

Abstract
Protective antigens of Pseudomonas aeruginosa (PcrV) and Yersinia pestis (LcrV) are key elements of specialized machinery, the type III secretion system (T3SS), which enables the injection of effector molecules into eukaryotic cells. Being positioned at the injectisome extremity, V proteins participate in the translocation process across the host cell plasma membrane. In this study, we demonstrate the assembly of V proteins into oligomeric doughnut-like complexes upon controlled refolding of the proteins in vitro. The oligomeric nature of refolded PcrV was revealed by size exclusion chromatography, native gel electrophoresis, and native mass spectrometry, which ascertain the capacity of the protein to multimerize into higher-order species. Furthermore, transmission electron microscopy performed on oligomers of both PcrV and LcrV revealed the presence of distinct structures with approximate internal and external diameters of 3-4 and 8-10 nm, respectively. The C-terminal helix, α12, of PcrV and notably the hydrophobic residues Val255, Leu262, and Leu276 located within this helix, were shown to be crucial for oligomerization. Moreover, the corresponding mutant proteins produced in P. aeruginosa were found to be non-functional in in vivo type III-dependent cytotoxicity assays by directly affecting the correct assembly of PopB/D translocon within the host cell membranes. The detailed understanding of structure-function relationships of T3SS needle tip proteins will be of value in further developments of new vaccines and antimicrobials.
The majority of human and animal Gram-negative bacterial pathogens use specialized nanomachinery, called type III secretion systems (T3SS),4 to deliver a subset of bacterial proteins into the host cell cytoplasm. The collection of T3SS-delivered “effectors” interferes with cell signaling, inflammation processes, and actin dynamics leading notably to breakdown of the immune system permitting the infection to be installed (1, 2).
The T3S apparatus is built up on the bacterial surface upon close contact with the target eukaryotic host cell. It is composed of more than 20 proteins that assemble into a syringe-like structure composed of a set of multimeric protein rings embedded within two bacterial membranes and of a “needle” section of approximately 80 nm protruding out from the surface (3-6). The whole structure is hollow and is presumed to serve as a conduit by which the substrate molecules travel in a partially unfolded state to reach their target localization (7). The first substrates supposed to pass through the needle channel are the so-called translocators, proteins that assemble at the needle extremity and/or insert within the host cell plasma membrane (8-10). The translocon is composed of three proteins, two of which possess hydrophobic domains and are found within host cell membranes after infection (11, 12). In addition, the translocators oligomerize in the presence of lipids to form rings with an internal diameter of 4 nm that permit the release of small molecules from lipid vesicles (13, 14), suggesting that these proteins participate in the breaching of the eukaryotic plasma membrane.
A key component of the effector delivery machinery and a part of the translocon is the well known protective antigen (15-18): LcrV in pathogenic Yersina spp. and PcrV in Pseudomonas aeruginosa. The localization of V proteins on the tip of the secretion needle has been elegantly visualized for the first time using scanning transmission electron microscopy on isolated native Yersinia needles (19). The position of the homologous protein IpaD of Shigella was also recently demonstrated (20, 21).
Functional studies by our group and others demonstrated that the V proteins are soluble and hydrophilic and they neither insert in nor interact with membranes (11, 14, 22). Moreover, V proteins of Yersinia and Pseudomonas fulfill their function by chaperoning the translocators (YopB/D in Yersinia and PopB/D in P. aeruginosa) to be correctly inserted into membranes (22-24), being thus required for the translocation process. In accordance with their position on the top of the secretion needle and with their function in translocation of effectors, antibodies raised against V proteins are protective both in cellular and animal models of infection (16, 22, 25-27), making these proteins attractive targets toward the development of vaccines.
In this work, we show that the V proteins, following controlled refolding in vitro, are able to multimerize into doughnut-like complexes consisting of at least four subunits. Oligomerization requires the C terminus of PcrV, notably the hydrophobic amino acids within the α12 helix. Mutations in the α12 helix have no effect on PcrV, PopB, and ExoS secretion in P. aeruginosa, but are drastic regarding bacterial cytotoxicity toward macrophages by influencing the incorporation of hydrophobic translocators within host membranes. These results reveal that V proteins, once located on the T3S needle tip, are oligomeric, and that the multimerization process probably requires unfolding of the protein during secretion.
