

Abstract
Rad52 plays essential roles in homology-dependent double-strand break repair. Various studies have established the functions of Rad52 in Rad51-dependent and Rad51-independent repair processes. However, the precise molecular mechanisms of Rad52 in these processes remain unknown. In the present study we have identified a novel DNA binding site within Rad52 by a structure-based alanine scan mutagenesis. This site is closely aligned with the putative single-stranded DNA binding site determined previously. Mutations in this site impaired the ability of the Rad52-single-stranded DNA complex to form a ternary complex with double-stranded DNA and subsequently catalyze the formation of D-loops. We found that Rad52 introduces positive supercoils into double-stranded DNA and that the second DNA binding site is essential for this activity. Our findings suggest that Rad52 aligns two recombining DNA molecules within the first and second DNA binding sites to stimulate the homology search and strand invasion processes.
The repair of DSBs4 is a critical process by which cells maintain genome integrity. In eukaryotic cells these breaks are repaired by homologous recombination (HR) and non-homologous end joining. In the homologous end joining pathway, broken DNA ends are rejoined by religation or annealing of short common sequences (microhomologies) near their ends. Because homologous end joining lacks a mechanism to reject erroneous pairings that occur by chance, recessed DNA, chromosome rearrangements, and mutations are possible outcomes of this repair pathway (1). By contrast, the HR pathway achieves high accuracy in DSB repair by utilizing mechanisms for a DNA homology search. In this pathway the ends of the DSBs are resected to generate 3′ single-stranded tails. The single-stranded region then either invades an undamaged homologous duplex DNA (strand invasion) or pairs with a complementary single strand (single-strand annealing). The importance of this pathway is underscored by the high conservation, from yeast to humans, of the factors that catalyze HR (2).
Rad52 is one of the key players in the yeast HR pathway, and its homologs have been found in higher eukaryotes. In yeast, Rad52 is essential in both the RAD51-dependent and -independent HR pathways and functions in several homology-dependent DSB repair events, including gene conversion, break-induced replication, and recombination between inverted repeats (3). Several biochemical studies have established that Rad52 functions as a mediator in RAD51-dependent HR pathways. These findings include facilitating the assembly of the Rad51 recombinase on replication protein A-coated ssDNA (4, 5) and stimulating the DNA strand exchange activity of the Rad51 recombinase (6-9). In vitro studies have also suggested more direct roles of Rad52 in HR. Rad52 catalyzes the annealing of complementary ssDNA and promotes D-loop formation and DNA strand exchange (10-14). The strand invasion promoted by Rad52 may be relevant to some RAD51-independent HR events such as break-induced replication, in which the recombination substrates are ssDNA and dsDNA (15). However, the detailed molecular mechanisms of these Rad52-mediated recombination reactions remain poorly understood.
Crystallographic studies of the N-terminal domain of the human Rad52 protein have provided a structural framework for explaining the Rad52-mediated recombination reactions. The isolated N-terminal half of the human Rad52 protein multimerizes into an undecameric ring structure, with a highly positively charged groove that runs around the ring (16, 17). Structure-based alanine scan mutagenesis studies have identified several residues in the groove that are essential for ssDNA binding (16), and these residues are also important for DNA binding by the full-length protein (18). A model for ssDNA bound to Rad52 has been proposed in which the bases face outward (17). Such base conformations could allow the ssDNA molecule to form base pairs with an incoming ssDNA during ssDNA annealing and strand exchange. In the D-loop formation reaction promoted by Rad52 in vitro, incubating Rad52 with dsDNA before the addition of ssDNA inhibits the reaction, suggesting that the initial formation of the Rad52-ssDNA complex is essential for the reaction (12). These observations indicated that the Rad52-ssDNA complex comprises the catalytic core in the Rad52-mediated recombination reactions.
Rad52 is also known to bind to dsDNA (7, 12). However, the detailed properties of dsDNA binding as well as its biological significance are unknown. Biochemical studies have demonstrated that the isolated N-terminal half of Rad52 forms a ternary complex with ssDNA and dsDNA and that the formation of this complex is essential for D-loop formation (16). Interestingly, ternary complex formation is also observed in structurally unrelated recombinases, such as RecA and RecT (19, 20). In particular, RecA possesses a second DNA binding site that binds either ssDNA or dsDNA to facilitate the search for DNA sequence homology (21). We, therefore, hypothesized that Rad52 may also have a second DNA binding site.
In this work we have identified novel DNA binding sites of Rad52. Mutations in the binding sites did not affect ssDNA binding but they significantly reduced ternary complex formation between a Rad52-ssDNA complex and dsDNA. The defects strongly correlated with the D-loop formation activities. Furthermore, we found that the dsDNA bound to this second DNA binding site was positively supercoiled. These results provide insights into how Rad52 interacts with DNA substrates to promote recombination reactions.
EXPERIMENTAL PROCEDURES
Protein Purification—The human Rad52 and Rad521-212 genes were cloned into the pET-15b vector (Novagen), as described elsewhere (12, 16). Expression vectors for Rad521-212 point mutants (Y51A, K62A, H69A, R70A, N76A, Y81A, K102A, R112A, K133A, K135A, K144A, T148A, C165A, Y171A, K102A/K133A, and K169A/K173A) were constructed using a QuikChange mutagenesis kit (Stratagene). Expression vectors of other point mutants were prepared previously as described (16).
