

Abstract
Mechanisms of transcriptional repression are important during cell differentiation. Mammalian heterochromatin protein 1 isoforms HP1α, HP1β, and HP1γ play important roles in the regulation of chromatin structure and function. We explored the possibility of different roles for the three HP1 isoforms in an integrated system, skeletal muscle terminal differentiation. In this system, terminal differentiation is initiated by the transcription factor MyoD, whose target genes remain mainly silent until myoblasts are induced to differentiate. Here we show that HP1α and HP1β isoforms, but not HP1γ, interact with MyoD in myoblasts. This interaction is direct, as shown using recombinant proteins in vitro. A gene reporter assay revealed that HP1α and HP1β, but not HP1γ, inhibit MyoD transcriptional activity, suggesting a model in which MyoD could serve as a bridge between nucleosomes and chromatin-binding proteins such as HDACs and HP1. Chromatin immunoprecipitation assays show a preferential recruitment of HP1 proteins on MyoD target genes in proliferating myoblasts. Finally, modulation of HP1 protein level impairs MyoD target gene expression and muscle terminal differentiation. Together, our data show a nonconventional interaction between HP1 and a tissue-specific transcription factor, MyoD. In addition, they strongly suggest that HP1 isoforms play important roles during muscle terminal differentiation in an isoform-dependent manner.
Mammalian heterochromatin protein 1 (HP1)5 isoforms are closely related non-histone proteins that are involved in transcriptional regulation and chromatin organization. In mammals, HP1 exists in three isoforms: α, β, and γ (Ref. 1 and references therein). Each is composed of a conserved chromodomain that is important for heterochromatin binding and a chromoshadow domain (CSD), whose structure is similar to that of the chromodomain that is involved in dimerization and interaction with proteins containing the consensus sequence PXVXL (2). The chromodomain and the CSD are separated by a less conserved region called the hinge region. HP1 isoforms exhibit different subnuclear localizations in interphasic nuclei: HP1α is mainly centromeric; HP1β is also centromeric but to a lesser extent; and HP1γ is located in both euchromatic and heterochromatic compartments (3). HP1 proteins are known to bind methylated histone H3 lysine 9 (H3K9) and heterodimerize, contributing to the formation and maintenance of heterochromatic structures (4, 5). HP1 isoforms interact with a wide variety of proteins, mainly chromatin-associated proteins (Refs. 1 and 6 and references therein).
HP1 proteins have been implicated in many differentiation pathways (7–10), although their individual roles are not always well understood. During terminal differentiation of cells, chromatin undergoes dramatic morphological changes (reviewed in Refs. 11–13), for example during muscle differentiation (14). These changes involve reorganization of constitutive heterochromatin (15) and selective silencing and activation of specific groups of genes (Refs. 7, 16, and 17; reviewed in Ref. 13 and references therein) and implicate both differential interactions of various proteins with chromatin and changes in the chromatin structure itself.
Skeletal muscle terminal differentiation begins with an irreversible withdrawal from the cell cycle, followed by musclespecific marker expression with fusion of myoblasts into multinucleated myotubes (18). Cell cycle withdrawal corresponds to a definitive silencing of proliferation associated genes, such as E2F targets (Ref. 16; reviewed in Ref. 19 and references therein). Terminal muscle differentiation is orchestrated by the myogenic basic helix loop helix transcription factors, such as MyoD, which is the master myogenic determination factor. In proliferating myoblasts, MyoD is expressed but is unable to activate its target genes even when it binds to their promoters (20, 21). Therefore, the requirement for MyoD to be continuously expressed in undifferentiated myoblasts is enigmatic. MyoD might have a repressive role at its target genes prior to initiating chromatin remodeling in differentiating cells (20, 22, 23). In proliferating myoblasts, MyoD is associated with histone deacetylases (HDACs) and might actively suppress expression of its targets by inducing a locally repressive chromatin structure (20, 24, 25). It is known that histone deacetylation contributes to H3K9 methylation on these same promoters (22). It has been shown that some MyoD target promoters, including p21 and myogenin, are methylated at H3K9 specifically in proliferating myoblasts (20, 22), a modification known to be bound by HP1 proteins.
Here we show that HP1 proteins associate directly with MyoD in an isoform-dependent manner and inhibit its transcriptional activity. Indeed, MyoD interacts preferentially with the isoforms HP1α and HP1β, but not HP1γ. In addition, HP1 proteins are recruited to MyoD target promoters preferentially in proliferating myoblasts. Finally, overexpression of HP1 isoforms interferes with muscle terminal differentiation in an isoform-dependent manner. Taken together, these data strongly suggest that, in addition to its role as an activator of differentiation-specific genes, MyoD can also act as a transcriptional repressor in proliferating myoblasts in cooperation with specific isoforms of HP1 protein.
EXPERIMENTAL PROCEDURES
Cell Culture and Transfection—C2C12, HEK 293, and HeLa cells were maintained using standard conditions. C2C12 cells were differentiated as described in Ref. 16. The biotin-streptavidin interaction studies were performed as described in Ref. 26.
Stable Cell Lines Establishment—A HeLa cell line stably expressing MyoD was established with a transgene encoding for full-length MyoD, and C2C12 cell lines expressing HP1 isoforms were established with transgenes encoding for full-length HP1α, HP1β, and HP1γ as described in Ref. 27. The transgenes were tagged with double hemagglutinin (HA) and double FLAG epitopes at the N terminus as described in Ref. 27. HeLa and C2C12 control cell lines transduced with the empty vector were also established.
Protein Complex Purification—MyoD complex characterization was performed as described in Ref. 27.
Protein Extraction, Coimmunoprecipitations, and Western Blotting—The biotin-streptavidin interaction studies were performed as described in Ref. 26. Expression vectors for biotinylatable proteins are kind gifts from Dr. V. Ogryzko (26).
For the study of the endogenous protein interactions, nuclear extracts were used for immunoprecipitation overnight, after which the immunoprecipitates were incubated with Ultralink immobilized protein A/G (Pierce) for 2 h at room temperature and washed with the dilution buffer 4–10 times.
Plasmids, GST Fusions, and GST Pulldown—Expression vectors derived from pGEX for GST fusions of wild type HP1α, β, γ, and HP1α deletion mutants GST-HP1α(1–119), GST-HP1α(1–66), and GST-HP1α(67–119) were described in Ref. 28. All of the plasmid constructs were expressed in Escherichia coli strain BL21 and purified using glutathione-Sepharose beads according to the manufacturer (Sigma). Purified proteins were quantified by Coomassie staining after SDS-PAGE separation and by the Bradford protein assay. BL21 cells were also used for bacterial expression of untagged MyoD as described in Ref. 29.
