

Abstract
The hepatitis C virus (HCV) RNA-dependent RNA polymerase (RdRp), encoded by nonstructural protein 5B (NS5B), is absolutely essential for the viral replication. Here we describe the development, characterization, and functional properties of the panel of monoclonal antibodies (mAbs) and specifically describe the mechanism of action of two mAbs inhibiting the NS5B RdRp activity. These mAbs recognize and bind to distinct linear epitopes in the fingers subdomain of NS5B. The mAb 8B2 binds the N-terminal epitope of the NS5B and inhibits both primer-dependent and de novo RNA synthesis. mAb 8B2 selectively inhibits elongation of RNA chains and enhances the RNA template binding by NS5B. In contrast, mAb 7G8 binds the epitope that contains motif G conserved in viral RdRps and inhibits only primer-dependent RNA synthesis by specifically targeting the initiation of RNA synthesis, while not interfering with the binding of template RNA by NS5B. To reveal the importance of the residues of mAb 7G8 epitope for the initiation of RNA synthesis, we performed site-directed mutagenesis and extensively characterized the functionality of the HCV RdRp motif G. Comparison of the mutation effects in both in vitro primer-dependent RdRp assay and cellular transient replication assay suggested that mAb 7G8 epitope amino acid residues are involved in the interaction of template-primer or template with HCV RdRp. The data presented here allowed us to describe the functionality of the epitopes of mAbs 8B2 and 7G8 in the HCV RdRp activity and suggest that the epitopes recognized by these mAbs may be useful targets for antiviral drugs.
Hepatitis C virus (HCV)2 is a small positive strand RNA virus of the Flaviviridae family that is associated specifically with non-A and non-B hepatitis post-transfusion blood infections in humans (1). HCV, a noncytopathic hepatotropic virus, is a major causative agent of acute and chronic hepatitis, liver cirrhosis, and hepatocellular carcinoma (2). Recently, the World Health Organization estimated the prevalence of HCV antibodies approximating 2%, indicating that 123 million persons worldwide are affected by this virus (3). In infected cells, HCV genomic single-stranded ∼9600-nucleotide RNA messenger directs the synthesis of the ∼3000-amino acid polyprotein precursor (4), which is co- and post-translationally cleaved by cellular and viral proteases producing mature structural and nonstructural proteins (5-7). The same genomic ssRNA serves as a template for the synthesis of the full-length minus strand, which is used for the overproduction of the virus-specific genomic ssRNA. The RNA-dependent RNA polymerase (RdRp), represented by nonstructural protein 5B (NS5B), is a single subunit catalytic component of the viral replication machinery responsible for both of these steps.
The catalytic domain of HCV RdRp has the “right-hand” configuration closely resembling those of HIV-1 reverse transcriptase (RT) (8) and the RdRps of poliovirus (9), reovirus (10), and phage ϕ6 (11). Similarly to these polymerases, HCV NS5B is divided into fingers, palm, and thumb functional subdomains. The fingers and thumb subdomains of the HCV RdRp interact extensively with each other. This interaction is mediated by two loops (Λ1 and Λ2) emanating from the fingers subdomain (12-14). The channel at the surface of HCV RdRp, bordered by fingers subdomain and Λ1 loop, is a putative RNA entry channel (14). The Λ1 loop of HCV RdRp has no structural counterparts in either reovirus polymerase or HIV-1 RT (13). Similarly to reovirus and ϕ6 polymerases, HCV RdRp has been crystallized in the “closed” form with the fingers conformation resembling that seen in HIV-1 RT (8, 10-14). The fingers subdomains of HCV, ϕ6, and reovirus polymerases are highly similar (10, 11). Remarkably, crystalline reovirus λ3 polymerase is able to catalyze phosphodiester bond formation, indicating that template and substrate binding occurs only with localized rearrangements of the closed polymerase form (10). Indeed, opening of the HCV RdRp closed form by indirect displacement of the Λ1 loop triggers inactivation of the polymerase (15). Thus, the fingers subdomain of NS5B is a central component for the overall HCV polymerase fold maintenance and is not amenable to large conformational changes.
Various small molecule HCV RdRp inhibitors such as nucleoside analogues (16, 17) and non-nucleoside inhibitors (NNI) (15, 18-22) were synthesized and reported to be efficient NS5B inhibitors. After conversion to nucleoside triphosphate by cell host machinery, nucleoside analogue competes with natural NTP at the catalytic site of RdRp and terminates the elongation on incorporation. The NNI class of compounds represents allosteric inhibitors that interfere with initiation of RNA synthesis. At least four binding sites for NNI on the HCV RdRp have been reported (23, 24). Surprisingly, all these binding sites are located exclusively in palm and thumb subdomains of HCV polymerase. Therefore, better understanding of the fingers subdomain role in the HCV RdRp function may provide new insights into viral RNA synthesis regulation and open new possibilities for antiviral drug design.
This study describes the isolation and characterization of the HCV RdRp fingers subdomain-specific monoclonal antibodies (mAbs). We used these mAbs as molecular probes for identifying functional determinants of the polymerase surface and to define new potential drug targets for the therapeutic intervention.
EXPERIMENTAL PROCEDURES
Materials—[α-32P]GTP, [α-32P]CTP, and poly(rC), were from Amersham Biosciences; [γ-32P]ATP was from PerkinElmer Life Sciences; nucleoside 5′-triphosphates, RNase inhibitor, phage T7 RNA polymerase, and RQ1 DNase were from Promega; (rG)12 oligonucleotide was from Proligo; heparin was from Sigma; and peptides were from JPT Peptide Technologies. All other reagents were of highest grade available from Sigma.
DNA and Site-directed Mutagenesis—DNA plasmids pFK-I389/neo/NS3-3′/5.1 (Con1/neo) (25), pFK-I341PI-Luc/NS3-3′/Con1/ET, and pFK-I341PI-Luc/NS3-3′/Con1/GND (26, 27) were described previously. The plasmid pET19b-NS5B/Con1 was constructed by inserting PCR-amplified HCV NS5B gene (GenBank™ accession number AJ242654) into the polylinker region of pET19b (Novagen and Merck) between NdeI and Bpu1102I sites. This plasmid was used for the Escherichia coli overexpression of fusion protein that contained N-terminal enterokinase-cleavable His10 tag and full-length HCV NS5B protein.
The PstI/ApaI NS5B DNA fragment, containing the DNA sequence encoding mAb 7G8 binding site, was derived from pET19b-NS5B/Con1 and cloned into the pBluescript II KS(+) (MBI Fermentas) polylinker region. PCR-based mutagenesis was carried out using this plasmid according to Mikaelian and Sergeant (28). Mutations were then transferred into pFK-I341PI-Luc/NS3-3′/Con1/ET (Con1/luc) and pET19b-NS5B/Con1 plasmids. All mutations were verified by DNA sequencing. For epitope mapping studies, corresponding NS5B gene fragments were PCR-amplified and inserted into the multiple cloning site of eukaryotic expression plasmid pQM-CMV-E2Tag-N (Quattromed, Estonia).
RNA Isolation, Synthesis, and Purification—Total cellular RNA was extracted with TRIzol reagent (Invitrogen). The 32P-labeled RNA probe, containing 681-nucleotide Bsp120I/KpnI fragment, complementary to NS5B coding sequence, was synthesized by runoff in vitro transcription in the presence of 50 μCi of [α-32P]GTP, purified as described below, and used for Northern hybridization. The specific activity of the 32P-labeled RNA was ∼2.0 × 109 cpm/μg.
HCV subgenomic RNA for RdRp assays and transient replication assays was produced from ScaI-linearized pFK-I341PI-Luc/NS3-3′/Con1/ET plasmids, containing different mutations by runoff in vitro transcription with phage T7 RNA polymerase and purified according to Lohmann et al. (29) with minor modifications. In brief, the 200-μl transcription reaction contained 5-10 μg of linearized DNA, 1 unit/μl T7 RNA polymerase, 2.5 mm NTPs, and 0.4 unit/μl RNase inhibitor in optimized transcription buffer (Promega) complemented by dithiothreitol. After 4 h of incubation at 37 °C, 1 unit of RQ1 DNase per 1 μg of DNA was added for 30 min. The samples were phenol/chloroform-extracted, RNA-precipitated with isopropyl alcohol, and pellet-washed several times with 70% ethanol. RNA pellet was dried and suspended in 10 mm Tris, pH 7.5, 1 mm EDTA. To remove contaminating NTPs, RNA templates were additionally purified on Sephadex G-25 columns (Amersham Biosciences). Resulting A260/A280 and A260/A230 ratios for HCV subgenomic RNA exceeded 2.1 as determined by ND-1000 spectrophotometer (NanoDrop Technologies, Inc.). Integrity of RNA was confirmed by denaturing formaldehyde gel electrophoresis.
To produce the 32P-labeled RNA for nitrocellulose filter binding assays 5′-phosphates of poly(rC) and HCV subgenomic RNA were removed with calf intestine alkaline phosphatase (Roche Applied Science) according to the manufacturer's protocol. The reaction was carried out for 3 h at 50 °C. The phage T4 polynucleotide kinase (Fermentas MBI) was used to label the dephosphorylated RNA in a forward reaction using 50 μCi of [γ-32P]ATP as a substrate at 37 °C for 20 min. The RNA was purified as described above. The specific activity of the 32P-labeled RNA was ∼1.8×106 cpm/μg for poly(rC) and ∼0.5 × 106 cpm/μg for HCV subgenomic RNA.
HCV RdRp and mAb Production and Purification—The E. coli) strain BL21(DE3) (Stratagene) was used for the expression of recombinant NS5B mutant proteins. Bacteria were grown at 37 °C in LB medium supplemented with 100 mg/ml ampicillin. When cultures reached A600 = 0.6, protein expression was induced by 0.6 mm isopropyl thiogalactoside and further incubated at 17 °C for 16-18 h. N-terminal His10-tagged fusion mutant proteins were affinity-purified to near homogeneity by immobilized metal-affinity chromatography on Ni2+-Sepharose (Qiagen) according to Binder et al. (30) with minor modifications. The NS5B was eluted with 1 ml of storage buffer containing 20 mm Tris, pH 7.5, 500 mm NaCl, 2% Triton X-100, 250 mm imidazole, pH 7.5, 30% glycerol, and 10 mm 2-mercaptoethanol. Protein concentrations were determined using Bradford assay (Bio-Rad) with bovine serum albumin standard (31). The protein was divided into smaller aliquots (NS5B concentrations were between 7.5 and 17.5 μm), frozen in liquid nitrogen, and stored at -70 °C.
The HCV RdRp-specific mAbs were purified from ascitic fluid by ammonium sulfate precipitation and protein G affinity chromatography on a standard fast protein liquid chromatography system (GE Healthcare) according to Juronen et al. (32). The IgG concentration was estimated at 280 nm using an extinction coefficient of 1.4 and using a Bradford protein assay (Bio-Rad) with IgG antibody standard. The purified mAbs were stored either as ammonium sulfate precipitates in phosphate-buffered saline at 4 °C or in phosphate-buffered saline containing 50% glycerol at -20 °C. For all functional assays, mAb buffer was exchanged to TN buffer (10 mm Tris, pH 7.5, 100 mm NaCl) by consecutive diluting and concentrating the samples using Amicon ultracentrifugal (Millipore) filter unit devices.