All proteins were expressed in the Escherichia coli strain JM109(DE3) carrying a plasmid (pArg3Arg4, a gift from K. Sakamoto, Systems and Structural Biology Center, Yokohama. Japan) to express low abundance tRNAs. All procedures after cell harvesting were carried out at 4 °C. For the purification of each protein, 2 liters of an LB culture were incubated at 30 °C, and protein expression was induced at an optical density (A600) of 0.6 with 1 mm isopropyl 1-thio-β-d-galactopyranoside (final concentration). After overnight induction, the cells were harvested, resuspended in 40 ml of buffer A (50 mm Tris-HCl, pH 7.8, 0.3 m KCl, 2 mm 2-mercaptoethanol, and 10% glycerol) containing 10 mm imidazole, and lysed by sonication. The cell lysate was cleared of insoluble material by centrifugation at 35,000 rpm for 30 min (Beckman Type 45 Ti rotor). The supernatant was mixed with 3 ml of nickel-nitrilotriacetic acid-agarose beads (Invitrogen or Qiagen) and was incubated with gentle mixing for 1 h. Afterward, the mixture was poured into an Econo-column, and the unbound cell lysate was drained out at a constant flow rate (2 ml/min). The packed, protein-bound, nickel-nitrilotriacetic acid-agarose beads were washed with 60 ml of buffer A containing 50 mm imidazole at a reduced flow rate (0.5 ml/min). Rad52 was eluted with a 60-ml linear gradient of 50-300 mm imidazole in buffer A. Rad521-212 and its point mutants were eluted with a 60-ml linear gradient of 50-400 mm imidazole in buffer A. Peak fractions were collected, and 3 units of thrombin protease (GE Healthcare) per mg of protein were added to remove the hexahistidine tag. The fractions were immediately dialyzed against 5 liters of buffer B (20 mm Hepes-KOH, pH 7.5, 0.2 m KCl, 0.5 mm EDTA, 2 mm 2-mercaptoethanol, and 10% glycerol). Dialysis was carried out for at least 1 day with one buffer exchange after 12 h of dialysis. The protein solutions were collected from the dialysis tubes and filtered through a 5-μm filter (Millipore). The removal of the hexahistidine tag was confirmed by SDS-PAGE. All proteins except for Rad521-212 Y81A, K133A, and K102A/K133A were loaded onto a 4 ml heparin-Sepharose column (GE Healthcare). After washing the column with 80 ml of buffer B, the proteins were eluted with an 80-ml linear gradient of 0.2-1 M KCl. Peak fractions were collected and concentrated to ∼2 mg/ml using an Amicon Ultra 30 ultrafiltration device (Millipore). Concentrated proteins were dialyzed against buffer C (20 mm Hepes-KOH, pH 7.5, 0.2 m KCl, 0.5 mm EDTA, 2 mm 2-mercaptoethanol, and 50% glycerol) and stored at -20 °C. For Rad521-212 Y81A and K133A, a 4-ml SP-Sepharose column (GE Healthcare) was used instead of the heparin-Sepharose column. For Rad521-212 K102A/K133A, the purification step using the heparin-Sepharose column was omitted. Protein concentrations were determined using a Bio-Rad protein assay kit, with bovine serum albumin (Nakalai Tesque) as the standard.
DNA Substrates—Negatively supercoiled plasmid DNA (pGsat4, 3.2 kilobases) was purified from sarkosyl-lysed cells as described (12). The closed, relaxed DNA used in the electron microscopic analysis was prepared in a 200-μl reaction mixture containing 80 μg of purified pGsat4, 80 units of wheat germ topoisomerase I (Promega), and 2/5 volume of buffer T (125 mm Tris-HCl, pH 7.5, 125 mm NaCl, 0.25 mm EDTA, 2.5 mm dithiothreitol, and 50% glycerol). This mixture was incubated at 37 °C for 1 h. The DNA was purified using a QIAquick PCR purification kit (Qiagen). For D-loop substrates, a 50-mer ssDNA oligonucleotide (5S-repeat-10-1, 5′-GGAATTCGGTATTCCCAGGCGGTCTCCCATCCAAGTACTAACCGAGCCCT-3′) and a pBlueScript II SK(+) vector containing 11 repeats of a sea urchin 5S rRNA gene (207-bp fragment) inserted into the SmaI site (pB5Sarray, 5.3 kilobases) were used. For the ssDNA annealing assay, two 50-mer ssDNA oligonucleotides (sense, SAT-1-50, 5′-ATTCCAACGTCCACCGACCAACTCTGAGTAACGTCTTGCTGCTGTGTGTA-3′, and antisense, SAT-1-50, 5′-TACACACAGCAGCAAGACGTTACTCAGAGTTGGTCGGTGGACGTTGGAAT-3′) were purified by electrophoresis using SequaGel 8 (National Diagnostics). All DNA concentrations are expressed in moles of nucleotides.
dsDNA Binding Assay—Reaction mixtures (8 μl), containing pGsat4 (50 ng) and 1 μl of buffer B were preincubated at 37 °C for 7 min. A 2-μl aliquot of Rad52 was then added to the reaction and incubated for 10 min. Complexes were fractionated through a 1% Seakem GTG-agarose (FMC BioProducts) gel in 1× TAE buffer for 3 h at 50 V using a GelMate apparatus (TOYOBO) and stained with 0.1 μg/ml ethidium bromide dissolved in 1× TAE buffer (40 mm Tris acetate, pH 7.5, 1 mm EDTA) for 15 min. Before visualization, the stained gel was washed with deionized water for 30 min. Complexes were visualized using a Molecular Imager FX (Bio-Rad).
ssDNA Binding, Ternary Complex Formation, and D-loop Formation Assays—For the ssDNA binding assay, a reaction mixture (8 μl) containing 1 μl of 10 μm 32P-labeled 5S-repeat-10-1 oligonucleotide and 2 μl of buffer D (250 mm Hepes-KOH, pH 7.5, 5 mm dithiothreitol, and 2.5 mg/ml bovine serum albumin (Roche Applied Science)) was preincubated at 37 °C for 5 min. A 2-μl aliquot of Rad52 was then added, and the reaction was incubated for 5 min. Complexes were fixed with 1 μl of 1% glutaraldehyde (0.1%) at 37 °C for 20 min. For the ternary complex formation and D-loop formation assays, a reaction mixture (7 μl) containing 1 μl of 10 μm 32P-labeled 5S-repeat-10-1 oligonucleotide and 2 μl of buffer D was preincubated at 37 °C for 5 min. A 2-μl aliquot of Rad52 was then added, and the reaction was incubated for 5 min. Afterward, 1 μl of 1 mm pB5Sarray (100 μm) was added, and the mixture was incubated at 37 °C for 10 min. Ternary complexes were fixed with 1 μl of 1% glutaraldehyde (0.1%) at 37 °C for 20 min. D-loops were deproteinized by adding 1 μl of a 0.25 m EDTA, 2.5% SDS solution and 1 μl of 20 mg/ml proteinase K (Roche Applied Science) and incubating the mixture at 37 °C for 15 min. For all three assays the products were fractionated through a 1% Seakem GTG-agarose (FMC BioProducts) gel in 0.5× TBE buffer for 2 h at 3.3 V/cm. The gels were dried, exposed to an imaging plate, and visualized using a BAS2500 image analyzer (Fuji).