EXPERIMENTAL PROCEDURES
Bacterial Strains and Growth Conditions
P. aeruginosa strains used in this study are listed in Table S1. All mutant strains derive from the cystic fibrosis isolate CHA (28, 29), which is referred to as the wild-type strain. P. aeruginosa was grown on Pseudomonas Isolation Agar plates (Difco) or in liquid Luria-Bertani (LB) medium at 37 °C with agitation. Carbenicillin was used for selection at 500 μg/ml for Pseudomonas Isolation Agar plates and 300 μg/ml in LB. An Escherichia coli Top10 strain was employed for standard cloning experiments using the pTOPO blunt-ended cloning kit (Invitrogen). E. coli BL21Star (DE3) (Invitrogen) was used for overproduction of proteins.
Cloning Procedures and Mutagenesis
Construction of Expression Vectors—A DNA fragment encoding full-length PcrV was PCR amplified with the Advantage 2 Polymerase mixture (Clontech), using oligonucleotides CG_VNdeI and CG_VAatII and pIApG-pcrV as template (supplemental Tables S1 and S2). The PcrVΔCter mutant (residue 1 to 254) was constructed by PCR with pIA60 as a template and oligonucleotides CG_VNdeI and CG_δVAat (supplemental Tables S1 and S2). The resulting PCR products were cloned into pTOPO vector (Invitrogen) and verified by double strand sequencing. The NdeI-AatII fragments were cloned into the second multicloning site of pET-Duet1 (Novagen) generating pET-Duet1-pcrV and pET-Duet1-pcrVdeltaCter.
Construction of Pseudomonas Complementation Vectors—The plasmid pIApG-pcrV was used to express wild-type PcrV in CHAΔV, a strain carrying a chromosomal deletion of pcrV (12). The truncated PcrV (PcrVΔCter, residues 1 to 254) was cloned from pIA60 (12) into the pTOPO vector as described above. The NdeI-HindIII-digested fragment was then cloned into the pIApG vector in replacement of the gfp cassette placing it under control of the pcrG promoter pG (as described in Ref. 12) generating pIApG-pcrVdeltaCter. Plasmids were introduced into P. aeruginosa CHAΔV strain by transformation as described elsewhere (30).
Site-directed Mutagenesis—PcrV mutants V255D, T259D, L262A, L262D, Y269D, V273D, L276A, L276D, R278A, F279A, Y283A, D284A, V286A, R288A, I290A, and V255D/L262D were generated by employing the QuikChange mutagenesis kit (Stratagene) using plasmids pIApG-pcrV or pET-Duet1-pcrV as templates. The oligonucleotides used are listed in supplemental Table S2. Mutated pIApG-pcrV plasmids were transformed into the CHAΔV strain for phenotypic studies. Mutated pET-Duet1-pcrV plasmids were transformed into E. coli BL21Star (DE3) for overproduction of proteins.
Expression and Purification of Wild-type and Mutant PcrV
Expression of PcrV and its mutants was performed in E. coli BL21Star (DE3) strain grown in 900 ml of Terrific broth (12 g/liter Bacto-tryptone, 24 g/liter Bacto-yeast extract, 4% w/v glycerol) buffered with 100 ml of potassium phosphate solution (0.17 m KH2PO4, 0.72 m K2HPO4, pH 7.4). Expression was induced with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside at an A600 of 0.7 and cells were additionally grown for 3 h at 37 °C at 210 rpm. Cells were harvested by centrifugation and lysed by three passages through a French press in lysis buffer (50 mm Tris/HCl, pH 8.8) supplemented with Protease Inhibitor Coctail (Complete Roche). The supernatant was cleared by ultracentrifugation at 50,000 × g at 4 °C for 45 min and applied to a 5-ml anion exchange column (HiTrap™ Q HP, GE Healthcare). The column was washed by 25 ml of wash buffer (50 mm Tris/HCl, pH 8.8) and the protein eluted with a 50-ml gradient ranging from 50 mm Tris/HCl, pH 8.8, to 50 mm Tris/HCl, pH 8.8, 350 mm NaCl. Fractions containing the protein were pooled and applied to a gel filtration column (HiLoad 16/60 Superdex™ 200, GE Healthcare) previously equilibrated in 25 mm Tris/HCl, pH 8, 100 mm NaCl.