Primer-dependent and de Novo RdRp Assays—For primer-dependent RdRp assay poly(rC) was mixed with (rG)12 primer, denatured at 95 °C for 2 min, and incubated 5 min at 4 °C prior to use. For primer-independent (de novo) RdRp assay, HCV subgenomic RNA was denatured similarly.
Primer-dependent RdRp assays containing 0.12 μm HCV NS5B, 0.4 μg of poly(rC) pre-annealed with 4 pmol of (rG)12 primer, and various mAbs concentrations (0.048-1.2 μm) were performed in RP buffer (10 mm Tris, pH 7.5, 12.5 mm KCl, 2.5 mm MgCl2, 0.5 mm dithiothreitol, 0.5 mm EGTA, pH 7.5, 10 units of RNasin (Promega)) in a total volume of 25 μl. Because of the addition of HCV RdRp and mAb, the reactions also contained 40 mm NaCl, 0.04% Triton X-100, 5 mm imidazole, 0.6% glycerol, and 0.2 mm 2-mercaptoethanol. If not otherwise indicated, in primer-dependent RdRp reactions the NS5B and mAbs were preincubated at 4 °C for 10 min, after that RNA was added and incubation was extended for another 10 min at 4 °C. The RdRp reactions were initiated by the addition of NTPs CTP, UTP, and ATP at a final concentration of 0.5 mm each and 1 μCi of [α-32P]GTP per reaction. The reactions were incubated at 25 °C for 1 h. For the single cycle RdRp assays, heparin (at 5 μg/ml final concentration) was added simultaneously with NTPs. The primer-dependent RdRp assays for different HCV NS5B mutant enzymes were performed similarly but without preincubations and in the absence of mAbs and heparin. Reactions were quenched by the addition of 1 ml of 10% trichloroacetic acid, 0.5% tetrasodium pyrophosphate (Na4PPi), and 100 μg of herring sperm DNA. After incubation at 4 °C for 30 min, the samples were filtered through glass microfiber GF/C filters (Whatman and Schleicher & Schuell), washed with 1% trichloroacetic acid, containing 0.1% Na4PPi, and dried for 15 min at 56 °C. The bound radioactivity was then measured by liquid scintillation counting of [32P]GMP incorporated into synthesized RNA.
Primer-independent (de novo) RdRp assays were performed exactly as primer-dependent assays with the following exceptions. HCV NS5B concentration was adjusted to 0.36 μm; HCV subgenomic RNA was used at a concentration 0.012 μm, and mAbs concentrations were between 0.144 and 3.6 μm. After NS5B, mAb, and RNA preincubations, the RdRp reactions were initiated by the addition of NTPs GTP, UTP, and ATP at a final concentration of 0.5 mm each and 10 μCi of [α-32P]CTP complemented with unlabeled 10 μm CTP per reaction. The reactions were incubated at 25 °C for 2 h. After addition of 20 μg of glycogen and 0.3 m sodium acetate, the samples were extracted with acidic phenol and chloroform mixture; RNA was precipitated with isopropyl alcohol, and the pellet was washed once with 70% ethanol. The RNA pellets were dissolved at 70 °C for 15 min in a 50% formamide, 0.25 mm EDTA, SDS, and 0.012% of bromphenol blue, xylene cyanol FF, ethidium bromide each. The samples were cooled at 4 °C prior to loading onto denaturing 6% formaldehyde agarose gel containing 40 mm MOPS, pH 7.0, 10 mm sodium acetate, and 1 mm EDTA. Electrophoresis was performed at 5 V/cm. Gels were dried, exposed, scanned with Typhoon Trio Variable Mode Imager (Amersham Biosciences), and analyzed using ImageQuant TL software (Amersham Biosciences) or SuperRX x-ray films (Fuji).
Inhibitory Concentration 50% (IC50) Determination—Because mAbs only partially blocked the HCV RdRp activity, both a residual activity and IC50 values were determined. IC50 (half-maximal effect) is expressed as the mean with a 95% confidence interval (CI). IC50 was estimated from the normalized HCV RdRp activity values plotted as a function of logarithmic mAb concentration fitted with a following four-parameter logistic Equation 1,
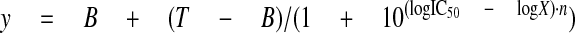 |
(Eq.1) |
where y is the response variable (cpm); X is the mAb concentration; IC50 is the concentration of mAb that provokes a response halfway between the residual activity response (B) and response in the absence of mAb (T), and n is the Hill slope factor. When the n value was set to 1, the three-parameter logistic equation was obtained. Both equations describe the sigmoidal dose-response curve, and the equation selection was based on the Akaike's Information Criteria (33) using the GraphPad 4.0 (GraphPad Software, San Diego). For statistical analyses, the equality and normality of the standard deviations and goodness of curve fit were tested.
Nitrocellulose Filter Binding Assays—RNA binding assays were performed identically to RdRp assays in 25 μl of RP buffer omitting the NTP addition. The concentration of HCV NS5B was set to 0.36 μm (600 ng), and mAbs concentrations were between 0.144 and 3.6 μm. After NS5B and mAb, a 10-min incubation at 4 °C, 100,000 cpm of 32P-labeled poly(rC)·(rG)12, or HCV subgenomic replicon RNAs were added, and incubation was extended for another 10 min at 4 °C. Then the reactions were supplemented with 75 μl of RP buffer containing 200 ng of respective analogous unlabeled RNA, yielding a 100-μl total reaction volume. Under these conditions ∼10% of the input RNA was bound. After a 1-h incubation at 25 °C, samples were filtered through 0.45-μm pore size nitrocellulose membrane filters (BA 85; Schleicher and Schuell), which had been presoaked in RP buffer for 1 h at 25 °C. Membrane filters were washed with 5 ml of RP buffer, and bound radioactivity was then measured by liquid scintillation counting. Background as determined by analogous titrations with NS5B omitted, and analogous titrations with NS5B substituted for bovine serum albumin were subtracted. The nonspecific binding of bovine serum albumin and RNA to the nitrocellulose membrane filters was below 0.1%.
ELISA—Indirect ELISAs with proteins and peptides were performed according to Pfaff et al. (34). Direct ELISAs with hybridomas were performed according to Juronen et al. (35).
Immunoprecipitation—Immunoprecipitation reactions were performed as described by Moradpour and colleagues (36).
Cell Lines and Transfection—Huh7 and Huh7-Lunet (37) cells were maintained in Dulbecco's modified Eagle's medium (DMEM), supplemented with penicillin, streptomycin, 10% fetal calf serum, 2 mm l-glutamine, and 10 μg/liter of Na2SeO3. The Huh7 cell lines harboring selectable HCV replicons contained 0.5 mg/ml G418 (PAA). RNA transfection experiments were carried out using electroporation in the GenePulser Xcell apparatus (Bio-Rad). Electroporation of RNA was performed as described previously (25-27), and capacitance was adjusted to 950 microfarads and voltage to 270 V. In brief, after addition of RNA to a 0.4-cm-wide cuvette (Bio-Rad) containing the 0.4 × 107 cells in 400 μl of cytomix buffer (38), the capacitor was discharged exponentially. The cells were immediately suspended, transferred to the DMEM containing 1.25% DMSO, and plated onto 60-mm dishes. After overnight incubation, the medium was changed to DMSO-free.
COS7 cells were maintained in Iscove's modified Dulbecco's medium, supplemented with 10% fetal calf serum, penicillin, and streptomycin. DNA was transfected into cells using electroporation in 250 μl of cytomix buffer. The capacitance was adjusted to 97 microfarads and voltage to 180 V. After discharging the capacitor, the cells were transferred to a fresh Iscove's modified Dulbecco's medium and plated onto 100-mm dishes.
Transient Replication Assays—Huh7 or Huh7-Lunet cells were transfected by electroporation with HCV subgenomic luciferase replicons at 1.5 μg per each transfection. Cells were harvested at different time points and lysed with a cell culture lysis reagent (Promega) according to the manufacturer's instructions. Subsequently, the lysate was mixed with luciferase assay reagent (Promega), and luminescence was measured. The values obtained with cells harvested 4 h post-electroporation were used to determine transfection efficiency. Total protein content in the lysate was measured in Bradford assay (Bio-Rad) for normalization of the replication signals.
RESULTS
Antibody Production, Characterization, and Epitope Mapping—Recombinant HCV NS5B RdRp was expressed in E. coli and purified to near homogeneity (Fig. 7A, lane WT) in its functionally active conformation. BALB/c mice were immunized with the functionally active nondenatured HCV NS5B protein, and 960 hybridomas were generated. We identified nine hybridomas that produced mAbs, which reacted strongly with NS5B protein in ELISA and immunoblot assays, suggesting that these mAbs recognize linear epitopes. Properties of these mAbs are summarized in Table 1. Importantly, all mAbs, except 10D7, were able to immunoprecipitate the HCV NS5B expressed in the context of the functional replication complex present in Huh7 human hepatocarcinoma cell line, carrying subgenomic HCV Con1/neo replicon (29) (Fig. 1, A-C). This suggests the utility of isolated mAbs for studying both HCV RdRp and its replication complex under native conditions.
FIGURE 7.

Purification of recombinant HCV NS5B proteins with single amino acid changes produced in E. coli and their conformational integrity. A, SDS-polyacrylamide gels (12% polyacrylamide) stained with Coomassie Brilliant Blue G-250. Consecutive lanes correspond to different mutant enzymes. 1/100 of each mutant enzyme produced from 300 ml of corresponding bacterial culture (the expression was induced with isopropyl thiogalactoside under constant conditions) was analyzed. Marker proteins sizes (in kDa) are indicated to the right from each gel. B, structural integrity of recombinant HCV NS5B mutant proteins depicted in A. Wells of MaxiSorp™ plate (Nunc) were coated with 200 ng of corresponding purified NS5B mutant protein in phosphate-buffered saline and blocked with bovine serum albumin. Equal amounts of HCV NS5B-specific and control (α-His and α-Bcl2) mAbs were incubated with the mutant proteins, and ELISA reactivity was detected by horseradish peroxidase-conjugated antibody. The normalized ELISA signal intensity was calculated as the logarithm of ELISA signals ratio obtained for the reaction with and without corresponding primary mAb. The experiment was performed in duplicate. Because of low variation, error bars are not visible on the graph.
TABLE 1.
Properties of HCV NS5B-specific mAbs
| mAb | Isotype | Epitope (aa)a | WBb | IPc | Effect on RdRp activity |
|---|---|---|---|---|---|
| 10D6 | IgG1 | 111-130 | + | + | NE |
| 10D7 | IgG1 | 92-105 | + | − | ND |
| 9A2 | IgG1 | 111-130 | + | + | ND |
| 7G8 | IgG1 | 92-105 | + | + | Inhibits |
| 6B12 | IgG1 | 92-105 | + | + | NE |
| 6G5 | IgG1 | 77-86 | + | + | NE |
| 7F12 | IgG1 | 77-86 | + | + | ND |
| 8G6 | IgG1 | 77-86 | + | + | ND |
| 8B2 | IgG2a | 1-9 | + | + | Inhibits |
Linear epitopes for mAbs are indicated; aa indicates amino acid region.