Beads coated with equal amounts of GST fusion proteins (1 μg) were incubated with 100 ng of recombinant MyoD in NTEN buffer (100 mm NaCl, 20 Mm Tris-HCl, pH 8, 0.5 mm EDTA, 0.1% Nonidet P-40) completed with protease inhibitors (Roche Applied Science) for 4 h at 30 °C. The beads were then washed four times with washing buffer (150 mm NaCl, 10 mm Tris-HCl, pH 7.5, 0.1% Nonidet P-40, 1 mm phenylmethylsulfonyl fluoride) and resuspended, and the proteins were resolved by SDS-PAGE gel for Western blot analysis.
For this GST pulldown assays using MyoD deletion mutants, HEK 293 cells were transfected with 10 μg of expression plasmids of tagged MyoD (wild type MyoD) or its deletion mutants (Cter, Nter, ΔCter, and ΔNter), using Lipofectamine (Qiagen). 48 h post-transfection, the cells were lysed in lysis buffer (300 mm NaCl, 50 mm Tris-HCl, pH 7.5, 0,4% Nonidet P-40, 10 mm MgCl2) to extract MyoD or its deletion mutants. GST pulldown assays were then performed as described above.
Gene Reporter Assays—HEK 293 cells at 60% confluence were cotransfected by calcium phosphate coprecipitation. 24 h post-transfection, the cells were lysed in a reporter lysis buffer (Promega, Charbonnières, France). Luciferase activity (Promega luciferase assay system) was determined and normalized to the level of β-galactosidase (Promega β-galactosidase enzyme assay system) and to the total protein amount. The plasmids used in the assay were pEMSV-MyoD, pEMSV, pcDNA3-HP1α, pcDNA3-HP1β, pcDNA3-HP1γ, pcDNA3, pMCK-Luciferase reporter gene, and pCMV-β-galactosidase.
Antibodies—The C-20 anti-MyoD, M-225 anti-myogenin, normal rabbit IgG, and normal mouse IgG antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-HP1 antibodies (2HP2G9, 1MOD1A9AS, and 2MOD1GC) were obtained from Euromedex (Souffelweyersheim, France). Rabbit polyclonal anti-Suv39h1 (suppressor of variegation 39h1) antibody (catalog number 07-550) was obtained from Upstate Biotechnology, Inc.. Rabbit polyclonal anti-MCK antibody was developed by Dr. H. Ito (30). The horseradish peroxidase-streptavidin conjugate (Sigma; catalog number S 2438), anti-FLAG and anti-α-tubulin antibodies were purchased from Sigma. Rat anti-HA antibody was purchased from Roche Applied Science.
Chromatin Immunoprecipitation (ChIP)—ChIP protocol and primers have been described in Ref. 26.
RESULTS
HP1 Proteins Interact with MyoD—To exhaustively characterize MyoD protein partners, we first purified MyoD complex from a HeLa cell line ectopically expressing a double tagged form of MyoD. To this end, we performed double affinity purification of the HA-FLAG-MyoD complex (Fig. 1A) from chromatin enriched in mononucleosomes (as described in Ref. 27). Mass spectrometry analysis of MyoD complex confirmed some “historical” partners of MyoD such as Id, E12/E47, and MEIS1, and other partners that have never been described to interact with MyoD, among them HP1 proteins, as confirmed by Western blotting (Fig. 1B). Thus, MyoD can coexist with HP1 proteins in the same complex, at least in an artificial cell system.
FIGURE 1.
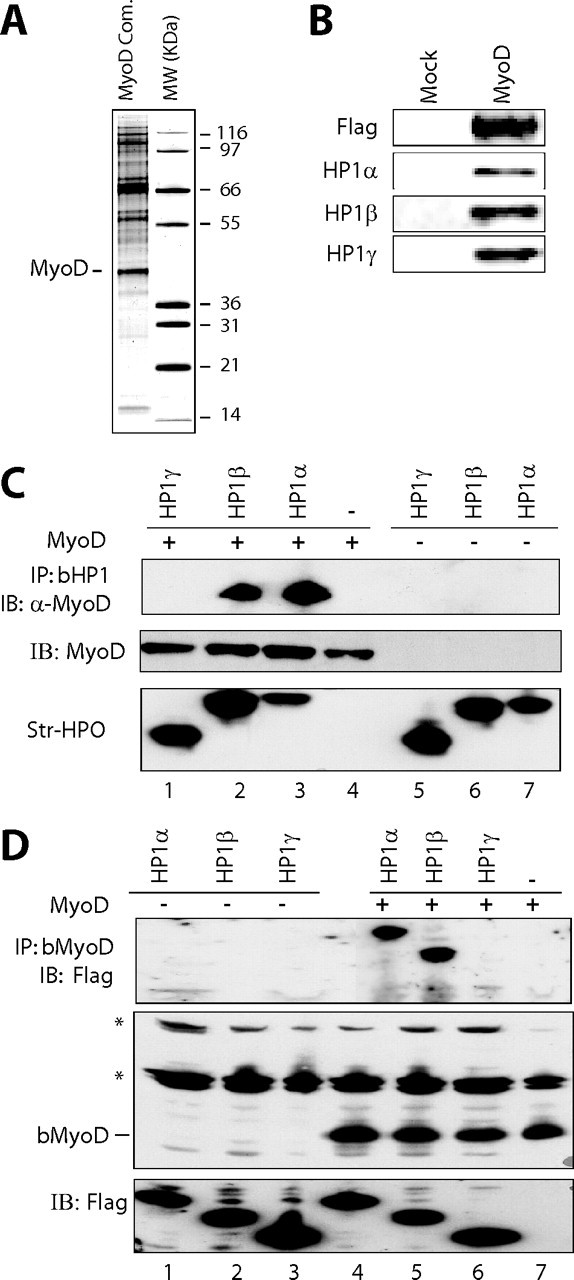
Ectopically expressed MyoD interacts with endogenous HP1 proteins. A, silver staining the double affinity-purified MyoD complex isolated from chromatin fractions in a HeLa cell line stably expressing FLAG-HA tagged MyoD. MW, protein molecular weight marker. The molecular masses of the markers are indicated. B, Western blot analysis of double purified FLAG-HA-MyoD (MyoD) or eluate from HeLa control cells (Mock). C, top panel, Western blotting with anti-MyoD on streptavidin-bead precipitates from HEK 293 cell extracts transfected with a MyoD expression vector (lanes 1–4) or with an empty vector (lanes 5–7), along with expression vectors for the biotinylated forms of HP1α (bHP1α)(lanes 3 and 7), HP1β (lanes 2 and 6), HP1γ (lanes 1 and 5), or the empty vector (lane 4). Middle and bottom panels, quantity control of MyoD detected with an anti-MyoD antibody (middle panel) and biotinylated HP1 detected using streptavidin-horseradish peroxidase conjugate (bottom panel) in the inputs. D, top panel, Western blotting of anti-FLAG-HP1 of streptavidin bead precipitates from HEK 293 cell extracts transfected with the expression vector for FLAG-HP1α (lanes 1 and 4), FLAG-HP1β (lanes 2 and 5), HP1γ (lanes 3 and 6), or with the empty vector (lane 7), along with expression vectors for the biotinylated form of MyoD (lanes 4–7) or the empty vector (lanes 1–3). Middle and bottom panels, quantity control of biotinylated MyoD (bMyoD) detected using streptavidin-horseradish peroxidase conjugate (middle panel) and that of FLAG-HP1 protein in the inputs as detected with an anti-FLAG antibody (bottom panel). *, endogenous biotinylated proteins. IB, immunoblot.