ssDNA Annealing Assay—The ssDNA annealing assay was essentially done as described previously (22). A 49-μl reaction mixture (sufficient for 6 reactions) containing 7 μl of 10 μm 32P-labeled sense SAT-1-50 and 14 μl of buffer S (150 mm Tris acetate, pH 7.5, 0.5 m NaCl, 25 mm magnesium acetate, and 5 mm dithiothreitol) was incubated at room temperature for 2 min. A 14-μl aliquot of 1.25 μm Rad521-212 (0.25 μm) was then added, and the mixture was incubated further for 10 min. To start the reaction a 6-μl aliquot of 10 μm antisense SAT-1-50 was added. At the times indicated, a 10-μl aliquot of the reaction mixture was mixed with 1 μl of 1 mm unlabeled sense SAT-1-50 to quench the reaction. For the zero-time conditions, the unlabeled sense SAT-1-50 was mixed together with antisense SAT-1-50 and Rad521-212. The DNA substrates and products were deproteinized by adding 2 μl of a 0.25 m EDTA, 2.5% SDS solution and 1 μl of 20 mg/ml proteinase K (Roche Applied Science) and incubating the mixture at 37 °C for 10 min. Products were fractionated by PAGE on 15-25% gradient gels in 0.5× TBE buffer. The gels were dried, exposed to an imaging plate, and visualized using a BAS2500 image analyzer (Fuji).
Electron Microscopic Visualization—A reaction mixture (20 μl) containing closed, relaxed pGsat4 (200 ng), 4 μl of 10 μm Rad521-212, and 8 μl of buffer T (125 mm Tris-HCl, pH 7.5, 125 mm NaCl, 0.25 mm EDTA, 2.5 mm dithiothreitol, and 50% glycerol) was incubated at 37 °C for 30 min. A freshly glow-discharged carbon support was floated on 4 μl of the sample placed on a sheet of Parafilm for 30 s. The support was removed from the sample drop, briefly wiped on a filter paper, and then floated on a 2% uranyl acetate solution (50 μl) placed on a Parafilm sheet for 1 min. The carbon support was wiped on a filter paper to remove the residual uranyl acetate solution. Specimens were imaged using a JEOL JEM 2000FX electron microscope.
Topoisomerase-mediated Relaxation Assay—To prepare covalently closed, relaxed DNA, a 25-μl reaction mixture (sufficient for 5 reactions) containing 1.6 μg of negatively supercoiled plasmid DNA (pGsat4, 3.2 kilobases), 30 units of wheat germ topoisomerase I (Promega), and 5 μl of buffer T was incubated at 37 °C for 30 min. Separate reaction mixtures (25.4 μl) containing 12 μl of buffer T and 6 μl of Rad52 were preincubated at 37 °C for 8 min. To this mixture a 4.6-μl aliquot of the previous reaction mixture containing the relaxed pGsat4 DNA and topoisomerase I was added to start the reaction. The incubation was performed at 37 °C for 30 min. To stop and deproteinize the reaction, either a 30-μl aliquot of a phenol/chloroform mixture or a 3-μl aliquot of a 0.25 m EDTA, 2.5% SDS solution was added to the reaction mixture, and the mixture was immediately vortexed for 10 s. The phenol/chloroform-extracted samples were further extracted once with chloroform. The reactions stopped with SDS were treated with 1 μl of 20 mg/ml proteinase K (Roche Applied Science) and incubated at 37 °C for 15 min.
All of the electrophoresis was performed at room temperature using 1× TAE buffer. For the one-dimensional gel electrophoresis, 10 μl of the reaction mixtures were fractionated through a 1% Seakem GTG agarose (FMC BioProducts) gel for 3 h at 50 V using a GelMate apparatus (TOYOBO). For the two-dimensional gel electrophoresis, 15 μl of the samples were fractionated through a 1.5% Seakem GTG-agarose gel for 3.5 h at 3.3 V/cm using the mini submarine gel electrophoresis apparatus with a 9 × 9-cm gel tray (Nihon Eido). Afterward, the running buffer was exchanged with 1× TAE buffer containing 5 μg/ml chloroquine. The gel was rotated 90° in a clockwise direction and soaked in this buffer for 2 h. The second dimension of electrophoresis was run at 1.3 V/cm for 20 h with buffer circulation.
DNA products were stained with GelStar (Cambrex) and diluted 10,000-fold in 1× TAE buffer for 40 min. After briefly washing the gel with deionized water, the products were visualized using a Molecular Imager FX (Bio-Rad).
DNA Supercoiling Inhibition Assay—A reaction mixture (23 μl, containing pGsat4 (300 ng), E. coli topoisomerase I (6 units, New England Biolabs), 0.3 μl of 100× bovine serum albumin (New England Biolabs), and 3 μl of 10× buffer 4 (New England Biolabs)) was incubated for 30 min at 37 °C. 1 μl of distamycin A, netropsin, or pentamidine (Sigma-Aldrich) was added to the reaction mixture, which was incubated for 10 min. Afterward, a 6-μl aliquot of 10 μm Rad521-212 was added, and the mixture was further incubated for 30 min at 37 °C. The samples were mixed with 30 μl of a phenol/chloroform mixture and were immediately vortexed for ∼10 s. A 10-μl aliquot of the aqueous phase was fractionated through a 1% Seakem GTG-agarose (FMC BioProducts) gel in 1× TAE buffer for 3 h at 50 V using a GelMate apparatus (TOYOBO), and the gel was stained with 0.1 μg/ml ethidium bromide dissolved in 1× TAE buffer for 15 min. Before visualization, the stained gel was washed with deionized water for 30 min. The complexes were visualized with a Molecular Imager FX (Bio-Rad). To analyze the Rad521-212-dsDNA complexes formed in the presence of distamycin A, netropsin, or pentamidine, the same reactions were repeated but without the addition of E. coli topoisomerase I and the deproteinization step. The complexes were fractionated and visualized as described above.