WB indicates Western blot.
IP indicates immunoprecipitation; NE indicates no effect; ND indicates not determined.
FIGURE 1.
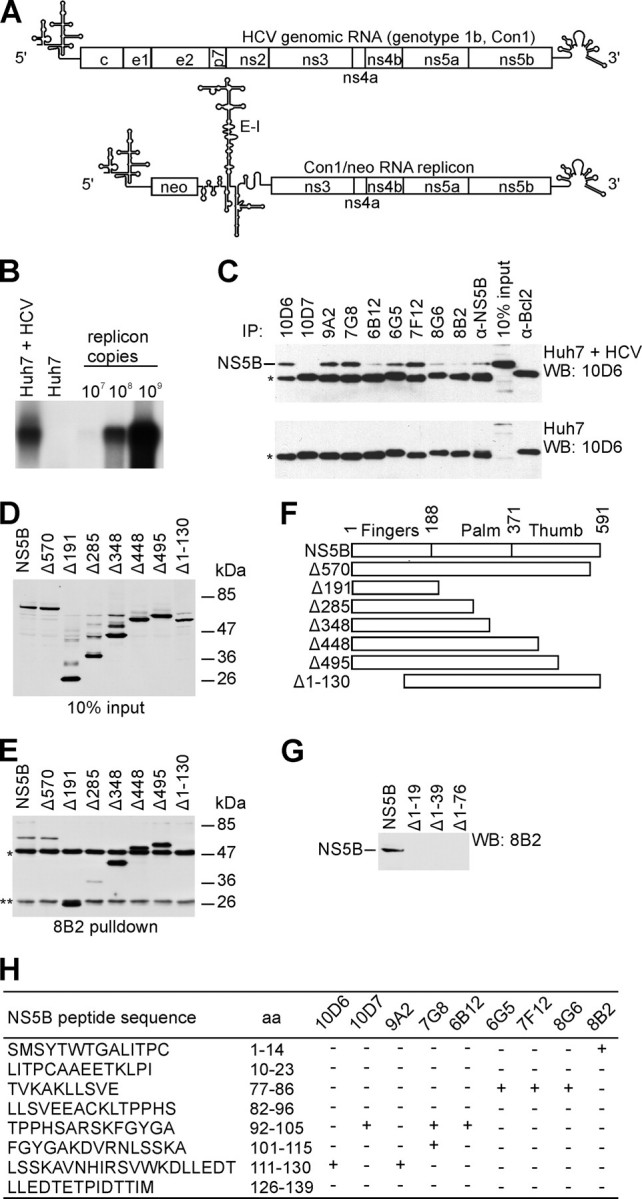
Features of mAbs directed against HCV NS5B RdRp. A, schematic representation of HCV genomic RNA and subgenomic replicon Con1/neo construct (25, 29) used for stable cell line selection (5′, HCV 5′-UTR; neo, neomycin phosphotransferase gene; E-I, encephalomyocarditis virus IRES; 3′, HCV 3′-UTR). Reported secondary structures for 5′-UTR (60), 3′-UTR (61), and encephalomyocarditis virus IRES (62) were used in the illustration. B, detection of Con1/neo subgenomic replicon in G418 selected Huh7 cells. Northern blot analysis was performed using total cellular RNA and antisense ns5b-specific probe. 32P-Labeled Con1/neo RNA was used as a marker. C, immunoprecipitation (IP)/Western blot (WB) analysis. Various mAbs were used to immunoprecipitate the NS5B either from the lysate of the Huh7 cell line containing selectable subgenomic HCV RNA replicon depicted in A (Huh7 + HCV) or from the Huh7 cell line lysate (Huh7). NS5B polyclonal antibodies (α-NS5B) and α-Bcl2 mAb, directed against cellular protein, were used as positive and negative controls, respectively. *, immunoglobulin heavy chain; protein blots were probed with NS5B-specific mAb 10D6, and reactivity was detected with secondary horseradish peroxidase-conjugated antibody of a murine origin. D, Western blot analysis of bovine papilloma virus E2 protein 3F12 epitope-tagged (63) NS5B truncated construct expression in COS7 cells. Molecular mass standards are indicated on the right. E, immunoprecipitation of the proteins shown in D using mAb 8B2. **, immunoglobulin light chain; protein blots were probed with bovine papilloma virus E2-specific mAb 3F12, and reactivity was detected as in C. F, schematic diagram of wild-type HCV NS5B (shown on top) and deletion constructs used for immunoprecipitation. Polymerase subdomains are indicated. Numbers refer to the first residue in a given subdomain. G, Western blot analysis of mAb 8B2 binding to N-terminal truncations of the NS5B protein. H, results of the epitope mapping for NS5B mAbs. Wells of MaxiSorp™ plate (Nunc) were coated with 5 μg of peptide and blocked with bovine serum albumin. HCV NS5B-specific mAbs were incubated with the peptides, and ELISA reactivity was detected by horseradish peroxidase-conjugated antibody; aa, amino acid residues.
However, mAbs 8B2, 6B12, and 8G6 reacted inefficiently in these immunoprecipitation experiments. For example, the mAb 8B2 performed strongly in ELISA when bacterially expressed and purified NS5B protein was used, but inefficiently in immunoprecipitation assays when crude cell extract from the Huh7 cells carrying HCV subgenomic replicon was used (Fig. 1C). Because of this we used the lysates of African green monkey COS7 cells transfected with expression constructs encoding tagged full-size and truncated NS5B proteins for following immunoprecipitation studies. The mAb 8B2 was able to immunoprecipitate all tagged proteins except N-terminally truncated NS5B (Fig. 1, D-F). However, the full-length NS5B was still immunoprecipitated inefficiently suggesting that in cells the mAb 8B2 epitope might be masked by protein/protein interactions. The C-terminally truncated NS5B mutants (Δ191, Δ348, Δ448, and Δ495) were at the same time immunoprecipitated efficiently from the COS7 cell lysates, which suggests that such deletions somehow expose the mAb 8B2 epitope by eliminating the interactions with other proteins. One of the reasons for the inefficient immunoprecipitation of the NS5B may also be the ability of the NS5B protein to oligomerize. Glu18 and His502 residues are critical for the oligomerization process of HCV RdRp (39).
In subsequent immunoblot experiments with tagged truncated NS5B proteins, the binding sites for mAbs were mapped to two nonoverlapping regions (amino acids 1-19 (Fig. 1G) and 77-139 (data not shown)), located within RdRp fingers subdomain. These regions were further divided into overlapping peptides that were used in ELISA to map the epitopes for mAbs more precisely (Fig. 1H and Table 1). Five peptides (amino acids 1-14, 77-86, 92-105, 101-115, and 111-130) reacted with mAbs in ELISA. Monoclonal antibodies, designated 8B2, 6G5, 6B12, 7G8, and 10D6, were further examined. The epitopes for mAbs 8B2 and 7G8 were of particular interest. The 8B2 mAb recognized and bound the N terminus of the HCV NS5B (amino acids 1-9). It has been shown that the activity of viral RdRps is highly sensitive to N-terminal truncations. By contrast, the mAb 7G8 recognized the C-terminal part of peptide consisting of amino acids 92-105, which contains the G motif sequence conserved in RdRps (40). The epitopes for mAbs 8B2 and 7G8 are interleaved with the Λ1 and Λ2 loops and depicted on the molecular model of the HCV RdRp (Fig. 2).
FIGURE 2.

mAbs 8B2 and 7G8 epitopes. The localization of mAbs 8B2 and 7G8 epitopes is shown on the three-dimensional x-ray structure of HCV RdRp complex with oligonucleotide RNA (Protein Data Bank accession code 1nb7, HCV J4 strain (45)). A and B, ribbon representations of the back view are shown. Solid molecular surface representations of back and top views are shown in C and D. The spatial orientation of HCV RdRp is identical for A-C. The epitopes of mAbs 8B2 (amino acids Ser1-Ala9) and 7G8 (amino acids Thr92-Ala105) are colored red-orange and magenta, respectively. Catalytic aspartate residues are represented as spheres, and their oxygen atoms are displayed as red. Manganese atoms are shown as spheres and depicted in purple. B, Fingers, Palm, and Thumb are depicted in cyan, gold, and green, respectively. Two loops emanating from Fingers subdomain and making extensive contacts with Thumb are designated Λ1 (amino acids Ile11-Ala45) and Λ2 (Met139-Ile160) and depicted in dark cyan. The RNA, colored blue, is represented as sticks embedded in transparent mesh molecular surface. Figures were prepared with UCSF, Chimera package (64, 65).
mAbs 8B2 and 7G8 Inhibit the Primer-dependent HCV RdRp Activity—The standard primer-dependent RdRp assay is a continuous polymerization reaction with multiple initiation and elongation cycles. RNA oligonucleotides composed of guanosine residues (12-mers) are annealed randomly to polycytidylic acid. The HCV RdRp binds this template-primer and initiates the elongation polymerization reaction. When RdRp dissociates from the template-primer, a new initiation and elongation cycle can take place. The potential of mAbs to inhibit the initiation or subsequent elongation activity of HCV RdRp was examined in primer-dependent RdRp assay with constant amounts of NS5B preincubated with different concentrations of mAbs. The template-primer RNA was subsequently added, and after preincubation the reaction was initiated by the addition of NTP. RNA synthesis was in a concentration-dependent manner inhibited by mAbs 8B2 and 7G8 (Fig. 3A). As could be determined from Fig. 3B, the half-maximal inhibitory concentrations for 8B2 and 7G8 were 0.111 μm (CI, 0.098 to 0.126 μm) and 0.268 μm (CI, 0.231 to 0.310 μm), respectively. The residual HCV RdRp activity was also different, 3832 ± 330 and 8599 ± 412 cpm for mAb 8B2 and 7G8, respectively. The action of mAbs 8B2 and 7G8 in combination was additive (IC50 = 0.096 μm; CI, 0.094 to 0.099 μm; Fig. 3B), indicating that there is no interference in binding to NS5B, which is in agreement with the different spatial location of the epitopes for these mAbs. The corresponding residual HCV RdRp activity value was 1756 ± 60 cpm. The effect of mAbs 8B2 and 7G8 was specific because of several reasons. First, as shown on Fig. 3A, other HCV RdRp-specific monoclonal antibodies 6G5, 6B12, and 10D6 had no effect on the RdRp activity, demonstrating that targeting a random peptide sequence in the fingers subdomain of HCV RdRp does not result in inhibition of the RdRp activity. Second, mAb α-(His)6, directed against histidine tag, had no effect on the RNA synthesis, indicating that targeting a heterologous peptide sequence N-terminally fused with NS5B does not alter RdRp activity.
FIGURE 3.

