

Abstract
Under various conditions, noxious stimuli damage mitochondria, resulting in mitochondrial fragmentation; however, the mechanisms by which fragmented mitochondria are eliminated from the cells remain largely unknown. Here we show that cytoplasmic vacuoles originating from the plasma membrane engulfed fragmented mitochondria and subsequently extruded them into the extracellular spaces in undergoing acute tumor necrosis factor α-induced cell death in a caspase-dependent fashion. Notably, upon fusion of the membrane encapsulating mitochondria to the plasma membrane, naked mitochondria were released into the extracellular spaces in an exocytotic manner. Mitochondrial extrusion was specific to tumor necrosis factor α-induced cell death, because a genotoxic stress-inducing agent such as cisplatin did not elicit mitochondrial extrusion. Moreover, intact actin and tubulin cytoskeletons were required for mitochondrial extrusion as well as membrane blebbing. Furthermore, fragmented mitochondria were engulfed by cytoplasmic vacuoles and extruded from hepatocytes of mice injected with anti-Fas antibody, suggesting that mitochondrial extrusion can be observed in vivo under pathological conditions. Mitochondria are eliminated during erythrocyte maturation under physiological conditions, and anti-mitochondrial antibody is detected in some autoimmune diseases. Thus, elucidating the mechanism underlying mitochondrial extrusion will open a novel avenue leading to better understanding of various diseases caused by mitochondrial malfunction as well as mitochondrial biology.
Normal mitochondria continuously undergo fission and fusion in a coordinated fashion (1, 2). Although mitochondria are major organelles to produce ATP through oxidative phosphorylation, apoptogenic factors such as cytochrome c, Smac/DIABLO, and Omi/HtrA are released from mitochondria during apoptosis (3, 4). Under physiological and pathological conditions, various stimuli damage mitochondria, resulting in drastic morphological changes of mitochondria including mitochondrial fragmentation (1, 2, 5). In response to apoptotic stimuli, cristae in mitochondria elongate and form vesicular structures. Then cristae become swollen, and finally the outer membrane of mitochondria is ruptured (6). Damaged mitochondria need to be eliminated in the cells; otherwise the cells eventually die because of activation of the mitochondrially dependent apoptotic pathway. One of such mechanisms might be autophagy. Autophagy is an evolutionally conserved and lysosome-mediated catabolic response that is induced under nutrient-poor conditions (7, 8). During autophagy, cytoplasmic components including mitochondria are engulfed by double-membrane structures known as autophagosomes in the cytoplasm, suggesting that autophagy might play a role in the elimination of damaged mitochondria under certain conditions. However, it remains to be solved whether autophagy plays a dominant role in elimination of damaged mitochondria during apoptosis.
Death-inducing ligands such as TNFα3 and Fas ligand induce mitochondrial fragmentation through activation of the caspase-dependent pathway in a cell type-dependent fashion (9). c-FLIP (cellular FLICE inhibitory protein) was identified as a homologue to an initiator caspase, caspase 8, and inhibits TNFα- and Fas-induced apoptosis by binding to and inhibiting activation of caspase 8 (10). A transcription factor, NF-κB has been shown to play a central role in suppressing genotoxin- and TNFα-induced apoptosis through up-regulating various antiapoptotic genes including members of the bcl-2 family, X chromosome-linked inhibitor of apoptosis, superoxide dismutase (sod)2, and c-Flip (11, 12). We and others have recently reported that c-FLIP plays a crucial role in protection of cells from TNFα- and Fas-induced cell death (13-15). During the course of electron microscopical analysis of dying c-Flip-/- murine embryonic fibroblasts (MEFs), we found that TNFα-induced mitochondrial fragmentation, and extensive cytoplasmic vacuoles have emerged in c-Flip-/- MEFs in a caspase-dependent fashion. Surprisingly, fragmented mitochondria were extruded from the cells through cytoplasmic vacuoles originating from the plasma membrane. Upon fusion of the membrane encapsulating mitochondria to the plasma membrane, naked mitochondria were released into the extracellular spaces. This process is qualitatively different from the previously well characterized shedding of apoptotic bodies that frequently encapsulate cytoplasmic organelles including mitochondria. Moreover, mitochondrial extrusion was specific to TNFα-induced cell death, because a genotoxic stress-inducing agent such as cisplatin did not elicit mitochondrial extrusion. Furthermore, fragmented mitochondria was engulfed by cytoplasmic vacuoles and extruded from hepatocytes of mice injected with anti-Fas antibody, suggesting that mitochondrial extrusion might be observed in vivo under pathological conditions. Together, these results suggest that fragmented mitochondria might be released from apoptotic cells under certain conditions; released mitochondria might be a source of antigens to elicit autoimmune diseases under some pathological conditions.
EXPERIMENTAL PROCEDURES
Reagents—TNFα, Z-VAD-fmk, and Ac-DEVD-AMC were purchased from BD Biosciences and Peptide Institute and used at final concentrations of 10 ng ml-1, 50 μm, and 20 μm, respectively. Cisplatin, cytochalasin D, butylated hydroxyanisole, 3-methlyadenine (3-MA), and paclitaxel were purchased from WAKO and Sigma-Aldrich and used at final concentrations of 400 μm, 1 μm, 100 μm, 10 mm, and 20 μm, respectively. CM-H2DCFDA, LysoTracker BlueDND-22, MitoTracker DeepRed, and FM1-43FX were purchased from Invitrogen and used at final concentrations of 1 μm, 1 μm, 100 nm, and 5 μg ml-1, respectively. Anti-cytochrome c oxidase IV (COX IV) (Abcam), anti-Lamp1 (BD Biosciences), anti-GM130 (BD Biosciences), and anti-mouse immunoglobulin (GE Healthcare) antibodies were purchased from the indicated sources. Anti-light chain 3 (LC3) antibody was previously described (16).