Expression and Purification of LcrV
Expression of the GST-LcrV fusion protein (22) was performed in E. coli BL21Star (DE3) in the same manner as for PcrV. Cells were harvested by centrifugation and lysed by three passages through a French press in phosphate-buffered saline (10 mm phosphate buffer, 2.7 mm KCl, 137 mm NaCl, pH 7.4) supplemented with Protease Inhibitor Coctail (Complete Roche). The supernatant was cleared by ultracentrifugation at 50,000 × g at 4 °C for 45 min and applied to a 1-ml glutathione affinity column (GST-Trap™ HP, GE Healthcare) equilibrated in phosphate-buffered saline. The column was washed with 5 ml of phosphate-buffered saline and then prepared for enzymatic digestion by equilibration with 10 ml of cleavage buffer (50 mm Tris/HCl, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1 mm dithiothreitol). Digestion was performed on the column with 160 units of PreScission Protease™ (GE Healthcare) incubated for 4 h at 4 °C. LcrV was eluted with elution buffer (50 mm Tris/HCl, pH 8, 10 mm reduced glutathione).
Intrinsic Fluorescence and Circular Dichroism Measurement
Tryptophan fluorescence emission spectra were measured on a Jasco FP-6500 fluorimeter with an excitation wavelength of 280 nm (10 nm slit). Emission was monitored from 300 to 400 nm (10 nm slit) at a 100 nm × min-1 rate and is the average of three accumulations. Proteins were analyzed in a 1-cm optical path cell at a concentration of 1 μm in 25 mm Tris/HCl, pH 8, 100 mm NaCl buffer. Spectra were corrected by subtraction of the buffer spectra using Jasco Spectra Analysis software. Far-UV and near-UV circular dichroism spectra were acquired on a Jasco J-810 spectrophotometer with a scan speed of 50 nm × min-1 at 20 °C. Spectra are the average of 15 scans corrected by subtraction of the spectra acquired on buffer (25 mm Tris/HCl, pH 8, 100 mm NaCl) alone. Measurements in the far-UV region were performed in 0.1-cm path length cells on a 1 μm protein sample in 25 mm Tris/HCl, pH 8, 100 mm NaCl buffer. The signal was recorded from 250 to 195 nm. Near-UV CD spectra were obtained from 320 to 250 nm using a 1-cm path length cell and a protein concentration of 10 μm in 25 mm Tris/HCl, pH 8, 100 mm NaCl buffer. CD measurements were normalized to protein concentration and presented as molecular ellipticity. To monitor the effect of the pH on PcrV, intrinsic fluorescence and CD spectra were obtained under the same conditions with 25 mm acetate buffers with pH values ranging from 6.0 to 3.0 and HCl solutions with pH values from 2.5 to 1.0. All buffers contained 100 mm NaCl.
Unfolding-Refolding of Proteins by pH Treatment
Pure PcrV or LcrV in 25 mm Tris/HCl, pH 8, 100 mm NaCl buffer was treated with HCl (12 M). The quantity of HCl necessary to reach pH 2.6 was previously determined on blank samples and was subsequently added to the protein samples while mixing. The pH was immediately neutralized to about 7.5 by adding a sufficient volume of 1 m Tris/HCl, pH 8. Samples were injected on HiLoad 16/60 Superdex™ 200 (GE Healthcare) to assess the efficiency of the oligomeric species formation. The ratio between the peak areas of the monomers and the multimers was calculated from size exclusion chromatography (SEC) chromatograms.