mAbs 8B2 and 7G8 inhibit the primer-dependent NS5B RdRp activity. RdRp assays were performed as specified under “Experimental Procedures” using poly(rC)/(rG)12 template-primer RNA. The order of reagent addition is indicated at the top of the graphs. The x axis displays mAb molar excess over NS5B, and the y axis shows the total amount of synthesized 32P-labeled RNA product in counts/min. The means of replicate (at least three) independent experiments with standard deviation values are indicated. A, selection of the mAbs inhibiting RdRp activity of HCV NS5B. B, NS5B RdRp activity inhibition by mAbs 8B2 and 7G8 separately and in combination. When applied in combination, mAbs 8B2 and 7G8 were used at the same concentration as for the individual mAb experiments. C, effect of NS5B and template-primer RNA preincubation on the inhibition of NS5B RdRp activity by mAbs 7G8 and 8B2. D, RdRp assay was initiated by the addition of NTP and incubated for 15 min prior to the addition of mAbs.
Next we evaluated the potential of mAbs 8B2 and 7G8 to inhibit the RNA synthesis from pre-formed RdRp-template-primer RNA complexes by modifying the order of reagent addition. First, HCV RdRp was preincubated with template-primer RNA. Second, different amounts of mAbs were added; incubation time was extended, and the reactions were initiated by supplying the NTP. In this assay format, the half-maximal inhibitory concentration of mAb 8B2 (IC50 = 0.029 μm; CI, 0.016 to 0.051 μm) decreased almost 4-fold compared with the standard RdRp assay. In contrast, the mAb 7G8 IC50 value remained virtually unchanged (0.258 μm; CI, 0.201 to 0.332 μm). Remarkably, the residual HCV RdRp activity increased for mAb 7G8 (13130 ± 410 cpm) and remained virtually unchanged for mAb 8B2 (4054 ± 1160 cpm) (Fig. 3C), compared with the standard RdRp assay (Fig. 3B). These data indicate that primer, template, or template-primer RNA can interfere with mAb 7G8 binding to HCV RdRp, but the binding is not blocked in this assay format.
To determine whether mAbs 8B2 and 7G8 are capable of interfering with the NTP-initiated RNA synthesis in a primer-dependent RdRp assay, we incubated the pre-bound RdRp-template-primer RNA complex with NTP prior to addition of different amounts of mAbs. Neither 8B2 nor 7G8 mAbs was able to inhibit the RdRp activity in this assay format (Fig. 3D). Consequently, when the RdRp-template-primer complex is formed and the elongative RNA synthesis reaction is triggered by NTP, the mAbs are unable to inhibit the RdRp activity. First, these data suggest that NS5B-mediated primer-dependent RNA synthesis is productive. Second, the NS5B epitopes in the synthesizing enzymes are not accessible to the mAbs in the RdRp working cycle.
Inhibition of HCV NS5B in a Single Cycle Primer-dependent RdRp Assay—The multiple initiation and elongation cycles of primer-dependent RdRp assay can be restricted to a single cycle RNA synthesis in the presence of heparin. Polyanion heparin functions as an analogue of the nucleic acid associating with the free RdRp molecules, including RdRp molecules that dissociate from RNA template during or after completion of elongation. Consequently, RNA synthesis reinitiation events should be blocked in the presence of heparin. Therefore RNA synthesis from the already initiated complexes and the complexes in elongation phase could be detected in the presence of heparin.
We first determined the heparin-response curve in our primer-dependent RdRp assays. Total amount of synthesized RNA decreased 5-10 times in the presence of heparin at concentrations equal to or greater than 5 μg/ml (Fig. 4A). This result implies that multiple initiation and elongation cycles contribute 80-90% of the total RNA synthesized in standard primer-dependent RdRp assays. Second, we used the heparin at a concentration of 5 μg/ml in primer-dependent RdRp assays with both 8B2 and 7G8 mAbs. A constant amount of NS5B was preincubated with different concentrations of mAbs. The template-primer RNA was subsequently added, and after preincubation the reaction was initiated by the addition of NTP in the presence of heparin. As a result, the mAb 8B2 IC50 value (0.015 μm; CI, 0.010 to 0.023 μm) decreased almost 8-fold compared with standard RdRp assay. As seen on Fig. 4B, the overall shape of dose-effect inhibition curve for mAb 8B2 was virtually unaffected compared with the standard RdRp assay results (Fig. 3B and Fig. 4C), whereas the dose-dependent inhibitory effect of mAb 7G8 disappeared in the presence of heparin. Our data suggest that mAb 8B2 inhibits the elongation of RNA chains, which is not blocked by heparin. In contrast, the disappearance of dose-dependent inhibitory effect for mAb 7G8 in the single cycle assay suggests that mAb 7G8 inhibitory effect is achieved through inhibition of multiple RNA synthesis initiations.
FIGURE 4.

In primer-dependent RdRp assay, RNA elongation and RNA synthesis initiation are inhibited by mAbs 8B2 and 7G8, respectively. A, determination of heparin concentration required for a single cycle primer-dependent RNA synthesis by HCV RdRp in vitro (representative dose-effect curve). After NS5B preincubation with template-primer RNA, RdRp assay was initiated by the simultaneous addition of NTP and various heparin concentrations (x axis). After NS5B preincubation with mAbs, the RNA template-primer was added, and RdRp assays were initiated by addition of NTP either in the presence (B) or in the absence (C) of a constant amount of heparin. The NTP and heparin were added simultaneously. The molar ratio of mAb to NS5B RdRp is plotted versus the total amount of synthesized 32P-labeled RNA product (counts/min). Note the scale of the y axis in B and C differs 10-fold.
Inhibition of HCV NS5B in Primer-independent (de Novo) RdRp Assay—It was previously demonstrated that HCV NS5B can initiate the RNA synthesis utilizing the de novo mechanism (41-43). Using the full-length ∼9500-nucleotide HCV subgenomic replicon RNA template, we examined the effect of mAbs in de novo RdRp assays. After NS5B and mAb preincubation, the HCV replicon RNA was added, and reaction was initiated by NTP. HCV RdRp synthesized a single RNA product that was equivalent in length to the HCV subgenomic replicon RNA (Fig. 5A, lane 13). This product was absent in the reaction with inactivated HCV NS5B containing the RdRp catalytic site mutation D318N (Fig. 5A, lane 14). The mAb 8B2 efficiently inhibited the HCV RdRp activity in a dose-dependent manner (Fig. 5A, lanes 7-12), whereas mAb 7G8 inhibited the polymerase activity very inefficiently (lanes 1-6). These results suggest that the inhibitory effect of mAb 7G8 depends strongly on the RNA template used.
FIGURE 5.
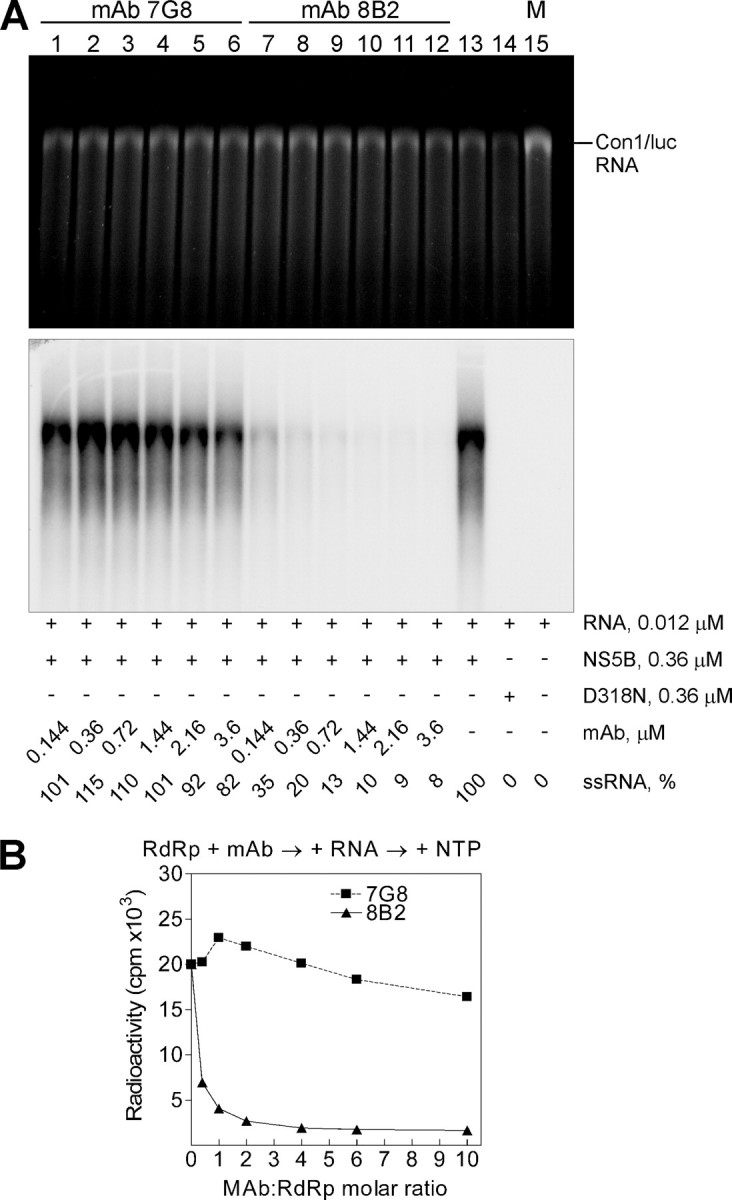
mAb 8B2 blocks primer-independent (de novo) RdRp activity of HCV NS5B in vitro. A, standard de novo RdRp assay was performed using an in vitro transcribed Con1/luc RNA template and increasing concentrations of mAbs 7G8 (lanes 1-6) and 8B2 (lanes 7-12). After a 2-h incubation at 25 °C, RNA was precipitated and analyzed on denaturing formaldehyde-agarose gel. Shown are ethidium bromide staining and the autoradiogram of the same gel. The critical additives and ssRNA yield normalized to the RdRp reaction without mAbs (lane 13) are indicated below the panels. The position of the Con1/luc RNA template is indicated on the right. M is the marker lane with transcribed and purified Con1/luc ssRNA (9510 nucleotides) used for the RdRp assay. D318N (lane 14) is an inactive form of the NS5B. The mAb:RdRp molar ratio value of 1 corresponds to 0.36 μm mAb concentration. B, quantitative PhosphorImager analysis of the reactions shown in A. The analysis of a single gel is presented. The curves show the efficiency of the ssRNA synthesis on Con1/luc RNA template by NS5B in the presence of mAbs 7G8 and 8B2.
Interestingly, residual HCV RdRp activity (1222 ± 47 cpm; see Fig. 5B) in the presence of mAb 8B2 in de novo RdRp assay was reduced more than 3-fold, when compared with the primer-dependent assay results. In addition, the dose-response curve for 8B2 effect in de novo RdRp assay format is very similar to the response curve for the combination of 8B2 and 7G8 mAbs in primer-dependent assay (Fig. 3B) but is steeper for the first quarter of the curve (IC50 = 0.062 μm; CI, 0.059 μm to 0.066 μm; Fig. 5B). Thus, the inhibitory effect of mAb 8B2 is unchanged regardless of the RNA template used.
