

Abstract
Prion protein-like protein/doppel is neurotoxic, causing ataxia and Purkinje cell degeneration in mice, whereas prion protein antagonizes doppel-induced neurodegeneration. Doppel is homologous to the C-terminal half of prion protein but lacks the amino acid sequences corresponding to the N-terminal half of prion protein. We show here that transgenic mice expressing a fusion protein consisting of the N-terminal half, corresponding to residues 1-124, of prion protein and doppel in neurons failed to develop any neurological signs for up to 730 days in a background devoid of prion protein. In addition, the fusion protein prolonged the onset of ataxia in mice expressing exogenous doppel. These results suggested that the N-terminal part of prion protein has a neuroprotective potential acting both cis and trans on doppel. We also show that prion protein lacking the pre-octapeptide repeat (Δ25-50) or octapeptide repeat (Δ51-90) region alone could not impair the antagonistic function against doppel.
The normal prion protein (PrPC)2 is a glycosylphosphatidylinositol (GPI)-anchored membrane glycoprotein expressed most abundantly in the central nervous system, particularly in neurons, and to a lesser extent in non-neuronal tissues, including the heart, lung, spleen, and kidney (1, 2). It is well known that conformational conversion of PrPC into the abnormally folded amyloidogenic isoform, PrPSc, plays a pivotal role in the pathogenesis of transmissible spongiform encephalopathies or prion diseases, including Creutzfeldt-Jakob disease in humans and bovine spongiform encephalopathy in cattle (1, 3). However, the physiological function of PrPC remains largely unknown.
We and others identified a novel gene, Prnd, that encodes a GPI-anchored PrP-like protein, termed Doppel (Dpl), 16 kb downstream of the murine PrP gene Prnp (4, 5). Dpl is expressed in the testis, heart, kidney, and spleen of wild-type mice but not in the brain where PrPC is actively expressed. Intriguingly, some lines of mice devoid of PrPC (Prnp0/0), including Ngsk, Rcm0, and Zrch II, ectopically expressed Dpl in their brains, particularly in neurons, because of an unusual intergenic splicing between Prnp and Prnd, developed ataxia, and Purkinje cell degeneration (5, 6). However, others, such as Zrch I and Npu, neither ectopically expressed Dpl nor exhibited ataxia and Purkinje cell degeneration (4, 5). It was finally confirmed that Dpl is neurotoxic, and PrPC antagonizes the neurotoxicity of Dpl by a demonstration that transgenically expressed Dpl caused ataxia and Purkinje cell degeneration in nonataxic Zrch I Prnp0/0 mice but not in wild-type mice (7-9). However, the exact mechanism of the antagonistic interaction of PrPC and Dpl remains unknown.
Dpl shares 23% identity in amino acid composition with PrP (4, 5) and bears conformational similarity to the C-terminal globular domain of PrPC, both comprising three α-strands and two short β-strands (10). However, Dpl lacks the amino acid sequences corresponding to the N-terminal half of PrPC (4, 5). Interestingly, it was shown that PrP with truncated N-terminal residues 32-121 or 32-134, termed PrPΔ32-121 or PrPΔ32-134, respectively, exhibited neurotoxicity similarly to that of Dpl, causing ataxia and cerebellar neurodegeneration in nonataxic Zrch I Prnp0/0 mice but not in wild-type mice (11, 12). Therefore, it might be possible that the neurotoxicity of Dpl is attributable to lack of the corresponding N-terminal part of PrPC. However, this remains to be elucidated.
We previously showed that the N-terminal residues 23-88 of PrPC are involved in the antagonistic function of PrPC against the Dpl neurotoxicity by demonstrating that PrP lacking the residues 23-88 completely lost the ability to rescue Ngsk Prnp0/0 mice from Dpl-induced Purkinje cell degeneration (13). Residues 23-88 include most of the PrP-specific octapeptide repeat (OR) region, which includes residues 51-90. Recent lines of evidence from cell culture experiments show that the OR may be involved in the neuroprotective function of PrPC (14-16). However, the biological relevance of OR in the neuroprotective function of PrPC against Dpl is not yet understood in vivo.
In this study, we generated transgenic (tg) mice, tg(PrPΔOR) and tg(PrPN-Dpl), expressing PrP lacking OR and Dpl fused with the N-terminal half of PrPC, respectively. We also produced tg(PrPΔpreOR) mice expressing PrP without the pre-OR region. By intercrossing these tg mice with mice transgenically overexpressing Dpl in neurons on the genetic background of nonataxic Zrch I Prnp0/0, we investigated whether or not these mutant molecules could antagonize Dpl neurotoxicity, rescuing mice from ataxia and Purkinje cell degeneration.
EXPERIMENTAL PROCEDURES
Construction of Transgenes—A DNA fragment corresponding to the N-terminal residues 1-124 of PrP was first amplified by PCR with primer a (5′-cccaagcttctcgagatggcgaaccttggc-3′, the underlined sequence corresponds to the HindIII and XhoI sites, and the boldface sequence represents a start codon) and primer f (5′-cttgatgaaggctccaaggccccccactac-3′, the underlined sequence corresponds to DNA encompassing residues 58-62 of Dpl, and the italic sequence corresponds to DNA encom-passing residues 120-124 of PrP) using PrP cDNA as a template. The resulting DNA fragment, containing the DNA sequence corresponding to residues 58-62 of Dpl at the 3′ site, was then utilized as a 5′ primer to amplify another DNA fragment corresponding to residues 58-179 of Dpl together with primer i (5′-cccaagcttctcgagttacttcacaatgaa-3′, the underlined sequence corresponds to the HindIII and XhoI sites, and the boldface sequence represents a stop codon) using Dpl cDNA as a template, resulting in amplification of a DNA fragment for the fusion protein PrPN-Dpl consisting of residues 1-124 of PrP and residues 58-179 of Dpl. After DNA sequence confirmation of the amplified fragment, it was inserted into a unique SalI site of the Syrian hamster PrP cosmid vector, CosSHa.tet (InPro Biotechnology, Inc. South San Francisco, CA), to construct the PrPN-Dpl transgene.
A DNA fragment corresponding to the N-terminal residues 1-24 of PrP was first amplified by PCR with primers a and c (5′-ggtgccaccctgaggctttttgcagaggcc-3′, the underlined and italic sequences correspond to DNAs encompassing residues 51-55 and 20-24 of PrP, respectively) using PrP cDNA as a template. The resulting DNA fragment containing the DNA sequence corresponding to residues 51-55 of PrP at the 3′ site was then utilized as a 5′ primer to amplify another DNA fragment corresponding to residues 51-254 of PrP together with primer g (5′-cccaagcttctcgagtcatcccacgatcag-3′, the underlined sequence corresponds to the HindIII and XhoI sites, and the boldface sequence represents a stop codon) using PrP cDNA as a template, resulting in amplification of a DNA fragment for the deletion protein PrPΔpreOR consisting of residues 1-24 and 51-254 of PrP. After DNA sequence confirmation of the amplified fragment, it was inserted into a unique SalI site of the Syrian hamster PrP cosmid vector, CosSHa.tet (InPro Biotechnology, Inc.), to construct the PrPΔpreOR transgene.
A DNA fragment corresponding to the N-terminal residues 1-50 of PrP was first amplified by PCR with primers a and d (5′-atgggtaccccctcctgggtaacggttgcc-3′, the underlined and italic sequences correspond to DNAs encompassing residues 91-95 and 46-50 of PrP, respectively) using PrP cDNA as a template. The resulting DNA fragment containing the DNA sequence corresponding to residues 91-95 of PrP at the 3′ site was then utilized as a 5′ primer to amplify another DNA fragment corresponding to residues 91-254 of PrP together with primer g using PrP cDNA as a template, resulting in amplification of a DNA fragment for the deletion protein PrPΔOR consisting of residues 1-50 and 91-254 of PrP. After DNA sequence confirmation of the amplified fragment, it was inserted into a unique SalI site of the Syrian hamster PrP cosmid vector, CosSHa.tet (InPro Biotechnology, Inc.), to construct the PrPΔOR transgene.
Generation of Transgenic Mice—The plasmid-derived sequences were removed from each of the transgene constructs, and the resulting DNAs were injected into the zygotes of C57BL/6 mice to generate tg mice as described elsewhere (17, 18).
Expression Vectors for Wild-type PrPC, PrPΔpreOR, PrPΔOR, and PrPΔ23-88—The DNA fragments encoding wild-type mouse PrPC and PrPΔ23-88 were amplified by PCR with a sense primer (5′-tcggatccagtcatcatggcgaaccttggc-3′; the underlined sequence corresponds to a BamHI site; the boldface sequence corresponds to a start codon) and an antisense primer (5′-cctctagacctcatcccacgatcaggaaga-3′; the underlined sequence corresponds to an XbaI site; the boldface sequence corresponds to a stop codon) using mouse genomic DNA extracted from wild-type mice and tg(PrPΔ23-88) mice (13). After confirmation of the DNA sequences, each DNA fragment was digested by BamHI and XbaI and introduced into a pcDNA3.1 vector (Invitrogen) to generate pcDNA3.1-moPrP and pcDNA3.1-PrPΔ23-88. pcDNA3.1-PrPΔpreOR and -PrPΔOR were constructed by digestion of each of the already cloned PCR products with HindIII and subsequent insertion of the digested fragments into a HindIII site of pcDNA3.1 vector.