Cell Culture and Generation of Transfectants—c-Flip-/- MEFs were cultured in Dulbecco's modified Eagle's medium with 10% fetal bovine serum. c-Flip-/- MEFs stably expressing a green fluorescent (GFP) fused to COX IV were generated using a retroviral vector (provided by D. Chan) as previously described (17). c-Flip-/- MEFs stably expressing c-FLIPL (we designated them as c-Flip-/- c-FLIPL MEFs hereafter) were previously described (13).
Immunofluorescence Analysis—c-Flip-/- MEFs stably expressing GFP-COX IV were cultured on a 60 μ-Dish (Ibidi) overnight. After TNFα stimulation, time lapse confocal microscopy was performed by using TCS SP5 (Leica). Movies and pictures were taken with the LAS AF confocal software (Leica). For Figs. 5B and 6E, c-Flip-/- MEFs were cultured on a 60 μ-Dish overnight. After TNFα stimulation, the cells were stained with MitoTracker DeepRed and/or LysoTracker BlueDND-22 for 30 min. Then the medium was replaced with ice-cold Hanks' balanced salt solution containing FM1-43FX. For Fig. 6 (A and F), c-Flip-/- MEFs were cultured on cover glasses in 24-well plate overnight. After TNFα stimulation, the cells were fixed with 4% paraformaldehyde in PBS for 30 min and permeabilized with 50 μgml-1 of digitonin-PBS for 10 min. The cells were immunostained with anti-LC3, anti-Lamp1, and anti-COX IV antibodies in 1% bovine serum albumin with TBST for 2 h and were visualized with Alexa 488- or 594-conjugated secondary antibodies (Invitrogen). Confocal microscopy was performed on FV1000 (Olympus). The pictures were analyzed by FV10-ASW (Olympus).
FIGURE 5.

Plasma membranes are involved in vacuole formation. A, c-Flip-/- MEFs were stimulated with TNFα for 90 min and analyzed by transmission electron microscopy. The red arrowheads indicate extruded mitochondria. Scale bar, 100 nm. B, c-Flip-/- MEFs were unstimulated or stimulated with TNFα for 90 min. Then the cells were stained with FM1-43FX (green) and MitoTracker (red). The white arrowheads indicate colocalization of the plasma membrane and mitochondria. Scale bars, 10 μm. C, c-Flip-/- MEFs were stimulated and analyzed as in A. The electron microscopical images corresponding to normal (top panel), typical apoptosis characterized by membrane blebbing (middle panel), and atypical apoptosis containing numerous cytoplasmic vacuoles (bottom panel) are shown. Scale bars, 1 μm.
FIGURE 6.

Autophagy does not play a major role in mitochondrial extrusion. A, c-Flip-/- MEFs were unstimulated or stimulated with TNFα for 90 min. Then the cells were fixed and immunostained with anti-LC3 antibody (green), and the nuclei were stained with Hoechst 33258 (blue). N, nucleus. Scale bar, 10 μm. B, c-Flip-/- MEFs were untreated or treated with TNFα, 3-MA, or TNFα plus 3-MA for 90 min. The cell lysates were analyzed by immunoblotting (IB) with anti-LC3 antibody. The arrows indicate LC3-I and LC3-II. The equal loading of the samples was verified by Western blotting with anti-tubulin antibody. The molecular mass markers are shown on the left. C, c-Flip-/- MEFs were stimulated with TNFα plus 3-MA for 90 min and analyzed by transmission electron microscopy. The enlarged image of the red box is presented in the right panel. The red arrowheads indicate extruded mitochondria. Scale bar, 1 μm. D, c-Flip-/- MEFs were untreated or treated as in B, and caspase 3 activities were measured by using fluorogenic substrates. The results are presented as the means ± S.D. of triplicate samples. E, c-Flip-/- MEFs were stimulated as in A. Then the cells were stained with LysoTracker (green) and MitoTracker (red). Scale bars, 10 μm. F, c-Flip-/- MEFs were stimulated as in A. Then the cells were fixed and immunostained with anti-Lamp1 (green) and anti-COX IV (red) antibodies. Scale bars, 10 μm.
Electron and Immunoelectron Microscopy—Wild-type, c-Flip-/- MEFs, and c-Flip-/- c-FLIPL MEFs were stimulated with the indicated agents for the indicated times. Preparation of samples for electron microscopy was previously described (18). To evaluate the percentages of cells showing typical apoptosis, necrosis, and atypical apoptosis characterized by numerous cytoplasmic vacuoles, sections were used for cell counting. For immunoelectron microscopy, the cells were fixed with periodate-lysine paraformaldehyde fixative, infiltrated with 40% polyvinylpyrrolidone, 2.3 m sucrose, 0.1 m phosphate buffer, pH 7.4, embedded on nails, and frozen in liquid nitrogen. Ultrathin cryosections were cut with the Ultracut UCT microtome equipped with the FC-4E cryoattachment (Leica) at -110 °C and transferred to nickel grids (150 mesh), blocked, and then incubated with anti-COX IV antibody at 4 °C overnight. Then the samples were incubated with secondary antibodies conjugated with 10-nm gold particles (British BioCell) for 2 h. The samples were refixed with 2.5% glutaraldehyde, 0.1 m phosphate buffer (PB), stained with 2% uranyl acetate solution for 30 min, and absorption-stained with 2% polyvinyl alcohol containing 0.2% methylcellulose and 0.2% uranyl acetate for 30 min. The samples were observed with a JEM-1230 electron microscope (JEOL).