The Binding of HCV Subgenomic RNA by NS5B Is Enhanced in the Presence of mAb 8B2—To analyze whether mAbs have an effect on the ability of NS5B to bind RNA, a radiolabeled RNA corresponding either to poly(rC)/(rG)12 or HCV subgenomic RNA was incubated with pre-formed RdRp-mAb complexes at different mAb concentrations as described under “Experimental Procedures.” The NS5B was immobilized onto nitrocellulose filters, and bound RNA was measured by liquid scintillation counting. When the primer-dependent RdRp assay RNA template [poly(rC)/(rG)12] was used, no difference in RNA binding was noticed (Fig. 6A). However, there was an evident and reproducible difference in RNA binding curve profiles for mAbs 8B2 and 7G8 when the HCV subgenomic RNA template was used. Therefore, in the presence of mAb 8B2 the binding of RNA by NS5B was increased ∼1.5-fold (Fig. 6B), whereas mAb 7G8 had virtually no effect on RNA binding.
FIGURE 6.

The binding of HCV subgenomic RNA by NS5B is enhanced in the presence of mAb 8B2. RNA binding assays were performed with a 32P-labeled poly(rC)/(rG)12 template-primer RNA (A) or HCV Con1/luc replicon RNA (B) in the presence of increasing concentrations of mAbs. The NS5B-RNA complexes were immobilized onto nitrocellulose filters (BA85, Schleicher & Schuell), and bound radioactivity was measured by liquid scintillation counting. The means of replicate (three) independent experiments with standard deviation values are indicated.
The lack of the RNA binding increase for template-primer RNA in the presence of mAb 8B2 suggests different RNA binding mechanisms for various RNA templates. It has been shown that HCV RdRp initiates the RNA synthesis preferentially from the 3′ terminus of the template RNA (41-43). The binding of template-primer RNA by NS5B RdRp seems to be a stochastic process. After binding of template-primer RNA. the polymerase, most likely, searches for the RNA 3′-hydroxyl group to initiate the RNA synthesis. Once the RNA is elongated, the RdRp dissociates and utilizes another substrate. This is corroborated by our primer-dependent RdRp assays in the absence and presence of heparin. Because there is more than one primer per template, the number of possible “replication initiation-competent” access sites for NS5B should be equal to the number of primers annealed plus the 3′-end of RNA template. Thus, there is more than one NS5B molecule per one template-primer RNA, and the effect of mAb in the RNA binding assay is masked by the out-titration of polymerase molecules. Alternatively, when the HCV subgenomic replicon RNA is used as a template, there is only one replication initiation-competent access site, the 3′-end of the RNA template. This is evident from the absence of prematurely terminated RNA in our de novo RdRp assays. Consequently, there should be one NS5B molecule per one template stably and productively associated at the replication initiation-competent site, allowing the effect of 8B2 mAb on RNA binding to be revealed.
Construction of Site-directed Mutants at the mAb 7G8 Epitope of HCV RdRp—Because mAb 7G8 inhibitory effect was demonstrated to be dependent on the RNA template used, but RNA binding was not affected, we decided to characterize the epitope of this mAb in more detail by carrying out site-directed mutagenesis. The objective of the mutagenesis was to reveal whether any of the mAb 7G8 epitope amino acid residues are important either for primer-dependent NS5B RdRp activity or the replication of HCV subgenomic replicons in the human hepatocellular carcinoma cell line (Huh7). Considering general suggestions for conservative substitutions (44), single changes of mAb 7G8 epitope residues were performed. Sixteen mutants were obtained (Table 2). Nonconservative changes of His95 to Ser and Phe101 to Ala were designed to remove respective aromatic ring structures. To remove the positive charge, Arg98 and Lys100 were changed to Ile. The remaining amino acid residue changes were conservative. Site-directed mutagenesis, overproduction, and purification of the mutant proteins were performed as described under “Experimental Procedures.” It should be pointed out that S99T expression level was significantly lower compared with other mutants, which is seen in Fig. 7A.
TABLE 2.
Summary of replication assays results
Mean values from each set of replicate experiments are shown, normalized to the median activity value of either wild-type HCV NS5B polymerase or adaptive Con1/luc HCV replicon controls. In each case, the standard deviation value was less than 10% of the median activity value.
| Mutation in RdRp | RdRp activitya | Mutation effect | Con1/luc activityb | Mutation effect | Phenotypec |
|---|---|---|---|---|---|
| %WT | %WT | ||||
| T92S | 18 | Down | 4 | Down | I |
| P93A | 90 | WT-like | 67 | WT-like | I |
| P94A | 115 | WT-like | 0 | Null | II |
| H95S | 104 | WT-like | 0 | Null | II |
| S96T | 82 | WT-like | 0 | Null | II |
| A97S | 88 | WT-like | 0 | Null | II |
| R98K | 157 | Up | 0 | Null | II |
| R98I | 68 | WT-like | 0 | Null | II |
| S99T | 0 | Null | 0 | Null | I |
| K100I | 20 | Down | 0 | Null | II |
| F101Y | 134 | Up | 0 | Null | II |
| F101A | 89 | WT-like | 0 | Null | II |
| G102A | 32 | Down | 3 | Down | I |
| Y103F | 89 | WT-like | 98 | WT-like | I |
| G104A | 146 | Up | 125 | WT-like | NA |
| A105S | 62 | WT-like | 60 | WT-like | I |
| WT | 100 | WT | 100 | WT | I |
| D318N | 0 | Null | 0 | Null | I |
The results of standard primer-dependent RdRp assays obtained with purified mutated enzymes are shown.
The results of transient replication assay obtained with mutated Con1/luc replicons (144 h post-transfection).
For phenotype I, the mutation results in the same effect in both assays (up/up, WT-like/WT-like, down/down, or null/null mutations). For phenotype II, the mutation is non-null in primer-dependent RdRp assay and null in transient replication assay; NA indicates not applicable.
Next, different NS5B-specific mAbs were used to test the structural integrity of mutated HCV polymerases in ELISA format. We assumed that if the protein fold was perturbed either locally or globally, then there should be a substantial shift in the ELISA signal intensity. As could be seen on Fig. 7B, only the mAbs 7G8, 6B12, and 10D7 (that bind the same NS5B peptide, Fig. 1H) demonstrate the fluctuation of signal intensity. These fluctuations were due to mutations in the amino acid residues important for binding mAbs. The results obtained indicate that mAbs 6B12 and 10D7 have very similar properties, whereas mAb 7G8 is different. The remaining mAbs, which had no binding sites in the mutated peptide (Fig. 1H), showed no fluctuation in ELISA signal intensity. This indicates that there were no global structural changes in the mutated enzymes, which justifies the selected mutagenesis strategy.
According to HCV NS5B RdRp x-ray crystal structures (13, 14, 45), the amino acid residues 93-105 of mAb 7G8 epitope are solvent-accessible. This mAb epitope contains a loop structure (residues 92-103) and a part of the helix structure (residues 104-105) (Fig. 8E).
FIGURE 8.
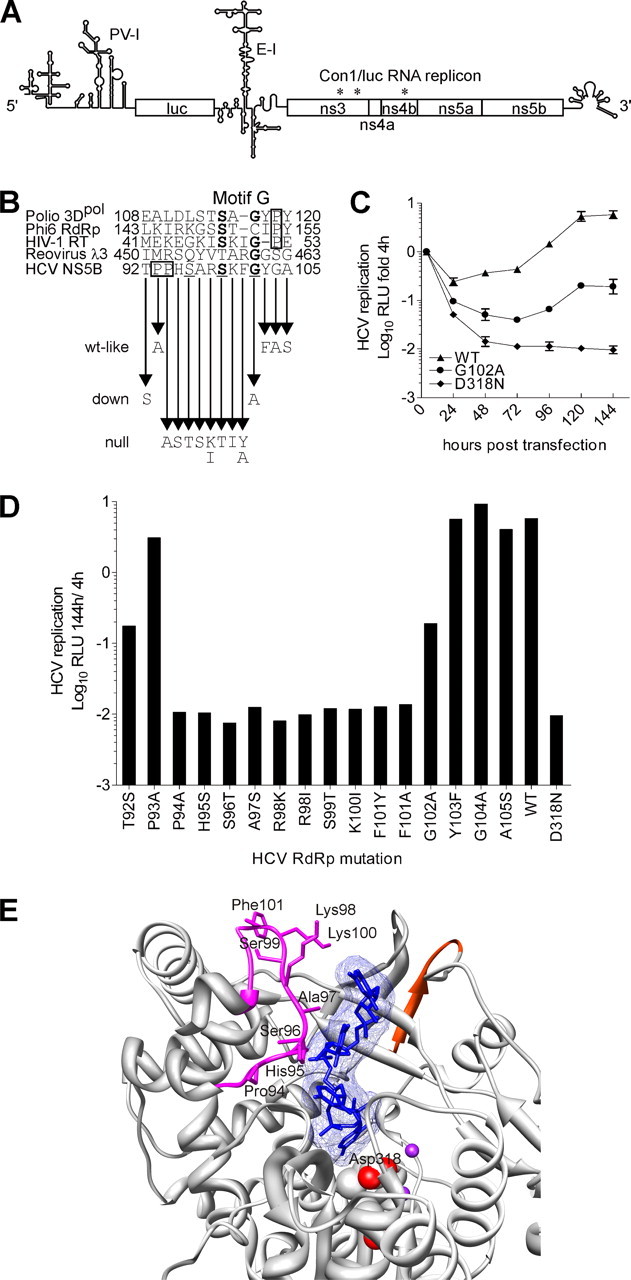
mAb 7G8 epitope site-directed mutations and their effect on HCV Con1/luc replicon transient replication. A, scheme of Con1/luc replicon (PV-I, poliovirus IRES; luc, firefly luciferase gene; E-I, encephalomyocarditis virus IRES; *, adaptive mutations (26, 27)). Reported secondary structure of poliovirus IRES (66) was used in the illustration. B, HCV NS5B peptide sequence, corresponding to mAb 7G8 epitope, was aligned based on protein secondary structure with the corresponding peptide sequences of HIV-1 RT and RdRps from poliovirus (3Dpol), phage ϕ6 (P2), and reovirus (λ3). Highly conserved residues of HCV NS5B and motif G are underlined and shown in boldface, respectively. The Pro residues flanking motif G are boxed. The arrows indicate NS5B amino acid residue changes that were made in Con1/luc replicon. The wt-like, down, and null refer to the resulting mutations features; wt-like, wild-type-like. C, representative Con1/luc replication kinetic curves in Huh7-Lunet cells characterizing wild-type (WT, WT-like), down (G102A), and null (D318N) mutations. Relative light units (RLU) were measured 4, 24, 48, 72, 96, 120, and 144 h after transfection. Relative light units were normalized to the input replication signal measured at 4 h post-transfection (logarithmic scale) and total protein content in the lysate. Note that, because of low variation, error bars are in most cases invisible. D, transient replication assay of mutant Con1/luc replicons in Huh7-Lunet cells. The HCV replication efficiency is given as luciferase activity (logarithmic scale) 144 h after transfection normalized to the input measured at 4 h and to the total protein content in the lysate. E, shown is the ribbon representation of a close-up view for mAb 7G8 epitope on the three-dimensional structure of HCV RdRp complex with RNA oligonucleotide (Protein Data Bank code 1nb7). The residues, which upon change produce the null mutations in transient replication assay, are depicted as sticks. The color codes are the same as in Fig. 2. The NS5B amino acid residue 98 of genotype 1b is Arg and Lys in the Con1 (this study) and J4 isolates, respectively. E was prepared with UCSF Chimera package (64, 65).