Native Gel Electrophoresis
Native gel electrophoresis was performed in an 8% acrylamide gel prepared in 0.4 m Tris/HCl, pH 8.9. Ten micrograms of the proteins were mixed with nondenaturating loading buffer (0.3 m Tris/HCl, pH 6.8, 50% glycerol, 0.5% bromphenol blue) and the samples were loaded and run at room temperature at 25 mA in 25 mm Tris base, 20 mm glycine. The gel was stained with Coomassie Blue.
Native Mass Spectrometry
Native mass spectrometry measurements were performed using a Q-TOF Micromass spectrometer (Micromass, Manchester, UK) equipped with an electrospray ion source. It operated with a needle voltage of 3,000 kV, and sample cone and extraction cone voltages of 30 and 0.5 V, respectively. The Backing Pirani pressure was set at 5 mbar. Mass spectra were recorded in the 1,500-6,000 mass to charge (m/z) range. Sample concentration was 10 μm in 25 mm ammonium acetate and was continuously infused at a flow rate of 5 μl/min. Mass spectra were acquired and data were processed with MassLynx 4.0 (Waters).
Transmission Electronic Microscopy
Samples for Transmission Electron Microscopy (TEM) analysis were loaded on carbon films at a concentration of 0.1 mg/ml and stained by 2% uranyl acetate. A grid was added on top of the carbon film and air dried. Micrographs were taken under low-dose conditions with a Philips CM12 microscope operating at 100 kV and a nominal magnification of 30,000-fold. Diameters of the ring-like structures were calculated using the conversion 1 pixel stands for 3.5 Å (conversion corresponding to the magnification used). The values are the mean of 10 structures evaluated on the photographs.
Macrophage Infection and Cytotoxicity Assays
Cytotoxicity assays were performed as previously described (31). Briefly, the macrophage cell line J774 (ATCC) was grown in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% of heat-inactivated fetal calf serum (Invitrogen). Cells were seeded at 2 × 105 per well in 48-well plates the day before infection. Overnight bacterial cultures were diluted in LB at an A600 of 0.1 and allowed to grow to A600 of 1 at 300 rpm and at 37 °C. Macrophages were infected at a multiplicity of infection of 5 and infection was performed for 3 h in 300 μl of Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum in a CO2 (5%) incubator at 37 °C. Cytotoxicity was assessed by determination of lactate dehydrogenase release into supernatants of the infected cells using a cytotoxicity detection kit (Roche) as described previously (32).
Analysis of Secreted T3SS Proteins
P. aeruginosa strains expressing wild-type or mutant PcrV were grown overnight at 37 °C with agitation in liquid LB with carbenicillin, when necessary. Overnight cultures were diluted to an A600 of 0.2 in LB containing 5 mm EGTA and 20 mm MgCl2 for T3SS induction. Incubation was prolonged for about 3 h until the culture reached an A600 of 1.0. Ten microliters of culture supernatants were directly analyzed by SDS-PAGE. Immunoblotting was performed using polyclonal antibodies raised against PcrV and PopB (12, 22) followed by a secondary antibody conjugated to horseradish peroxidase (Sigma). Membranes were developed by using an enhanced chemiluminescence kit (ECL, GE Healthcare).