Caspase Activity—Caspase 3 activity was measured by a fluorometric assay as previously described (13). Briefly, MEFs were stimulated with TNFα in the absence or presence of inhibitors for the indicated times, and then the cells were lysed in radioimmune precipitation assay buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1% Nonidet P-40, 0.5% deoxycholate, 0.1% SDS, 25 mm β-glycerophosphate, 1 mm sodium orthovanadate, 1 mm sodium fluoride, 1 mm phenylmethylsulfonyl fluoride, 1 μgml-1 aprotinin, and 1 μgml-1 leupeptin). The cell lysates were incubated with Ac-DEVD-AMC, and the release of fluorescent 7-amino-4-methylcoumarin was measured on a fluorometer (Labsystems).
Measurement of ROS Accumulation—MEFs (4 × 105 cells) were plated onto 6-well plates and stimulated with TNFα in the absence or presence of various inhibitors for 2 h. After stimulation, the cells were washed with Opti-MEM (Invitrogen) and then incubated with CM-H2DCFDA in the dark for 30 min at 37 °C. Then the cells were harvested and analyzed on a flow cytometer (FACSCalibur; BD Biosciences). The data were processed by using the CellQuest program (BD Biosciences).
Anti-Fas Antibody Injection—Eight-week-old female C57BL/6 mice were purchased from Charles River. The mice were injected intravenously with 50 μg of anti-Fas antibody (Jo-2) (provided by S. Nagata) or PBS. At 4 h after injection, the sera were collected, and the livers were fixed by in vivo perfusion with 2.5% glutaraldehyde for electron microscopy. As a positive control for GM130, hepatocytes from untreated mice were lysed in radioimmune precipitation assay buffer. After centrifugation, the sera and the lysates were subjected to SDS-PAGE and transferred onto polyvinylidene difluoride membranes. The membranes were immunoblotted with anti-COX IV, anti-GM130, and anti-mouse immunoglobulin antibodies. The membranes were developed with ECL Western blotting Detection System Plus (GE Healthcare).
RESULTS
Fragmented Mitochondria Are Engulfed by the Cytoplasmic Vacuoles and Extruded from c-Flip-/- MEFs during Cell Death—c-Flip-/- MEFs died rapidly after TNFα stimulation, enabling us to efficiently monitor the fate of damaged mitochondria during apoptosis. To visualize mitochondria, we first generated c-Flip-/- MEFs stably expressing a GFP fused to cytochrome oxidase IV (COX IV), a mitochondrial inner membrane protein. We then stimulated c-Flip-/- MEFs expressing GFP-COX IV with TNFα and monitored the mitochondrial morphology by time lapse confocal microscopy. Mitochondria displayed an elongated and tubular morphology and actively underwent fusion and fission in unstimulated cells (Fig. 1A and supplemental Movie S1). In contrast, mitochondrial fragmentation was rapidly induced after TNFα stimulation (Fig. 1B and supplemental Movie S1).
FIGURE 1.

Fragmented mitochondria are engulfed by the cytoplasmic vacuoles and extruded from c-Flip-/- MEFs during cell death. A and B, c-Flip-/- MEFs stably expressing GFP-COX IV were unstimulated (A) or stimulated with TNFα for 60 min (B). Scale bars, 25 μm. C and D, c-Flip-/- MEFs were stimulated with TNFα for 60 (C) or 90 (D) min and analyzed by transmission electron microscopy. The enlarged images of the boxed areas are shown in the right panels. The arrowheads and arrows indicate extruded and fragmented mitochondria, respectively. Scale bars, 500 nm. E, the percentages of cells showing typical apoptosis, necrosis, and atypical apoptosis characterized by numerous cytoplasmic vacuoles and mitochondrial extrusion were calculated by counting randomly selected areas (total 100-200 cells/sample). Three independently prepared samples were counted and are presented as the means ± S.D. F, c-Flip-/- MEFs were stimulated with TNFα for 90 min and then analyzed by immunoelectron microscopy using anti-COX IV antibody, followed by 10-nm colloidal gold-conjugated secondary antibody. Scale bar, 200 nm.
We next investigated the mitochondrial morphology by transmission electron microscopy in more detail. Consistent with our previous study using c-Flip knockdown cells (14), TNFα induced rapid activation of the caspase cascade, resulting in typical apoptosis and necrosis in c-Flip-/- MEFs (supplemental Fig. S1). In addition, we found that fragmented mitochondria were detected in the cytoplasmic vacuoles at 60 min after TNFα stimulation (Fig. 1C). Surprisingly, some fragmented mitochondria appeared to be released into the extracellular spaces. Because the nuclei of these cells were slightly pyknotic, and the plasma membrane integrity was preserved, these morphological changes might be early events of apoptosis but not secondary necrosis. Along with the progression of apoptosis, numerous cytoplasmic vacuoles appeared and gathered around the periphery of the cytoplasm, and some vacuoles similarly contained fragmented mitochondria at 90 min after TNFα stimulation (Fig. 1D, upper panels). Upon fusion of the membrane encapsulating mitochondria to the plasma membrane, naked mitochondria were released into the extracellular spaces in an exocytotic manner. Notably, fragmented but not intact mitochondria were encapsulated by the vacuoles, suggesting that mitochondrial fragmentation is a prerequisite of mitochondrial extrusion (Fig. 1D, lower panels). This process is qualitatively different from a previously well characterized shedding of apoptotic bodies that frequently encapsulate cytoplasmic organelles including mitochondria (Fig. 1D and supplemental Fig. S1). Based on transmission electron microscopical analysis, this atypical apoptosis characterized by numerous cytoplasmic vacuoles and mitochondrial extrusion was observed in nearly 30% of c-Flip-/- MEFs (Fig. 1E and supplemental Fig. S1).
