco, C., Bradley, P. B., Pastor, N., Cowan, J. A., and Stemmler, T. L. (2007) Chem. Commun. (Camb.) 1798–1800 [DOI] [PMC free article] [PubMed]
- 60.He, Y., Alam, S. L., Proteasa, S. V., Zhang, Y., Lesuisse, E., Dancis, A., and Stemmler, T. L. (2004) Biochemistry 43 16254–16262 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.