Hemolysis Assay and Purification of Red Blood Cell Membranes
Hemolysis assays were performed on sheep red blood cells (RBCs) (ELITech, France) as described (11, 12, 22). The membranes of RBCs were purified after the infection with P. aeruginosa according to the protocol described before (12, 22) with some modifications. Briefly, sheep RBCs (50% suspension) were washed three times in RPMI 1640 medium (Sigma) and resuspended at a concentration of 3 to 6 × 109 RBCs/ml at 4 °C. P. aeruginosa were grown to an A600 of 1, and 3 × 109 colony forming units/assay were centrifuged and resuspended in 500 μl of RPMI 1640 medium. In a final volume of 1.5 ml, 3 × 109 RBCs were mixed with P. aeruginosa at a multiplicity of infection of 1 and centrifuged at 2,000 × g for 10 min at 4 °C to maximize the bacterial-RBC contact. After a 1-h incubation at 37 °C in the presence of a Protease Inhibitor Coctail (Complete; Roche), the RBCs were lysed by adding 2 ml of MilliQ water and vortexing. Bacteria and cell debris were removed by centrifugation, and the supernatant was brought to a final concentration of 62% sucrose. The sample was overlaid with a discontinuous sucrose gradient consisting of 4 ml of 44% and 3 ml of 25% sucrose in Tris/saline solution (30 mm Tris/HCl, 150 mm NaCl, pH 7.5). All solutions were supplemented with protease inhibitor mixture and centrifuged in a SW41 rotor at 28,000 × g for 16 h at 4 °C. The membranes were recovered from the interface of the 25 and 44% sucrose layers, diluted in Tris/saline, and concentrated by ultracentrifugation at 372,000 × g for 20 min at 4 °C. The pellets were resuspended in Laemmli buffer, and the proteins were separated by SDS-PAGE. The presence of PopB, PopD, and PcrV in the membranes was analyzed by immunoblot detection using the antibodies as described above.
Structure Analysis
The atomic structure of LcrV (Protein Data Bank number 1RGF) was analyzed with PyMol (33). The C-terminal α-helices of LcrV (Y. pestis) and PcrV was aligned using ClustalW.
RESULTS
Refolding of PcrV Promotes the Formation of Multimers—Recombinant PcrV and its homologues were previously shown to behave as monomers in solution (14, 23, 34-36), although some reports showed that LcrV may exist as a dimer (37, 38). The crystal structure of the LcrV monomer revealed that it folds into 6 β-sheets and 12 α-helices with a unique intramolecular coiled-coil, thus resembling an elongated dumbbell-shaped molecule (36). However, the monomeric state of V antigens is incompatible with the position and shape of LcrV and PcrV at the extremity of the native needles of Yersinia recently visualized by scanning transmission electron microscopy (19). To biochemically demonstrate the ability of PcrV to oligomerize, we postulated that the protein should be unfolded and refolded to reach its final structure. Indeed, it is thought that the secreted proteins should be partially unfolded to travel through the 3-nm narrow T3SS needle and then refold when they reach the needle extremity or their final destination (39).
Native, non-tagged P. aeruginosa PcrV was obtained by a two-step purification including anion exchange chromatography followed by a SEC as described under “Experimental Procedures.” To mimic in vitro the secretion of PcrV that occurs in a partially unfolded conformation, different conditions were tested and the variation of pH was chosen, because lowering the pH to 2.6 drives the protein into a molten globule conformation (see below). This flexible conformation corresponds to an intermediate state in protein folding that facilitates structural rearrangement (40).
The unfolding and refolding of PcrV in vitro was followed by intrinsic fluorescence and circular dichroism (CD) experiments (Fig. 1). The intrinsic fluorescence of PcrV was monitored by determining the emission maxima wavelengths (λmax) and the ratio of the intensities at 320 and 365 nm (Fig. 1A). These measurements give an indication of the compactness of the protein and solvent exposure of tryptophan residues because the fluorescence of these residues depends on the hydrophobicity of their environment. PcrV harbors three tryptophan residues (Trp61, Trp92, and Trp186), and the intrinsic fluorescence of the protein reflects the average contribution of each tryptophan. Based on the LcrV crystal structure, Trp61 and Trp186 are relatively exposed to the solvent, whereas Trp92 is involved in the interaction between α-helices 7 and 12. This is in agreement with the λmax of 337 nm observed at neutral pH, corresponding to an overall moderate solvent exposure. Lowering the pH in a range from 7.2 to 1.0 induces a reversible exposure of PcrV tryptophan residues, noticed by a red-shift of the λmax that reached a maximum at pH 2.6. CD spectra were collected in far- and near-UV to assess secondary and tertiary structures, respectively. At pH 7.2, the PcrV CD spectrum in the near-UV region exhibited a minimum at 284 nm reflecting the existence of a stable tertiary structure. Bringing down the pH to 2.6 resulted in a significant decrease of the signal, indicating a loss of rigid tertiary structure. The unfolding can be reversed by the rapid adjustment to neutral pH (Fig. 1B). The CD spectra of PcrV in the far-UV region are identical at pH 7.2 and 2.6 (Fig. 1C) and display minima at 208 and 222 nm, characteristics of a large predominance of α-helices, indicating that the secondary structure of the protein is not modified even at pH 2.6. Thus, the protein adopts at this pH a molten globule conformation characterized by a native-like secondary structure and a loss of rigid tertiary structure. Taken together, these data show that pH may be used to reversibly manipulate the PcrV folding.