To confirm that the extruded organelles are mitochondria, we performed immunoelectron microscopy using anti-COX IV antibody. Immunogold labeling for COX IV accumulated in the organelles in the extra cellular spaces (Fig. 1F), suggesting that fragmented mitochondria are actually extruded from the cells. Together, these results suggest that fragmented mitochondria are extruded from c-Flip-/- MEFs during apoptosis.
A Genotoxic Stress-inducing Agent Does Not Elicit Mitochondrial Extrusion in c-Flip-/- MEFs—To investigate whether other cytotoxic stimuli similarly induce mitochondrial extrusion in c-Flip-/- MEFs, we stimulated c-Flip-/- MEFs with a genotoxic stress-inducing agent such as cisplatin. Cisplatin has been shown to induce apoptosis of various types of cells through direct inhibition of DNA synthesis. Upon treatment of c-Flip-/- MEFs with cisplatin, 40% cells died at 16 h after stimulation (data not shown). In contrast to TNFα stimulation, cisplatin moderately induced cytoplasmic vacuolic changes but did not induce mitochondrial extrusion (Fig. 2). Collectively, mitochondrial extrusion is specific to TNFα-induced cell death.
FIGURE 2.

A genotoxic stress-inducing agent does not elicit mitochondrial extrusion in c-Flip-/- MEFs. c-Flip-/- MEFs were stimulated with cisplatin for 16 h and analyzed by transmission electron microscopy. The enlarged image of the boxed area is shown in the right panel. N, nucleus. M, mitochondria. Scale bars, 2 μm.
TNFα-induced Mitochondrial Extrusion, but Not Mitochondrial Fragmentation, Is Suppressed in the Presence of Cytochalasin D or Paclitaxel—Previous studies have shown that an intact actin cytoskeleton is required for membrane blebbing (19, 20). Thus, we investigated whether the actin- and tubulin-destabilizing compounds such as cytochalasin D and paclitaxel inhibit mitochondrial extrusion. As expected, cytochalasin D or paclitaxel substantially inhibited cytoplasmic vacuole formation and subsequent mitochondrial extrusion as well as membrane blebbing (Fig. 3, A and B). However, these agents did not inhibit mitochondrial fragmentation or activation of caspase 3 (Fig. 3C), indicating that mitochondrial extrusion does not appear to affect the final outcome of cell survival or death. Moreover, these results suggest that an intact actin and tubulin polymerization are required for mitochondrial extrusion.
FIGURE 3.

TNFα-induced mitochondrial extrusion, but not mitochondrial fragmentation, is suppressed in the presence of cytochalasin D or paclitaxel. A and B, c-Flip-/- MEFs were stimulated with TNFα for 60 min in the presence of cytochalasin D (A) or paclitaxel (B) and analyzed by transmission electron microscopy. The enlarged images of the boxed areas are presented in the right panels. N, nucleus. M, mitochondria. Scale bars, 2 μm. C, TNFα-induced caspase 3 activities are not inhibited in the presence of cytochalasin D or paclitaxel. c-Flip-/- MEFs were stimulated with TNFα for 60 min in the absence or presence of cytochalasin D or paclitaxel, and caspase 3 activities were measured by using fluorogenic substrates. The results are presented as the means ± S.D. of triplicate samples.
The Caspase-dependent Pathway Plays a Crucial Role in Mitochondrial Extrusion—Mitochondrial extrusion was induced in TNFα-treated c-Flip-/- MEFs (Fig. 1), in which TNFα rapidly induced caspase 3 activation (supplemental Fig. S1), prompting us to test whether the caspase-dependent pathway is essential for mitochondrial extrusion. As expected, TNFα-induced mitochondrial fragmentation and extrusion were completely inhibited in the presence of a broad caspase inhibitor, Z-VAD-fmk, suggesting that these events are indeed induced in a caspase-dependent fashion (Fig. 4A). To confirm that the caspase-dependent pathway is required for mitochondrial extrusion under different experimental conditions, we stimulated wild-type and c-Flip-/- MEFs stable expressing c-FLIPL (c-Flip-/- c-FLIPL MEFs) with TNFα and examined the morphology of TNFα-treated cells. As shown in Fig. 4B, TNFα did not activate or very weakly activated caspase 3 in wild-type and c-Flip-/- c-FLIPL MEFs, respectively. Consistent with these results, TNFα did not induce mitochondrial extrusion in wild-type or c-Flip-/- c-FLIPL MEFs (Fig. 4, C and D).
FIGURE 4.
