

Abstract
The telomere is a functional chromatin structure that consists of G-rich repetitive sequences and various associated proteins. Telomeres protect chromosomal ends from degradation, provide escape from the DNA damage response, and regulate telomere lengthening by telomerase. Multiple proteins that localize at telomeres form a complex called shelterin/telosome. One component, TRF1, is a double-stranded telomeric DNA binding protein. Inactivation of TRF1 disrupts telomeric localization of other shelterin components and induces chromosomal instability. Here, we examined how the telomeric localization of shelterin components is crucial for TRF1-mediated telomere-associated functions. We found that many of the mTRF1 deficient phenotypes, including chromosomal instability, growth defects, and dysfunctional telomere damage response, were suppressed by the telomere localization of shelterin components in the absence of functional mTRF1. However, abnormal telomere signals and telomere elongation phenotypes were either not rescued or only partially rescued, respectively. These data suggest that TRF1 regulates telomere length and function by at least two mechanisms; in one TRF1 acts through the recruiting/tethering of other shelterin components to telomeres, and in the other TRF1 seems to play a more direct role.
The telomere is a chromosomal end structure comprised of G-rich tandem repeat sequences and various telomere localizing proteins. This structure prevents chromosome end degradation, escapes from the DNA damage response, and controls telomerase-mediated elongation (1, 2). In mammals, six telomere-localizing proteins, TRF1, TRF2, TIN2, RAP1, TPP1 (previously known as TINT1/PTOP/PIP1), and POT1 form a large complex termed the “shelterin/telosome,” which is important for regulating telomeric structure and function (3-5). In addition, the 3′ end of telomeric DNA is single-stranded, and this G-rich overhang is integrated into the double-stranded telomeric DNA region to form a loop structure called the t-loop; this loop prevents DNA damage recognition and telomerase access (5, 6).
Previous studies have elucidated the role(s) of each shelterin component in telomere regulation as follows; TRF1 and TRF2 are telomeric double-stranded DNA-binding proteins. TRF1 is similar to TRF2 in structure and forms homodimers to bind telomeric DNA, but the major functions of these proteins are distinct (7, 8). TRF1 negatively regulates telomerase-dependent elongation (9), and TRF1 deletion either in knock-out mouse ES cells or by small interfering RNA-mediated deletion revealed that TRF1 regulates telomeric localization of other shelterin components and maintains the functional telomere structure (3, 10-13). On the other hand TRF2 is a telomere capping molecule, and deletion of TRF2 immediately induces end-to-end fusion, cell senescence, or cell death via the activation of telomere dysfunctional DNA damage responses (14-16). These proteins are associated with TIN2, which contributes to the stabilization of telomeric localization of them (17, 18). Furthermore, TIN2 also interacts with TPP1, which tethers to the single-stranded DNA-binding protein POT1, and TIN2 recruitment to telomere is essential for TPP1-POT1 telomeric localization (13, 17, 19, 20). POT1 functions to inhibit telomerase access to the telomere, suggesting POT1 is a terminal transducer of TRF1 telomere length regulation (10, 21). In addition, there are two homologues of human POT1 in the mouse, POT1a and POT1b, and their knock-out studies revealed that POT1 is also crucial for protecting telomere single-stranded from end-to-end fusion (22-24).
As described above, each shelterin component plays a crucial role in telomere length regulation and the maintenance of the functional telomere structure. However, it is still not well understood how shelterin components functionally interact in terms of telomere dynamics and the regulation of chromosomal stability. To address this issue we analyzed how shelterin components interact and contribute to the telomere-associated phenotypes of mTRF1 deficient (mTRF1Δ) ES cells.
EXPERIMENTAL PROCEDURES
Establishment of TRF1 Cond-KO ES Cell Lines Stably Expressing Transgenes—the linearized plasmids of pCAG-FLAG-IRES-Puro containing mTRF1, cmTRF1, mTRF2, or Tin2-cmTRF2 cDNAs were transfected into mTRF1 cond-KO ES cells, and the established stably expressed cell lines were selected by puromycin. In addition, linearized plasmids of pCAG-MYC-cmTRF1-IRES-HisD or pCAG-MYC-Tin2-cmTRF1-IRES-HisD were transfected into mTRF1-cond KO, FLAG-mTRF2+ ES cells, and the established stable cell lines were selected by l-histidinol dihydrochloride (Sigma). In all the established mTRF1 cond-KO ES cell lines, the 4-hydroxytamoxifen (OHT)3 treatment induced efficient loxP-mTRF1 deletion but had no effect on other transgenes expression (supplemental Figs. 5, A and C, and 7).
Immunoprecipitation—Cells were harvested and lysed in ice-cold lysis buffer (50 mm Tris-HCl (pH 7.5), 1 mm EDTA, 400 mm NaCl, 1% Triton X-100, 0.1% SDS, 1 mm dithiothreitol, and a protease inhibitor mixture (Roche Applied Science)). After incubation on ice for 30 min, an equal volume of cold-water was added and mixed. The treated samples were centrifuged, and supernatants were collected as total cell extracts. Extracts were immunoprecipitated with anti-FLAG monoclonal antibody (M2, Sigma) and immunoblotted with anti-mTIN2 antibody (a kind gift of Sahn-Ho Kim).
Chromatin Immunoprecipitation (ChIP) Assay—ChIP assay was performed as described previously (11). Briefly, the cross-linked cell lysates were immunoprecipitated with 1 μg of anti-mTRF2 antibody. The immuno-DNA complexes were collected by protein G-coupled Sepharose beads, then incubated at 65 °C overnight to reverse the cross-linking and treated with proteinase K to remove the mTRF2 protein. The remaining mTRF2-associated ChIP DNA was slot-blotted and hybridized with 32P-labeled (CCCTAA)4 probe. Autoradiogram signals were quantified with BAS 5000.