Breeding Procedures—Zrch I Prnp0/0 mice on the C57BL/6 ×129Sv mixed background and tg(Dpl32) mice on the C57BL/6 background were generated as described (8, 19). tg(Dpl32)/Prnp0/0 mice were previously produced by serially mating tg(Dpl32) mice (C57BL/6) with Zrch I Prnp0/0 mice, which were obtained by mating pairs of Zrch I Prnp+/0 mice that had been generated by crossing Zrch I Prnp0/0 mice (C57BL/6 × 129Sv) with FVB wild-type mice. Thus, tg(Dpl32)/Prnp0/0 mice have a mixed genetic background of C57BL/6 × 129Sv × FVB. Tg(PrPN-Dpl), tg(PrPΔpreOR), and tg(PrPΔOR) mice were successively mated with Zrch I Prnp0/0 mice, which had been backcrossed with C57BL/6 mice at least nine times, to produce each line of tg mice with the Zrch I Prnp0/0 genetic background. The resulting tg mice with the Zrch I Prnp0/0 genetic background were then mated with tg(Dpl)/Prnp0/0 mice (C57BL/6 × 129Sv × FVB) to produce each line of double tg (dtg) mice co-expressing each of the respective transgenes and Dpl on the Zrch I Prnp0/0 genetic background. Therefore, all dtg mice have a mixed genetic background of C57BL/6 × 129Sv × FVB. Animals were cared for in accordance with the Guidelines for Animal Experimentation of Nagasaki University.
Diagnosis of Ataxia—The behavior of mice was inspected at least every 3 days evaluating difficulties for walking straight or trembling in their hindquarters on initiation of movement and during walking. When mice showed such abnormal behaviors, they were subjected to a second inspection at least 3 days later. At this time, if the same or exacerbated symptoms were obvious, mice were diagnosed with ataxia, and the date of the first recognition of the abnormal behaviors was registered as the onset of the ataxia. If the symptoms were trivial or difficult to diagnose as ataxia by an investigator, another investigator also inspected the mice to confirm the symptoms. In this case, mice were not diagnosed as ataxia until the two investigators independently confirmed the symptoms.
Western Blotting—Homogenates (10%, w/v) were prepared in a lysis buffer containing 150 mm NaCl, 50 mm Tris-HCl, pH 7.5, 0.5% Triton X-100, 0.5% sodium deoxycholate, 1 mm EDTA, and protease inhibitor mixture (Nakalai Tesque Co., Kyoto, Japan) and centrifuged at low speed. Protein concentrations of the resulting supernatant were determined using the BCA protein assay kit (Pierce). Total proteins were electrophoresed through a 12% SDS-polyacrylamide gel and electrically transferred to an Immobilon-P polyvinylidene difluoride membrane (Millipore Corp.). The membrane was immersed in 5% nonfat dry milk containing TBST (0.1% Tween 20, 100 mm NaCl, 10 mm Tris-HCl, pH 7.6) for 1 h at room temperature and incubated with M20 goat polyclonal antibodies (Santa Cruz Biotechnology, Santa Cruz, CA), SAF32 mouse monoclonal antibody (SPI-BIO, Montigny le Bretonneux, France), or FL176 rabbit polyclonal antibodies against human Dpl (Santa Cruz Biotechnology) for 2 h at room temperature in 1% nonfat dry milk containing TBST. The membrane was washed once in TBST for 15 min and three times for 5 min. Signals were visualized using horseradish peroxidase-conjugated secondary antibodies (Amersham Biosciences) and the ECL system (Amersham Biosciences).
PNGase F Digestion—PNGase F digestion was performed according to the manufacturer's protocol (New England Biolabs, Inc., Ipswich, MA). Briefly, mouse brain homogenates were denatured by boiling for 10 min in the presence of 0.5% SDS and 1% mercaptoethanol and then treated with PNGase F (500 units/liter) in 1% Nonidet P-40 and 0.05 m sodium phosphate, pH 7.5, for 60 min at 37 °C.
In Situ Hybridization—In situ hybridization was performed as described elsewhere (8). Briefly, mouse brains were fixed in 4% paraformaldehyde, embedded in paraffin, and sliced to 5 μm thickness. The tissue sections were then deparaffinized, digested with 10 mg/ml proteinase K for 10 min at 37 °C, and soaked in 0.25% acetic anhydride, 0.1 mm triethanolamine hydrochloride, pH 8.0, 0.9% NaCl for 10 min. After this, the sections were hybridized with PrP cRNA probes labeled with digoxigenin-UTP (Roche Diagnostics) in buffer (50% formamide, 10 mm Tris-HCl, pH 7.5, 1 mm EDTA, 0.6 m NaCl, 0.5 mg/ml yeast tRNA, 0.25% SDS, 5× Denhardt's solution) at 50 °C for 16 h, and followed by several washes in 4× SSC and immersion in 50% formamide, 2× SSC at 50 °C for 30 min. The probe used for PrP was derived from the PCR product corresponding to PrP residues 26-187. The hybridized sections were then digested with 20 μg/ml RNase A at 37 °C for 30 min and finally washed in 0.2× SSC at 50 °C for 20 min. Signals were detected by enzyme-linked immunosorbent assay using alkaline phosphatase-conjugated anti-digoxigenin Fab fragments (1:500, Roche Diagnostics) and nitro blue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate.
Immunohistochemistry—Deparaffinized sections were placed in 3% H2O2 in methanol for 30 min at room temperature to abolish endogenous peroxidase activity. The tissue sections were incubated overnight at 4 °C with anti-spot 35 (calbindin) polyclonal antibodies, IBL-N rabbit antibodies against the N-terminal peptide of PrP (Immuno Biological Laboratories, Gunma, Japan), and ICSM-18 monoclonal antibody recognizing residues 146-159 of murine PrP. To detect immunoreactivities, we used the EnVision+ system in accordance with the manufacturer's recommendations (Dako, Glostrup, Denmark). The antibody-bound peroxidase was detected with 0.04% diaminobenzidine (Sigma).
Flow Cytometry—African green monkey kidney COS-7 cells were transiently transfected by pcDNA3.1 vector alone, pcDNA3.1-moPrP, pcDNA3.1-PrPΔpreOR, pcDNA3.1-PrPΔOR, and pcDNA3.1-PrPΔ23-88 using Lipofectamine 2000 (Invitrogen). The cells were harvested with phosphate-buffered saline containing 20 mm EDTA 48 h after transfection, suspended in 5% fetal bovine serum-containing BSS buffer (140 mm NaCl, 5.4 mm KCl, 0.8 mm MgSO4, 0.3 mm Na2HPO4, 0.4 mm KH2PO4, 1 mm CaCl2, pH 7.0), and incubated with 100-fold diluted SAF61 antibodies for 1 h on ice. The treated cells were then washed twice with 5% fetal bovine serum-containing BSS buffer, incubated with Alexa Fluor 488 goat anti-mouse IgG (H+L) (Invitrogen), and analyzed by EPICS XL (Beckman Coulter Inc., Fullerton, CA).
RESULTS
Generation and Characterization of tg(PrPN-Dpl), tg(PrPΔpreOR), and tg(PrPΔOR) Mice—The amino acid alignment of PrP and Dpl depicts the homology between the C-terminal regions of the two proteins, corresponding to the residues 125-254 of PrP and 58-179 of Dpl, both of which form a neurotoxic globular structure with three α-helices and two β-strands (Fig. 1A). Therefore, to evaluate the effects of the N-terminal region of PrP on Dpl in cis, the PrP N-terminal residues 1-124 were fused to the Dpl residues 58-179 to make the PrPN-Dpl transgene (Fig. 1A). The PrPΔpreOR and PrPΔOR, PrP deletion mutants lacking the N-terminal residues 25-50 and 51-90, respectively, were also constructed to examine the involvement of each region in protection from the Dpl-induced neurodegeneration (Fig. 1A). We introduced each corresponding DNA into the Syrian hamster PrP cosmid vector, CosSHa.tet (20), allowing each of the mutant proteins to be expressed under the control of the hamster PrP promoter (Fig. 1B). These transgenes were then microinjected into fertilized eggs of C57BL/6 mice, yielding four founders from the PrPN-Dpl transgene and two from each of the PrPΔpreOR and PrPΔOR transgenes. All of these founders successfully transferred the transgenes into their offspring. These tg mice were successively intercrossed with nonataxic Zrch I Prnp0/0 mice to eliminate endogenous PrPC.