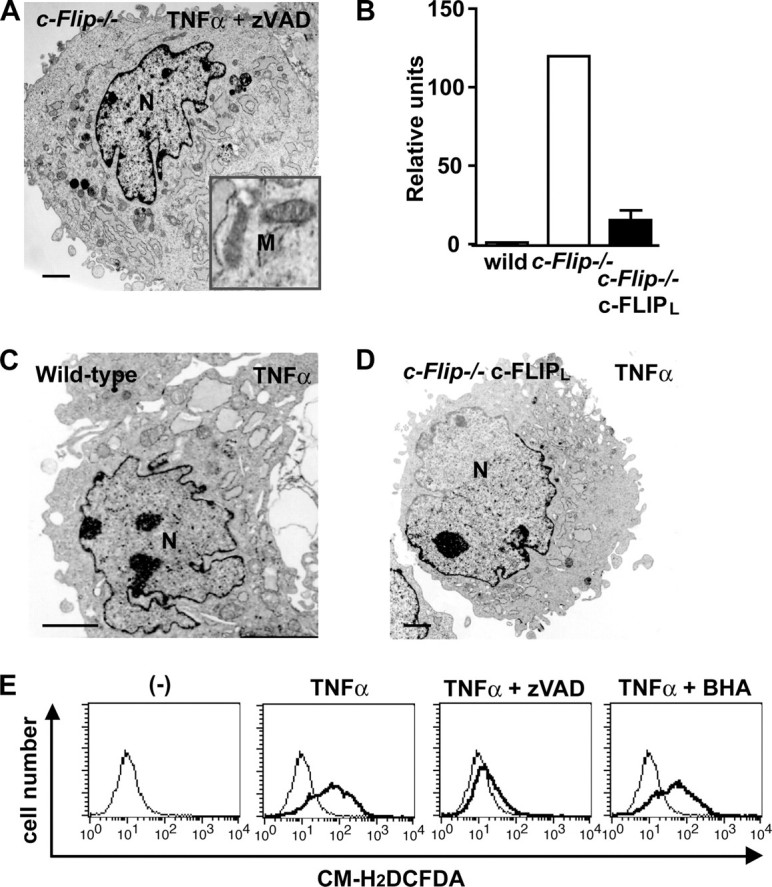
The caspase-dependent pathway plays a crucial role in mitochondrial extrusion. A, c-Flip-/- MEFs were stimulated with TNFα for 60 min in the presence of Z-VAD-fmk and analyzed by transmission electron microscopy. Mitochondria showing normal structures are shown in the boxed area. N, nucleus. M, mitochondria. Scale bars, 2 μm. B, wild-type, c-Flip-/- MEFs, and c-Flip-/- c-FLIPL MEFs were stimulated with TNFα for 90 min, and caspase 3 activities were measured by using fluorogenic substrates. The results are presented as the means ± S.D. of triplicate samples. C and D, wild-type and c-Flip-/- c-FLIPL MEFs were stimulated as in B and analyzed by transmission electron microscopy. N, nucleus. E, c-Flip-/- MEFs were unstimulated (thin lines) or stimulated (bold lines) with TNFα in the absence or presence of Z-VAD-fmk or butylated hydroxyanisole for 2 h, and then the cells were labeled with CM-H2DCFDA and analyzed by flow cytometry.
A very recent study has shown that ROS might be involved in mitochondrial extrusion (21). As we previously reported (13), TNFα induced robust ROS accumulation in c-Flip-/- MEFs (Fig. 4E). Notably, Z-VAD-fmk substantially inhibited TNFα-induced ROS accumulation as well as caspase activation in c-Flip-/- MEFs (Fig. 4E and supplemental Fig. S1), suggesting that ROS accumulate in the cells in a caspase-dependent fashion. In contrast, antioxidants such as butylated hydroxyanisole or N-acetyl cysteine did not inhibit TNFα-induced ROS accumulation in c-Flip-/- MEFs (Fig. 4E and data not shown), although these agents almost completely suppressed TNFα-induced ROS accumulation in traf2-/- traf5-/- and relA-/- MEFs (18). At this moment, we could not selectively suppress TNFα-induced ROS accumulation in c-Flip-/- MEFs without inhibiting caspase activation; therefore, we cannot formally exclude the possibility that the ROS-dependent pathway might also be involved in mitochondrial extrusion under our experimental conditions. Further study will be required to address this issue.
Plasma Membranes Are Involved in Vacuole Formation—We next investigated the origin of the vacuoles engulfing fragmented mitochondria. We found that some vacuoles fused to the plasma membrane (Fig. 5A), prompting us to test whether these vacuoles might originate from the plasma membrane. Then we labeled c-Flip-/- MEFs with a membrane-impermeable dye, FM1-43FX that is incorporated into the plasma membrane lipids (22). c-Flip-/- MEFs stained with FM1-43FX displayed a polygonal shape before stimulation (Fig. 5B, upper panels). TNFα-stimulated cells shrank and displayed multiple globular structures (Fig. 5B, middle panels), reflecting membrane blebbing detected by transmission electron microscopy (Fig. 5C, middle panel). In these typical apoptotic cells, FM1-43FX did not merge with MitoTracker, a mitochondrial marker (Fig. 5B, middle panels). In sharp contrast, in ∼30% of cells, the plasma membrane appeared to intrude into the cytoplasm and merged with MitoTracker (Fig. 5B, bottom panels). These results suggest that vacuoles originating from the plasma membrane might engulf fragmented mitochondria.
Autophagy Does Not Play a Major Role in Mitochondrial Extrusion—Recent studies have shown that autophagy is associated with cell death under certain conditions (23, 24). Autophagosomes are cytoplasmic vacuoles and characterized by a punctate staining pattern with anti-LC3 antibody, prompting us to test whether autophagy might contribute to generation of the cytoplasmic vacuoles in c-Flip-/- MEFs upon TNFα stimulation. However, TNFα stimulation neither increased a punctate staining pattern of LC3 compared with unstimulated cells nor induced, but rather suppressed, the conversion of LC3-I to LC3-II, a hallmark of autophagy induction (Fig. 6, A and B). To further exclude the possibility that autophagy participates in mitochondrial extrusion, we treated c-Flip-/- MEFs with TNFα in the presence of 3-MA, an inhibitor for autophagy induction, and examined the morphology. Treatment of c-Flip-/- MEFs with 3-MA substantially inhibited the conversion of LC3-I to LC3-II (Fig. 6B). Under these experimental conditions, treatment of the cells with 3-MA did not inhibit TNFα-induced mitochondrial extrusion or caspase 3 activation in c-Flip-/- MEFs (Fig. 6, C and D). Moreover, mitochondrial markers (MitoTracker or COX IV) did not merge with lysosomal markers (LysoTracker or Lamp1) in TNFα-stimulated c-Flip-/- MEFs, excluding the possibility that engulfed mitochondria in the cytoplasmic vacuoles eventually fuse into lysosomes, resulting in degradation in an autophagy-dependent manner (Fig. 6, E and F). Together, these results suggest that autophagy might not play a major role in the formation of the cytoplasmic vacuoles in c-Flip-/- MEFs.