Fluorescence in Situ Hybridization (FISH) and Immuno-FISH Analysis—FISH assay was performed as described previously (11). Colcemid-treated cells were harvested and incubated in 0.03 mm sodium citrate and fixed with calnoy solution (MeOH: acetic acid = 3:1). Metaphase cells were spread out and fixed with 4% paraformaldehyde, treated with 1 mg/ml pepsin, and fixed again. Slides were dehydrated with 70, 90, and 100% EtOH. The Cy3-Telomere peptide nucleic acid probe (0.5 ng/ml) was mounted on the slides, heated for 3 min at 80 °C, and incubated at room temperature. Slides were washed in solution I (70% formamide, 10 mm Tris-HCl (pH 7.5), 0.1% bovine serum albumin) and TBS-T and dehydrated again. DNA was stained with 4′, 6-diamidino-2-phenylindole and examined under fluorescence microscopy.
In the case of Immuno-FISH, cells suspended in hypotonic buffer (10 mm Tris-HCl (pH 7.5), 20 mm NaCl, 5 mm KCl, 1 mm CaCl2, 0.5 mm MgCl2, and 50% glycerol) were spotted onto glass slides by centrifugation and fixed with 4% paraformaldehyde and with 70% ethanol. Then cells were permeabilized with 0.5% Nonidet P-40 and heated with 0.1 m citrate buffer at 120 °C for 5 min. Preheated Cye3-Tel peptide nucleic acid was hybridized, and cells were permeabilized again and incubated with anti-FLAG, anti-mTIN2 or anti-phospho histone H2A.X (Ser-139) (upstate) antibody. Alexa-488-conjugated goat IgG were used as the secondary antibody. Labeled cells were examined by fluorescence microscopy (Axioplan II, Zeiss). For all of the immunofluorescence comparison data, the exposure time remained the same in the same set of experiments.
Preparation of Genomic DNA and the Telomere Restriction Fragment (TRF) Analysis—The genomic DNA was prepared using the DNeasy Blood and Tissue kit (Qiagen). Purified genomic DNA was digested with HinfI and separated by pulse-field gel electrophoresis. The gel was dried and denatured in buffer containing 1.5 m NaCl and 0.4 n NaOH and then neutralized in 1.5 m NaCl and 0.5 m Tris-HCl (pH 8.0) buffer. The telomere probe (CCCTAA)4 was end-labeled with γ-[32P]ATP and used for hybridization at 42 °C in five times SSC and five times in Denhardt's solution. The hybridized gel was washed in a 0.1× SSC solution at 25 °C. TRF signals were detected with BAS 5000 and Median telomere length and semi-interquartile range was calculated using Telometric software, Version 1.2 (Fox Chase Cancer Center, Philadelphia, PA) (25, 26).
RESULTS
Creation of a Chicken-Mouse TRF1 Chimera That Can Localize to Telomeres but Does Not Associate with mTIN2—We previously described the phenotype of a murine TRF1 conditional knock-out model (mTRF1 cond-KO) in ES cells in which both alleles of the endogenous mTRF1 gene were inactivated but exogenous mTRF1 cDNA flanked by two loxP sequences and a transgene encoding a Cre-estrogen receptor fusion molecule, Mer-Cre-Mer, were expressed (11). The deletion of mTRF1 by the OHT-mediated lox/Cre recombination caused growth defects and chromosomal instability. Furthermore, mTIN2 and mTRF2 resulted in lost or decreased telomeric association in these cells, respectively, and abnormal telomere structures (doublet, broken, and loss of telomeres) were frequently observed in metaphase-chromosome spreads. To further examine whether telomere localization of other shelterin components, mTPP1 and mPOT1, are affected in mTRF1Δ ES cells, we introduced the GFP-mTPP1, GFP-mPOT1a, and GFP-mPOT1b expression vectors. All of these GFP fusion molecules formed nuclear foci and also overlapped with FLAG-mTRF1 signals, indicating their telomere localization (supplemental Fig. 1A). In mTRF1 cond-KO ES cells, after mTRF1 was deleted by OHT treatment, all of these fusion molecules lost telomeric foci formation, and only diffused nuclear signals were detected (supplemental Fig. 1B). These data support that the telomere localization of shelterin components is severely affected by the absence of mTRF1 in mTRF1Δ ES cells (3, 7, 10-12).
To elucidate the importance of mTRF1 in shelterin regulation of telomere function, we aimed to investigate the role of each shelterin component in the mTRF1Δ phenotypes. First, we designed a mTRF1 mutant molecule that could bind to telomeric DNA but could not interact with mTIN2 to clarify whether telomeric localization of mTIN2 was crucial for the TRF1Δ phenotype. We deleted the N-terminal domain of mTRF1 that is responsible for homodimerization and its interaction with mTIN2 (TRFH domain) (27). However, this mTRF1 deletion mutant did not localize to telomeres (data not shown), probably because it was unable to form dimers, an essential requisite for telomere DNA binding (5, 7).
On the other hand, we obtained evidence that a chicken TRF1 homologue (cTRF1) localized to telomeres in a chicken B cell line, DT40 and that a GFP-tagged mTIN2 did not form telomeric foci in the same cells (data not shown). Furthermore, the chicken telomeric DNA sequences are the same as those of mammals (28). Thus, we reasoned that the cTRF1 would not interact with mTIN2. To demonstrate this, we expressed MYC-mTIN2 and either FLAG-cTRF1 or FLAG-mTRF1 in HEK293T cells. The immunoprecipitation analyses showed that the interaction of cTRF1 with mTIN2 was greatly reduced in transient expression, although a significant amount of mTIN2 associated with mTRF1 (supplemental Fig. 2). Then, we established stable clone expressing FLAG-cTRF1 in mTRF1 cond-KO ES cell lines and examined FLAG-cTRF1 telomere localization. Although chicken and mouse telomere DNA sequences are the same, immunostaining with anti-FLAG antibodies showed only faint cTRF1 signals localized at the telomeres even before mTRF1 was deleted (Fig. 1B, middle panel). After the OHT treatment mTIN2 telomere localization was undetected, and none of the TRF1Δ phenotypes could be rescued by FLAG-cTRF1 expression (data not shown).
FIGURE 1.