FIGURE 1.
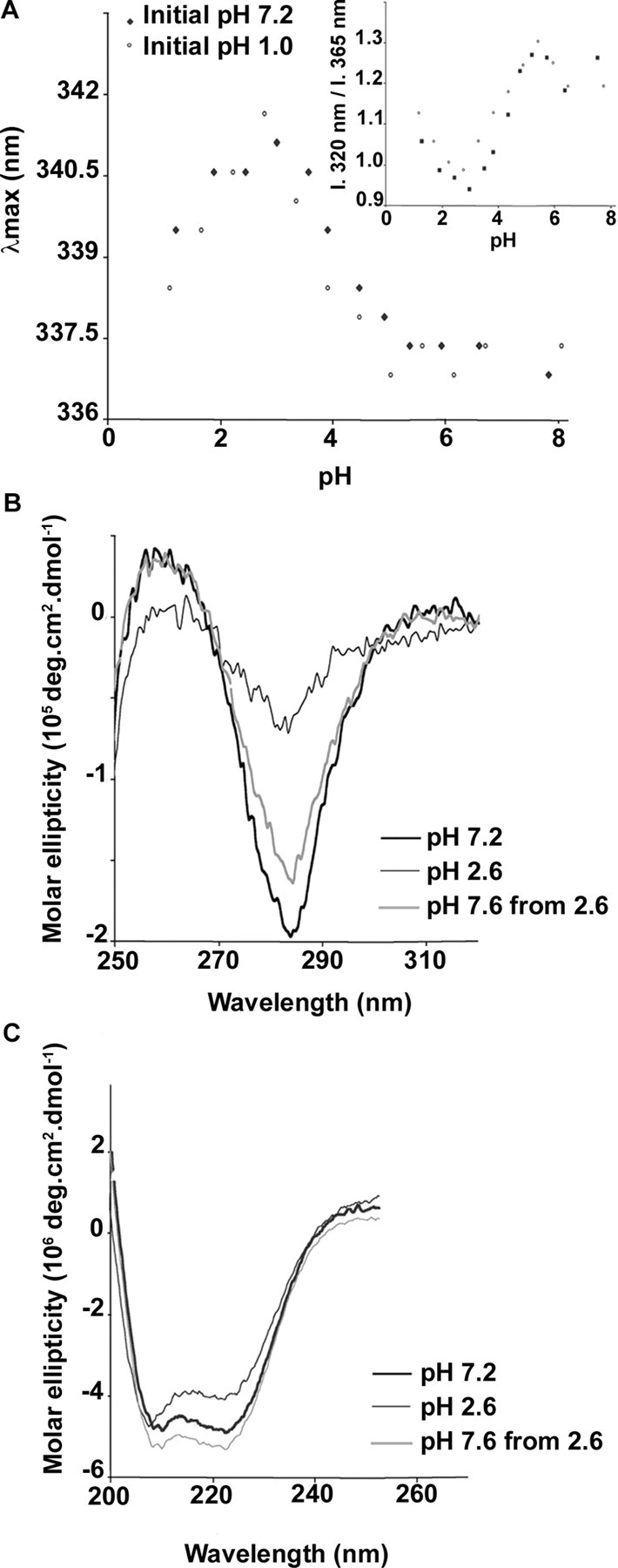
Reversible PcrV unfolding induced by pH decrease. Conformational changes were assessed by monitoring tryptophan fluorescence emission and circular dichroism (CD). A, fluorescence emission spectra were recorded upon excitation at 280 nm at pH ranging from 1.0 to 7.0 and a protein concentration of 1 μm. The initial pH of the PcrV solution, either 7.2 (closed diamonds) or 1.0 (open circles), was promptly changed by a 10-fold rapid dilution into buffer solutions with lower or higher pH values, respectively. For each spectrum, the maximum wavelength of emission as well as the ratio of the emission intensity (I) at 320 and 365 nm (inset) was determined. CD spectra of PcrV at pH 7.2 (bold line), 2.6 (thin line), and 7.6 after incubation at pH 2.6 (gray line), were recorded in near-UV (B) and far-UV (C) to assess tertiary and secondary structure, respectively. The pH was lowered to 2.6 by rapid dilution, as described above, and subsequently neutralized by the addition of 1 m Tris/HCl, pH 8.0. Spectra are the average of 15 scans. Protein concentrations were 10 and 1 μm in 25 mm Tris/HCl, pH 7.2, containing 100 mm NaCl for near- and far-UV, respectively.
SEC analysis of the untreated PcrV (Vuntreated) revealed a single peak eluting at 72 ml, which corresponds to a globular protein with a molecular mass of ∼34 kDa (Fig. 2A). Native mass spectrometry (32,354.60 Da; predicted 32,461 Da) and native PAGE confirmed the monomeric state of PcrV (Fig. 2C), in agreement with previous reports (14, 35). The unfolded-refolded PcrV mixture contained three major protein species clearly separated by SEC: the monomeric protein Vmono eluting from the column at the same volume as Vuntreated, species eluting in the void volume corresponding to aggregates, and novel form(s), Voligo, eluting as a broad peak between 54 and 67 ml (Fig. 2A). This large peak contains a mixture of oligomers ranging from dimers to hexamers, as suggested by preliminary SEC-Multi-angle Laser Light Scattering studies (not shown). The quantification of peak areas from SEC chromatograms showed that oligomeric species represent 30% of the amount of monomer after pH treatment.