Fragmented Mitochondria Are Engulfed in the Cytoplasmic Vacuoles and Extruded from Hepatocytes after Anti-Fas Antibody Injection—We finally examined whether mitochondrial extrusion is observed in vivo. To mimic TNFα-induced rapid cell death of c-Flip-/- MEFs, we induced fulminant hepatitis of mice by injection of anti-Fas antibody (25). We examined the morphology of the liver by transmission electron microscopy at 4 h after anti-Fas antibody injection. Consistent with a previous study (25), anti-Fas antibody, but not PBS, injection induced massive hepatocyte cell death accompanied by severe hemorrhage (Fig. 7A). Interestingly, mitochondrial fragmentation and cytoplasmic vacuoles containing fragmented mitochondria were also observed in hepatocytes (Fig. 7, B and C), and some fragmented mitochondria were detected in the space of Disse (Fig. 7C). Moreover, COX IV was detected in the sera of four of five mice injected with anti-Fas antibody, but not PBS (Fig. 7D). In contrast, GM130, a cis-Golgi marker did not present in the sera after anti-Fas antibody injection at least under our experimental conditions (Fig. 7E), suggesting that mitochondria but not the Golgi apparatus were extruded from hepatocytes into the sera. These results strongly suggest that fragmented mitochondria are extruded in vivo at least under pathological conditions.
FIGURE 7.

Fragmented mitochondria are engulfed in the cytoplasmic vacuoles and extruded from hepatocytes after anti-Fas antibody injection. The mice were injected with PBS or anti-Fas antibody and sacrificed at 4 h after injection. A, histological analysis of the livers. The sections were stained with toluidine blue (×100). B and C, ultrastructural analysis of the liver. Normal and fragmented mitochondria are shown in the red boxes (B). The enlarged image of the boxed area is shown in C. The arrowheads and arrow indicate fragmented and extruded mitochondria, respectively. N, nucleus. S, sinusoid. Scale bars, 500 nm. D, release of COX IV in the sera of mice injected with anti-Fas antibody. The sera were collected at 4 h after injection and analyzed by immunoblotting (IB) with anti-COX IV (upper panel) and anti-mouse Ig (lower panel) antibodies. IgH and IgL indicate Ig heavy and light chains, respectively. The numbers indicate individual mice. The arrow indicates COX IV. The asterisks indicate nonspecific bands. The molecular mass markers are shown on the left. E, GM130, a cis-Golgi marker does not present in the sera. The same sera used in D were analyzed by Western blotting with anti-GM130 antibody. As a positive control, total lysates of the liver were applied to the same gel (L). The numbers indicate individual mice. The arrow indicates GM130. The asterisks indicate nonspecific bands. The molecular mass markers are shown on the left.
DISCUSSION
In the present study, we have shown that fragmented mitochondria are extruded from apoptotic cells in vitro and in vivo. In response to TNFα or anti-Fas antibody treatment, cytoplasmic vacuoles originating from the plasma membrane engulf fragmented mitochondria and eventually release naked mitochondria into the extracellular spaces. Given that the actin- and tubulin-destabilizing compounds inhibit cytoplasmic vacuole formation and mitochondrial extrusion, intact actin and tubulin cytoskeletons are required for mitochondrial extrusion.
Mitochondrial extrusion is tightly associated with activation of the caspase-dependent pathway based on the following observations. First, mitochondrial extrusion was induced in TNFα-treated c-Flip-/- MEFs (Fig. 1), in which activation of caspase was rapidly induced (supplemental Fig. S1). Second, Z-VAD-fmk suppressed mitochondrial extrusion (Fig. 4A). Third, TNFα did not induce mitochondrial extrusion in wild-type or c-Flip-/- cFLIPL MEFs (Fig. 4, C and D). More importantly, only fragmented, but not intact mitochondria were engulfed by cytoplasmic vacuoles, followed by the extrusion (Fig. 1), suggesting that fragmentation of mitochondria is a prerequisite process for mitochondrial extrusion. In this respect, it is intriguing to speculate that signal(s) similar to the “eat me” signals (26) may turn out to be expressed on the outer membrane of fragmented mitochondria to facilitate their engulfment by cytoplasmic vacuoles. Further study will be required to address this possibility.
Mitochondrial extrusion and the shedding of apoptotic bodies encapsulating mitochondria appear to be qualitatively different processes. The most striking difference is that naked but not encapsulated mitochondria were released into the extracellular spaces during mitochondrial extrusion (Fig. 1). Currently, we have no data as to whether released naked or encapsulated mitochondria elicit different biological responses in vivo. However, it is reasonable to speculate that clearance of naked mitochondria by nearby phagocytes may be delayed as compared with encapsulated mitochondria because of altered expression levels of the eat me signals (26). Therefore, mitochondrial extrusion may have biological significance following acute and massive cell death in vivo, although mitochondrial extrusion itself does not appear to affect the final outcome of dying cells (Fig. 3C).