RESULTS
Mapping of the dsDNA Binding Site of Rad52—To define the dsDNA binding site on the core N-terminal domain of Rad52, a series of alanine mutants (supplemental Table 1s) were prepared, and their dsDNA binding activities were examined by an electrophoretic mobility shift assay (EMSA). Negatively supercoiled plasmid DNA was incubated with increasing concentrations of each mutant protein, and the resulting complexes were fractionated on an agarose gel. We found that the K102A, K133A, K144A, E145A, K169A, Y171A, and R173A mutants formed faster migrating complexes than the wild-type and other mutant Rad521-212 proteins (Fig. 1A). Rad521-212 also bound to topologically relaxed DNA. When an EMSA was performed using plasmid DNA that was linearized with a blunt-cutting restriction endonuclease, DNA binding defects of the K102A, K133A, K144A, and E145A mutants were observed (supplemental Fig. 1s). Because Rad521-212 forms oligomeric ring structures, we tested whether the mutants were proficient in oligomerization. As judged from the gel filtration analysis, the wild-type and mutant proteins had the same apparent molecular weights, suggesting that the undecameric ring formation was not affected by the mutations (supplemental Fig. 2s). These results indicate that the Lys-102, Lys-133, Lys-144, Glu-145, Lys-169, Tyr-171, and Arg-173 residues play important roles in dsDNA binding. Except for Tyr-171, all of these residues are exposed on the surface of the crystal structure of the N-terminal domain of Rad52 and, therefore, could potentially interact with dsDNA.
FIGURE 1.

Alanine scan mutagenesis of Rad521-212. A, EMSA. Alanine point mutants (0, 0.5, 1, 1.5, or 2 μm) of Rad521-212 were incubated with 15 μm negatively supercoiled plasmid DNA (pGsat4, 3.2 kilobases), and complexes were fractionated through an agarose gel. WT, wild type. B, surface view of the Rad521-212 protomer. The stem and domed cap regions are colored light brown and light blue, respectively. Lys-102 and Lys-133 (magenta) are located at the rim of the stem region, whereas Lys-169, Tyr-171, and Arg-173 (also magenta) are located at the edge of the domed cap region. The previously identified ssDNA binding residues (Arg-55, Tyr-65, Lys-152, Arg-153, Arg-156, dark blue) are clustered at the bottom of the groove formed between the stem and domed cap regions. C, surface view of the Rad521-212 undecameric ring. All structural figures were created using the PyMOL program.
Interestingly, the Lys-102, Lys-133, Lys-169, and Arg-173 residues are clustered at two separate locations and are outside the previously found DNA binding site that binds to ssDNA (Figs. 1, B and C). To investigate whether the presently identified sites constitute a second DNA binding site, we examined the ssDNA binding activities of the K102A, K133A, K169A, and R173A mutants. The wild-type Rad521-212 protein saturated the ssDNA at an ∼1:4 molar ratio of protein to nucleotides (Fig. 2B, first row, far left), suggesting that the Rad521-212 protomers on average bind to four nucleotides of ssDNA. This is consistent with the conclusions drawn in previous studies (17, 23). Under the same reaction conditions, the K102A, K133A, and K169A mutants showed similar electrophoretic mobilities as compared with that of the wild-type Rad521-212 protein (Fig. 2B, first row). We also prepared double point mutants, K102A/K133A and K169A/R173A, in an attempt to completely disrupt either one of the DNA binding sites. These mutants also bound to ssDNA with similar affinities, as compared with the wild-type and other mutant proteins. These results indicate that Lys-102, Lys-133, Lys-169, and Lys-173 comprise a second DNA binding site of Rad52.
FIGURE 2.

D-loop formation activities of Rad521-212 mutants. A, a schematic diagram of the D-loop formation reaction promoted by Rad521-212. B, increasing concentrations (0, 0.03, 0.06, 0.12, 0.25, 0.5, 1, 2 μm) of wild-type (WT) or mutant Rad521-212 proteins were initially complexed with 1 μm ssDNA (5S-repeat-10-1, 50-mer) (first row). Afterward, negatively supercoiled plasmid DNA (pB5Sarray, 5.3 kilobases) was added, and the reactions were either treated with glutaraldehyde (ternary complexes, second row) or proteinase K (D-loops, third row).
Residues in the Second DNA Binding Site Essential for D-loop Formation—Rad52 promotes the formation of D-loops between an ssDNA oligonucleotide and a negatively super-coiled plasmid DNA containing the complementary sequence (12). The isolated N-terminal half of Rad52 was found to form a ternary complex with ssDNA and dsDNA in this reaction (16). To determine whether the presently identified dsDNA binding sites are involved in ternary complex formation and D-loop formation, the mutants defective in dsDNA binding were subjected to ternary complex formation and D-loop formation assays. In both assays, Rad52-ssDNA complexes were initially formed, and then dsDNA was added (Fig. 2A). Before electrophoretic separation of the products, the reaction mixtures were treated with either a protein-DNA cross-linking reagent (glutaraldehyde) to stabilize the ternary complexes or a protease to deproteinize the D-loops. We found that all of the mutants with the exception of K169A and R173A were defective in ternary complex formation (Fig. 2B, second row). The mutants were also defective in catalyzing D-loop formation, and the decreases in this activity closely paralleled the decreases in the ternary complex formation activities (Fig. 2B, third row). These results indicate that the second DNA binding site is required for Rad52-mediated D-loop formation. Furthermore, the K102A/K133A double mutant was nearly devoid of D-loop formation activity, whereas the K169A/R173A double mutant still had residual activity, which suggests that Lys-102 and Lys-133 play more critical roles in D-loop formation than Lys-169 and Arg-173.
Involvement of the Second DNA Binding Site in ssDNA Annealing—To further characterize the roles of Lys-102 and Lys-133 in the Rad52-mediated recombination reactions, we examined the ssDNA annealing activities of the mutants. As in the D-loop formation assay, a Rad521-212-ssDNA complex was initially formed, and the second ssDNA oligonucleotide was subsequently added to start the annealing reaction (Fig. 3A). As compared with the spontaneous reaction, the wild-type Rad521-212 protein significantly catalyzed strand annealing, particularly within the first 3 min (Fig. 3B). By contrast, we failed to observe any catalytic activity of the K102A, K133A, and K102A/K133A mutants in the annealing reaction, as shown by the lower annealing rates of the mutants than that of the spontaneous reaction (Fig. 3B). These results indicate that the second DNA binding site, formed by Lys-102 and Lys-133, also plays a critical role in ssDNA annealing.