To further characterize these interactions, we used HEK 293 human cells to express biotinylatable forms of either MyoD or HP1 proteins α, β, or γ (as described in Ref. 26). The system is based on the coexpression of the target protein fused to a short biotin acceptor domain together with the biotinylating enzyme BirA from E. coli. The strength of the biotin-streptavidin interaction allows a robust characterization of protein-protein interactions, even in stringent conditions. Using biotinylated HP1 proteins, detected with streptavidin-horseradish peroxidase (Fig. 1C, bottom panel), we could precipitate MyoD with HP1α and HP1β but not with HP1γ (Fig. 1C, lanes 1–3). The signal is specific for HP1α and β; we did not detect MyoD in the HP1γ precipitate, nor in the control cells, which do not express any biotinylatable protein (Fig. 1C, lanes 1 and 4, respectively), nor in the cells that express biotinylated HP1 proteins but not MyoD (Fig. 1C, lanes 5–7). The level of ectopic MyoD was normalized by Western blotting, and that of biotinylated HP1 proteins was normalized by streptavidin-horseradish peroxidase (Fig. 1C, bottom panel). The reverse experiment using biotinylated MyoD and FLAG-tagged HP1 proteins (Fig. 1D, top panel) led to the same conclusion: a preferential interaction of MyoD with HP1α and HP1β, but not with HP1γ. The precipitation of biotinylated MyoD by streptavidin beads coprecipitated HP1α and HP1β but never HP1γ (Fig. 1D, lanes 4–6). The coprecipitation of HP1α and HP1β with biotinylated MyoD was specific, because there was no signal in cells not transfected with the biotinylated MyoD expressing vector (Fig. 1D, lanes 1–3). The absence of an interaction between MyoD and HP1γ is not due to a low expression of this isoform, because this is shown in the bottom panel of Fig. 1D (normalization of the inputs). Taken together, these results confirm, in cells, the specific interaction of MyoD with HP1α and HP1β, but not HP1γ.
MyoD Interacts with HP1α and HP1β, but Not with HP1γ, in Proliferating Myoblasts—To confirm the interaction between MyoD and HP1 proteins in a more physiological context, we used C2C12 myoblasts. In this system, MyoD is expressed at a low level in proliferating myoblasts, and its expression increases upon initiation of terminal differentiation. Immunoprecipitation (IP) experiments were performed using isoform-specific anti-HP1 or anti-MyoD antibodies or an anti-Suv39h1 antibody as a positive control, because this H3K9 methylase is known to interact with HP1. Indeed, Suv39h1 coprecipitated the three isoforms of HP1 as expected (Fig. 2A). Endogenous MyoD specifically coprecipitated endogenous HP1α and HP1β, but not HP1γ (Fig. 2A), and the IgG control did not show any HP1 signal (Fig. 2A). The results of the reciprocal IP confirmed these conclusions: IP of HP1α or HP1β, but not HP1γ, coprecipitated MyoD (Fig. 2B), and the control IP did not give any detectable signal (Fig. 2B). The absence of the interaction between MyoD and HP1γ is not due to a low expression of the latter, because this isoform is highly detected by Western blotting. Taken together, these results show that MyoD interacts preferentially with the α and β isoforms of HP1, but not with the γ isoform, in myoblasts.
FIGURE 2.
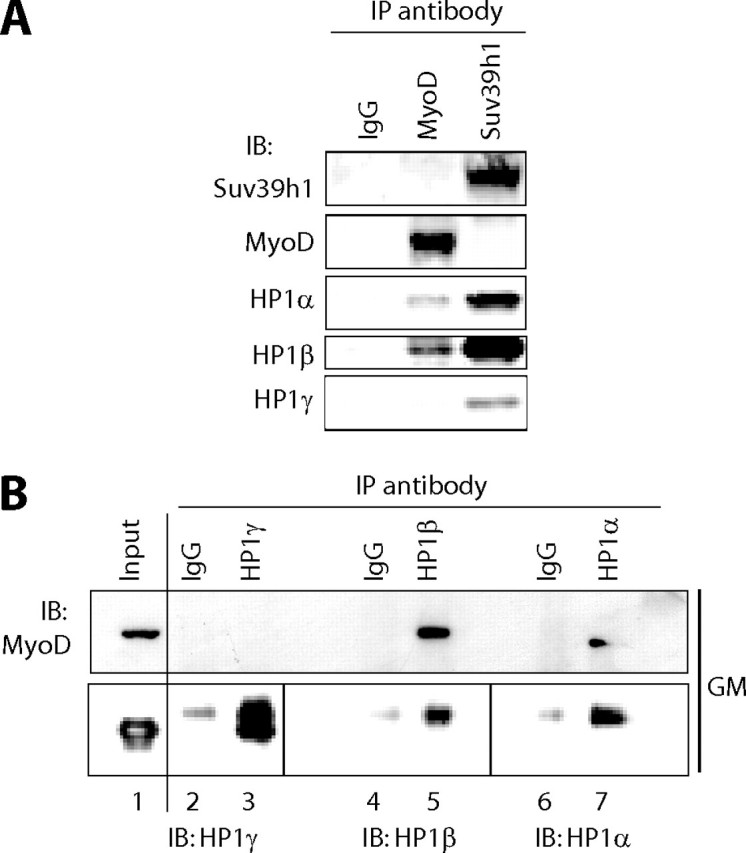
HP1α and HP1β, but not HP1γ, coprecipitate with MyoD in myoblasts. Nuclear extracts from growing myoblasts were used for immunoprecipitation with antibodies raised against MyoD, Suv39h1, or control beads (A) or against HP1α, HP1β, HP1γ, or normal mouse IgG as a negative control (B). The resulting precipitates were then subjected to Western blotting (immunoblot, IB) analysis for the presence of Suv39h1, MyoD, HP1α, HP1β, and HP1γ as indicated in A and B. Total input lysate was loaded in B to show that HP1γ is present in the inputs, even if it is not present in the IP. Lower panel of B, the faint signal obtained with anti-HP1 Western blotting in the control IgG IP corresponds to IgG light chain (noise).