FIGURE 1.
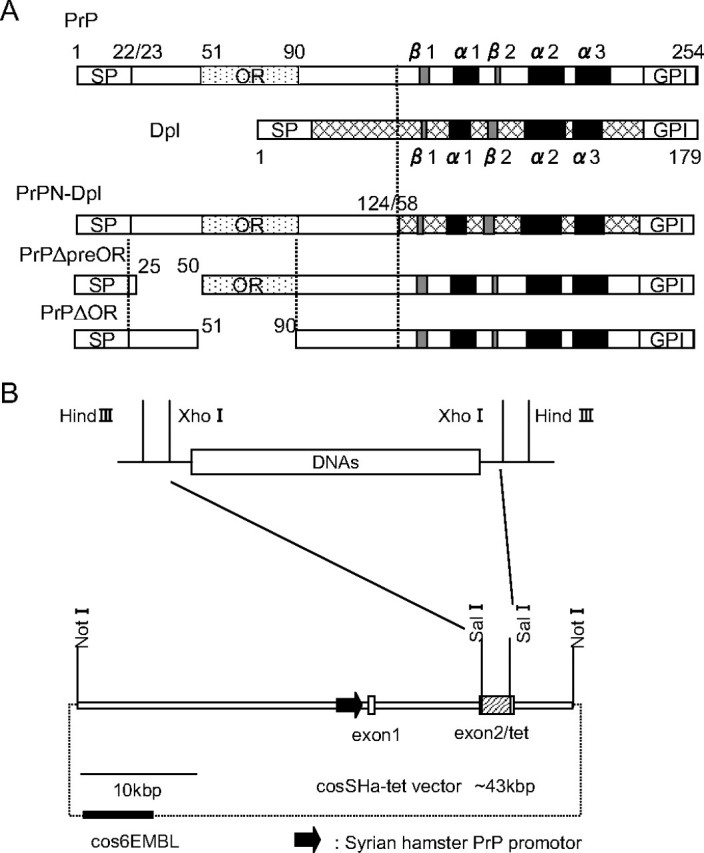
A, schematic representations of the mutant proteins, PrPN-Dpl, PrPΔpreOR, and PrPΔOR. PrPN-Dpl is a fusion protein consisting of the N-terminal residues 1-124 of PrP and the residues 58-179 of Dpl. PrPΔpreOR and PrPΔOR lack the residues 25-50 (preOR) and 51-90 (OR) of PrP, respectively. Arabic numbers represent the codon numbers. SP, signal peptide; OR, octapeptide repeat; GPI, GPI anchor signal; α, α-helix; β, β-strand. B, configuration of the transgenes. Each transgene was constructed by replacing the DNA fragment encoding PrPN-Dpl, PrPΔpreOR, or PrPΔOR with a Sal-Sal fragment of the cosSHa-tet vector carrying the Syrian hamster PrP promoter. The vector-derived DNAs were removed by digestion with NotI, and the purified fragments were used as transgenes.
The expression of mutant proteins was confirmed in 1/2 tg(PrPΔpreOR)/Prnp0/0, 2/2 tg(PrPΔOR)/Prnp0/0, and 1/4 tg(PrPN-Dpl)/Prnp0/0 mice by Western blotting. As shown in Fig. 2A, goat M-20 antibodies against the C-terminal PrP peptide visualized bands in the cerebellar tissue homogenates from tg(PrPΔpreOR)/Prnp0/0 and tg(PrPΔOR)/Prnp0/0 mice (left panel, lanes 4 and 5). These bands migrated slightly faster than authentic PrPC of wild-type mice. But M20 antibodies did not detect any immunoreactivities in tg(PrPN-Dpl) mice (Fig. 2A, lane 7). On the other hand, SAF32 anti-OR antibodies revealed signals in the cerebellum of tg(PrPΔpreOR)/Prnp0/0 and tg(PrPN-Dpl)/Prnp0/0 mice, but not in tg(PrPΔOR)/Prnp0/0 mice (Fig. 2A, right panel). In situ hybridization showed that each transgene was ubiquitously expressed over the brain, with the strongest signals being detectable in Purkinje cells (Fig. 2B) and hippocampal neurons (data not shown). In Zrch I Prnp0/0 mice, some Purkinje cells were faintly stained because of nonspecific hybridization of the probe because no PrP could be detected by Western blotting (Fig. 2, A, lane 1, and B).
FIGURE 2.
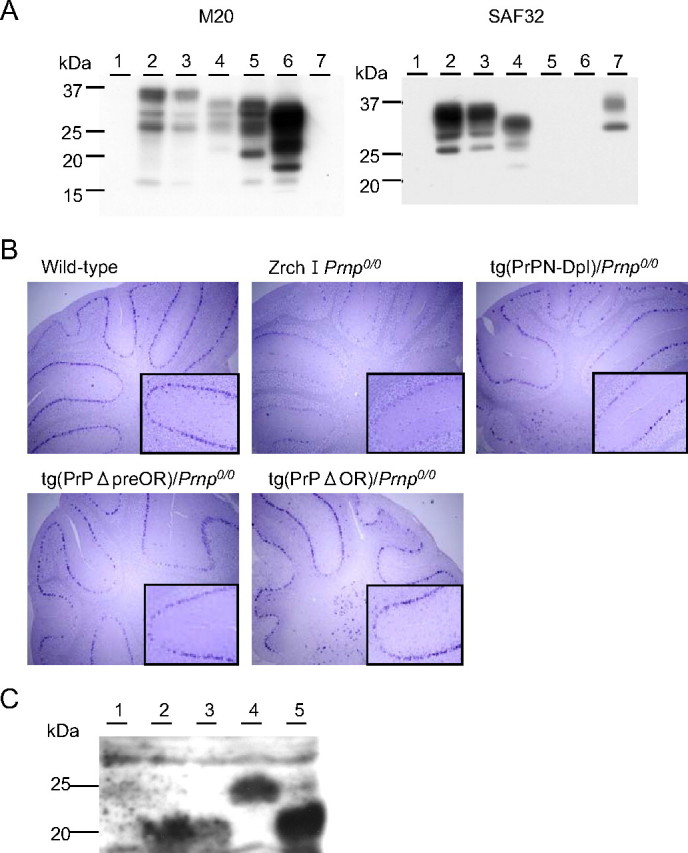
A, Western blotting of the cerebella of tg(PrPN-Dpl)/Prnp0/0, tg(PrPΔpreOR)/Prnp0/0, and tg(PrPΔOR)/Prnp0/0 mice. 30 μg of the total proteins were loaded onto each lane. Lane 1, Zrch I Prnp0/0 mice; lane 2, wild-type mice; lane 3, Zrch I Prnp0/+ mice; lane 4, tg(PrPΔpreOR)/Prnp0/0 mice; lane 5, tg(PrPΔOR)/Prnp0/0 mice; lane 6, tg(MHM2Δ23-88)/Prnp0/0 mice; lane 7, tg(PrPN-Dpl)/Prnp0/0 mice. B, in situ hybridization of the cerebella of wild-type, Zrch I Prnp0/0, tg(PrPN-Dpl)/Prnp0/0, tg(PrPΔpreOR)/Prnp0/0, and tg(PrPΔOR)/Prnp0/0 mice. Purkinje cells in Zrch I Prnp0/0 mice show background staining with the PrP cRNA probe. In contrast, strongly stained Purkinje cells are observed in wild-type, tg(PrPN-Dpl)/Prnp0/0, tg(PrPΔpreOR)/Prnp0/0, and tg(PrPΔOR)/Prnp0/0 mice. Magnification, ×10; inset magnification, ×50. C, Western blotting of the PNGase F-treated homogenates of the cerebella from wild-type (lane 1, 100 μg of the total proteins), Ngsk Prnp0/0 (lane 2, 100 μg), Ngsk Prnp0/+ (lane 3, 100 μg), tg(PrPN-Dpl)/Prnp0/0 (lane 4, 200 μg), and tg(Dpl32)/Prnp0/0 mice (lane 5, 100 μg) using anti-Dpl FL176 antibodies.