Telomere localization of cmTRF1, but there was no TIN2 binding. A, schematics of DNA constructs of mouse (m), chicken (c), and chicken-mouse fusion (cm) TRF1. For the cmTRF1 construct, the TRFH domain of mTRF1 (1-247) was replaced with that of chicken TRF1 (1-214). Myb, Myb-type DNA binding motif. B, fluorescent images show the telomeric localization of FLAG-tagged mTRF1 constructs in TRF1 cond-KO ES cells without OHT treatment. FLAG-cmTRF1 is co-localized with telomeric DNA, indicating that cmTRF1 localized to the telomere. The two immunofluorescence images (red and green) for each cell type were superimposed (Merge) to evaluate co-localization (yellow). C, immunoprecipitation (IP) analysis of the association of cmTRF1 with mTIN2 in mTRF1 cond-KO ES cells. mTRF1 cond-KO ES cells (lane 1) and the ES cells that stably expressed FLAG-mTRF1 (lane 2) or FLAG-cmTRF1 (lane 3) were subjected to immunoprecipitation using an anti-FLAG antibody. Endogenous mTIN2 (detected with anti-mTIN2 antibody) was co-immunoprecipitated with FLAG-mTRF1 (lane 5) but not with FLAG-cmTRF1 (lane 6) by Western blotting (WB).
Next, we made a chicken-mouse TRF1 chimera (cmTRF1) in which the N terminus of the mouse TRF1, including the TRFH domain (amino acids 1-247) was replaced with that of the chicken TRF1 (amino acids 1-214) (Fig. 1A and supplemental Fig. 3). Because it possessed the mouse Myb domain and the chicken TRFH domain, cmTRF1 was expected to firmly bind to telomeres by forming a homodimer but not to interact with mTIN2. Therefore, we hoped that cmTRF1 would complement for the mTRF1 telomere functions other than those which are mTIN2-mediated or -regulated. To investigate whether cmTRF1 interacts with mTIN2 and also other telomeric proteins, cell lysates of HEK293T cells co-expressing FLAG-cmTRF1 and GFP-fused mTIN2, mTRF2, mTPP1, mPOT1a, or mPOT1b were immunoprecipitated with anti-GFP antibody. As shown in supplemental Fig. 4, there is no evident interaction of FLAG-cmTRF1 with mTIN2 and other shelterin components. Then, we established stable clone expressing FLAG-cmTRF1 (supplemental Fig. 5A) and confirmed again that association of cmTRF1 with mTIN2 was not detected (Fig. 1C). Furthermore, the co-localization of cmTRF1 foci with telomeric DNA indicated that cmTRF1 was localized at telomeres (Fig. 1B, bottom panel).
cmTRF1 Could Not Rescue Any of the Phenotypes Observed in mTRF1Δ ES Cells—To investigate whether any mTRF1Δ phenotypes were rescued by FLAG-cmTRF1 expression, we first examined localization of telomere components in mTRF1-KO ES cells and those expressing either FLAG-mTRF1 or FLAG-cmTRF1 with or without OHT (supplemental Fig. 5A). Telomere localization of endogenous mTIN2 and exogenous GFP-mTPP1, GFP-mPOT1a, and GFP-mPOT1b were examined by immunostaining. The binding of mTRF2 to telomeres was studied with a ChIP assay. All the data indicated that cmTRF1 could not rescue the telomere localization defects of any of the shelterin components in mTRF1Δ ES cells, although telomeric localization in all of them was rescued in ES cells expressing FLAG-mTRF1 (Fig. 2, A and B, and supplemental Figs. 1, C and D, and 6A). Growth defects and abnormal telomere signals were not changed by the presence of FLAG-cmTRF1 (Fig. 2, C and D, and Table 1). In addition, accumulation of H2A.X phosphorylation (γH2AX) at telomeres, referred to as telomere dysfunction-induced foci (TIFs) (29, 30), was observed after OHT treatment in mTRF1 cond-KO ES cells with or without FLAG-cmTRF1 expression (supplemental Fig. 5B, top and bottom panels). The TIF formation was not induced in TRF1Δ ES cells expressing FLAG-mTRF1, confirming that TIF formation is another mTRF1Δ phenotype (supplemental Fig. 5B, middle panel). In conclusion, these results suggest that cmTRF1 can localize to telomeres but cannot rescue any of the phenotypes induced in the mTRF1Δ ES cells. These results further support the possibility that the inability to recruit TIN2 and other shelterin components to telomeres is the cause of the mTRF1Δ phenotypes.
FIGURE 2.