MyoD Interacts Directly with HP1α and HP1β in Vitro—We studied the possible direct interaction between MyoD and HP1 proteins. GST-HP1 fusion proteins produced in bacteria were immobilized on agarose-glutathione beads, and an untagged recombinant form of MyoD (purification protocol described in Ref. 29) was detected by Western blotting using an anti-MyoD antibody. GST pulldown experiments showed that MyoD interacts with HP1α and HP1β but not with HP1γ (Fig. 3A, lanes 1–3). The interaction of MyoD with HP1α and HP1β was specific; we did not detect any MyoD signal in the presence of GST protein alone (Fig. 3A, lane 4). The quantity of each GST-HP1 protein was checked by Western blotting using an anti-GST antibody (Fig. 3A, lower panel). The interaction of HP1 proteins with MyoD was specific and was not seen with myogenin, another myogenic basic helix loop helix factor (Fig. 3B). These results show that, very interestingly, MyoD, but not myogenin, interacts specifically and directly with the isoforms α and β of HP1.
FIGURE 3.
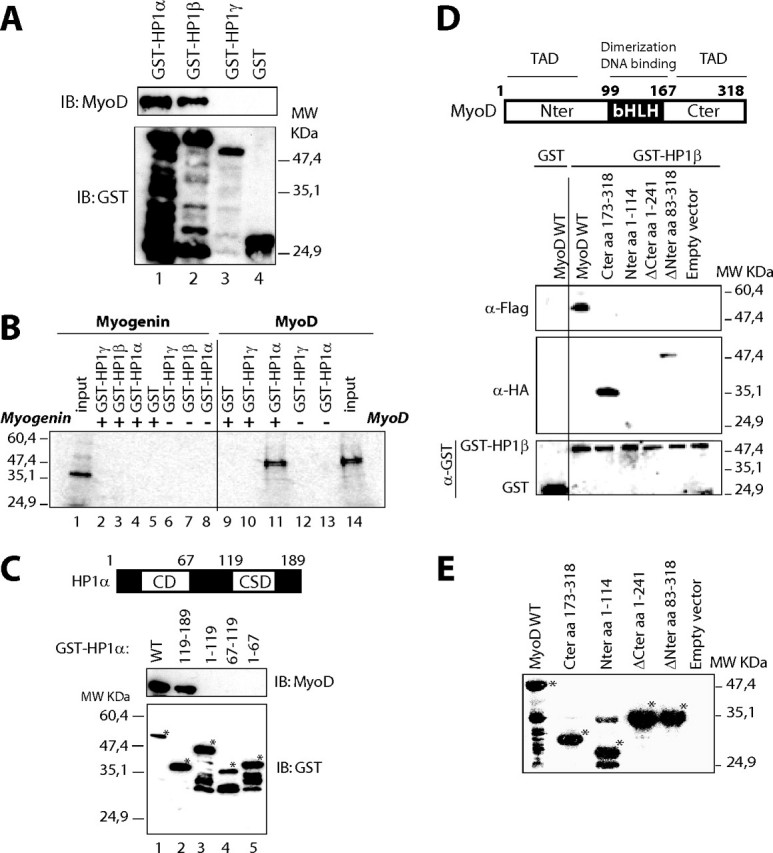
HP1α and HP1β, but not HP1γ, interact directly with MyoD in vitro via their chromoshadow domain and MyoD transactivating domain. A, HP1α and HP1β interact directly with MyoD in a GST pulldown assay. Equivalent amounts of bacterially produced untagged MyoD were incubated with GST beads (lane 4) or GST-HP1 beads (lanes 1–3). MyoD was detected by Western blotting using an anti-MyoD antibody (upper panel), and GST fusions using an anti-GST antibody (lower panel). IB, immunoblot. B, unlike MyoD, myogenin does not interact with HP1 proteins. Myogenin and MyoD were in vitro translated in the presence of radioactive [35S] methionine (inputs on lanes 1 and 14, respectively). MyoD or myogenin were incubated with equivalent amounts of either GST or GST-HP1 proteins, as indicated. GST pulldown was then conducted as described under “Experimental Procedures,” except that at the end of the experiment, the radiolabeled proteins were detected by autoradiography. Lanes 1–8, GST pulldown experiment with [35S]myogenin; lanes 9–14, with [35S]MyoD. C, the chromoshadow domain of HP1α is required for interaction with MyoD. Equivalent amounts of bacterially produced MyoD were incubated with GST-HP1α deletion mutants on beads (lanes 2–5) or GST-HP1α wild type (lane 1). MyoD was detected by Western blotting using an anti-MyoD antibody (upper panel) and GST fusions using an anti-GST antibody (lower panel). D, HP1β interacts with MyoD wild type and with MyoD deletion mutants containing the C-terminal domain. Top panel, schematic diagram of MyoD functional domains. TAD, transactivation domain. Lower panels, wild type MyoD was detected using anti-FLAG antibody, and its deletion mutants were detected using anti-HA antibody (upper panels). GST and GST-HP1β were detected using anti-GST antibody (lower panel). E, Western blotting using anti-FLAG and anti-HA to verify the expression level of MyoD and its deletion mutants in HeLa cells used in D*, specific bands.
The Chromoshadow Domain of HP1α and the C-terminal Domain of MyoD Are Required for Their Interaction—In an attempt to delimit the domain of HP1 responsible for interaction with MyoD, we used deletion mutants of HP1α fused to GST. A GST pulldown experiment was performed using bacterially produced MyoD as described above. GST pulldown revealed an interaction of MyoD with the wild type HP1α as expected (Fig. 3C, lane 1) and with a HP1α 119–189 mutant, which retains the CSD (Fig. 3C, lane 2). MyoD failed to interact with truncated HP1α versions lacking the CSD, i.e. mutants 1–119, 67–119, and 1–67 (Fig. 3C, lanes 3–5). The amounts of the different GST-HP1α mutants were checked by Western blotting (Fig. 3C, bottom panel). These experiments clearly show that the CSD of HP1α is required for interaction with MyoD.
The same experiments were performed using tagged deletion mutants of MyoD ectopically expressed in HEK 293 cells. The results show that the C-terminal domain of MyoD is required for the interaction with HP1 (Fig. 3, D and E).
HP1 Represses MyoD Transcriptional Activity—To investigate the effects of HP1 proteins on MyoD transcriptional activity, we used a Luciferase reporter gene under the control of the muscle creatine kinase (MCK) promoter, which is a direct target promoter of MyoD. Cotransfection experiments were performed in different nonmuscle cells lines that do not express MyoD endogenously. We observed that the cotransfection of either HP1α- or HP1β-expressing plasmid together with MyoD expression vector resulted in the inhibition of MCK promoter activity in a dose-dependent manner (Fig. 4, A and B, black bars). Interestingly, cotransfection of a HP1γ expression vector had no effect on the activity of MyoD (Fig. 4C, black bars). The expression of transfected MyoD was not influenced by cotransfection with HP1 expression vectors as determined by Western blotting (data not shown). The inhibitory effect of HP1α and HP1β is specific and is MyoD-dependent. Indeed, it was not seen with β-galactosidase expression under a cytomegalovirus promoter (used as a normalization control for transfection efficiency) nor with a pMCK-luciferase vector in the absence of MyoD (Fig. 4, gray bars). Thus, HP1α and HP1β, but not HP1γ, directly inhibit MyoD activity.