We also performed immunohistochemical analysis of cerebella from these tg mice with the Zrch I Prnp0/0 background using two different antibodies, rabbit polyclonal IBL-N and mouse monoclonal ICSM-18 antibodies, which are directed against residues 24-37 and 146-159 of murine PrP, respectively. Both antibodies showed no immunoreactivities in the cerebella of Zrch I Prnp0/0 mice (Fig. 3, E-H). In contrast, the molecular and granule cell layers of normal C57BL/6 mice were clearly stained with both antibodies (Fig. 3, A-D). However, there seemed to be no immunoreactivity in the Purkinje cell layer (Fig. 3, A-D). These staining patterns of PrPC in the cerebellum of normal mice were consistent with previous reports (21-23). PrPΔpreOR mutant protein was expressed in the cerebella of tg(PrPΔpreOR)/Prnp0/0 mice indistinguishably from PrPC in C57BL/6 mice, detectable in the molecular and granule cell layers but not in the Purkinje cell layer (Fig. 3, K and L). PrPΔOR and PrPN-Dpl mutant proteins were also expressed in the molecular and granule cell layers of tg(PrPΔOR)/Prnp0/0 and tg(PrPN-Dpl)/Prnp0/0 mice, respectively (Fig. 3, M-R). However, the mutant proteins were more abundant in the granule cell layer than in the molecular layer (Fig. 3, M-R). Moreover, in tg(PrPΔOR)/Prnp0/0 mice, the Purkinje cell layer was devoid of the signal, but the basolateral area surrounding some but not all Purkinje cells was strongly stained (Fig. 3, M and N and Fig. 7). In tg(PrPN-Dpl)/Prnp0/0 mice, the cell bodies of Purkinje cells appeared positive, and some cells scattered in the granule cell layer were strongly stained in the cell bodies (Fig. 3, Q and R). These cells are currently unidentified. Moreover, cortical neurons of tg(PrPN-Dpl)/Prnp0/0 mice but not wild-type and tg(PrPΔOR)/Prnp0/0 mice were positively stained in the cell bodies by IBL-N antibodies (data not shown).
FIGURE 3.
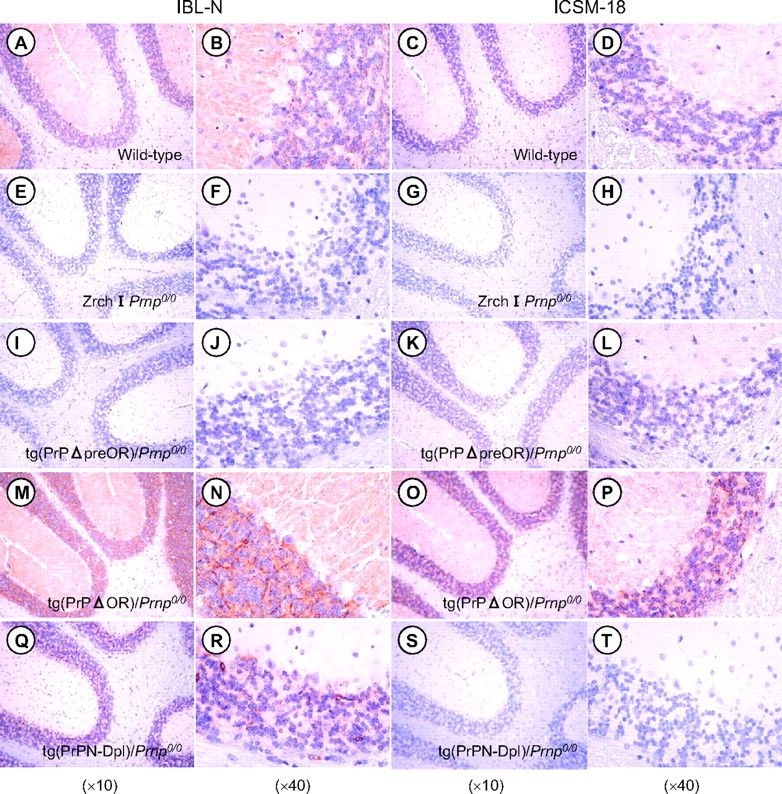
Cytological distribution of PrPΔpreOR, PrPΔOR, and PrPN-Dpl in the cerebella of tg mice. The cerebellar sections from C57BL/6 (A-D), Zrch I Prnp0/0 mice (E-H), tg(PrPΔpreOR)/Prnp0/0 mice (I-L), tg(PrPΔOR)/Prnp0/0 mice (M-P), and tg(PrPN-Dpl)/Prnp0/0 mice (Q-T) were subjected to immunohistochemistry using IBL-N and ICSM-18 antibodies, which are directed against PrP residues 24-37 and 146-159, respectively.
FIGURE 7.

Immunohistochemical analysis of PrPΔ23-88 in the cerebella of mice. The cerebellar sections from Zrch I Prnp0/0 mice, tg(PrPΔ23-88)/Prnp0/0 mice, and tg(PrPΔOR)/Prnp0/0 mice were subjected to immunohistochemistry using IBL-N and ICSM-18 antibodies, which are directed against PrP residues 24-37 and 146-159, respectively. Magnification, ×20.
PrPN-Dpl Delays Onset of Dpl-induced Ataxia and Purkinje Cell Degeneration in Mice—No tg(PrPN-Dpl)/Prnp0/0 mice showed any abnormal symptoms, including ataxia, up to 730 days after birth, at the time of writing (Fig. 4A). Purkinje cells were also unaffected in these mice (data not shown). The signals visualized by anti-Dpl antibodies on a Western blot of brain homogenates from tg mice was about 35% that of Ngsk Prnp0/0 mice, ectopically expressing Dpl in neurons under the control of the PrP promoter (Fig. 2C). These results indicate that, unlike wild-type Dpl, the fusion protein PrPN-Dpl might be nontoxic to Purkinje cells even in the absence of PrPC, although we could not completely rule out the possibility that the lack of neurotoxicity of PrPN-Dpl is because of its lower expression.
FIGURE 4.
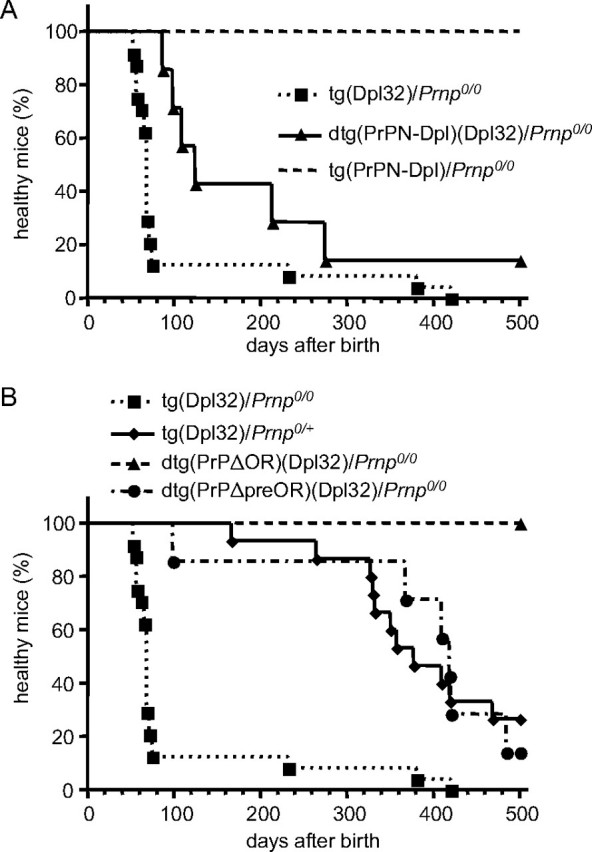
A, rescue from ataxia in dtg(PrPN-Dpl)(Dpl32)/Prnp0/0 mice. No ataxic symptoms were observed in tg(PrPN-Dpl)/Prnp0/0 mice for up to at least 500 days after birth. In contrast, tg(Dpl32)/Prnp0/0 mice developed ataxia 99 ± 20 days after birth. The PrPN-Dpl transgene delayed the onset of ataxia in tg(Dpl32) mice to 200 ± 52 days, as observed in dtg(PrPN-Dpl)(Dpl32)/Prnp0/0 mice. B, rescue of the ataxia in dtg(PrPΔpreOR)(Dpl32)/Prnp0/0 and dtg(PrPΔOR)(Dpl32)/Prnp0/0 mice. No ataxic symptoms were observed in dtg(PrPΔOR)(Dpl32)/Prnp0/0 mice for up to at least 500 days after birth. dtg(PrPΔpreOR)(Dpl32)/Prnp0/0 mice developed delayed onset of ataxia at 385 ± 47 days after birth similarly to tg(Dpl32)/Prnp0/+ mice, those developing ataxia at 387 ± 25 days after birth.
We next generated dtg mice by intercrossing tg(PrPN-Dpl)/Prnp0/0 mice with tg(Dpl32)/Prnp0/0 mice, expressing the full-length Dpl in neurons, including Purkinje cells under the control of the neuron-specific enolase promoter at a level higher than that in Ngsk Prnp0/0 mice (Fig. 2C), which develop ataxia because of Purkinje cell degeneration 99 ± 20 days after birth (Fig. 4A, Table 1, and Fig. 5). The times of onset of ataxia in tg(Dpl32)/Prnp0/0 mice were slightly prolonged compared with those reported previously (8). This is probably because we employed more strict criteria for diagnosis of ataxia in this study. The resulting dtg(PrPN-Dpl)(Dpl32)/Prnp0/0 mice eventually suffered from ataxia, but their onsets were significantly delayed to 200 ± 52 days after birth (Fig. 4A and Table 1). Consistent with this, immunohistochemistry using antibodies against calbindin, a Purkinje cell-specific marker, revealed well preserved Purkinje cells in the dtg mice 90 days after birth (Fig. 5), when Purkinje cells had been significantly lost in tg(Dpl32)/Prnp0/0 mice (Fig. 5). No decreased expression of Dpl could be detected in the brains of dtg(PrPN-Dpl)(Dpl32)/Prnp0/0 mice, compared with tg(Dpl32)/Prnp0/0 mice (Fig. 6). These results indicate that the fusion protein PrPN-Dpl antagonizes the Dpl-induced neurotoxicity, similar to PrPC.