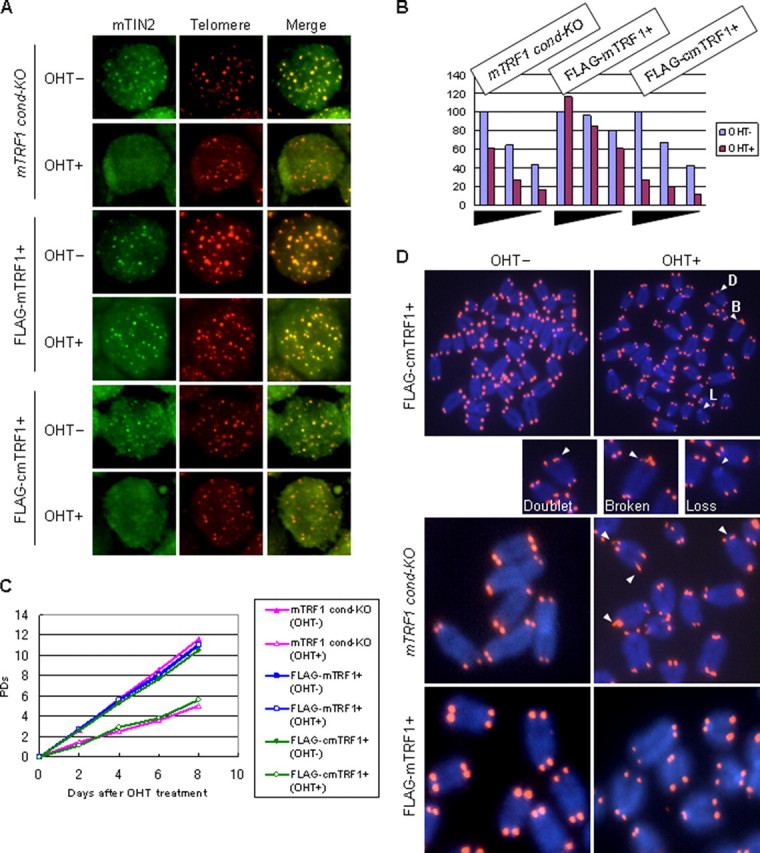
mTRF1Δ phenotypes in mTRF1 cond-KO ES cells that express FLAG-cmTRF1. A, telomere localization of mTIN2 in mTRF1 cond-KO ES cells that expressed FLAG-cmTRF1 with or without OHT treatment (OHT+ and OHT-, respectively) by Immuno-FISH analysis. As a negative or positive control, mTRF1 cond-KO ES cells and mTRF1 cond-KO ES cells expressing FLAG-mTRF1 with or without OHT treatment was used. The two fluorescence images (red and green) for each cell type were superimposed (Merge) to evaluate co-localization (yellow). B, mTRF2-associated telomere DNA was measured in mTRF1 cond-KO ES cells and in the same cells expressing FLAG-cmTRF1 or FLAG-mTRF1, with or without OHT treatment. mTRF2-associated telomere DNA (ChIP DNA) was serially diluted (equivalents of 1.3 × 106, 6.5 × 105, and 3.3 × 105 cells); the dilutions are indicated by black triangle bars along the abscissa. The values on the ordinate represent the signal intensities from each sample normalized to the ChIP telomere signals/input telomere signals detected in 1.3 × 106 cells without OHT (100%). The raw autoradiogram data is shown in supplemental Fig. 4A. C, growth characteristics. Cells were treated with or without OHT (OHT+ and OHT-, respectively) for 2 days and then re-plated without OHT. The population doublings (PDs) after OHT treatment were measured every 2 days (up to 8 days). D, FISH analysis using the Cy3-(CCCTAA)4 peptide nucleic acid probe (red) in metaphase chromosome spreads of TRF1 cond-KO ES cells, TRF1 cond-KO, FLAG-mTRF1+ ES cells, and mTRF1 cond-KO, FLAG-cmTRF1+ ES cells with or without OHT treatment. The arrowheads show abnormal telomere signals. D, doublet; two signals from one chromosomal end; B, broken; more than two or diffused telomere signals; L, loss; no telomere signals.
TABLE 1.
End-to-end fusions and aberrant telomere signals in metaphase chromosome spreads of mTRF1 cond-KO ES cells
The numbers in parentheses indicate the percentage of abnormal signals in all the chromosomal ends.
|
mTRF1 cond-KO |
FLAG-mTRF1+ |
FLAG-cmTRF1+ |
FLAG-mTRF2+ |
FLAG-TIN2-cmTRF1+ |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| OHT− | OHT+ | OHT− | OHT+ | OHT− | OHT+ | OHT− | OHT+ | OHT− | OHT+ | |
| No. of metaphase | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 |
| No. of the chromosomes per metaphase | 40.5 | 40.6 | 40.7 | 40.7 | 40.4 | 40.9 | 40.2 | 41.1 | 40.7 | 40.8 |
| Doublet and broken telomere signals | 7 (0.43) | 258 (15.9) | 28 (1.16) | 19 (1.16) | 24 (1.48) | 185 (11.3) | 16 (3.94) | 240 (14.6) | 15 (0.92) | 165 (10.1) |
| Loss of telomere signals | 5 (0.31) | 54 (3.3) | 1 (0.06) | 4 (0.04) | 4 (0.25) | 79 (4.8) | 1 (0.25) | 85 (5.17) | 1 (0.06) | 30 (1.84) |
| End-to-end fusions of telomere | 0 (0) | 2 (0.12) | 3 (0.18) | 0 (0) | 0 (0) | 9 (0.55) | 0 (0) | 2 (0.12) | 0 (0) | 1 (0.06) |
Aberrant Telomere Structure Is Independent of TRF2 and TIN2-TPP1-POT1 Telomere Localization Defects—To clarify further whether the telomeric localization of mTIN2 was essential for the mTRF1Δ phenotypes, we established ES cells in which mTIN2 localized to the telomere in the absence of functional mTRF1. We applied two different strategies for creating these cells. First, we overexpressed exogenous mTRF2 in mTRF1 cond-KO ES cells. Previous work had shown that some mTRF2 remained on the telomeres in the absence of mTRF1 (Ref. 11) and Fig. 2B) and also that mTIN2 interacted with mTRF2 in vivo (17, 18). Therefore, we expected that overproduction of exogenous mTRF2 would suppress the reduction of mTRF2 from telomeres in mTRF1Δ ES cells and that some mTIN2 would be recruited to telomeres through the TRF2-TIN2 interaction. The second strategy was to utilize a fusion molecule that combined mTIN2 and cmTRF1 (TIN2-cmTRF1) (Fig. 3A). We reasoned that TIN2-cmTRF1 should localize to telomeres by cmTRF1-mediated telomere binding. Thus, we established the stable clones expressing either FLAG-mTRF2 or FLAG-TIN2-cmTRF1 in mTRF1 cond-KO ES cell lines (mTRF1 cond-KO, FLAG-mTRF2+ or mTRF1 condKO, FLAG-TIN2-cmTRF1+, respectively) (supplemental Fig. 5C). We found that TIN2-cmTRF1 localized to telomeres in the presence or absence of mTRF1 and that it led GFP-mTPP1, GFP-mPOT1a, and GFP-mPOT1b to form telomeric foci (Fig. 3B and supplemental Fig. 1F), and mTRF2 telomere binding was not also diminished in the absence of mTRF1 (Fig. 3C and supplemental Fig. 6B), corresponding to previous reports in that the telomeric localization of mTPP1 and its associated protein mPOT1 was dependent on mTIN2 (13, 17, 19). However, the overproduction of exogenous mTRF2 rescued mTRF2 telomere binding but did not maintain endogenous mTIN2 localization at telomeres in mTRF1Δ ES cells (Fig. 3, B and C, and supplemental Fig. 6B). Furthermore, GFP-mTPP1, GFP-mPOT1a, and GFP-mPOT1b could not form telomere foci in mTRF1Δ, FLAG-mTRF2+ ES cells (supplemental Fig. 1E). These data clearly indicate that mTRF2 telomere localization does not serve as a scaffold for mTIN2, mTPP1, or mPOT1 recruitment to telomeres.