Mitochondrial extrusion is specific to TNFα- and anti-Fas antibody-induced cell death (Figs. 1 and 7), because a genotoxic stress-inducing agent such as cisplatin does not elicit mitochondrial extrusion in c-Flip-/- MEFs (Fig. 2). Given that TNFα and anti-Fas antibody induce rapid cell death compared with cisplatin in c-Flip-/- MEFs (2-4 h versus 16 h), we might have overlooked a small amount of mitochondria extruded from the cells during cell death.
Extensive cytoplasmic vacuole formation observed in c-Flip-/- MEFs prompted us to test whether autophagy is induced in c-Flip-/- MEFs upon TNFα stimulation. However, LC3-II, a marker for autophagy induction, was not increased but rather decreased in c-Flip-/- MEFs upon TNFα stimulation (Fig. 6B), suggesting that TNFα stimulation did not elicit autophagy. Moreover, 3-MA did not inhibit TNFα-induced mitochondrial extrusion or caspase 3 activation (Fig. 6, C and D), suggesting that autophagy is not involved in mitochondrial extrusion or TNFα-induced cell death under our experimental conditions. Interestingly, our preliminary experiments showed that expression levels of LC3-II were increased in unstimulated c-Flip-/- MEFs in the presence of a cysteine protease inhibitor, E64d (data not shown). This result suggests that conversion of autophagosomes to autolysosomes at the basal levels might be enhanced in c-Flip-/- MEFs. Currently, we have no idea how to explain the reason why deletion of the c-Flip gene might enhance conversion of autophagosomes to autolysosomes, which will be investigated in the future study.
Some cell types contain specialized lysosomal compartments that store newly synthesized secretory proteins, which are referred to as secretory lysosomes (27). These include melanosomes, basophilic granules, mast cell secretory granules, and cytotoxic T lymphocyte lytic granules. Upon various stimuli, the secretory granules are transported along microtubules toward the plasma membrane, resulting in the release of contents of secretory lysosomes. Vacuoles originating from the plasma membrane engulf fragmented mitochondria, and actin polymerization is required for mitochondrial extrusion (Figs. 3 and 5), prompting us to investigate whether secretory lysosomes might participate in mitochondrial extrusion. However, lysosomal markers such as Lamp1 or LysoTracker did not colocalize with MitoTracker or COX IV in TNFα-stimulated c-Flip-/- MEFs (Fig. 6, E and F). These results suggest that fragmented mitochondria might not be engulfed by lysosomes or extruded in a secretory lysosome-dependent fashion.
In addition to under pathological conditions, it is reasonable to speculate that mitochondrial extrusion might constantly operate to eliminate a small amount of fragmented mitochondria under physiological conditions. Such a small amount of extruded mitochondria might have been engulfed by nearby phagocytes, explaining the reason why mitochondrial extrusion has been overlooked in vivo. Interestingly, mitochondria have been reported to disappear during the maturation of erythrocytes (28). This suggests that mitochondria might be degraded or extruded from erythroblasts during maturation, although its detailed molecular mechanism remains largely unknown. In addition, anti-mitochondrial antibody is frequently detected in patients suffering from primary biliary cirrhosis (29). Considering that the release of nuclear fragments from apoptotic cells is considered to be the source of antigens in autoimmune diseases such as systemic lupus erythematosus (30), mitochondria released from apoptotic cells might also be a source of antigens to elicit autoimmune diseases under some pathological conditions. Together, elucidating the signaling pathways leading to mitochondrial extrusion might provide novel target(s) to treat various diseases caused by mitochondrial malfunction and some autoimmune diseases.
Supplementary Material
Acknowledgments
We thank W.-C. Yeh for providing c-Flip-/- MEFs, D. Chan for providing an expression vector for GFP-COX IV, S. Nagata for providing anti-Fas antibody, and T. Ueno for providing anti-LC3 antibody and helpful suggestions. We also thank S. Tanaka and H. Kato for support of time lapse microscopy; Y. Kojima, M. Yoshida, J. Nakamoto, and K. Takahashi for support of transmission electron microscopy; and H. Ohno, K. Takeda, H. Ushio, and M. Nakayama for suggestions.
This work was supported in part by grants-in-aid for 21st Century Center of Excellence Research, High-Tech Research Center Project for Private Universities; a matching fund subsidy from the Ministry of Education, Culture, Sports, Science and Technology, Japan, and Scientific Research (B) and (C), and Young Scientists (B) from Japan Society for the Promotion of Science, Japan; and grants from the Tokyo Biochemical Research Foundation, the Mitsubishi Pharma Foundation, and NOVARTIS Foundation (Japan) for the Promotion of Science. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Movie S1 and Fig. S1.
Footnotes
The abbreviations used are: TNF, tumor necrosis factor; CM-H2DCFDA, 5-(and-6)-carboxy-2′,7′-dichlorodihydrofluorescein diacetate; COX IV, cytochrome oxidase IV; c-FLIP, cellular FLICE inhibitory protein; GFP, green fluorescent protein; LC3, light chain 3; MEF, murine embryonic fibroblast; 3-MA, 3-methlyadenine; Z, benzyloxycarbonyl; fmk, fluoromethyl ketone; PBS, phosphate-buffered saline; ROS, reactive oxygen species.