TABLE 1.
Antagonistic effects of mutant proteins on Dpl-induced neurotoxicity in tg mice
| tg or dtg lines | PrP genetic background | Expression level of mutant or wild-type forms of PrPa(fold) | No. of ataxic mice/No. of total mice | Times to the onset of ataxiab(days) | p value log rank test |
|---|---|---|---|---|---|
| tg(Dpl32) | Zrch I Prnp0/0 | 0 | 24/24c | 99 ± 20d | |
| Zrch I Prnp0/+ | 0.5 | 11/15e | 387 ± 25d | <0.0001 | |
| dtg(PrPN-Dpl)(Dpl32) | Zrch I Prnp0/0 | 0.21 | 6/7 | 200 ± 52 | 0.016 |
| tg(PrPN-Dpl) | 0/7 | >730 | <0.0001 | ||
| dtg(PrPΔpreOR)(Dpl32) | Zrch I Prnp0/0 | 0.42 | 6/7 | 385 ± 47 | 0.0004 |
| tg(PrPΔpreOR) | 0/9 | >500 | <0.0001 | ||
| dtg(PrPΔOR)(Dpl32) | Zrch I Prnp0/0 | 1.7 | 0/6 | >500 | <0.0001 |
| tg(PrPΔOR) | 0/6 | >500 | <0.0001 |
Expression levels were compared with those of PrP in wild-type mice using Western blotting.
The times were expressed as mean ± S.E. days after birth.
These 24 mice were produced by breeding of tg(Dpl32)/Prnp0/0 mice with tg(PrPN-Dpl)/Prnp0/0 mice, tg(PrPΔpreOR)/Prnp0/0 mice, and tg(PrPΔOR)/Prnp0/0 mice.
These times were slightly different from those previously reported (8) probably due to more strict diagnostic criteria for ataxia.
These 15 mice were produced by breeding tg(Dpl32) mice with Zrch I Prnp0/0 mice.
FIGURE 5.
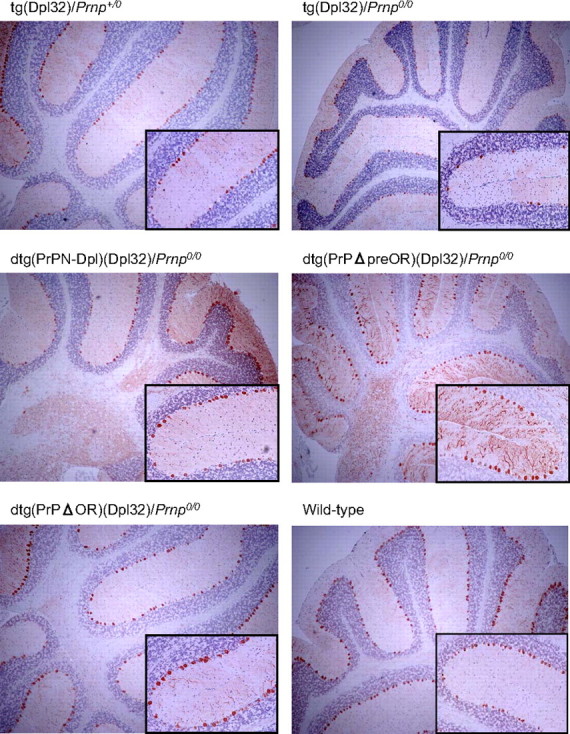
Purkinje cells in the cerebella of dtg mice. Purkinje cells were immunohistochemically stained using anti-calbindin antibodies and the EnVision+ system. Magnification, ×20; inset magnification, ×100.
FIGURE 6.

Western blotting of the PNGase F-treated homogenates (75μg of total proteins) of the cerebella from Zrch I Prnp0/0, tg(Dpl32)/Prnp0/0, tg(Dpl32)/Prnp+/0, dtg(PrPΔpreOR)(Dpl32)/Prnp0/0, dtg(PrPΔOR)(Dpl32)/Prnp0/0, and dtg(PrPN-Dpl)(Dpl32)/Prnp0/0 mice using anti-Dpl FL176 antibodies.
PrPΔpreOR and PrPΔOR Inhibit Dpl-induced Ataxia and Purkinje Cell Degeneration in Mice—To evaluate the potential of PrPΔpreOR and PrPΔOR to antagonize the neurotoxicity of Dpl, tg(PrPΔpreOR)/Prnp0/0 and tg(PrPΔOR)/Prnp0/0 mice were intercrossed with tg(Dpl32)/Prnp0/0 mice. We previously showed that the onset of ataxia by Dpl-induced Purkinje cell degeneration depended on the expression levels of wild-type PrPC, and neither ataxia nor Purkinje cell degeneration occurred in tg(Dpl32) mice on the wild-type (Prnp+/+) background (8). The expression levels of PrPΔOR and PrPΔpreOR in each of the dtg mouse lines, dtg(PrPΔOR)(Dpl32)/Prnp0/0 and dtg(PrPΔpreOR)(Dpl32)/Prnp0/0, were 1.7- and 0.4-fold, respectively, of the level of PrPC in wild-type mice (Fig. 2A). The dtg(PrPΔOR)(Dpl32)/Prnp0/0 mice showed no ataxic symptoms up to 500 days after birth (Fig. 4B and Table 1). On the other hand, dtg(PrPΔpreOR)(Dpl32)/Prnp0/0 mice developed ataxia 385 ± 47 days after birth, which was very delayed compared with the onset in tg(Dpl32)/Prnp0/0 mice (99 ± 20 days) and similar to that in tg(Dpl32) mice on the heterozygous Prnp background (Prnp+/0), 387 ± 25 days (Fig. 4B and Table 1). In contrast to tg(Dpl32)/Prnp0/0, Purkinje cells were unaffected in both dtg mouse lines 90 days after birth on the PrP-null background (Fig. 5). Moreover, Dpl was not decreased in the brains of these dtg mice, compared with tg(Dpl32) mice (Fig. 6). These results indicate that PrPΔpreOR and PrPΔOR preserve the potential to protect from Dpl-induced Purkinje cell degeneration.
PrPΔ23-88 Is Expressed in the Cerebellum of Mice and on the Surface of Cultured Cells Similarly to Wild-type PrPC—We previously showed that PrPΔ23-88 was incompetent to rescue Ngsk Prnp0/0 mice from the Dpl-induced Purkinje cell degeneration, indicating that the region comprising the residues 23-88 is important for PrPC to be protective against Dpl (13). To further gain insights into the role of the residues 23-88 in the neuroprotective function of PrPC, we investigated cytological expression of PrPΔ23-88 in the cerebellum of mice. The cerebella from tg(PrPΔ23-88) mice on the Ngsk Prnp0/0 background as well as from Zrch I Prnp0/0 and tg(PrPΔOR)/Prnp0/0 mice were subjected to immunohistochemistry using IBL-N and ICSM-18 antibodies. Consistent with the results shown in Fig. 3, no signals could be detected in Zrch I Prnp0/0 mice, and tg(PrPΔOR)/Prnp0/0 mice showed abundant expression of PrPΔOR in the molecular and granule cells layers but not in the Purkinje cell layer (Fig. 7). PrPΔ23-88 was detected in the molecular and granule cell layers but not in the Purkinje cell layer (Fig. 7), similarly to PrPΔOR (Fig. 7) and wild-type PrPC (Fig. 3, A-D). We also investigated the cell surface expression of PrPΔ23-88 using cultured cells in comparison with that of wild-type PrPC and two other neuroprotective mutants, PrPΔpreOR and PrPΔOR. COS-7 monkey kidney cells were transiently transfected with each expression vector and then subjected to flow cytometry analysis using SAF61 monoclonal antibodies against PrP-(142-160) residues. PrPΔ23-88 was detected on the cell surface of COS-7 cells similarly to that of wild-type PrPC, PrPΔpreOR, and PrPΔOR (Fig. 8). These results indicate that lack of the residues 23-88 neither alter cell types for PrPΔ23-88 to be expressed in the cerebellum of mice nor impair the cell surface expression of PrPΔ23-88.
FIGURE 8.