FIGURE 4.
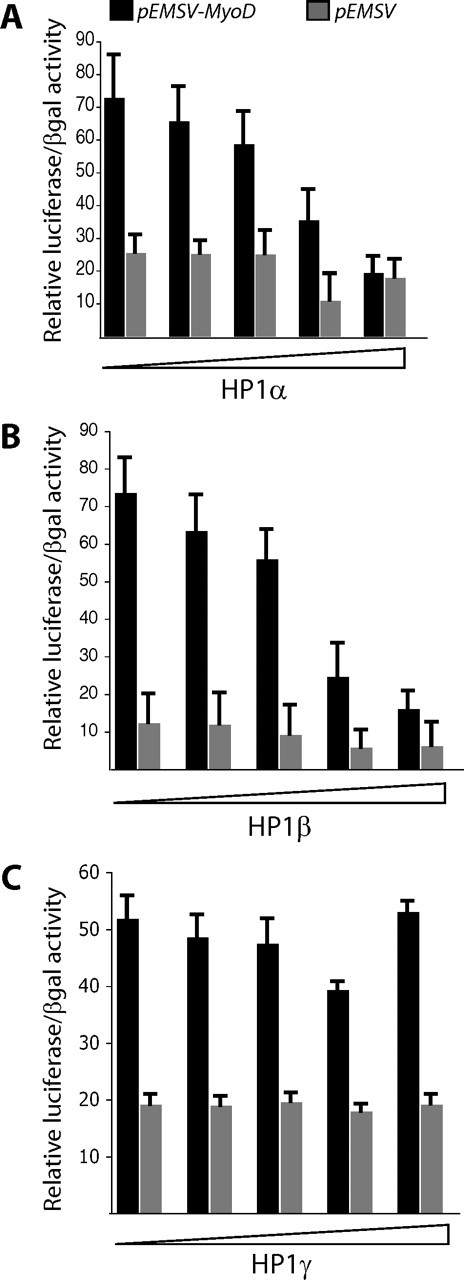
HP1α and HP1β, but not HP1γ, repress MyoD-mediated transcription. Cotransfection into HEK 293 cells of an MCK promoter-driven luciferase reporter plasmid with a fixed amount of MyoD expression vector (800 ng), and increasing amounts of pcDNA3 vector expressing HP1α (A), HP1β (B), or HP1γ (C). The quantities of different pcDNA3-HP1 expression vectors used were: 0, 100, 200, 300, and 500 ng. The total amount of pcDNA3 was normalized when necessary to 500 ng. Expression of MyoD and different HP1s was analyzed by Western blotting (data not shown). The inhibitory effect of HP1α and HP1β was specific and MyoD-dependent (compare black bars to gray bars). Indeed, it was not seen with β-galactosidase expression under a cytomegalovirus promoter (used as a normalization control for the transfection), nor with the MCK-luciferase vector in the absence of MyoD (gray bars). Black bars correspond to transfections with pEMSV-MyoD vector, and gray bars correspond to the empty vector control pEMSV.
HP1 Protein Isoforms Are Preferentially Recruited to MyoD Target Genes in Proliferating Myoblasts—To test the recruitment of HP1 isoforms into MyoD target promoters, we performed ChIP experiments using specific antibodies to each HP1 isoform. Our results show a preferential enrichment in all the three HP1 isoforms on both p21 and MCK promoters, which are MyoD targets activated early and late in differentiating cells, respectively, in proliferating myoblasts compared with differentiating myotubes (Fig. 5). Thus, even HP1γ, which does not interact directly with MyoD, is recruited on MyoD target promoters, suggesting that HP1γ may regulate myogenesis independently of any interaction with MyoD (see below). These results suggest that all the three HP1 isoforms regulate MyoD target genes in proliferating myoblasts using different mechanisms. Note that HP1 proteins are not found on coding regions of MyoD target genes (data not shown).
FIGURE 5.

Preferential recruitment of HP1 on MyoD target promoters in proliferating myoblasts. Chromatin fractions from either proliferating C2C12 myoblasts (black bars) or cultured 48 h in differentiating medium (gray bars) were immunoprecipitated using anti-HP1 antibodies (HP1) and analyzed by quantitative PCR. We quantified copy numbers of the p21 and MCK promoters regions harboring the MyoD-binding site. gapdh and 36B4 genes were used as negative controls to normalize our ChIP results. The results are the means of two independent experiments and six PCR measurements.
HP1 Levels Are Crucial for Muscle Terminal Differentiation—To test more generally the role of HP1 proteins in muscle terminal differentiation, we employed gain-of-function strategy. To ectopically express HP1 protein isoforms, we used a retrovirus expressing HA and FLAG-tagged isoforms of HP1 or an empty vector as a negative control to transduce C2C12 cells (see “Experimental Procedures”). Immunofluorescence studies using anti-HA antibody showed that the exogenous HP1 isoforms had normal subnuclear localization (Fig. 6A). Overexpression of any of the HP1 isoforms, α, β, or γ, resulted generally in the inhibition of differentiation compared with the control cell line. HP1-overexpressing cells did not express the differentiation markers myogenin and MCK (Fig. 6B) and did not fuse into myotubes (data not shown). The faint expression of MCK and myogenin in HP1α-expressing cells cultured in differentiation medium was due to the fact that this cell line was not pure for the expression of tagged HP1α, ∼90%; during culture, some cells lost tagged HP1α expression. We presume that these cells differentiated normally and expressed myogenin and MCK.
FIGURE 6.