FIGURE 3.
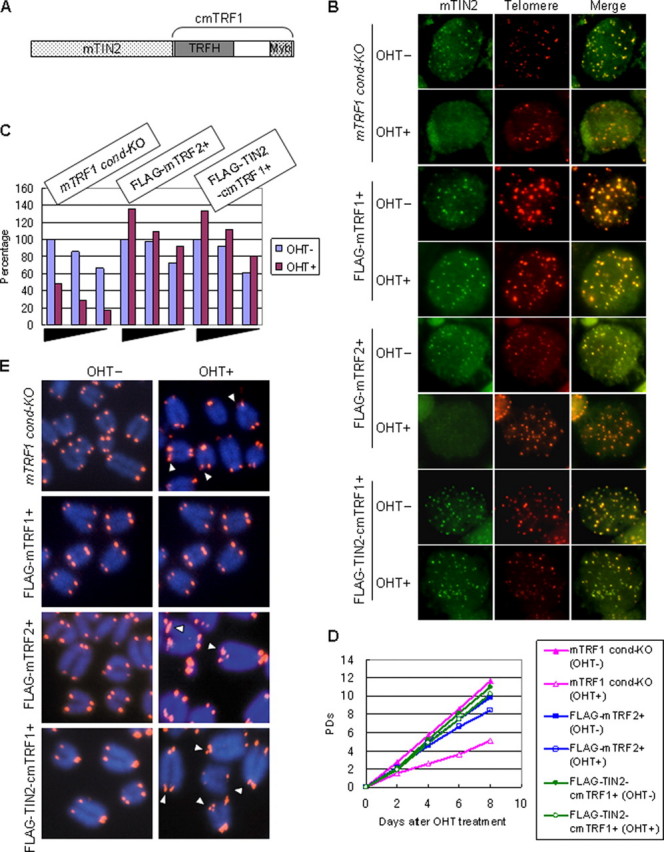
TRF1Δ phenotypes in cells that expressed FLAG-mTRF2 or FLAG-TIN2-cmTRF1. A, schematic of the structure of the FLAG-tagged TIN2-cmTRF1 fusion molecule. The C terminus of mTIN2 was combined with the N terminus of cmTRF1. Telomeric localization of mTIN2 (B) and mTRF2 (C), growth characteristics (D), and abnormal telomere signals in metaphase chromosomal spreads (E) were investigated in mTRF1 cond-KO ES cells that expressed FLAG-mTRF2 or FLAG-TIN2-cmTRF1 with or without OHT treatment (OHT+ and OHT-, respectively). The arrowheads in E show abnormal telomeres. The raw autoradiogram data of C are shown in supplemental Fig. 4B. PDs, population doublings.
Growth defects and TIF formation (γH2AX) were not induced in either mTRF1Δ, FLAG-mTRF2+ or mTRF1Δ, FLAG-TIN2-cmTRF1+ ES cells (Fig. 3D and supplemental Fig. 5D). However, abnormal telomere signals in the metaphase spreads were not suppressed in either type of mTRF1Δ ES cells (Fig. 3E). Doublets and broken telomeres and loss of telomeres were observed in 15.9 and 3.3% of the analyzed chromosomal ends of mTRF1Δ cells, respectively, as well as 14.6 and 5.17% of mTRF1Δ, FLAG-mTRF2+ cells, 10.1 and 1.84% of mTRF1Δ, FLAG-TIN2-cmTRF1+ cells, and 1.16 and 0.24% of mTRF1Δ, FLAG-mTRF1+ cells (Table 1). These data strongly suggest that aberrant telomere signals in the metaphase spreads are not caused by defects in telomere localization of the shelterin components mTIN2, mTRF2, mTPP1, or mPOT1.
Abnormal Telomere Signals Are Maintained without Chromosomal Instability after Long Term Culture—Even without TIF formation and growth defects, abnormal telomere signals were maintained in the mTRF1Δ ES cells that expressed mTRF2 or TIN2-cmTRF1. Our previous studies demonstrate that in long term cultures, mTRF1Δ ES cells accumulated many end-to-end fusions. (Ref. 11 and Table 2). To examine the relationship between chromosomal instability and the abnormal telomere signals we subcloned mTRF1Δ, FLAG-mTRF2+, mTRF1Δ, FLAG-TIN2-cmTRF1+, and mTRF1Δ, FLAG-mTRF1+ ES cells (supplemental Fig. 7A) and examined whether end-to-end fusion or aneuploid chromosomes accumulated after ∼60 days culture. We found that none of these ES cell clones showed growth defects during the culture period (data not shown), but abnormal telomere signals were observed in mTRF1Δ cells that expressed FLAG-mTRF2 or FLAG-TIN2-cmTRF1 after ∼60 days culture. Nevertheless, there were no obvious alterations of chromosomal number or accumulation of end-to-end fusions (summarized in Table 2). These results indicate that abnormal telomere signals in the metaphase spreads did not correlate with chromosomal instability in TRF1Δ ES cells.
TABLE 2.