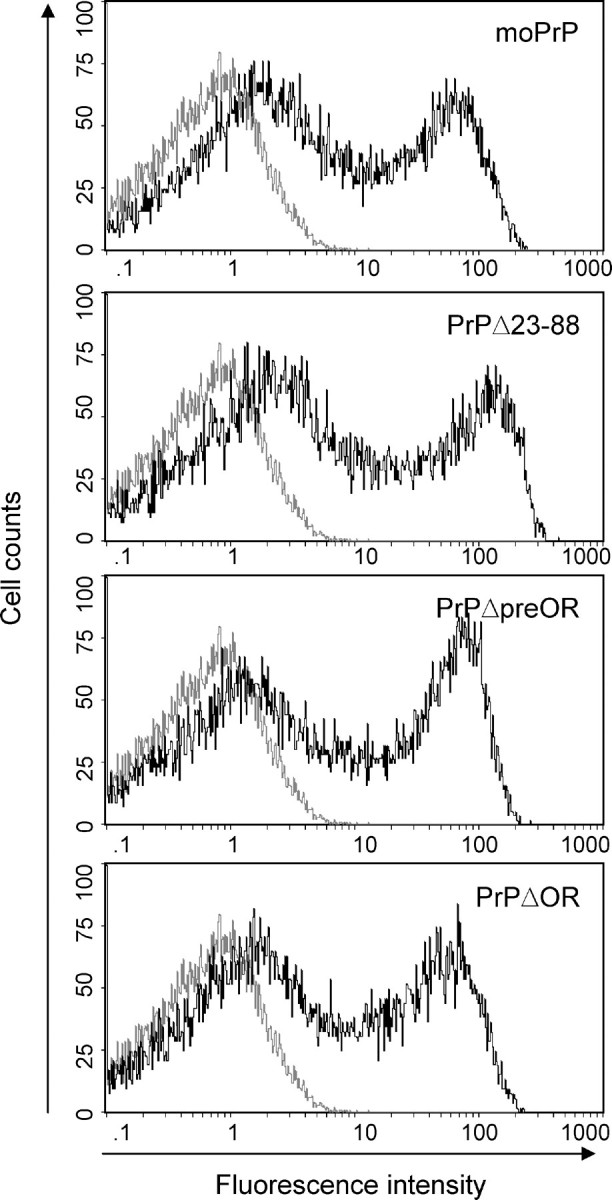
Cell surface expression of PrP mutants. COS-7 cells were transiently transfected with expression vectors encoding wild-type mouse PrPC, PrPΔpreOR, PrPΔOR, and PrPΔ23-88 and subjected to flow cytometry analysis using SAF61 antibodies 48 h after transfection. All PrP mutants were expressed on the cell surface similarly to wild-type PrPC. Gray and black lines indicate cells transfected with pcDNA3.1 vector alone and pcDNA3.1 carrying cDNA corresponding wild-type PrPC, PrPΔpreOR, PrPΔOR, or PrPΔ23-88.
DISCUSSION
Accumulating evidence indicates a neuroprotective role for PrPC. For instance, Prnp0/0 mice are highly sensitive to ischemic or traumatic brain damage, developing more severe pathological changes than in wild-type mice (24-27). In contrast, Dpl, the first identified structural homologue of the C-terminal domain of PrPC, is neurotoxic causing ataxia and Purkinje cell degeneration in mice (7-9). Interestingly, PrPC functionally antagonizes the neurotoxicity of Dpl, preventing the neurodegeneration (7-9). However, the mechanism of the antagonistic interaction between PrPC and Dpl or the truncated PrPs remains to be elucidated.
trans and cis Neuroprotection by the N-terminal Domain of PrPC against Dpl in Mice—In this study, we showed that PrPN-Dpl (the N-terminal residues 1-124 of PrPC fused with the residues 58-179 of Dpl) was itself nontoxic and could mitigate the neurotoxicity of wild-type Dpl in Zrch I Prnp0/0 mice, prolonging the times to the onset of ataxia and Purkinje cell degeneration. Residues 58-179 of Dpl are homologous to residues 125-254 of PrP (10), which encompasses the neurotoxic PrPΔ32-134 peptide. Drisaldi et al. (16) showed that Dpl lacking the N-terminal residues 29-49 or 50-90 was still neurotoxic to primary granule cells from Zrch I Prnp0/0 mice. It is therefore very likely that Dpl-(58-179) is neurotoxic, similarly to the wild-type Dpl in mice devoid of PrPC. Thus, these results indicate that the N-terminal region of PrP might have neuroprotective potential acting both cis and trans on Dpl in mice. Interestingly, Rossi et al. (28) showed that Zrch II Prnp0/0 mice, which develop ataxia and Purkinje cell degeneration because of the ectopic expression of Dpl in Purkinje cells, could be rescued by breeding with tga20 mice expressing PrPC abundantly in the molecular and granule cells but not in Purkinje cells. This suggests that PrPC expressed by neighboring cells, such as molecular and granule cells, is able to counteract the neurotoxicity of Dpl that is expressed on Purkinje cells and that the trans neuroprotection of PrPC might involve intercellular counteraction against Dpl.
OR Is Dispensable for Neuroprotective Function of PrPC against Dpl in Mice—In this study, we also showed that PrPΔOR, PrP lacking the OR alone, rescued mice from the ataxia and Purkinje cell degeneration induced by Dpl. This clearly indicates that the OR is unnecessary for PrPC to antagonize the neurotoxicity of Dpl in mice. Interestingly, Shmerling et al. (11) described that the OR is also unnecessary for PrPC to antagonize the neurotoxicity of truncated PrPs. They showed that granule cell death induced by PrPΔ32-134 could be abrogated by PrPΔ32-93, which lacks the entire OR and about 2/3 of the pre-OR in mice (11). In contrast, in primary cultures of granule cells from Zrch I Prnp0/0 mice, apoptotic cell death induced by transient overexpression of Dpl could be successfully rescued by wild-type PrPC but not by PrP lacking the OR (16). Dpl was preferentially toxic to Purkinje cells and not to granule cells in mice (8, 28, 29). Therefore, Dpl toxicity may vary in primary cultured granule cells and mouse models. However, why PrP lacking the OR has differential activity against Dpl in primary cultured granule cells and mice is unknown.
Kuwahara et al. (31) showed that hippocampal neuronal cell lines established from Prnp0/0 mice easily succumbed to apoptosis after serum withdrawal. Furthermore, expression of the anti-apoptotic molecule Bcl-2 could rescue cell lines from apoptosis (31). Bounhar et al. (14) also showed that PrPC prevented human primary neurons from Bax-induced apoptosis. This suggests that the neuroprotective function of PrPC might involve anti-apoptotic activities. Interestingly, PrP lacking OR failed to rescue the cells from serum withdrawal- and Bax-induced apoptosis, indicating that the OR plays an important role in the anti-apoptotic function of PrPC (14, 32). Furthermore, our present results showing that PrPΔOR antagonized Dpl in mice clearly indicates that neuroprotection by PrPC against Dpl is not associated with OR-mediated anti-apoptotic activities.
The anti-apoptotic activity of PrPC may also be associated with anti-oxidative responses (32, 33). Binding of PrPC to copper may be important for the anti-oxidative function of PrPC by either chelating copper or by activating anti-oxidant enzymes, such as Cu,Zn-superoxide dismutase, via transfer of the bound copper to the enzymes, or both (34-36). Six conserved histidine residues have been identified as copper-binding sites in human PrPC, with four in the OR and two at positions 96 and 111 (37). As PrPΔOR blocked Dpl-mediated neurotoxicity, OR-mediated copper binding might not be involved in the neuroprotection of PrPC against Dpl. In addition, our previous result that PrPΔ23-88, in which two other histidine residues are preserved, failed to rescue mice from ataxia and Purkinje cell degeneration, indicate that copper binding at these sites might not be relevant to the antagonistic function of PrPC against Dpl. Taken together, these suggest that the copper binding-mediated function of PrPC, including anti-oxidative activity, is not associated with its neuroprotective function against Dpl. However, we cannot rule out copper binding to all histidine residues simultaneously for PrPC to have anti-oxidative function.