FIGURE 2.
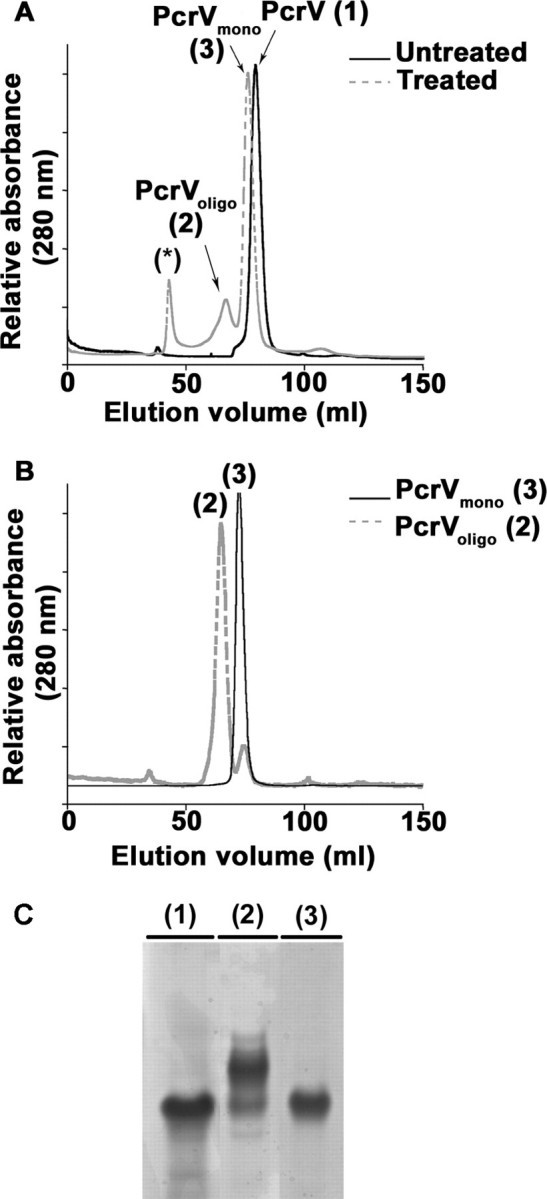
Formation of novel, oligomeric species of PcrV. A, SEC analysis of pH-treated and untreated PcrV protein shows the formation of two novel protein peaks after refolding. Untreated PcrV was recovered as a single peak corresponding to a monomer (PcrVuntreated (1)). After pH treatment, two novel peaks arise in addition to the one corresponding to the monomer (PcrVmono (3)). One peak corresponds to multimeric species (PcrVoligo (2)) and the other probably to aggregated species eluting in void volume (asterisk). B, proteins present either in peak 2 (PcrVoligo) or peak 3 (PcrVmono) were analyzed again by SEC showing that the oligomers and monomers are stable species. C, the different protein peaks were submitted to native gel electrophoresis followed by Coomassie Blue staining. Lanes 1-3 correspond to 10 μg of protein contained in the peaks eluted from SEC: PcrVuntreated, PcrVoligo, and PcrVmono, respectively. PcrVuntreated (1) runs as a unique band as does the PcrVmono (3), whereas PcrVoligo (2) separates into additional bands with lower electrophoretic mobilities.
When Vmono or Voligo species were re-submitted to SEC analysis, peaks with an elution volume corresponding mainly to Vmono or Voligo species, respectively, could be detected (Fig. 2B). Indeed, less than 20% of monomers could be observed in the reinjected oligomeric fraction, most probably originating from the partially overlapping fractions. This observation suggests that the newly formed oligomers are stable complexes.
To further determine the nature of this novel form of PcrV, the protein peak was analyzed by native gel electrophoresis, which revealed one major band migrating slower than Vuntreated, suggesting the presence of multimeric PcrV (Fig. 2C). Moreover, the native mass spectrometry performed on Voligo identified at least three multimeric species of molecular masses of 64,707.60, 97,061.74, and 129,409.77 Da, corresponding to dimers, trimers, and tetramers of PcrV, respectively.