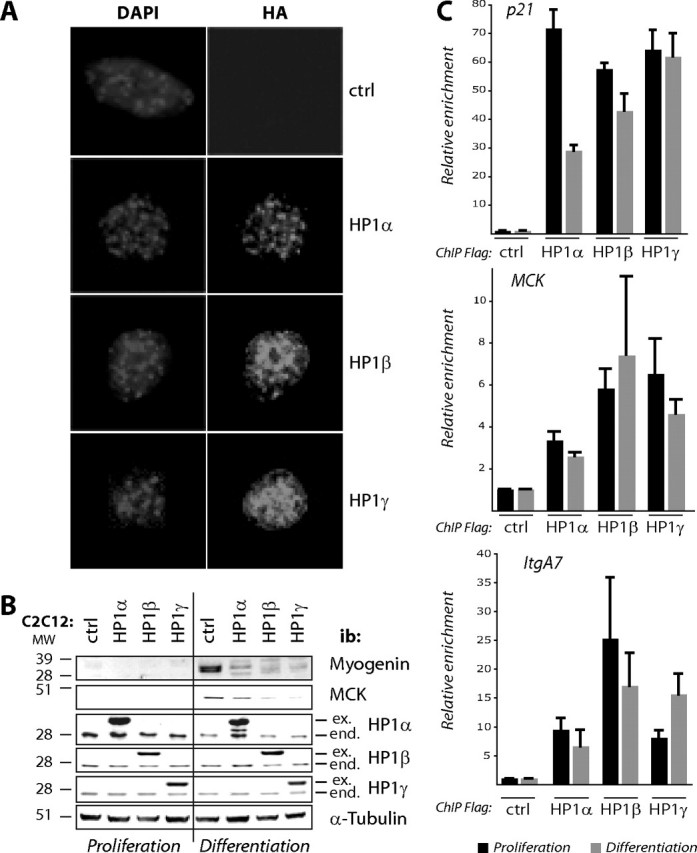
Modulating the levels of HP1 isoforms affects muscle terminal differentiation. A and B, C2C12 cells stably expressing HA-FLAG tagged HP1 isoforms as indicated or control cells (ctrl) were cultured under proliferation conditions or in differentiation medium. Proliferating cells were tested by immunofluorescence using anti-HA antibody (Roche Applied Science) and 4′,6′-diamino-2-phenylindole (DAPI) to stain DNA (A) or analyzed by Western blotting with isoform-specific anti-HP1, anti-myogenin, anti-MCK, and anti-α-tubulin as a loading control (B). Note that the kinetic studies were carried out in the same 10-cm-diameter cell culture dish for each sample. C, FLAG-HA tagged HP1 isoforms are recruited to MyoD target promoters regardless of differentiation. ChIP experiments using anti-FLAG resin were performed from myoblasts stably expressing the tagged HP1 isoforms (or from the control cell line, ctrl) either proliferating (black bars) or cultured in differentiation conditions (gray bars). We quantified copy numbers of the MCK, ItgA7, and p21 promoter regions harboring the MyoD target sequence. The 36B4 gene was used as a negative control. The results are the means of three independent experiments.
Overexpression of HP1 proteins could have broad effects (31). Thus, differentiation inhibition we have seen in cells overexpressing HP1 proteins could be a result of these broad effects. To check the specificity of the effects seen when we overexpress HP1 isoforms (Fig. 6, A and B), we performed ChIP experiments using anti-FLAG resin to test whether the tagged HP1 isoforms are recruited to MyoD target genes. Our results show that the exogenous HP1 isoforms are indeed physically recruited into MyoD target genes (Fig. 6C), but unlike the endogenous HP1 (Fig. 5), these exogenous isoforms remain on MyoD target promoters even in myoblasts cultured in differentiation conditions (Fig. 6C). This result suggests that the differentiation defect seen in HP1-overexpressing myoblasts could be due, at least in part, to a lack of de-repression of MyoD target genes. Taken together, these results suggest that overexpression of HP1 proteins impairs MyoD target genes expression and thus muscle terminal differentiation, regardless of the HP1 isoform.
DISCUSSION
Repression of MyoD target genes in proliferating myoblasts involves H3K9 methylation (20, 22), which is normally recognized by HP1 proteins (4, 5). This suggests the formation of local facultative heterochromatin on MyoD target promoters, insuring their stable repression until the cells undergo terminal differentiation. We tested the hypothesis that HP1 proteins are involved in the repression of MyoD target genes and explored their role in muscle terminal differentiation in general.
Physical and Functional MyoD/HP1 Interaction—Immunoprecipitation assays showed that MyoD interacts with HP1α and HP1β, but not with HP1γ in myoblasts. In vitro assays demonstrated that MyoD interacts directly with HP1α and HP1β via their chromoshadow domains and the MyoD transactivating domain. A reporter gene assay indicated that this interaction contributes to MyoD-mediated transcriptional repression. Our results provide evidence for a physical interaction between MyoD and HP1α/β and suggest a direct role for these two HP1 isoforms in regulating the repressive function of MyoD. A similar result has been described for HP1α and muscle enhancer factor 2C, which belongs to the muscle enhancer factor 2 family of myogenic transcription factors (22). HP1α has also been shown to interact and cooperate with two critical hematopoietic transcription factors, PU.1/GATA-1 (32) and the differentiation factor C/EBPα (33). In the case of MyoD, it has been shown to bind target promoters in proliferating myoblasts, where it acts as a repressor (20). This repression involves HDACs, among other factors (25). MyoD binds HDAC1 via its helix-loop-helix domain (25) and HP1 via its C-terminal domain, which contains a pseudo-PXVXL motif (PALLL) at position 266, found in many HP1-interacting proteins (34). Such a motif is not found in myogenin, which does not interact with any of the HP1 isoforms. Thus, MyoD could form a ternary complex with HDAC1 and HP1. MyoD could thus serve as a bridge between nucleosomes and chromatin-binding proteins. The first enzymes to be recruited by MyoD could be HDACs that deacetylate H3K9 to allow its subsequent methylation by a histone methyltransferase (20, 22). Methylated H3K9 could then be recognized and stabilized by HP1 proteins. Thus, there would be a complex composed of MyoD-HDAC-histone methyltransferase-HP1, which ensures the stable silencing of MyoD target genes by preventing H3K9 acetylation before the initiation of differentiation, as previously suggested in: (22). In conclusion, the equilibrium between H3K9 acetylation and methylation at MyoD target promoters may represent a switch that determines the timing of differentiation.
Specificity of the Interaction of MyoD with HP1α and HP1β—We report differential associations of HP1α/β with MyoD, whereas HP1γ does not associate with MyoD, at least in our hands. The only system in which HP1γ was found in association with MyoD was in HeLa cells. Because this interaction was not found in myoblasts, it could be due to the large amount of HeLa cells we used to purify MyoD complex. We concluded that MyoD does not directly interact with HP1γ.
HP1 proteins share extensive structural identity and several characteristics (35–40). Despite these structural and biochemical similarities, HP1 proteins differ in some properties. For example, they differ in their subnuclear distribution (28, 41, 42). In addition, HP1γ is the only isoform that has been linked to transcription activation (43). Finally, among the HP1 binding proteins, several have been reported to interact with all HP1 isoforms, such as SP100; in contrast, TAFII130 binds to HP1α and HP1γ, but not to HP1β (41), and BRG1 binds only to HP1α (28). Thus, the absence of interaction between MyoD and HP1γ could be due to structural and subnuclear localization differences compared with the two other HP1 isoforms. Together, these results strongly support the notion that HP1 proteins form distinct complexes in cells (28, 44) and suggests both shared and distinctive roles for these proteins in muscle development.
HP1 Level Is Crucial for Skeletal MyoD Target Genes Expression and Muscle Terminal Differentiation—It was shown that HP1 isoform expression levels do not change during muscle differentiation (45), and our results show that HP1 isoform levels do not significantly change in regenerating muscle, with only a slight increase in HP1α and HP1γ levels (not shown). As observed with overexpression of Suv39h1 (Refs. 42 and 46) and unpublished results), overexpression of any of the three HP1 isoforms resulted in the loss of differentiation capacity. Our results show that exogenous HP1 isoforms remain on MyoD target promoters even in differentiation conditions. Thus, overexpression of HP1 or Suv39h1 could preclude de-repression of MyoD target genes, thus inhibiting terminal differentiation.