Mutations in the mAb 7G8 Epitope Exert Differential Effect on the Primer-dependent HCV NS5B RdRp Activity—We evaluated the efficiency of purified mutant RdRps in our in vitro replication assays. Four groups could be distinguished, based on the performance of NS5B mutant enzymes in primer-dependent RdRp assay format. (i) Three mutant polymerases were able to synthesize RNA better than the wild-type polymerase and were termed up mutations. These RdRps contained R98K, F101Y, and G104A mutations with a relative value of ≥134% in comparison with the wild-type RdRp activity (Table 2). (ii) Three mutant polymerases containing G102A, K100I, and T92S are termed down mutations and displayed reduced RNA synthesis capacity (32, 20, and 18% respectively). (iii) Mutant polymerases containing P94A, H95S, P93A, F101A, Y103F, A97S, S96T, R98I, and A105S changes displayed wild-type-like activity (115, 104, 90, 89, 89, 88, 82, 68, and 62% respectively). (iv) Single S99T mutation abolished the RdRp primer-dependent in vitro activity of HCV NS5B and was termed null mutation.
The epitope of mAb 7G8 contains three amino acid residues (Ser96, Ser99, and Gly102) that are conserved in all HCV genotypes and subtypes available from European HCV data base euHCVdb® (46). On the other hand, residues Ser99 and Gly102 compose the G motif ((T/S)X1-2G) of HCV RdRp, a conserved motif in viral RdRps that is important for template-primer RNA recognition (40, 47). The null polymerization of the S99T mutant and the reduced activity of NS5B with G102A down mutation in our primer-dependent RdRp assays corroborate the importance of motif G for the template-primer recognition and overall enzyme activity (Table 2).
Effect of mAb 7G8 Epitope Mutations on the HCV Subgenomic Replicon Transient Replication—Next we transferred the polymerase mutations into the context of adaptive HCV replicon Con1/luc (26, 27) and examined the replication efficiency of the resulting replicons using the wild-type Con1/luc replicon as a reference (Fig. 8 and Table 2). For the transient replication assays we used Huh7 (data not shown) and Huh7-Lunet (37) cells, an enhanced replication-permissive derivative of Huh7 cells, which gave essentially the same results. The mutants were classified to three groups as follows. (i) Two down mutations were identified. The changes T92S and G102A severely impaired replication yielding nearly 30 times less efficient replicons. (ii) Wild-type-like activity was obtained with G104A, Y103F, P93A, and A105S (125, 98, 67, and 60% respectively). (iii) The majority (10 of 16) of mutant replicons exhibited null replication as follows: P94A, H95S, S96T, A97S, R98K, R98I, S99T, K100I, F101Y, and F101A.
Transient replication assay results demonstrate the presence of a stretch of eight consecutive amino acid residues (Pro94-Phe101) in the mAb 7G8 epitope, which is highly sensitive to any modification either conservative or nonconservative (Fig. 8, B and D, and Table 2). Interestingly, based on the structural alignment of HCV NS5B and HIV-1 reverse transcriptase catalytic complex (8), Bressanelli et al. (13) predicted that this peptide stretch would be involved in the primer and template strand binding. Most important, in the HCV RdRp complex with oligonucleotide RNA x-ray structure, three amino acids of mAb 7G8 epitope (residues 95, 97, and 98) were reported to contact RNA directly (Fig. 8E) (45).
mAb 7G8 Epitope Amino Acid Residues Are Involved in the Interaction of Template-Primer or Template with HCV RdRp—During the HCV RNA replication, the NS5B RdRp utilizes the virus-specific positive-sense RNA genome to produce the full-length minus-strand RNA, which in turn is used for the overproduction of HCV genomic RNA. Because the copy-back or self-priming RNA synthesis by itself is incapable of generating the precise viral genome 5′ and 3′ termini, de novo initiation of viral replication is the most likely mechanism for HCV in infected cells (41, 48, 49). Moreover, in cells harboring HCV subgenomic replicons (e.g. Huh7 or Huh7-Lunet), exclusively negative and positive HCV replicon (Fig. 1B) unit size RNA can be detected (50). Consequently, the comparison of single mutation effects in primer-dependent RdRp assay and transient replication assay may illuminate the differences between two replication initiation mechanisms, mediated by HCV RdRp.
We compared the effects of every single mutation in both primer-dependent RdRp assay and transient replication assay. As a result of this analysis, all mutations except one were assigned to two different phenotypes. Six mutations that resulted in the same effect in both assays were classified as phenotype I. Other nine non-null mutations that had a differential effect in primer-dependent RdRp assay, but were null mutations in transient replication assay, were assigned as phenotype II. The mutation G104A was clearly an up mutation in the primer-dependent RdRp assay; however, the enhancement in the replication efficiency was too low to classify it as an up mutation in transient replication assay. Consequently, we were unable to associate the G104A mutation with any of the phenotypes.
The mutations grouped in phenotype I demonstrate that the catalytic activity of the HCV NS5B polymerase is the primary determinant for the observed effects in both assays. For example, the null mutation S99T produces catalytically inactive enzyme and renders the respective HCV replicon replication-deficient. On the other hand, mutations A105S and T92S are wild-type-like and down mutations, respectively, in both assays.
The phenotype II demonstrates the abruptness of HCV putative de novo initiation replication, which takes place in Huh7-Lunet cells, compared with the gradual modulation of replication signal in primer-dependent in vitro RdRp assay. The removal of the positive charge (R98K, R98I, and K100I) or enhancement of the side chain polarity by introducing a hydroxyl group (A97S, H95S, and F101Y) leads to replicon null replication. Such mutations may interfere with the binding of negatively charged phosphate backbone of the HCV replicon RNA. The remaining three mutations show that modification of the loop (Fig. 2A and Fig. 8E), recognized by mAb 7G8, by either removing bulky side chains (P94A and F101A) or introducing an extra methyl group (S96T) in the 94-101-residue region also results in abolishment of HCV replicon replication.
In the primer-dependent RdRp assay, phenotype II mutations produce either wild-type-like (P94A, H95S, S96T, and A97S) or reduced (K100I) RdRp activity. Interestingly, opposite effects can be obtained by mutating a single amino acid, as is the case with residues Arg98 and Phe101 (Table 2). This fact suggests that alteration of the RdRp activity is not a direct effect on the catalysis of polymerization reaction. On the contrary, it seems to be a consequence of alterations in the stabilization of the common substrate, the primer, and template RNA strands.
The whole spectrum of effects is revealed for phenotype II mutations in the primer-dependent RdRp assay. This indicates that compared with the fine-tuned transient replication system, primer-dependent in vitro assay is essentially a surrogate purely artificial system for measuring of the RdRp activity. In the primer-dependent RdRp assay, the HCV polymerase is taken out of the HCV replication complex context, which is the key component of the transient replication system. However, taken together both systems imply that the residues in mAb 7G8 epitope are important for the interaction of either template or template-primer with HCV RdRp.
DISCUSSION
HCV RdRp, the central enzyme responsible for the replication of viral genome in infected cells, is a promising target for antiviral drug development. Numerous small molecule inhibitors were reported for the HCV RdRp. The rational design of polymerase substrate analogues resulted in the identification of different nucleoside analogue inhibitors competing for the catalytic site and interfering with the elongation of nascent RNA synthesized by NS5B. On the other hand, high throughput drug screening studies identified a diversity of NNI, blocking the initiation of the viral RNA synthesis. To our knowledge, the binding sites for NNI molecules are dispersed in the palm and thumb subdomains of the HCV polymerase. These circumstances validate the approach of using mAbs as molecular probes for studies aimed at the elucidation of the HCV RdRp fingers subdomain function. In this study, we have functionally characterized HCV RdRp fingers subdomain-specific mAbs. Specifically, we have demonstrated that mAbs 8B2 and 7G8 inhibit the elongation and initiation reactions, respectively, carried out by HCV RdRp.
In our initial attempt to understand the molecular mechanism of mAb 8B2 function, we have performed several experiments. First, the variation of the order of addition of reagents in the primer-dependent RdRp assay demonstrates virtually no difference in the 8B2 inhibitory effect (Fig. 3, B and C). This is an indication that RNA template-primer binding to NS5B does not affect subsequent mAb 8B2 binding. Second, mAb 8B2 inhibits both standard and heparin single cycle primer-dependent RdRp assay with equal efficiency (Fig. 4, B and C). This demonstrates that 8B2 inhibits specifically the elongation of RNA chains, carried out by NS5B. Third, the 8B2 inhibitory effect in de novo RdRp assay is even more potent compared with that in the primer-dependent assay (Fig. 5). This evidence suggests that the inhibitory effect of 8B2 does not depend on the RNA substrate used. Because RNA binding to NS5B had not affected the inhibition efficiency of mAb 8B2, we wanted to know whether mAb 8B2 itself is able to influence the RNA binding by NS5B. The results of the filter binding assays suggested that the RNA binding by NS5B is enhanced in the presence of mAb 8B2 (Fig. 6B). Taken together, these findings lead us to suggest that mAb 8B2 inhibits the NS5B RdRp activity by “freezing” the NS5B-RNA complex and impeding the efficient RNA chain elongation.
In this context, it should be noted that the NS5B affinity to the RNA template and the efficiency of RNA synthesis using such a template are tightly linked, i.e. there is an inverse correlation between the RNA template binding and RdRp activity. Lohmann et al. (51) reported that strong RNA binding interferes with enzyme processivity. This is essentially what we are observing in our experiments with the mAb 8B2. In addition, it was reported that the removal of 19 and 22 amino acids from the N terminus of HCV RdRp and poliovirus 3Dpol RdRp, respectively, produces virtually inactive polymerases in the in vitro elongation assays (51, 52). Remarkably, the mAb 8B2 target epitope is located on the NS5B N terminus (amino acids Ser1-Ala9). Thus, mAb binding to this epitope produces a similar effect to that seen using N-terminally truncated NS5B. Moreover, there is an ∼2-fold decrease in the RNA binding for the 3Dpol lacking 22 N-terminal residues (52). This evidence, obtained by Thompson et al. (52), suggests that the N-terminal modification of the enzyme, triggering its inactivation, is linked to its ability to bind the RNA. It seems that the change in the RNA binding state should not be profound enough to interfere with the RdRp activity highlighting the unique properties of mAb 8B2.