FIGURE 3.

ssDNA annealing activities of Rad521-212 mutants. A, a schematic diagram of the ssDNA annealing reaction promoted by Rad521-212. B, percentages of DNA annealing catalyzed by the wild-type (WT) Rad521-212 (closed square), K102A (open diamond), K133A (open triangle), and K102A/K133A (open square) proteins. Proteins (0.25 μm) were first complexed with 1 μm ssDNA (sense SAT-1-50, 50-mer) followed by the addition of a complementary ssDNA (antisense SAT-1-50, 50-mer). The spontaneous reaction is indicated by closed circles.
Electron Microscopic Visualization of Rad521-212-dsDNA Complexes—Presently little is known about the molecular mechanism that underlies the interaction between Rad52 and dsDNA. The EMSAs suggest that the discrete Rad521-212-dsDNA complexes associate to form large networks (Fig. 1A and supplemental Fig. 1s). To analyze the structure of the Rad521-212-dsDNA complexes, Rad521-212 was complexed with covalently closed, relaxed plasmid DNA and was visualized by electron microscopy. We observed large networks (Fig. 4, middle panel), which closely resembled the full-length Rad52 protein-dsDNA complexes (24). In the absence of DNA these large networks were not observed (Fig. 4, left panel), indicating that the networks consist of multiple Rad52 rings or ring-like structures closely assembled on dsDNA. Strikingly, in contrast to the wild-type Rad521-212 protein, we failed to observe similar networked structures of the K102A mutant complexed with dsDNA (Fig. 4, right panel). The K102A-dsDNA complexes resembled the structure of Rad521-212 without DNA even though the formation of the K102A-dsDNA complex was confirmed by an EMSA (supplemental Fig. 3s). These observations indicate that Rad52 forms large networked structures on dsDNA and that the Lys-102 residue is responsible for stabilizing the higher order structures of the Rad52-dsDNA complex, through interactions with dsDNA.
FIGURE 4.

Electron micrographs of Rad521-212 complexed with covalently closed, relaxed plasmid DNA. Negatively stained samples of Rad521-212, the Rad521-212-DNA complex, and the Rad521-212 K102A-DNA complex (from left to right) are shown. The black bars denote 10 nm.
Positive Supercoils Generated by Rad52 Binding—The networked structures formed by Rad52 and dsDNA raise the possibility that Rad52 induces conformational changes in dsDNA. To test this possibility, topological changes in covalently closed, relaxed plasmid DNA were monitored in the presence of Rad52 and wheat germ topoisomerase I (Fig. 5A). If Rad52 is able to change the conformation of the DNA, then compensating changes should occur in the DNA regions not bound by Rad52 because the DNA is topologically constrained. These compensatory changes are removed by the topoisomerase, which can relax either positive or negative supercoils in DNA. The topology of the DNA substrate after deproteinization will reflect the changes in the DNA conformation caused by the Rad52 binding.
FIGURE 5.
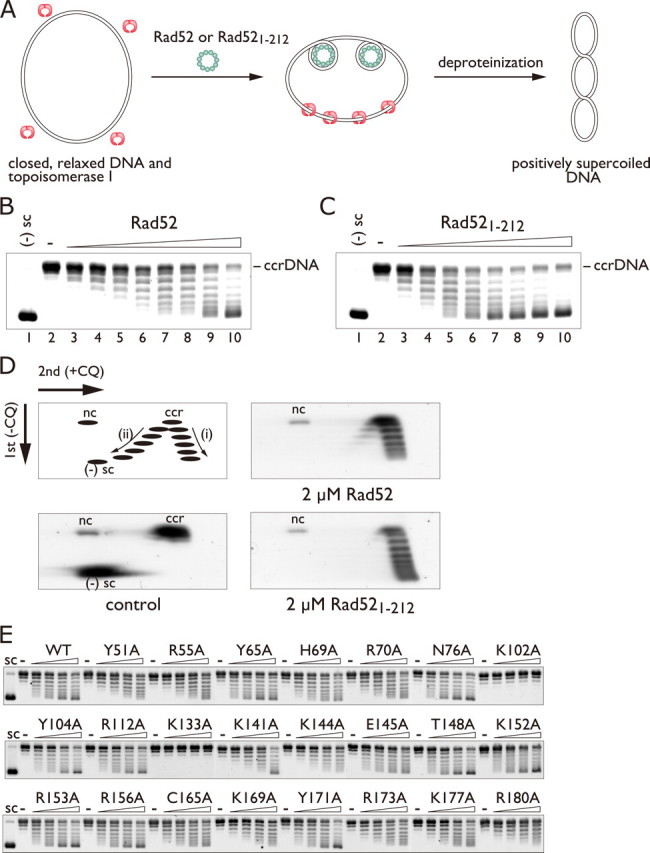
Positive supercoiling activity of Rad52 and Rad521-212. A, schematic diagram of the topoisomerase I-mediated relaxation assay. B and C, one-dimensional gel electrophoresis of DNA topoisomers generated in the presence of Rad52 (B) and Rad521-212 (C). In these reactions the protein concentrations were increased from 0 to 4 μm in 0.5 μm increments. ccrDNA denotes covalently closed, relaxed DNA. D, two-dimensional gel electrophoresis of DNA topoisomers generated in the presence of Rad52 and Rad521-212. The 2 μm Rad52 reaction (B, lane 6), resolved in a two-dimensional gel, is shown in D, top right. Similarly, the 2 μm Rad521-212 reaction (C, lane 6), resolved in a two-dimensional gel, is shown in D, bottom right. The top left figure in D is a schematic diagram of the relative positions of covalently closed, relaxed DNA (ccr), nicked circular DNA (nc), positively supercoiled topoisomers (i), and negatively supercoiled topoisomers (ii) resolved by two-dimensional gel electrophoresis. E, alanine scan mutagenesis of Rad521-212. The mutant proteins (0, 0.5, 1, 1.5, or 2 μm) were incubated with 30 μm of covalently closed, relaxed plasmid DNA. CQ, chloroquine.