However, down-regulation of HP1 isoforms with small interfering RNAs resulted in a generally poor differentiation efficiency (supplemental data). This result was unexpected, because we have shown that HP1α and HP1β are involved in the regulation of MyoD activity. This apparent contradiction could be explained several ways. First of all, down-regulation of only one HP1 isoform might not be sufficient to activate MyoD target genes. One can also hypothesize that H3K9 remains methylated in the absence of one HP1 isoform and could be sufficient per se to silence transcription as described by others (47). In addition, other repressor proteins, like the histone H3 lysine 27 methylase EZH2 (48) and HDAC1 (25), are recruited to silent MyoD target genes in myoblasts, and this could be sufficient to inhibit gene expression per se in the absence of HP1. Secondly, down-regulation of one HP1 isoform may be sufficient to interfere with (or inhibit) general chromatin reorganization, which occurs in differentiating myoblasts (11, 12, 14), thereby inhibiting terminal differentiation. More likely, HP1 proteins are required to silence proliferation-associated genes in differentiating cells, a step that is obligatory to initiate terminal differentiation (see supplemental data). Indeed, we previously found comparable results with Suv39h1, a H3K9 histone methyltransferase, which is involved in muscle terminal differentiation by silencing proliferation genes, i.e. E2F target genes (16). It has also been shown that HP1 proteins associate with proliferation genes only in differentiated cells, where these genes are irreversibly silenced (Ref. 7) and unpublished results) and upon triggering irreversible cell cycle exit in senescent cells (17). Thus, when we down-regulate HP1 protein expression in myoblasts and try to induce differentiation, E2F target genes are not correctly silenced in the absence of HP1, cells do not undergo irreversible cell cycle exit, and differentiation cannot start (supplemental data). We have observed a similar phenotype when we down-regulate the Suv39h1 expression in myoblasts (Ref. 16 and unpublished results).
Concerning HP1γ, ChIP experiments showed its preferential enrichment on MyoD target genes in proliferating myoblasts. Although we observed that HP1γ does not interact directly with MyoD, its overexpression interferes with muscle terminal differentiation. Thus, HP1γ might regulate myogenesis independently of any direct interaction with MyoD. Because it is known that HP1 proteins heterodimerize, HP1γ could be recruited by HP1α/β. In addition, HP1γ might be necessary for the general nuclear reorganization occurring in differentiating myoblasts. Indeed, mutation in the Su(var)205 gene, which encodes HP1, in Drosophila melanogaster causes lethality at larval stages (49), suggesting a general role of HP1 as a non-histone chromatin protein. However, to our knowledge, no HP1 knock-out mice have been described to date.
In conclusion, our results provide additional evidence that members of the HP1 protein family are functionally distinct and point to a novel role for MyoD in the organization of heterochromatin-like local structures on its targets in proliferating myoblasts. These data strongly suggest that, in addition to the role of MyoD as an activator of differentiation-specific genes, it can also act as an active transcriptional repressor in proliferating myoblasts, in cooperation with specific isoforms of HP1 proteins.
Supplementary Material
Acknowledgments
We warmly thank Dr. S. A. Leibovitch for providing MyoD expressing vectors, Dr. H. Ito for the kind gift of anti-MCK antibody, Drs. D. Trouche and A. Hamiche for providing crucial reagents, and Drs. V. Ogryzko and A. Viens for sharing plasmids and technical help. We thank Dr. L. L. Pritchard and A. Marchand for critical reading of the manuscript.
This work was supported by the Association Française contre les Myopathies, the Fondation Bettencourt-Schueller, the Ligue Nationale contre le Cancer, the Association pour la Recherche sur le Cancer, Ministèredela Recherche ACI-Jeunes Chercheurs Décision 04375, the CNRS, Grant LSHG-CT-2004-502950 from the European Union 6th Framework program (to A. H.-B.), and the Université Paris-Sud Orsay. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains four supplemental figures. We dedicate this work to the memory of Hélène Richard-Foy.
Footnotes
The abbreviations used are: HP1, heterochromatin protein 1; CSD, chromoshadow domain; H3K9, histone H3 lysine 9; HDAC, histone deacetylase; MCK, muscle creatine kinase; HA, hemagglutinin; GST, glutathione S-transferasel; ChIP, chromatin immunoprecipitation; IP, immunoprecipitation.
References
- 1.Hediger, F., and Gasser, S. M. (2006) Curr. Opin. Genet. Dev. 16 143–150 [DOI] [PubMed] [Google Scholar]
- 2.Cavalli, G., and Paro, R. (1998) Curr. Opin. Cell Biol. 10 354–360 [DOI] [PubMed] [Google Scholar]
- 3.Dialynas, G. K., Terjung, S., Brown, J. P., Aucott, R. L., Baron-Luhr, B., Singh, P. B., and Georgatos, S. D. (2007) J. Cell Sci. 120 3415–3424 [DOI] [PubMed] [Google Scholar]
- 4.Lachner, M., O'Carroll, D., Rea, S., Mechtler, K., and Jenuwein, T. (2001) Nature 410 116–120 [DOI] [PubMed] [Google Scholar]
- 5.Bannister, A. J., Zegerman, P., Partridge, J. F., Miska, E. A., Thomas, J. O., Allshire, R. C., and Kouzarides, T. (2001) Nature 410 120–124 [DOI] [PubMed] [Google Scholar]
- 6.Lomberk, G., Wallrath, L., and Urrutia, R. (2006) Genome Biol. 7 228–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Panteleeva, I., Boutillier, S., See, V., Spiller, D. G., Rouaux, C., Almouzni, G., Bailly, D., Maison, C., Lai, H. C., Loeffler, J. P., and Boutillier, A. L. (2007) EMBO J. 26 3616–3628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meshorer, E., Yellajoshula, D., George, E., Scambler, P. J., Brown, D. T., and Misteli, T. (2006) Dev. Cell 10 105–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cammas, F., Herzog, M., Lerouge, T., Chambon, P., and Losson, R. (2004) Genes Dev. 18 2147–2160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cammas, F., Oulad-Abdelghani, M., Vonesch, J. L., Huss-Garcia, Y., Chambon, P., and Losson, R. (2002) J. Cell Sci. 115 3439–3448 [DOI] [PubMed] [Google Scholar]