Next, we analyzed the mAb 7G8 in vitro properties. Analogous experiments were performed. First, in the primer-dependent RdRp assays, the variation of order of the addition of reagents demonstrated that the inhibitory effect of 7G8 is maximal when the RdRp is preincubated with mAb (Fig. 3B). However, this effect is alleviated when the RdRp is preincubated with template-primer RNA (Fig. 3C). This indicates that RNA binding by NS5B interferes with the mAb 7G8 to some extent. Second, in the single cycle primer-dependent RdRp assay, the inhibitory effect of mAb 7G8 is lost (Fig. 4B). Consequently, the mAb 7G8 inhibitory effect is gained by the inhibition of multiple RNA synthesis initiations, which are very frequent in the primer-dependent RdRp assay (Fig. 4A). Third, in de novo RdRp assay with HCV subgenomic RNA templates, the mAb 7G8 inhibitory effect is reduced nearly 3-fold compared with the primer-dependent assay (Fig. 5). This evidence suggests that the mAb 7G8 inhibitory effect directly depends on the RNA substrate used. In addition, we performed filter binding studies and found that RNA binding by NS5B is not affected in the presence of mAb 7G8 (Fig. 6B). On the basis of these results, we concluded that mAb 7G8 is interfering with the initiation of RNA synthesis and depends largely on the RNA template. However, we could not rule out directly the precise mechanism of mAb 7G8 action, and additional experiments had to be performed.
One question that may arise from the experimental results for mAb 7G8 is why the template-primer RNA synthesis is inhibited regardless of RNA binding not affected. One of the possible answers is certainly the numerous RNA contact points on the HCV polymerase molecule that were deduced by modeling and revealed experimentally (12-14, 45, 53, 54). To address that issue in more detail, we performed the site-directed mutagenesis of the mAb 7G8 epitope. Comparison of single mutation effects in both primer-dependent and transient replication cellular assays (Table 2) suggested that the amino acid residues of the mAb 7G8 epitope on NS5B surface are involved in the interaction with template-primer or template RNA. Interestingly, the mAb 7G8 epitope contains the newly recognized motif G ((T/S)X1-2G) conserved in viral RdRps. It has been proposed that this motif serves to enforce the recognition and proper orientation of adjacent basic residues relative to the primer (40, 55). Indeed, in our primer-dependent assays the conservative mutations of the Ser99 and Gly102 result in total inactivation and reduction of HCV RdRp activity, respectively. Moreover, the profile of adjacent residue mutations (Arg98 and Phe101) suggested that these residues are involved in the stabilization of the common substrate, viz. the primer strand. Contrary to the remaining mutations modulating the RdRp activity in primer-dependent assay, corresponding mutations produced null replication in transient replication assay. In this way we have identified the stretch of eight consecutive amino acid residues (Pro94-Phe101) in the mAb 7G8 epitope, which is highly sensitive to any modification either conservative or nonconservative (Fig. 8, B and D). Taken together, our mutagenesis results indicate that mAb 7G8 epitope contains an amino acid stretch, which includes motif G, which is important for both template-primer and template-proper interaction.
Another question that may arise is why the inhibitory effect of the mAb 7G8 is not so pronounced as the mAb 8B2 effect. A more careful examination of the epitope mapping studies (Fig. 1H) indicates that, in ELISA, mAb 7G8 reacts with two peptides, which overlap at residues Phe101-Ala105. However, the ELISA reactivity obtained with amino acids 92-105 was several orders of magnitude higher (data not shown). Consequently, the minimal epitope for mAb 7G8 includes the Phe101-Ala105 peptide and probably one or two residues upstream of this sequence. Examination of the mAb 7G8 epitope in the context of HCV RdRp complex with oligonucleotide RNA x-ray structure reveals that Phe101 is the last amino acid involved in the interaction with RNA, according to our data (Fig. 8E). Thus, the epitope of mAb 7G8 is located downstream of this interaction platform or has a minor overlap with it. This explains why the efficiency of mAb 7G8 is not very pronounced. Interestingly, in the structures of RT and 3Dpol, ϕ6 and λ3 RdRps the motif G is flanked downstream by a Pro residue. However, in the HCV RdRp, there are two consecutive Pro residues upstream of the motif G (Fig. 8B). It has been proposed that proline in the protein loop structure controls the structural flexibility of the loop. The proline is capable of intrinsic cis/trans isomerization and could serve as a molecular switch for adopting different conformations by the enzyme (56). The flexibility of the loop harboring mAb 7G8 epitope (Fig. 2A and Fig. 8E) would make it a more difficult to target by the mAb. If the Pro residue of motif G is indeed involved in loop conformation control, then it is possible that HCV NS5B, overproduced in E. coli, is a mixture of both cis- and trans-enzyme forms, which are both replication-competent, but only one form is recognized and inhibited by mAb 7G8.
Interestingly, in the poliovirus 3Dpol, the change of G motif Pro residue to Ala or Gly totally abolished the elongation RdRp activity in primer-dependent assay (47). Thompson and Peersen (47) hypothesized that the proline isomerization locks the enzyme-substrate complex into an elongation-competent state. In our primer-dependent RdRp assays, it seems that mAb 7G8 still allows the first round of initiation to occur, but subsequent RNA synthesis re-initiation reactions are inhibited (Fig. 4B). Thus, another interesting possibility for the mAb 7G8 action is the stabilization of the elongation-competent state of the HCV RdRp after one round of RNA synthesis, which could prevent the enzyme dissociation from template and subsequent initiation. Even though mutations P93A and P94A produce wild-type-like RdRp activity in the primer-dependent RdRp assay, the latter mutation abolishes transient replication, indicating the importance of Pro94 for Con1/luc replicon propagation in Huh7 cells (Table 2). Interestingly, it has been reported that cyclophilin B, a peptidyl-prolyl isomerase, interacts with the HCV NS5B via residues 521-591 (57). Further studies will be required to clarify these issues.
It should be noted that one group produced a large set of clustered mutants, with one mutant centering on Phe101 by simultaneously substituting the amino acid residues 97-103 with a flexible linker sequence (AAASAAA). Qin et al. (58) found that in the (rU)14-directed RNA synthesis on polyadenylic acid, the RdRp activity is not compromised by this substitution. In contrast, the nonconservative site-directed changes of the basic residues 98 and 100 to acidic Glu residues resulted in 95% loss in activity (53). Our results are in accordance with this latter study, namely R98I and K100I nonconservative mutations reduced the HCV RdRp activity by 34 and 80%, respectively (Table 2). Milder effect of our mutations is easily explained, namely our substitutions just removed the basicity instead of making the environment more acidic as reported by Deval et al. (53). According to Lohmann et al. (51), the polyadenylic acid is bound ∼4-fold tighter than polycytidylic acid by HCV RdRp, which also correlates with the efficiency of RNA synthesis. The efficiency of RNA synthesis is ∼4-fold higher for poly(rC)/(rG)12 (used in this study) compared with poly(rA)/(rU)12 (51). Because peptide residues 97-103 of the polymerase contain motif G and are involved in template-primer proper binding, the question of RNA affinity can be a decisive issue. These discrepancies between studies highlight the importance of testing the mutation phenotypes in different experimental systems.
Interestingly, recently two NS5B-specific mAbs were isolated. These mAbs recognized a single epitope in the fingers subdomain amino acid residues 67-88 and were capable of inhibiting RdRp activity in vitro (59). However, the molecular mechanism of RdRp inhibition was not explained. Because the mAbs isolated in this study, recognizing the 77-86-amino acid peptide, had no inhibitory effect on the RdRp activity, it is likely that epitopes for the mAbs isolated by Kang et al. (59) are located in the 67-76 NS5B peptide sequence.
In this study we have characterized the mechanism of action of mAbs 8B2 and 7G8 on the HCV RdRp. mAbs 8B2 and 7G8 bind different epitopes in the fingers subdomain of NS5B and interfere with the elongation of the RNA chain and initiation of RNA synthesis, respectively. In addition, we have extensively characterized motif G of HCV RdRp. The results presented here illuminate the fingers subdomain function of the viral RdRp during RNA synthesis. Our findings may have importance for the development of potent small molecule inhibitors targeting HCV RdRp.
Acknowledgments
We gratefully acknowledge V. Lohmann and R. Bartenschlager for providing the Huh7-Lunet cell line and subgenomic HCV replicons pFK-I389/neo/NS3-3′, pFK-I341PI-Luc/NS3-3′/Con1/ET, and the inactive analogue of the latter; J. Parik for performing animal immunizations; M. Pooga for stimulating discussions on protein handling and reagents; A. Merits, P. Spuul, and K. Wilfong for their comments on the manuscript; S. Sõber for the help with statistical analysis; G. Halus for technical assistance, and A. Kalling for cell culture assistance.
This work was supported by Grants 5998 and 5999 from Estonian Science Foundation and by Target Financial Project SF0182566 from Basic Science Committee. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: HCV, hepatitis C virus; CI, 95% confidence interval; ELISA, enzyme-linked immunosorbent assay; HIV-1, human immunodeficiency virus type 1; IRES, internal ribosome entry site; mAb, monoclonal antibody; NNI, non-nucleoside inhibitor; NS5B, nonstructural protein 5B; RdRp, RNA-dependent RNA polymerase; RT, reverse transcriptase; ssRNA, single strand RNA; UTR, untranslated region; WT, wild type; MOPS, 4-morpholinepropanesulfonic acid.