When increasing amounts of Rad52 were added to the reaction mixture containing topoisomerase I, higher proportions of the DNA exhibited increased mobility through the agarose gel (Fig. 5B, lanes 3-10), indicating Rad52 binding altered the conformation of the DNA. To confirm that they are topoisomers, the products were further resolved in the second dimension in the presence of chloroquine. Chloroquine intercalates between base pairs and unwinds DNA and, therefore, influences the topology of covalently closed molecules but not those of nicked or linear DNA substrates. Under controlled conditions chloroquine-containing agarose gel electrophoresis of topoisomers results in the slower migration of negatively supercoiled topoisomers and the faster migration of positively supercoiled topoisomers as shown schematically in Fig. 5D, top left. The DNA products migrated faster than the nicked circular DNA in the second dimension (Fig. 5D, top right), indicating that they are topoisomers. These topoisomers were positively supercoiled, as demonstrated by their faster migration than the covalently closed, relaxed DNA in the second dimension. From these results we conclude that Rad52 introduces positive supercoils in dsDNA.
The N-terminal half of Rad52 is highly conserved from yeast to human and is essential for DNA binding and self-association (25). Biochemical studies have shown that the isolated N-terminal half of Rad52 retained the self-association, DNA binding, and D-loop formation activities (12, 26). To further validate the positive supercoiling activity of Rad52, we analyzed the supercoiling activity of a Rad52 fragment (Rad521-212) lacking the C-terminal half (amino acid residues 213-418) using the topoisomerase-mediated relaxation assay described above. As expected, Rad521-212 introduced positive supercoils in dsDNA (Fig. 5D, bottom right). This result indicates that positive supercoiling is mediated by the N-terminal half of Rad52. Furthermore, to define the dsDNA binding residues essential for positive supercoiling, we performed alanine scan mutagenesis using the topoisomerase-mediated relaxation assay described above. We found that the K102A and K133A mutants were severely defective in introducing positive supercoils in DNA (Fig. 5E).
Notably, the isolated, conserved domain of Rad52 (Rad521-212) was significantly more efficient in promoting positive supercoils than the full-length protein. The different activities could arise from the difference between the oligomeric structures of Rad521-212 and the full-length protein (16, 17, 27). Previous electron microscopic studies revealed that the full-length and the C terminal-truncated Rad52 form similar sized rings. Hence, the distance between the conserved N-terminal domains of Rad52 in the undecameric form is likely to be shorter than that in the heptameric form. The closer alignment of these residues in the undecameric form may be essential for promoting more extensive DNA-positive supercoiling, which the heptameric form may not be able to accomplish.
Inhibition of Supercoiling Activity by DNA Minor Groove Binders—To further investigate the mechanism by which Rad521-212 binds to dsDNA and promotes positive supercoiling, we focused on minor groove binding peptides that are known to introduce positive supercoils in DNA (28, 29). In particular, netropsin and distamycin A have lysine-like structures at one end, and to some extent the structures are similar to that of the dsDNA binding site of Rad521-212 containing Lys-102 and Lys-133 (Fig. 6A). Based on these observations, we postulated that Rad521-212 may recognize the DNA minor groove to promote positive supercoils. To test this hypothesis we examined whether Rad521-212 would compete with the peptides to promote positive supercoiling. In the assay the peptides were preincubated with covalently closed, relaxed plasmid DNA, and then Rad521-212 and E. coli topoisomerase I were added to the complex. At concentrations of distamycin A and netropsin in which the positive supercoiling induced by these peptides was not detectable (Fig. 6B, lanes 3-6 and 15-18), the super-coiling activity of Rad521-212 was potently inhibited (Fig. 6B, lanes 8-11 and 20-23). Netropsin was more efficient than distamycin A in inhibiting the positive supercoiling activity of Rad521-212, which correlates with a previous study showing that netropsin promotes positive supercoils more effectively than distamycin A (29). Consistent with this view, pentamidine, a DNA minor groove binder that was previously shown to be nearly ineffective in introducing positive supercoils (29), required a 10-fold higher concentration than that of distamycin A for the inhibition (Fig. 6B, lanes 32-35). We also found that these ligands did not completely inhibit the dsDNA binding activity of Rad521-212 at concentrations in which positive supercoiling was completely inhibited (Fig. 6C). These observations suggest that the inhibition was caused by the peptides binding to the DNA minor groove and blocking the essential interactions between Rad521-212 and DNA required for positive supercoiling.
FIGURE 6.

DNA minor groove binders inhibit the positive supercoiling activity of Rad521-212. A, structures of distamycin A, netropsin, and pentamidine. B, topoisomerase I-mediated relaxation assay in the presence of minor groove binding ligands. Increasing concentrations of the ligands with or without 2 μm Rad521-212. Lanes 12, 24, and 36 show the addition of the ligands before E. coli topoisomerase I. C, DNA binding activity of Rad521-212 in the presence of the ligands. Negatively supercoiled plasmid DNA (pGsat4, 3.2 kilobases) was used.
Predicted dsDNA Binding Path across the Rad52 Structure—In the crystal structure of Rad52, Lys-102 and Lys-133 are clustered along the rim of the stem region. Based on this observation, we propose a dsDNA binding path that runs around the ring structure near these residues (Figs. 1, B and C, and 7). dsDNA bound to this site may be further stabilized by Lys-169 and Arg-173, which are also essential for ternary complex formation but to lesser extents than Lys-102 and Lys-133. We note that Lys-169 and Arg-173 in the uncomplexed Rad521-212 structure are too far from the modeled dsDNA. Thus, significant movement of these residues is predicted for the interaction to occur (supplemental Fig. 4s). This conformational change appears possible, since the α helix (residues 169-177) and the flanking loop regions are relatively mobile in the crystal structure. Furthermore, a previous computational sequence analysis suggested that the α helix of Rad52, containing Lys-169 and Arg-173, is part of a small nucleic acid binding motif found in many repair and recombination proteins (30-32). These observations suggest that Lys-169 and Arg-173 are DNA binding residues and that the dsDNA bound to the second DNA binding site may be clamped between the edge of the domed cap region (Lys-169, Arg-173) and the rim of the stem region (Lys-102, Lys-133) (Figs. 1, B and C, and 7).
FIGURE 7.