N-terminal Residues and the Neuroprotective Function of PrPC against Dpl in Mice—In this study, we also showed that PrPΔpreOR, PrP lacking residues 25-50, prevented Dpl-induced ataxia and Purkinje cell degeneration in mice as efficiently as PrPΔOR. This indicates that N-terminal residues 25-50 are not required for PrPC to antagonize Dpl in mice. The two deletions, Δ25-50 and Δ51-90, almost entirely cover the region deleted in PrPΔ23-88, which failed to rescue mice from the neurotoxicity of Dpl (13). PrPΔ23-88 is a chimeric protein of mouse and hamster PrPs, containing two methionines at 108 and 111 in mouse PrP instead of leucine and valine. No such substitutions were present in PrPΔpreOR and PrPΔOR. However, we previously showed that Ngsk Prnp0/0 mice were successfully rescued from ataxia and Purkinje cell degeneration by full-length chimeric PrP with these methionine substitutions (13), clearly indicating that the incompetence of PrPΔ23-88 to antagonize Dpl is because of lack of residues 23-88 and not to the amino acid substitutions. We also showed here that PrPΔ23-88, PrPΔpreOR, and PrPΔOR were similarly expressed in the cerebellum of mice, consistent with these mutant molecules being expressed under the control of the same hamster PrP promoter/enhancer. Moreover, in this study, we used tg(Dpl32)/Prnp0/0 mice for the rescue experiments instead of Ngsk Prnp0/0 mice because tg mice develop ataxia and Purkinje cell degeneration on the Zrch I Prnp0/0 background much earlier than Ngsk Prnp0/0 mice because of higher expression of Dpl in their brains (8). Dpl was expressed in tg(Dpl32) mice from the neuron-specific enolase promoter and in Ngsk Prnp0/0 mice from the residual PrP promoter (4, 8). However, Dpl was similarly expressed in neurons of tg(Dpl32) mice and Ngsk Prnp0/0 mice with the highest expression in Purkinje cells and hippocampal neurons (4, 8). Therefore, Dpl is toxic to Purkinje cells in the same way in both tg (Dpl32)/Prnp0/0 mice and Ngsk Prnp0/0 mice. Taken together, these results indicate that PrPΔpreOR and PrPΔOR but not PrPΔ23-88 can antagonize Dpl neurotoxicity in mice.
PrPΔpreOR and PrPΔOR but not PrPΔ23-88 have the N-terminal two amino acids (residues 23 and 24) conserved adjacent to the junction with the signal peptide. Thus, the two amino acids may be important for the neuroprotection of PrPC against Dpl. This may be consistent with the observation that PrPΔ32-93 protected against the truncated PrPs (11). Interestingly, in PrPΔpreOR the two amino acids are followed by residues starting from 51, generating a new N-terminal sequence (KKPQGGTWG), which is very similar to the N-terminal 9 residues (KKRPKPGGW) of wild-type PrPC and PrPΔ32-93. Six out of 9 of these amino acids are identical. Therefore, this new N-terminal sequence might mimic the function of wild-type PrPC. In PrPΔOR, the N-terminal sequence is intact. Thus, these N-terminal residues might be important for the neuroprotection of PrPC against Dpl. However, it is possible that the antagonistic function of PrPC against Dpl is impaired only by a large deletion of the N-terminal domain with or without the N-terminal residues, as observed in PrPΔ23-88.
Interestingly, PrP with only the central residues 105-125 or 94-134 deleted was reported to be neurotoxic, causing cerebellar degeneration or demyelination in mice, respectively (38, 39). These results suggest that these central residues are essential for PrPC to be neuroprotective. However, PrPΔ23-88 contains these central residues but has no protective activity against Dpl (13). Therefore, the central residues alone might not be enough for PrPC neuroprotectivity, and other region(s), present among the N-terminal residues 23-88, may also be necessary for neuroprotection. These region(s) might be located in the N-terminal 2 or 9 residues. However, unrelated region(s) to the N-terminal 2 or 9 residues may also be necessary.
Possible Mechanisms for N-terminal Region Neuroprotectivity of PrPC against Dpl—There are reports showing that the N-terminal domain is involved in the subcellular trafficking of PrPC (40-44). In this study, we found that PrPΔ23-88, PrPΔpreOR, and PrPΔOR were expressed in the molecular and granule cell layers of the cerebellum and on the cell surface of COS-7 monkey kidney cells similarly to that in wild-type PrPC. This indicates that the cellular expression and cell surface transport of these mutant molecules may be unchanged. It is therefore unlikely that the cell surface localization of PrPΔ23-88 is different from that of PrPΔpreOR and PrPΔOR because of the large deletion of the N-terminal domain, thus impairing the neuroprotective function of PrPC. The N-terminal part is also involved in efficiency of PrPC endocytosis. PrPΔ23-90 and PrPΔ48-93, which lacks the OR region, were shown not to be efficiently internalized in mouse neuroblastoma N2a cells (44), indicating that lack of the OR alone might affect the internalization of PrPC. However, we showed here that PrPΔOR was neuroprotective against Dpl in mice, indicating that the internalization may not be relevant to the neuroprotective activity of PrPC. Recently, Santuccione et al. (45) showed that PrPC activates p59Fyn to enhance neurite outgrowth via recruitment of the neuronal cell adhesion molecule to lipid rafts, indicating that the proper localization at lipid rafts could be important for PrPC function. Interestingly, PrPΔ23-90 but not PrP lacking the OR region was not properly targeted to lipid rafts (44). Thus, PrPΔ23-88 but not PrPΔOR and PrPΔpreOR may not properly localize at lipid rafts either because of lack of the N-terminal 2 or 9 residues or because of large scale deletion of the N-terminal domain with or without the N-terminal residues, resulting in unsuccessful rescue of mice from Dpl neurotoxicity.
Alternatively, the N-terminal region may be involved in the neuroprotective function of PrPC by eliciting a neuroprotective signal through an associated molecule, as in the models proposed so far (11, 30, 38, 39, 46). Among them, Weissmann and Aguzzi (46) proposed that PrPC binds to an as yet unidentified molecule and elicits a Purkinje cell survival signal through the N-terminal domain. Dpl can bind to the molecule but cannot generate the signal because of lack of the N-terminal domain, resulting in Purkinje cell degeneration. However, PrPC competes with Dpl for the molecule, thereby preventing Dpl-induced Purkinje cell degeneration. The results showing that PrPN-Dpl, PrPΔpreOR, and PrPΔOR but not PrPΔ23-88 antagonize the neurotoxicity of Dpl suggests that the former three molecules bind the molecule and produce the survival signal through the N-terminal domain of PrPC, preventing neurodegeneration. This may be because they have a part of or the whole N-terminal domain. It might be also possible that Dpl itself may bind to its own unidentified cognate molecule to elicit a neurotoxic signal and PrPC, PrPN-Dpl, PrPΔpreOR, and PrPΔOR but not PrPΔ23-88 may compete for the molecule via a part of or the whole N-terminal domain, thereby preventing Dpl-mediated neurotoxicity. However, these models can be verified only if the hypothetical molecules are identified.
In this study, we showed that the N-terminal domain mediates the neuroprotective function of PrPC against Dpl in trans and cis and that the OR region and residues 25-50 (pre-OR) are dispensable for the neuroprotective function of PrPC. However, to understand the exact molecular mechanism how the N-terminal domain is involved in the neuroprotective function of PrPC, further studies are required.
Acknowledgments
We thank Prof. Stanley B. Prusiner and Dr. Patrick Tremblay for providing Zrch I Prnp0/0 mice.
This study was supported in part by a Research on Specific Diseases grant from the Ministry of Health, Labor and Welfare, Japan. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: PrP, prion protein; Dpl, doppel; OR, octapeptide repeat; GPI, glycosylphosphatidylinositol; tg, transgenic; dtg, double transgenic; SOD, superoxide dismutase; PNGase, peptide:N-glycosidase.