References
- 1.Choo, Q. L., Kuo, G., Weiner, A. J., Overby, L. R., Bradley, D. W., and Houghton, M. (1989) Science 244 pa359-362 [DOI] [PubMed] [Google Scholar]
- 2.Hoofnagle, J. H. (2002) Hepatology 36 Suppl. 1, 21-29 [Google Scholar]
- 3.Perz, J. P., Farrington, L. A., Pecoraro, C., Hutin, Y. J., and Armstrong, G. L. (2004) 42nd Annual Meeting of the Infectious Diseases Society of America, September 30-October 3, p. 214, Boston, MA
- 4.Grakoui, A., Wychowski, C., Lin, C., Feinstone, S. M., and Rice, C. M. (1993) J. Virol. 67 1385-1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bartenschlager, R., Ahlborn-Laake, L., Mous, J., and Jacobsen, H. (1993) J. Virol. 67 3835-3844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grakoui, A., McCourt, D. W., Wychowski, C., Feinstone, S. M., and Rice, C. M. (1993) J. Virol. 67 2832-2843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Selby, M. J., Glazer, E., Masiarz, F., and Houghton, M. (1994) Virology 204 114-122 [DOI] [PubMed] [Google Scholar]
- 8.Huang, H., Chopra, R., Verdine, G. L., and Harrison, S. C. (1998) Science 282 1669-1675 [DOI] [PubMed] [Google Scholar]
- 9.Hansen, J. L., Long, A. M., and Schultz, S. C. (1997) Structure (Lond.) 5 1109-1122 [DOI] [PubMed] [Google Scholar]
- 10.Tao, Y., Farsetta, D. L., Nibert, M. L., and Harrison, S. C. (2002) Cell 111 733-745 [DOI] [PubMed] [Google Scholar]
- 11.Butcher, S. J., Grimes, J. M., Makeyev, E. V., Bamford, D. H., and Stuart, D. I. (2001) Nature 410 235-240 [DOI] [PubMed] [Google Scholar]
- 12.Ago, H., Adachi, T., Yoshida, A., Yamamoto, M., Habuka, N., Yatsunami, K., and Miyano, M. (1999) Structure (Lond.) 7 1417-1426 [DOI] [PubMed] [Google Scholar]
- 13.Bressanelli, S., Tomei, L., Roussel, A., Incitti, I., Vitale, R. L., Mathieu, M., De Francesco, R., and Rey, F. A. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 13034-13039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lesburg, C. A., Cable, M. B., Ferrari, E., Hong, Z., Mannarino, A. F., and Weber, P. C. (1999) Nat. Struct. Biol. 6 937-943 [DOI] [PubMed] [Google Scholar]
- 15.Biswal, B. K., Cherney, M. M., Wang, M., Chan, L., Yannopoulos, C. G., Bilimoria, D., Nicolas, O., Bedard, J., and James, M. N. (2005) J. Biol. Chem. 280 18202-18210 [DOI] [PubMed] [Google Scholar]
- 16.Carroll, S. S., Tomassini, J. E., Bosserman, M., Getty, K., Stahlhut, M. W., Eldrup, A. B., Bhat, B., Hall, D., Simcoe, A. L., LaFemina, R., Rutkowski, C. A., Wolanski, B., Yang, Z., Migliaccio, G., De Francesco, R., Kuo, L. C., MacCoss, M., and Olsen, D. B. (2003) J. Biol. Chem. 278 11979-11984 [DOI] [PubMed] [Google Scholar]
- 17.Dutartre, H., Boretto, J., Guillemot, J. C., and Canard, B. (2005) J. Biol. Chem. 280 6359-6368 [DOI] [PubMed] [Google Scholar]
- 18.Dhanak, D., Duffy, K. J., Johnston, V. K., Lin-Goerke, J., Darcy, M., Shaw, A. N., Gu, B., Silverman, C., Gates, A. T., Nonnemacher, M. R., Earnshaw, D. L., Casper, D. J., Kaura, A., Baker, A., Greenwood, C., Gutshall, L. L., Maley, D., DelVecchio, A., Macarron, R., Hofmann, G. A., Alnoah, Z., Cheng, H. Y., Chan, G., Khandekar, S., Keenan, R. M., and Sarisky, R. T. (2002) J. Biol. Chem. 277 38322-38327 [DOI] [PubMed] [Google Scholar]
- 19.Gu, B., Johnston, V. K., Gutshall, L. L., Nguyen, T. T., Gontarek, R. R., Darcy, M. G., Tedesco, R., Dhanak, D., Duffy, K. J., Kao, C. C., and Sarisky, R. T. (2003) J. Biol. Chem. 278 16602-16607 [DOI] [PubMed] [Google Scholar]
- 20.Di Marco, S., Volpari, C., Tomei, L., Altamura, S., Harper, S., Narjes, F., Koch, U., Rowley, M., De Francesco, R., Migliaccio, G., and Carfi, A. (2005) J. Biol. Chem. 280 29765-29770 [DOI] [PubMed] [Google Scholar]
- 21.Love, R. A., Parge, H. E., Yu, X., Hickey, M. J., Diehl, W., Gao, J., Wriggers, H., Ekker, A., Wang, L., Thomson, J. A., Dragovich, P. S., and Fuhrman, S. A. (2003) J. Virol. 77 7575-7581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang, M., Ng, K. K., Cherney, M. M., Chan, L., Yannopoulos, C. G., Bedard, J., Morin, N., Nguyen-Ba, N., Alaoui-Ismaili, M. H., Bethell, R. C., and James, M. N. (2003) J. Biol. Chem. 278 9489-9495 [DOI] [PubMed] [Google Scholar]
- 23.Pauwels, F., Mostmans, W., Quirynen, L. M., van der Helm, L., Boutton, C. W., Rueff, A. S., Cleiren, E., Raboisson, P., Surleraux, D., Nyanguile, O., and Simmen, K. A. (2007) J. Virol. 81 6909-6919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koch, U., and Narjes, F. (2006) Infect. Disord. Drug Targets 6 31-41 [DOI] [PubMed] [Google Scholar]
- 25.Krieger, N., Lohmann, V., and Bartenschlager, R. (2001) J. Virol. 75 4614-4624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lohmann, V., Hoffmann, S., Herian, U., Penin, F., and Bartenschlager, R. (2003) J. Virol. 77 3007-3019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Windisch, M. P., Frese, M., Kaul, A., Trippler, M., Lohmann, V., and Bartenschlager, R. (2005) J. Virol. 79 13778-13793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mikaelian, I., and Sergeant, A. (1992) Nucleic Acids Res. 20 376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lohmann, V., Korner, F., Koch, J., Herian, U., Theilmann, L., and Bartenschlager, R. (1999) Science 285 110-113 [DOI] [PubMed] [Google Scholar]
- 30.Binder, M., Quinkert, D., Bochkarova, O., Klein, R., Kezmic, N., Bartenschlager, R., and Lohmann, V. (2007) J. Virol. 81 5270-5283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bradford, M. M. (1976) Anal. Biochem. 72 248-254 [DOI] [PubMed] [Google Scholar]
- 32.Juronen, E., Parik, J., and Toomik, P. (1991) J. Immunol. Methods 136 103-109 [DOI] [PubMed] [Google Scholar]
- 33.Akaike, H. (1992) No to Hattatsu 24 127-133 [PubMed] [Google Scholar]
- 34.Pfaff, E., Mussgay, M., Bohm, H. O., Schulz, G. E., and Schaller, H. (1982) EMBO J. 1 869-874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Juronen, E., Tasa, G., Uuskula, M., Pooga, M., and Mikelsaar, A. V. (1996) Hybridoma 15 77-82 [DOI] [PubMed] [Google Scholar]
- 36.Moradpour, D., Bieck, E., Hugle, T., Wels, W., Wu, J. Z., Hong, Z., Blum, H. E., and Bartenschlager, R. (2002) J. Biol. Chem. 277 593-601 [DOI] [PubMed] [Google Scholar]
- 37.Friebe, P., Boudet, J., Simorre, J. P., and Bartenschlager, R. (2005) J. Virol. 79 380-392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van den Hoff, M. J., Moorman, A. F., and Lamers, W. H. (1992) Nucleic Acids Res. 20 2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qin, W., Luo, H., Nomura, T., Hayashi, N., Yamashita, T., and Murakami, S. (2002) J. Biol. Chem. 277 2132-2137 [DOI] [PubMed] [Google Scholar]
- 40.Gorbalenya, A. E., Pringle, F. M., Zeddam, J. L., Luke, B. T., Cameron, C. E., Kalmakoff, J., Hanzlik, T. N., Gordon, K. H., and Ward, V. K. (2002) J. Mol. Biol. 324 47-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kao, C. C., Yang, X., Kline, A., Wang, Q. M., Barket, D., and Heinz, B. A. (2000) J. Virol. 74 11121-11128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luo, G., Hamatake, R. K., Mathis, D. M., Racela, J., Rigat, K. L., Lemm, J., and Colonno, R. J. (2000) J. Virol. 74 851-863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oh, J. W., Sheu, G. T., and Lai, M. M. (2000) J. Biol. Chem. 275 17710-17717 [DOI] [PubMed] [Google Scholar]
- 44.Bordo, D., and Argos, P. (1991) J. Mol. Biol. 217 721-729 [DOI] [PubMed] [Google Scholar]
- 45.O'Farrell, D., Trowbridge, R., Rowlands, D., and Jager, J. (2003) J. Mol. Biol. 326 1025-1035 [DOI] [PubMed] [Google Scholar]
- 46.Combet, C., Garnier, N., Charavay, C., Grando, D., Crisan, D., Lopez, J., Dehne-Garcia, A., Geourjon, C., Bettler, E., Hulo, C., Le Mercier, P., Bartenschlager, R., Diepolder, H., Moradpour, D., Pawlotsky, J. M., Rice, C. M., Trepo, C., Penin, F., and Deleage, G. (2007) Nucleic Acids Res. 35 D363-D366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thompson, A. A., and Peersen, O. B. (2004) EMBO J. 23 3462-3471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bressanelli, S., Tomei, L., Rey, F. A., and De Francesco, R. (2002) J. Virol. 76 3482-3492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bartenschlager, R., and Lohmann, V. (2000) J. Gen. Virol. 81 1631-1648 [DOI] [PubMed] [Google Scholar]
- 50.Quinkert, D., Bartenschlager, R., and Lohmann, V. (2005) J. Virol. 79 13594-13605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lohmann, V., Korner, F., Herian, U., and Bartenschlager, R. (1997) J. Virol. 71 8416-8428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thompson, A. A., Albertini, R. A., and Peersen, O. B. (2007) J. Mol. Biol. 366 1459-1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Deval, J., D'Abramo, C. M., Zhao, Z., McCormick, S., Coutsinos, D., Hess, S., Kvaratskhelia, M., and Gotte, M. (2007) J. Biol. Chem. 282 16907-16916 [DOI] [PubMed] [Google Scholar]
- 54.Kim, Y. C., Russell, W. K., Ranjith-Kumar, C. T., Thomson, M., Russell, D. H., and Kao, C. C. (2005) J. Biol. Chem. 280 38011-38019 [DOI] [PubMed] [Google Scholar]
- 55.Imbert, I., Guillemot, J. C., Bourhis, J. M., Bussetta, C., Coutard, B., Egloff, M. P., Ferron, F., Gorbalenya, A. E., and Canard, B. (2006) EMBO J. 25 4933-4942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Andreotti, A. H. (2003) Biochemistry 42 9515-9524 [DOI] [PubMed] [Google Scholar]
- 57.Watashi, K., Ishii, N., Hijikata, M., Inoue, D., Murata, T., Miyanari, Y., and Shimotohno, K. (2005) Mol. Cell 19 111-122 [DOI] [PubMed] [Google Scholar]
- 58.Qin, W., Yamashita, T., Shirota, Y., Lin, Y., Wei, W., and Murakami, S. (2001) Hepatology 33 728-737 [DOI] [PubMed] [Google Scholar]
- 59.Kang, S. M., Choi, S. H., Park, C. Y., Kim, M. H., Kim, T. K., Park, J. M., Koh, M. S., Kang, H. J., and Hwang, S. B. (2008) J. Viral Hepat. 15 305-313 [DOI] [PubMed] [Google Scholar]
- 60.Pestova, T. V., Shatsky, I. N., Fletcher, S. P., Jackson, R. J., and Hellen, C. U. (1998) Genes Dev. 12 67-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bradrick, S. S., Walters, R. W., and Gromeier, M. (2006) Nucleic Acids Res. 34 1293-1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pilipenko, E. V., Blinov, V. M., Chernov, B. K., Dmitrieva, T. M., and Agol, V. I. (1989) Nucleic Acids Res. 17 5701-5711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kaldalu, N., Lepik, D., Kristjuhan, A., and Ustav, M. (2000) BioTechniques 28 456-460, 462 [DOI] [PubMed] [Google Scholar]
- 64.Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., and Ferrin, T. E. (2004) J. Comput. Chem. 25 1605-1612 [DOI] [PubMed] [Google Scholar]
- 65.Sanner, M. F., Olson, A. J., and Spehner, J. C. (1996) Biopolymers 38 305-320 [DOI] [PubMed] [Google Scholar]
- 66.Borman, A. M., Deliat, F. G., and Kean, K. M. (1994) EMBO J. 13 3149-3157 [DOI] [PMC free article] [PubMed] [Google Scholar]