A ternary complex model depicting the first and second DNA binding sites on Rad521-212. The Lys-102 and Lys-133 residues are colored magenta. The ssDNA molecule (yellow) binds inside the groove, whereas the dsDNA molecule (orange) runs around the rim of the stem region. The DNA model was created using the NAB program (40), with ΔLk =+2. Lk, linking number.
DISCUSSION
Several lines of evidence have indicated that Rad52 catalyzes the pairing of homologous ssDNA and dsDNA, which is one of the critical steps in HR. However, the precise mechanism by which Rad52 promotes this reaction is not well understood. Here, we have identified a second DNA binding site of Rad52 and have shown that it is essential for mediating D-loop formation and ternary complex formation between the Rad52-ssDNA complex and dsDNA. Structure-based mutagenesis studies revealed the location of the second DNA binding site on the Rad52 ring structure, which is near the entrance of the positively charged groove that houses the first DNA binding site. We have previously shown that when Rad52 is complexed with dsDNA before ssDNA, both the ternary complex and D-loop formation activities are inhibited (12, 16). Consistent with this finding, the spatial locations of the two binding sites suggest that the dsDNA molecule bound to the second DNA binding site can sterically hinder the access of ssDNA to the first DNA binding site (Fig. 7). Hence, initial ssDNA binding is essential for the D-loop formation promoted by Rad52. These findings underscore the view that Rad52 promotes DNA strand invasion by binding to both ssDNA and dsDNA (Fig. 8).
FIGURE 8.

A unifying view of the ssDNA annealing and strand invasion reactions promoted by Rad52. For simplification, a Rad52 protomer (blue) is shown. Both ssDNA annealing and D-loop formation require the initial formation of the Rad52-ssDNA complex. The second DNA substrate then binds to the second DNA binding site, and the reactions take place at the entrance of the groove.
The second DNA binding site of Rad52 promotes DNA positive supercoiling, a novel activity of Rad52 that may be involved in the DNA strand invasion process. Mutations in either Lys-102 or Lys-133, which both map to the second DNA binding site, severely impaired the DNA positive supercoiling activity of Rad52. The fact that Rad52 promotes positive supercoils in dsDNA implies a specific dsDNA binding mode exhibited by Rad52 either involving a right-handed wrapping of DNA or an over-winding of the DNA double helix. The location of the second DNA binding site on the Rad52 ring structure suggests partial or full wrapping of the DNA around the ring structure. Our electron microscopic analysis of Rad52-dsDNA complexes revealed large networked structures that probably contain multiple Rad52 rings or ring-like structures. These observations suggest the possibility of Rad52 rings associating with each other in a specific manner in the presence of dsDNA, with the dsDNA molecule either wrapped around the higher order Rad52 complexes in a right-handed fashion or over-wound along its double-helical axis.
Based on the above observations, one possible mechanism for Rad52-mediated strand invasion is the unwinding of dsDNA promoted by the right-handed wrapping of dsDNA around multiple Rad52 rings. The dsDNA region bound to Rad52 becomes unwound due to a compensatory effect and is aligned with the ssDNA molecule bound to the first DNA binding site of Rad52. In this model the homology search and the strand invasion occur via the RecA-type strand invasion mechanism (19, 33, 34). The present data, however, cannot exclude the possibility that Rad52 promotes D-loop formation via an annealing between ssDNA and single-stranded regions in dsDNA. The second DNA binding site is also critical for ssDNA annealing and may associate with a second, incoming ssDNA. Regardless of the underlying mechanism, the present results demonstrate the important roles of the second DNA binding site in Rad52-mediated recombination reactions.
The positive supercoiling activity of Rad52 could play an essential role during recombinational repair. A previous in vivo study showed that yeast cells with mutations in Rad52 corresponding to the second DNA binding site display synergistic sensitivity toward γ-rays in the Rad59Δ background (35). Rad59, a yeast Rad52 paralog, has homology to the N-terminal half of Rad52 and catalyzes ssDNA annealing. These observations suggest that the functions exhibited by the second DNA binding site of Rad52 are shared between Rad52 and Rad59 and are an important component in the Rad52-mediated DSB repair process. Although a homolog of Rad59 has not been found in higher eukaryotes, a functional counterpart, perhaps the splicing variant of Rad52 (36, 37), may play a similar role to that of Rad59.
Finally, although our present findings suggest possible roles for the second DNA binding site in Rad52-mediated recombination reactions, it may also play essential roles in other processes. For example, Rad52 has been shown to work in conjunction with the Mus81-Eme1 complex (38) as well as to interact physically with the WRN helicase (39). These proteins are essential for restoring the collapsed replication fork caused by a DSB and probably function in the later stages of HR. Our results raise the possibility that Rad52 may exert topological effects on the replication fork during replication repair. Further studies are required to understand the molecular mechanisms underlying the function of Rad52 on dsDNA in HR-dependent repair pathways.
Supplementary Material
Acknowledgments
We thank N. Sarai for critically reading the manuscript.
This work was supported by the Targeted Proteins Research Program and the RIKEN Structural Genomics/Proteomics Initiative of the National Project on Protein Structural and Functional Analyses, Ministry of Education, Culture, Sports, Science, and Technology of Japan. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Table 1s and Figs. 2s-4s.
Footnotes
The abbreviations used are: DSB, double-strand break; HR, homologous recombination; ssDNA, single-stranded DNA; dsDNA, double-stranded DNA; EMSA, electrophoretic mobility shift assay.