End-to-end fusions and aberrant telomere signals in metaphase chromosome spreads of mTRF1-positive and -negative ES cell clones after ~60 days culture
The numbers in parentheses indicate the percentage of abnormal signals in all the chromosomal ends.
| loxP-mTRF1 cDNA |
mTRF1 ΔES cells (clone
81-24)a |
FLAG-mTRF1+ |
FLAG-mTRF2+ |
FLAG-TIN2-cmTRF1+ |
|||
|---|---|---|---|---|---|---|---|
| (−) | (+) | (−) | (+) | (−) | (+) | (−) | |
| No. of metaphases | 12 | 10 | 10 | 10 | 10 | 10 | 10 |
| No. of the chromosomes per metaphase | 37.5 | 40.7 | 40.3 | 41.1 | 40.7 | 40.8 | 41.3 |
| Doublet and broken telomere signals | 26 (1.6) | 2 (0.12) | 22 (1.34) | 367 (22.5) | 21 (1.29) | 541 (32.7) | |
| Loss of telomere signals | 0 (0) | 0 (0) | 0 (0) | 72 (4.42) | 0 (0) | 15 (0.91) | |
| End-to-end fusion of telomere | 68 (15.1) | 0 (0) | 0 (0) | 0 (0) | 1 (0.06) | 0 (0) | 5 (0.3) |
Indicates data reproduced with permission from Iwano et al. (11).
POT1-TPP1-independent Telomere Length Regulation of TRF1—According to the current understanding, shelterin components and the dynamics of higher-order telomere structures, including t-loop formation, are crucial for the regulation of telomerase-mediated telomere lengthening (5). The aberrant telomere signals that we observed in mTRF1Δ ES cells represent unknown qualitative defects of telomere structure that are independent of chromosomal instability. Therefore, we examined the relationship between those abnormal telomere signals and telomere length regulation. We cultured mTRF1-present and -absent ES clones that expressed FLAG-mTRF1, FLAG-mTRF2, or FLAG-TIN2-cmTRF1 for ∼60 days and monitored their telomere lengths during this culture period (supplemental Fig. 7A). Telomere lengths were examined by the TRFs analysis (Fig. 4A). Fig. 4B shows each median telomere length. In contrast to mTRF1 cond-KO, FLAG-mTRF1+ and mTRF1 condKO, FLAG-TIN2-cmTRF1+ ES cells, we found that mTRF1 cond-KO, FLAG-TRF2+ ES cells had a much shorter median telomere length (less than 20 kb) and remained this size during the culture period, in agreement with previous reports of TRF2-mediated telomere shortening (31-33) (Fig. 4A, lanes 7-9). Interestingly, this exogenous mTRF2-mediated telomere shortening was canceled out by the mTRF1 deletion; in the absence of mTRF1, telomeres rapidly elongated to a median telomere length of 21-27.5 kb (Fig. 4A, lanes 10-12). These data suggest that mTRF2-mediated telomere length regulation may be primarily dependent on mTRF1 expression. No telomere elongation or shortening was observed in either mTRF1-positive or -negative ES cells that expressed either FLAG-mTRF1 or FLAG-TIN2-cmTRF1 in ∼60 day cultures (Fig. 4A, lanes 1-6 and 13-18).
FIGURE 4.

Long term monitoring of Telomere elongation dynamics. mTRF1 cond-KO ES cells that expressed FLAG-mTRF1, FLAG-mTRF2, or FLAG-TIN2-cmTRF1 were examined after 1-2, 3-5, and 9-10 weeks in culture. A, telomere restriction fragment analysis of genomic DNA (20 μg). (+) and (-) show the presence or absence of mTRF1 cDNA flanked by loxP sites. Molecular weight markers are shown at the left. B, telomere lengths are represented as median telomere length ± semi-interquartile range (indicating extent of length heterogeneity) (kb). mTRF1Δ, FLAG-mTRF2+ ES cells (open triangles, middle pair) displayed telomere elongation from 21 to 27.5 kb.
To examine the relationship between the telomere elongation phenotype of mTRF1Δ, FLAG-mTRF2+ ES cells and the inability of telomeres to recruit mTIN2, mTPP1, or mPOT1, we further introduced either MYC-cmTRF1 or MYC-TIN2-cmTRF1 expression vectors into mTRF1 cond-KO, FLAG-mTRF2+ ES cells and subcloned mTRF1Δ cells (supplemental Fig. 7B). We then monitored alterations of telomere length in mTRF1-present and -absent ES clones during ∼60 days culture (Fig. 5A).
FIGURE 5.

Long term monitoring of telomere elongation dynamics in ES cells that overexpress mTRF2. mTRF1 cond-KO, FLAG-mTRF2+ ES cells that expressed MYC-cmTRF1 or MYC-TIN2-cmTRF were examined after 1-2, 3-4, 6-7, and 8-10 weeks in culture. A, TRF analysis of the genomic DNA (20 μg). (+) and (-) show the presence or absence of mTRF1 cDNA flanked by loxP sites. Molecular weight markers are shown at the left. B, telomere lengths are represented as the median ± SIR (semi-interquartile range; kb). Telomeres from MYC-cmTRF1 cells were elongated from 20 to 26 kb, and those from MYC-TIN2-cmTRF1 cells were elongated from 18 to 21 kb in the absence of mTRF1 during ∼60 days of culture (open squares and triangles). Two independent clones of each transfectant showed similar TRFs profiles.
In mTRF1 cond-KO, FLAG-mTRF2+ ES cells that expressed MYC-cmTRF1 or MYC-TIN2-cmTRF1, the median telomere lengths were under 20 kb (Fig. 5B), similar to that of the parental phenotype (Fig. 4B). After OHT treatment and subcloning of mTRF1 deficient cells, the median telomere lengths were slightly elongated; that is, 20 kb for cells that expressed MYC-cmTRF1, 18 kb for cells that expressed MYC-TIN2-cmTRF1, and 15-16 kb for mTRF1 cond-KO, FLAG-mTRF2+ ES cells (parental phenotype) (Fig. 5A, lanes 5 and 13). However, during ∼60 days of culture, mTRF1 cond-KO cells that expressed MYC-cmTRF1 and MYC-TIN2-cmTRF1 maintained their telomere lengths under 20 kb, but the mTRF1Δ clones displayed progressive elongation of their telomeres. Although the mTRF1 deletion-induced telomere elongation in both MYC-cmTRF1 and MYC-TIN2-cmTRF1-expressing clones, the telomere lengthening in the MYC-TIN2-cmTRF1-expressing clones was significantly diminished (3-kb telomere DNA elongation/60 days) compared with that in the MYC-cmTRF1-expressing clone (6-kb telomere DNA elongation/60 days) (Fig. 5, A and B, lanes 5-8 and 13-16, respectively, and not shown). These data suggest that TRF1 has two molecular pathways involved in negative telomere length regulation; one is mediated by the recruitment of the TIN2-TPP1-POT1 complex to telomeres (10, 19), and the other is independent of the TIN2-TPP1-POT1 recruitment. The latter pathway may be mediated by other TRF1 functions that include its DNA bending activity or its regulation or recruitment of a novel telomere-associating protein(s).