References
- 1.Chan, D. C. (2006) Cell 125 1241-1252 [DOI] [PubMed] [Google Scholar]
- 2.Cereghetti, G. M., and Scorrano, L. (2006) Oncogene 25 4717-4724 [DOI] [PubMed] [Google Scholar]
- 3.Wang, X. (2001) Genes Dev. 15 2922-2933 [PubMed] [Google Scholar]
- 4.Green, D. R., and Kroemer, G. (2004) Science 305 626-629 [DOI] [PubMed] [Google Scholar]
- 5.Youle, R. J., and Karbowski, M. (2005) Nat. Rev. Mol. Cell Biol. 6 657-663 [DOI] [PubMed] [Google Scholar]
- 6.Sun, M. G., Williams, J., Munoz-Pinedo, C., Perkins, G. A., Brown, J. M., Ellisman, M. H., Green, D. R., and Frey, T. G. (2007) Nat. Cell Biol. 9 1057-1065 [DOI] [PubMed] [Google Scholar]
- 7.Mizushima, N., Noda, T., Yoshimori, T., Tanaka, Y., Ishii, T., George, M. D., Klionsky, D. J., Ohsumi, M., and Ohsumi, Y. (1998) Nature 395 395-398 [DOI] [PubMed] [Google Scholar]
- 8.Ohsumi, Y. (2001) Nat. Rev. Mol. Cell Biol. 2 211-216 [DOI] [PubMed] [Google Scholar]
- 9.Wallach, D., Varfolomeev, E. E., Malinin, N. L., Goltsev, Y. V., Kovalenko, A. V., and Boldin, M. P. (1999) Annu. Rev. Immunol. 17 331-367 [DOI] [PubMed] [Google Scholar]
- 10.Budd, R. C., Yeh, W. C., and Tschopp, J. (2006) Nat. Rev. Immunol. 6 196-204 [DOI] [PubMed] [Google Scholar]
- 11.Nakano, H., Nakajima, A., Sakon-Komazawa, S., Piao, J. H., Xue, X., and Okumura, K. (2006) Cell Death Differ. 13 730-737 [DOI] [PubMed] [Google Scholar]
- 12.Karin, M., and Lin, A. (2002) Nat. Immunol. 3 221-227 [DOI] [PubMed] [Google Scholar]
- 13.Nakajima, A., Komazawa-Sakon, S., Takekawa, M., Sasazuki, T., Yeh, W. C., Yagita, H., Okumura, K., and Nakano, H. (2006) EMBO J. 25 5549-5559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakajima, A., Kojima, Y., Nakayama, M., Yagita, H., Okumura, K., and Nakano, H. (2008) Oncogene 27 76-84 [DOI] [PubMed] [Google Scholar]
- 15.Chang, L., Kamata, H., Solinas, G., Luo, J. L., Maeda, S., Venuprasad, K., Liu, Y. C., and Karin, M. (2006) Cell 124 601-613 [DOI] [PubMed] [Google Scholar]
- 16.Asanuma, K., Tanida, I., Shirato, I., Ueno, T., Takahara, H., Nishitani, T., Kominami, E., and Tomino, Y. (2003) FASEB J. 17 1165-1167 [DOI] [PubMed] [Google Scholar]
- 17.Sasazuki, T., Sawada, T., Sakon, S., Kitamura, T., Kishi, T., Okazaki, T., Katano, M., Tanaka, M., Watanabe, M., Yagita, H., Okumura, K., and Nakano, H. (2002) J. Biol. Chem. 277 28853-28860 [DOI] [PubMed] [Google Scholar]
- 18.Sakon, S., Xue, X., Takekawa, M., Sasazuki, T., Okazaki, T., Kojima, Y., Piao, J. H., Yagita, H., Okumura, K., Doi, T., and Nakano, H. (2003) EMBO J. 22 3898-3909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sebbagh, M., Hamelin, J., Bertoglio, J., Solary, E., and Breard, J. (2005) J. Exp. Med. 201 465-471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coleman, M. L., Sahai, E. A., Yeo, M., Bosch, M., Dewar, A., and Olson, M. F. (2001) Nat. Cell Biol. 3 339-345 [DOI] [PubMed] [Google Scholar]
- 21.Lyamzaev, K. G., Nepryakhina, O. K., Saprunova, V. B., Bakeeva, L. E., Pletjushkina, O. Y., Chernyak, B. V., and Skulachev, V. P. (2008) Biochim. Biophys. Acta 1777 817-825 [DOI] [PubMed] [Google Scholar]
- 22.Brooks, C., Wei, Q., Feng, L., Dong, G., Tao, Y., Mei, L., Xie, Z. J., and Dong, Z. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 11649-11654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Edinger, A. L., and Thompson, C. B. (2004) Oncogene 23 5654-5663 [DOI] [PubMed] [Google Scholar]
- 24.Levine, B., and Yuan, J. (2005) J. Clin. Investig. 115 2679-2688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ogasawara, J., Watanabe-Fukunaga, R., Adachi, M., Matsuzawa, A., Kasugai, T., Kitamura, Y., Itoh, N., Suda, T., and Nagata, S. (1993) Nature 364 806-809 [DOI] [PubMed] [Google Scholar]
- 26.Erwig, L. P., and Henson, P. M. (2008) Cell Death Differ. 15 243-250 [DOI] [PubMed] [Google Scholar]
- 27.Blott, E. J., and Griffiths, G. M. (2002) Nat. Rev. Mol. Cell Biol. 3 122-131 [DOI] [PubMed] [Google Scholar]
- 28.Simpson, C. F., and Kling, J. M. (1968) J. Cell Biol. 35 237-245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaplan, M. M., and Gershwin, M. E. (2005) N. Engl. J. Med. 353 1261-1273 [DOI] [PubMed] [Google Scholar]
- 30.Stollar, B. D., and Stephenson, F. (2002) Lupus 11 787-789 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.