- 11.Francastel, C., Schubeler, D., Martin, D. I., and Groudine, M. (2000) Nat. Rev. Mol. Cell Biol. 1 137–143 [DOI] [PubMed] [Google Scholar]
- 12.Meshorer, E., and Misteli, T. (2006) Nat. Rev. Mol. Cell Biol. 7 540–546 [DOI] [PubMed] [Google Scholar]
- 13.Guasconi, V., Souidi, M., and Ait-Si-Ali, S. (2005) Cancer Biol. Ther. 4 134–138 [DOI] [PubMed] [Google Scholar]
- 14.Moen, P. T., Jr., Johnson, C. V., Byron, M., Shopland, L. S., de la Serna, I. L., Imbalzano, A. N., and Lawrence, J. B. (2004) Mol. Biol. Cell 15 197–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brero, A., Easwaran, H. P., Nowak, D., Grunewald, I., Cremer, T., Leonhardt, H., and Cardoso, M. C. (2005) J. Cell Biol. 169 733–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ait-Si-Ali, S., Guasconi, V., Fritsch, L., Yahi, H., Sekhri, R., Naguibneva, I., Robin, P., Cabon, F., Polesskaya, A., and Harel-Bellan, A. (2004) EMBO J. 23 605–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Narita, M., Nunez, S., Heard, E., Narita, M., Lin, A. W., Hearn, S. A., Spector, D. L., Hannon, G. J., and Lowe, S. W. (2003) Cell 113 703–716 [DOI] [PubMed] [Google Scholar]
- 18.Buckingham, M. (1996) Biochem. Soc. Trans. 24 506–509 [DOI] [PubMed] [Google Scholar]
- 19.Walsh, K., and Perlman, H. (1997) Curr. Opin. Genet. Dev. 7 597–602 [DOI] [PubMed] [Google Scholar]
- 20.Mal, A., and Harter, M. L. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 1735–1739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ohkawa, Y., Marfella, C. G., and Imbalzano, A. N. (2006) EMBO J. 25 490–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang, C. L., McKinsey, T. A., and Olson, E. N. (2002) Mol. Cell Biol. 22 7302–7312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de la Serna, I. L., Carlson, K. A., and Imbalzano, A. N. (2001) Nat. Genet. 27 187–190 [DOI] [PubMed] [Google Scholar]
- 24.Fulco, M., Schiltz, R. L., Iezzi, S., King, M. T., Zhao, P., Kashiwaya, Y., Hoffman, E., Veech, R. L., and Sartorelli, V. (2003) Mol. Cell 12 51–62 [DOI] [PubMed] [Google Scholar]
- 25.Mal, A., Sturniolo, M., Schiltz, R. L., Ghosh, M. K., and Harter, M. L. (2001) EMBO J. 20 1739–1753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Viens, A., Mechold, U., Lehrmann, H., Harel-Bellan, A., and Ogryzko, V. (2004) Anal. Biochem. 325 68–76 [DOI] [PubMed] [Google Scholar]
- 27.Robin, P., Fritsch, L., Philipot, O., Svinarchuk, F., and Ait-Si-Ali, S. (2007) Genome Biol. 8 R270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nielsen, A. L., Sanchez, C., Ichinose, H., Cervino, M., Lerouge, T., Chambon, P., and Losson, R. (2002) EMBO J. 21 5797–5806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Polesskaya, A., Duquet, A., Naguibneva, I., Weise, C., Vervisch, A., Bengal, E., Hucho, F., Robin, P., and Harel-Bellan, A. (2000) J. Biol. Chem. 275 34359–34364 [DOI] [PubMed] [Google Scholar]
- 30.Ito, H., Kamei, K., Iwamoto, I., Inaguma, Y., and Kato, K. (2001) Exp. Cell Res. 266 213–221 [DOI] [PubMed] [Google Scholar]
- 31.Sharma, G. G., Hwang, K. K., Pandita, R. K., Gupta, A., Dhar, S., Parenteau, J., Agarwal, M., Worman, H. J., Wellinger, R. J., and Pandita, T. K. (2003) Mol. Cell Biol. 23 8363–8376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stopka, T., Amanatullah, D. F., Papetti, M., and Skoultchi, A. I. (2005) EMBO J. 24 3712–3723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Demarco, I. A., Periasamy, A., Booker, C. F., and Day, R. N. (2006) Nat. Methods 3 519–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lechner, M. S., Schultz, D. C., Negorev, D., Maul, G. G., and Rauscher, F. J., III (2005) Biochem. Biophys. Res. Commun. 331 929–937 [DOI] [PubMed] [Google Scholar]
- 35.Minc, E., Allory, Y., Courvalin, J. C., and Buendia, B. (2001) Methods Cell Sci. 23 171–174 [DOI] [PubMed] [Google Scholar]
- 36.Minc, E., Allory, Y., Worman, H. J., Courvalin, J. C., and Buendia, B. (1999) Chromosoma 108 220–234 [DOI] [PubMed] [Google Scholar]
- 37.Minc, E., Courvalin, J. C., and Buendia, B. (2000) Cytogenet. Cell Genet. 90 279–284 [DOI] [PubMed] [Google Scholar]
- 38.Nielsen, A. L., Ortiz, J. A., You, J., Oulad-Abdelghani, M., Khechumian, R., Gansmuller, A., Chambon, P., and Losson, R. (1999) EMBO J. 18 6385–6395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nielsen, A. L., Oulad-Abdelghani, M., Ortiz, J. A., Remboutsika, E., Chambon, P., and Losson, R. (2001) Mol. Cell 7 729–739 [DOI] [PubMed] [Google Scholar]
- 40.Nielsen, S. J., Schneider, R., Bauer, U. M., Bannister, A. J., Morrison, A., O'Carroll, D., Firestein, R., Cleary, M., Jenuwein, T., Herrera, R. E., and Kouzarides, T. (2001) Nature 412 561–565 [DOI] [PubMed] [Google Scholar]
- 41.Vassallo, M. F., and Tanese, N. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 5919–5924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Czvitkovich, S., Sauer, S., Peters, A. H., Deiner, E., Wolf, A., Laible, G., Opravil, S., Beug, H., and Jenuwein, T. (2001) Mech. Dev. 107 141–153 [DOI] [PubMed] [Google Scholar]
- 43.Vakoc, C. R., Mandat, S. A., Olenchock, B. A., and Blobel, G. A. (2005) Mol. Cell 19 381–391 [DOI] [PubMed] [Google Scholar]
- 44.Aagaard, L., Laible, G., Selenko, P., Schmid, M., Dorn, R., Schotta, G., Kuhfittig, S., Wolf, A., Lebersorger, A., Singh, P. B., Reuter, G., and Jenuwein, T. (1999) EMBO J. 18 1923–1938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Agarwal, N., Hardt, T., Brero, A., Nowak, D., Rothbauer, U., Becker, A., Leonhardt, H., and Cardoso, M. C. (2007) Nucleic Acids Res. 35 5402–5408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mal, A. K. (2006) EMBO J. 25 3323–3334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stewart, M. D., Li, J., and Wong, J. (2005) Mol. Cell Biol. 25 2525–2538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Caretti, G., Di Padova, M., Micales, B., Lyons, G. E., and Sartorelli, V. (2004) Genes Dev. 18 2627–2638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eissenberg, J. C., James, T. C., Foster-Hartnett, D. M., Hartnett, T., Ngan, V., and Elgin, S. C. (1990) Proc. Natl. Acad. Sci. U. S. A. 87 9923–9927 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.