DISCUSSION
By exploring different ways to tether shelterin components to telomeres in the absence of functional TRF1, we clarified that the mTRF1Δ phenotypes can be categorized as dependent or independent of TIN2-TPP1-POT1 telomere localization. Thus, the growth defects, TIF formation, and chromosomal instability were dependent, the abnormal telomere signals in the metaphase spreads were independent, and the telomere elongation phenotypes were a mixture of dependent and independent.
TRF1-mediated Telomere Length Regulation—It has been shown that TRF1 is a regulator for telomerase-dependent telomere elongation. In addition, it was also shown that telomeric TPP1-POT1 functions as a transducer of the telomere length regulation of TRF1 (10, 19). However, although some of our results confirm this mechanism (Fig. 5A, lane 1-8, and B), our studies with mTRF1Δ FLAG-mTRF2+ ES cells that expressed MYC-cmTRF1 or MYC-TIN2-cmTRF1 suggested that a TPP1-POT1-independent pathway also exists.
What is the nature of the TPP1-POT1-independent telomere length regulation of TRF1? There are several different possibilities. In some studies we utilized mTRF1 cond-KO ES cells that overexpressed mTRF2. In these cells the telomere length was considerably shortened (median telomere length <20 kb) compared with those in control mouse ES cells (median telomere length >30 kb). Telomere shortening with mTRF2 overexpression has been reported previously, and it was shown that endonuclease protein XPF was involved in this process (31-33). Our subsequent results showed that mTRF1 deletion blocked the TRF2-mediated telomere shortening. Thus, the first hypothesis is that the TPP1-POT1-independent TRF1 regulation of telomere elongation may be mediated by the regulation of XPF telomere localization. Our second hypothesis is that TRF1 acts in an intrinsic manner, and its regulation of telomere elongation may be TIN2-mediated. As described, TRF1 can bend telomere DNA (7), and TIN2 enhances the TRF1-telomere DNA interaction; this suggests that TIN2 may alter the conformation of TRF1 and induce a structural change in telomeres that is less accessible to telomerase (34). Our results showed that the fusion of TIN2 to cmTRF1 localized exogenous TIN2-(-cmTRF1) to telomeres and recruited other shelterin components, but this might not induce conformational changes of TRF1 (cmTRF1). Thus, we speculate that the TPP1-POT1-independent telomere length regulation of TRF1 depends on the ability to induce a telomeric structural change. Our last hypothesis acknowledges the possibility that a novel mTRF1-associated molecule(s) exists that does not bind to the cmTRF1. Thus, in this hypothetical scenario we speculate that the TPP1-POT1-independent TRF1 regulation of telomere elongation depends on the association of this putative molecule with TRF1.
The Nature of Abnormal Telomere Signals in mTRF1Δ Metaphase Spreads—Abnormal telomere signals in the TRF1Δ metaphase spreads were not suppressed by the telomere localization of other shelterin components. In general, telomere loss or telomere shortening below a critical length resulted in an inability to maintain the requisite end protection and promoted chromosomal instabilities, primarily induced by end-to-end fusion. Suppression of growth defects, TIF formation, and chromosomal instability (fusions and aneuploidy) in mTRF1Δ, FLAG-TIN2-cmTRF1+ ES cells indicated that the DNA damage response pathways were not activated. Therefore, we assumed that the observed abnormal telomere signals in mTRF1Δ ES cells did not reflect telomere disruption in vivo. In mTRF1Δ ES cells, telomere chromatin condensation may have been incomplete, and thus, telomeres were highly susceptible to DNA breakage in the physically disruptive process of FISH analysis. Telomere regions are usually heterochromatic (35). In addition, it is reported that telomere RNA localizing at telomeres appears to be essential for heterochromatinization, and TRF1 is involved in accumulation of telomere RNA (36). The TRF1 function, which is not complemented with cmTRF1, may be involved in this telomere heterochromatinization. In any event, further elucidation of the nature of the abnormal telomere signals observed in mTRF1Δ ES cells will be crucial for obtaining insight into the mechanism of the TPP1-POT1-independent TRF1-mediated telomere length regulation.
Supplementary Material
Acknowledgments
We thank Sahn-ho Kim for providing anti-mTIN2 antibody.
This work was supported by a grant-in-aid from the Ministry of Education, Science, Technology, and Culture of Japan (to Y. S. and M. T.) and by the 21st Century COE Program of the Ministry of Education, Culture, Sports, Science, and Technology to Graduate School of Biostudies and Institute for Virus Research, Kyoto University (to K. O. and T. I.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1-7.
Footnotes
The abbreviations used are: OHT, 4-hydroxytamoxifen; ChIP, chromatin immunoprecipitation; FISH, fluorescence in situ hybridization; TIF, telomere dysfunction-induced foci; m, mouse; c, chicken; cm, chicken-mouse; TRF, telomere restriction fragment; TRFH, TRF homology domain; GFP, green fluorescent protein; kb, kilobase(s); cond-KO, conditional knock-out.