References
- 1.Daley, J. M., Palmbos, P. L., Wu, D., and Wilson, T. E. (2005) Annu. Rev. Genet. 39 431-451 [DOI] [PubMed] [Google Scholar]
- 2.Sung, P., and Klein, H. (2006) Nat. Rev. Mol. Cell Biol. 7739 -750 [DOI] [PubMed] [Google Scholar]
- 3.Symington, L. S. (2002) Microbiol. Mol. Biol. Rev. 66630 -670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Song, B., and Sung, P. (2000) J. Biol. Chem. 27515895 -15904 [DOI] [PubMed] [Google Scholar]
- 5.Sugiyama, T., and Kowalczykowski, S. C. (2002) J. Biol. Chem. 27731663 -31672 [DOI] [PubMed] [Google Scholar]
- 6.Sung, P. (1997) J. Biol. Chem. 27228194 -28197 [DOI] [PubMed] [Google Scholar]
- 7.Benson, F., Baumann, P., and West, S. C. (1998) Nature 391401 -404 [DOI] [PubMed] [Google Scholar]
- 8.Shinohara, A., and Ogawa, T. (1998) Nature 391404 -407 [DOI] [PubMed] [Google Scholar]
- 9.New, J. H., Sugiyama, T., Zaitseva, E., and Kowalczykowski, S. C. (1998) Nature 391407 -410 [DOI] [PubMed] [Google Scholar]
- 10.Mortensen, U. H., Bendixen, C., Sunjevaric, I., and Rothstein, R. (1996) Proc. Natl. Acad. Sci. U. S. A. 9310729 -10734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reddy, G., Golub, E. I., and Radding, C. M. (1997) Mutat. Res. 37753 -59 [DOI] [PubMed] [Google Scholar]
- 12.Kagawa, W., Kurumizaka, H., Ikawa, S., Yokoyama, S., and Shibata, T. (2001) J. Biol. Chem. 27635201 -35208 [DOI] [PubMed] [Google Scholar]
- 13.Kumar, J. K., and Gupta, R. C. (2004) Proc. Natl. Acad. Sci. U. S. A. 1019562 -9567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bi, B., Rybalchenko, N., Golub, E. I., and Radding, C. M. (2004) Proc. Natl. Acad. Sci. U. S. A. 1019568 -9572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kraus, E., Leung, W.-Y., and Haber, J. E. (2001) Proc. Natl. Acad. Sci. U. S. A. 988255 -8262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kagawa, W., Kurumizaka, H., Ishitani, R., Fukai, S., Nureki, O., Shibata, T., and Yokoyama, S. (2002) Mol. Cell 10359 -371 [DOI] [PubMed] [Google Scholar]
- 17.Singleton, M. R., Wentzell, L. M., Liu, Y., West, S. C., and Wigley, D. B. (2002) Proc. Natl. Acad. Sci. U. S. A. 9913492 -13497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lloyd, J. A., McGrew, D. A., and Knight, K. L. (2005) J. Mol. Biol. 345239 -249 [DOI] [PubMed] [Google Scholar]
- 19.Shibata, T., Cunningham, R. P., DasGupta, C., and Radding, C. M. (1979) Proc. Natl. Acad. Sci. U. S. A. 765100 -5104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Noirot, P., Gupta, R. C., Radding, C. M., and Kolodner, R. D. (2003) EMBO J. 22 324-334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mazin, A. V., and Kowalczykowski, S. C. (1996) Proc. Natl. Acad. Sci. U. S. A. 9310673 -10678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sugiyama, T., New, J. H., and Kowalczykowski, S. C. (1998) Proc. Natl. Acad. Sci. U. S. A. 956049 -6054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parsons, C. A., Baumann, P., Van Dyck, E., and West, S. C. (2000) EMBO J. 194175 -4181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Van Dyck, E., Hajibagheri, N. M. A., Stasiak, A., and West, S. C. (1998) J. Mol. Biol. 2841027 -1038 [DOI] [PubMed] [Google Scholar]
- 25.Park, M. S., Ludwig, D. L., Stigger, E., and Lee, S.-H. (1996) J. Biol. Chem. 27118996 -19000 [DOI] [PubMed] [Google Scholar]
- 26.Ranatunga, W., Jackson, D., Lloyd, J. A., Forget, A. L., Knight, K. L., and Borgstahl, G. E. O. (2001) J. Biol. Chem. 27615876 -15880 [DOI] [PubMed] [Google Scholar]
- 27.Stasiak, A. Z., Larquet, E., Stasiak, A., Muller, S., Engel, A., Van Dyck, E., West, S. C., and Egelman, E. H. (2000) Curr. Biol. 10337 -340 [DOI] [PubMed] [Google Scholar]
- 28.Snounou, G., and Malcolm, A. D. B. (1983) J. Mol. Biol. 167211 -216 [DOI] [PubMed] [Google Scholar]
- 29.Störl, K., Burckhardt, G., Lown, J. W., and Zimmer, C. (1993) FEBS Lett. 334 49-54 [DOI] [PubMed] [Google Scholar]
- 30.Iyer, L. M., Koonin, E. V., and Aravind, L. (2002) BMC Genomics 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shao, X., and Grishin, N. V. (2000) Nucleic Acids Res. 282643 -2650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Doherty, A. J., Serpell, L. C., and Ponting, C. P. (1996) Nucleic Acids Res. 242488 -2497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Folta-Stogniew, E., O'Malley, S., Gupta, R., Anderson, K. S., and Radding, C. M. (2004) Mol. Cell 15 965-975 [DOI] [PubMed] [Google Scholar]
- 34.Nishinaka, T., Shinohara, A., Ito, Y., Yokoyama, S., and Shibata, T. (1998) Proc. Natl. Acad. Sci. U. S. A. 9511071 -11076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feng, Q., During, L., de Mayolo, A. A., Lettier, G., Lisby, M., Erdeniz, N., Mortensen, U. H., and Rothstein, R. (2007) DNA Repair 627 -37 [DOI] [PubMed] [Google Scholar]
- 36.Kito, K., Wada, H., Yeh, E. T., and Kamitani, T. (1999) Biochim. Biophys. Acta 1489303 -314 [DOI] [PubMed] [Google Scholar]
- 37.Thorpe, P. H., Marrero, V. A., Savitzky, M. H., Sunjevaric, I., Freeman, T. C., and Rothstein, R. (2006) Mol. Cell. Biol. 263752 -3763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Doe, C. L., Osman, F., Dixon, J., and Whitby, M. C. (2004) Nucleic Acids Res. 325570 -5581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baynton, K., Otterlei, M., Bjoras, M., von Kobbe, C., Bohr, V. A., and Seeberg, E. (2003) J. Biol. Chem. 27836476 -36486 [DOI] [PubMed] [Google Scholar]
- 40.Macke, T., and Case, D. A. (1998) Modeling Unusual Nucleic Acid Structures (Leontes, N. B. and SantaLucia, J., Jr., eds) pp. 379-393, American Chemical Society, Washington, DC
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.