References
- 1.Oesch, B., Westaway, D., Walchli, M., McKinley, M. P., Kent, S. B., Aebersold, R., Barry, R. A., Tempst, P., Teplow, D. B., Hood, L. E., Prusiner, S. B., and Weissmann, C. (1985) Cell 40 735-746 [DOI] [PubMed] [Google Scholar]
- 2.Prusiner, S. B. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 13363-13383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prusiner, S. B. (1997) Science 278 245-251 [DOI] [PubMed] [Google Scholar]
- 4.Li, A., Sakaguchi, S., Atarashi, R., Roy, B. C., Nakaoke, R., Arima, K., Okimura, N., Kopacek, J., and Shigematsu, K. (2000) Cell. Mol. Neurobiol. 20 553-567 [DOI] [PubMed] [Google Scholar]
- 5.Moore, R. C., Lee, I. Y., Silverman, G. L., Harrison, P. M., Strome, R., Heinrich, C., Karunaratne, A., Pasternak, S. H., Chishti, M. A., Liang, Y., Mastrangelo, P., Wang, K., Smit, A. F., Katamine, S., Carlson, G. A., Cohen, F. E., Prusiner, S. B., Melton, D. W., Tremblay, P., Hood, L. E., and Westaway, D. (1999) J. Mol. Biol. 292 797-817 [DOI] [PubMed] [Google Scholar]
- 6.Li, A., Sakaguchi, S., Shigematsu, K., Atarashi, R., Roy, B. C., Nakaoke, R., Arima, K., Okimura, N., Kopacek, J., and Katamine, S. (2000) Am. J. Pathol. 157 1447-1452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moore, R. C., Mastrangelo, P., Bouzamondo, E., Heinrich, C., Legname, G., Prusiner, S. B., Hood, L., Westaway, D., DeArmond, S. J., and Tremblay, P. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 15288-15293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamaguchi, N., Sakaguchi, S., Shigematsu, K., Okimura, N., and Katamine, S. (2004) Biochem. Biophys. Res. Commun. 319 1247-1252 [DOI] [PubMed] [Google Scholar]
- 9.Anderson, L., Rossi, D., Linehan, J., Brandner, S., and Weissmann, C. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 3644-3649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mo, H., Moore, R. C., Cohen, F. E., Westaway, D., Prusiner, S. B., Wright, P. E., and Dyson, H. J. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 2352-2357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shmerling, D., Hegyi, I., Fischer, M., Blattler, T., Brandner, S., Gotz, J., Rulicke, T., Flechsig, E., Cozzio, A., von Mering, C., Hangartner, C., Aguzzi, A., and Weissmann, C. (1998) Cell 93 203-214 [DOI] [PubMed] [Google Scholar]
- 12.Flechsig, E., Hegyi, I., Leimeroth, R., Zuniga, A., Rossi, D., Cozzio, A., Schwarz, P., Rulicke, T., Gotz, J., Aguzzi, A., and Weissmann, C. (2003) EMBO J. 22 3095-3101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Atarashi, R., Nishida, N., Shigematsu, K., Goto, S., Kondo, T., Sakaguchi, S., and Katamine, S. (2003) J. Biol. Chem. 278 28944-28949 [DOI] [PubMed] [Google Scholar]
- 14.Bounhar, Y., Zhang, Y., Goodyer, C. G., and LeBlanc, A. (2001) J. Biol. Chem. 276 39145-39149 [DOI] [PubMed] [Google Scholar]
- 15.Sakudo, A., Lee, D. C., Saeki, K., Nakamura, Y., Inoue, K., Matsumoto, Y., Itohara, S., and Onodera, T. (2003) Biochem. Biophys. Res. Commun. 308 660-667 [DOI] [PubMed] [Google Scholar]
- 16.Drisaldi, B., Coomaraswamy, J., Mastrangelo, P., Strome, B., Yang, J., Watts, J. C., Chishti, M. A., Marvi, M., Windl, O., Ahrens, R., Major, F., Sy, M. S., Kretzschmar, H., Fraser, P. E., Mount, H. T., and Westaway, D. (2004) J. Biol. Chem. 279 55443-55454 [DOI] [PubMed] [Google Scholar]
- 17.Brinster, R. L., Chen, H. Y., Trumbauer, M. E., Yagle, M. K., and Palmiter, R. D. (1985) Proc. Natl. Acad. Sci. U. S. A. 82 4438-4442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilmut, I., Hooper, M. L., and Simons, J. P. (1991) J. Reprod. Fertil. 92 245-279 [DOI] [PubMed] [Google Scholar]
- 19.Bueler, H., Fischer, M., Lang, Y., Bluethmann, H., Lipp, H. P., DeArmond, S. J., Prusiner, S. B., Aguet, M., and Weissmann, C. (1992) Nature 356 577-582 [DOI] [PubMed] [Google Scholar]
- 20.Scott, M. R., Kohler, R., Foster, D., and Prusiner, S. B. (1992) Protein Sci. 1 986-997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu, T., Zwingman, T., Li, R., Pan, T., Wong, B. S., Petersen, R. B., Gambetti, P., Herrup, K., and Sy, M. S. (2001) Brain Res. 896 118-129 [DOI] [PubMed] [Google Scholar]
- 22.Barmada, S., Piccardo, P., Yamaguchi, K., Ghetti, B., and Harris, D. A. (2004) Neurobiol. Dis. 16 527-537 [DOI] [PubMed] [Google Scholar]
- 23.Watts, J. C., Drisaldi, B., Ng, V., Yang, J., Strome, B., Horne, P., Sy, M. S., Yoong, L., Young, R., Mastrangelo, P., Bergeron, C., Fraser, P. E., Carlson, G. A., Mount, H. T., Schmitt-Ulms, G., and Westaway, D. (2007) EMBO J. 26 4038-4050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McLennan, N. F., Brennan, P. M., McNeill, A., Davies, I., Fotheringham, A., Rennison, K. A., Ritchie, D., Brannan, F., Head, M. W., Ironside, J. W., Williams, A., and Bell, J. E. (2004) Am. J. Pathol. 165 227-235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sakurai-Yamashita, Y., Sakaguchi, S., Yoshikawa, D., Okimura, N., Masuda, Y., Katamine, S., and Niwa, M. (2005) Neuroscience 136 281-287 [DOI] [PubMed] [Google Scholar]
- 26.Weise, J., Crome, O., Sandau, R., Schulz-Schaeffer, W., Bahr, M., and Zerr, I. (2004) Neurosci. Lett. 372 146-150 [DOI] [PubMed] [Google Scholar]
- 27.Hoshino, S., Inoue, K., Yokoyama, T., Kobayashi, S., Asakura, T., Teramoto, A., and Itohara, S. (2003) Acta Neurochir. Suppl. 86 297-299 [DOI] [PubMed] [Google Scholar]
- 28.Rossi, D., Cozzio, A., Flechsig, E., Klein, M. A., Rulicke, T., Aguzzi, A., and Weissmann, C. (2001) EMBO J. 20 694-702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sakaguchi, S., Katamine, S., Nishida, N., Moriuchi, R., Shigematsu, K., Sugimoto, T., Nakatani, A., Kataoka, Y., Houtani, T., Shirabe, S., Okada, H., Hasegawa, S., Miyamoto, T., and Noda, T. (1996) Nature 380 528-531 [DOI] [PubMed] [Google Scholar]
- 30.Behrens, A., and Aguzzi, A. (2002) Trends Neurosci. 25 150-154 [DOI] [PubMed] [Google Scholar]
- 31.Kuwahara, C., Takeuchi, A. M., Nishimura, T., Haraguchi, K., Kubosaki, A., Matsumoto, Y., Saeki, K., Yokoyama, T., Itohara, S., and Onodera, T. (1999) Nature 400 225-226 [DOI] [PubMed] [Google Scholar]
- 32.Sakudo, A., Lee, D. C., Nishimura, T., Li, S., Tsuji, S., Nakamura, T., Matsumoto, Y., Saeki, K., Itohara, S., Ikuta, K., and Onodera, T. (2005) Biochem. Biophys. Res. Commun. 326 600-606 [DOI] [PubMed] [Google Scholar]
- 33.Brown, D. R., Nicholas, R. S., and Canevari, L. (2002) J. Neurosci. Res. 67 211-224 [DOI] [PubMed] [Google Scholar]
- 34.Haigh, C. L., and Brown, D. R. (2006) J. Neurochem. 98 677-689 [DOI] [PubMed] [Google Scholar]
- 35.Brown, D. R., Qin, K., Herms, J. W., Madlung, A., Manson, J., Strome, R., Fraser, P. E., Kruck, T., von Bohlen, A., Schulz-Schaeffer, W., Giese, A., Westaway, D., and Kretzschmar, H. (1997) Nature 390 684-687 [DOI] [PubMed] [Google Scholar]
- 36.Brown, D. R., Schulz-Schaeffer, W. J., Schmidt, B., and Kretzschmar, H. A. (1997) Exp. Neurol. 146 104-112 [DOI] [PubMed] [Google Scholar]
- 37.Jackson, G. S., Murray, I., Hosszu, L. L., Gibbs, N., Waltho, J. P., Clarke, A. R., and Collinge, J. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 8531-8535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li, A., Christensen, H. M., Stewart, L. R., Roth, K. A., Chiesa, R., and Harris, D. A. (2007) EMBO J. 26 548-558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baumann, F., Tolnay, M., Brabeck, C., Pahnke, J., Kloz, U., Niemann, H. H., Heikenwalder, M., Rulicke, T., Burkle, A., and Aguzzi, A. (2007) EMBO J. 26 538-547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pauly, P. C., and Harris, D. A. (1998) J. Biol. Chem. 273 33107-33110 [DOI] [PubMed] [Google Scholar]
- 41.Shyng, S. L., Moulder, K. L., Lesko, A., and Harris, D. A. (1995) J. Biol. Chem. 270 14793-14800 [DOI] [PubMed] [Google Scholar]
- 42.Taylor, D. R., Watt, N. T., Perera, W. S., and Hooper, N. M. (2005) J. Cell Sci. 118 5141-5153 [DOI] [PubMed] [Google Scholar]
- 43.Nunziante, M., Gilch, S., and Schatzl, H. M. (2003) J. Biol. Chem. 278 3726-3734 [DOI] [PubMed] [Google Scholar]
- 44.Walmsley, A. R., Zeng, F., and Hooper, N. M. (2003) J. Biol. Chem. 278 37241-37248 [DOI] [PubMed] [Google Scholar]
- 45.Santuccione, A., Sytnyk, V., Leshchyns'ka, I., and Schachner, M. (2005) J. Cell Biol. 169 341-354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weissmann, C., and Aguzzi, A. (1999) Science 286 914-915 [DOI] [PubMed] [Google Scholar]