References
- 1.Riha, K., Heacock, M., and Shippen, D. (2006) Annu. Rev. Genet. 40 237-277 [DOI] [PubMed] [Google Scholar]
- 2.Smogorzewska, A., and de Lange, T. (2004) Annu. Rev. Biochem. 73 177-208 [DOI] [PubMed] [Google Scholar]
- 3.Liu, D., O'Connor, M. S., Qin, J., and Songyang, Z. (2004) J. Biol. Chem. 279 51338-51342 [DOI] [PubMed] [Google Scholar]
- 4.O'Connor, M. S., Safari, A., Xin, H., Liu, D., and Songyang, Z. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 11874-11879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Lange, T. (2005) Genes Dev. 19 2100-2110 [DOI] [PubMed] [Google Scholar]
- 6.Griffith, J. D., Comeau, L., Rosenfield, S., Stansel, R. M., Bianchi, A., Moss, H., and de Lange, T. (1999) Cell 97 503-514 [DOI] [PubMed] [Google Scholar]
- 7.Bianchi, A., Smith, S., Chong, L., Elias, P., and de Lange, T. (1997) EMBO J. 16 1785-1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Broccoli, D., Smogorzewska, A., Chong, L., and de Lange, T. (1997) Nat. Genet. 17 231-235 [DOI] [PubMed] [Google Scholar]
- 9.van Steensel, B., and de Lange, T. (1997) Nature 385 740-743 [DOI] [PubMed] [Google Scholar]
- 10.Loayza, D., and De Lange, T. (2003) Nature 423 1013-1018 [DOI] [PubMed] [Google Scholar]
- 11.Iwano, T., Tachibana, M., Reth, M., and Shinkai, Y. (2004) J. Biol. Chem. 279 1442-1448 [DOI] [PubMed] [Google Scholar]
- 12.Ye, J. Z., Donigian, J. R., van Overbeek, M., Loayza, D., Luo, Y., Krutchinsky, A. N., Chait, B. T., and de Lange, T. (2004) J. Biol. Chem. 279 47264-47271 [DOI] [PubMed] [Google Scholar]
- 13.Liu, D., Safari, A., O'Connor, M. S., Chan, D. W., Laegeler, A., Qin, J., and Songyang, Z. (2004) Nat. Cell Biol. 6 673-680 [DOI] [PubMed] [Google Scholar]
- 14.Smogorzewska, A., van Steensel, B., Bianchi, A., Oelmann, S., Schaefer, M. R., Schnapp, G., and de Lange, T. (2000) Mol. Cell. Biol. 20 1659-1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Celli, G. B., and de Lange, T. (2005) Nat. Cell Biol. 7 712-718 [DOI] [PubMed] [Google Scholar]
- 16.Bae, N. S., and Baumann, P. (2007) Mol. Cell 26 323-334 [DOI] [PubMed] [Google Scholar]
- 17.Houghtaling, B. R., Cuttonaro, L., Chang, W., and Smith, S. (2004) Curr. Biol. 14 1621-1631 [DOI] [PubMed] [Google Scholar]
- 18.Kim, S. H., Beausejour, C., Davalos, A. R., Kaminker, P., Heo, S. J., and Campisi, J. (2004) J. Biol. Chem. 279 43799-43804 [DOI] [PubMed] [Google Scholar]
- 19.Ye, J. Z., Hockemeyer, D., Krutchinsky, A. N., Loayza, D., Hooper, S. M., Chait, B. T., and de Lange, T. (2004) Genes Dev. 18 1649-1654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baumann, P., and Cech, T. (2001) Science 292 1171-1175 [DOI] [PubMed] [Google Scholar]
- 21.Kelleher, C., Kurth, I., and Lingner, J. (2005) Mol. Cell. Biol. 25 808-818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He, H., Multani, A. S., Cosme-Blanco, W., Tahara, H., Ma, J., Pathak, S., Deng, Y., and Chang, S. (2006) EMBO J. 25 5180-5190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hockemeyer, D., Daniels, J. P., Takai, H., and de Lange, T. (2006) Cell 126 63-77 [DOI] [PubMed] [Google Scholar]
- 24.Wu, L., Multani, A. S., He, H., Cosme-Blanco, W., Deng, Y., Deng, J. M., Bachilo, O., Pathak, S., Tahara, H., Bailey, S. M., Deng, Y., Behringer, R. R., and Chang, S. (2006) Cell 126 49-62 [DOI] [PubMed] [Google Scholar]
- 25.Zhong, Z. H., Jiang, W. Q., Cesare, A. J., Neumann, A. A., Wadhwa, R., and Reddel, R. R. (2007) J. Biol. Chem. 282 29314-29322 [DOI] [PubMed] [Google Scholar]
- 26.Grant, J., Broccoli, D., Muquit, M., Manion, F., Tisdall, J., and Ochs, M. (2001) Biotechniques 31 1314-1316 and 1318 [DOI] [PubMed] [Google Scholar]
- 27.Kim, S. H., Kaminker, P., and Campisi, J. (1999) Nat. Genet. 23 405-412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nanda, I., and Schmid, M. (1994) Cytogenet. Cell Genet. 65 190-193 [DOI] [PubMed] [Google Scholar]
- 29.Takai, H., Smogorzewska, A., and de Lange, T. (2003) Curr. Biol. 13 1549-1556 [DOI] [PubMed] [Google Scholar]
- 30.d'Adda di Fagagna, F., Reaper, P., Clay-Farrace, L., Fiegler, H., Carr, P., Von Zglinicki, T., Saretzki, G., Carter, N., and Jackson, S. (2003) Nature 426 194-198 [DOI] [PubMed] [Google Scholar]
- 31.Muñoz, P., Blanco, R., Flores, J., and Blasco, M. (2005) Nat. Genet. 37 1063-1071 [DOI] [PubMed] [Google Scholar]
- 32.Wu, Y., Zacal, N., Rainbow, A., and Zhu, X. (2007) DNA Repair (Amst) 6 157-166 [DOI] [PubMed] [Google Scholar]
- 33.Karlseder, J., Smogorzewska, A., and de Lange, T. (2002) Science 295 2446-2449 [DOI] [PubMed] [Google Scholar]
- 34.Kim, S., Han, S., You, Y., Chen, D., and Campisi, J. (2003) EMBO Rep. 4 685-691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blasco, M. A. (2007) Nat. Rev. Genet. 8 299-309 [DOI] [PubMed] [Google Scholar]
- 36.Schoeftner, S., and Blasco, M. (2008) Nat. Cell Biol. 10 228-236 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.