Oligomerization Leads to Doughnut-like Species without Any Changes in Global Tertiary Structure—Vuntreated, Vmono, and Voligo fractions were further analyzed by TEM (Fig. 3A). Although preparations of Vuntreated and Vmono were uniform with no distinct elements, the Voligo fraction contained numerous doughnut-like species with a visible hole in the middle. The size of Voligo (averaged over 10 elements obtained in three independent experiments) could be estimated at 9.7 ± 1.4 and 3.9 ± 1.4 nm for external and internal diameters, respectively. Some structures were irregular or even disrupted, suggesting that the in vitro oligomerization process was not complete for all the molecules.
FIGURE 3.

Oligomeric V species exhibit doughnut-like shapes. Transmission electronic microscopy photographs of fractions obtained after size exclusion chromatography of untreated (A), oligomeric (B), and monomeric (C) PcrV or LcrV. All samples were coated on carbon/formar grids at a protein concentration of 0.1 mg/ml and stained with 2% uranyl acetate. No distinct structures could be observed in peaks corresponding to monomeric forms (A and C), whereas doughnut-like structures are readily observed in samples corresponding to oligomeric PcrV or LcrV (B). Scale bar corresponds to 20 nm.
Y. pestis LcrV shares a high degree of identity (41%) with P. aeruginosa PcrV and their roles in translocon assembly and translocation have been proposed to be similar (22, 41). A small-scale expression and purification was performed on LcrV to test the behavior of the protein in the folding assay. Notably, when LcrV was partially unfolded and refolded by rapid pH changes, two novel protein peaks appeared on SEC (data not shown). The first, corresponding to the aggregated protein, eluted in the void volume of SEC, and the second one matched to a presumable LcrVoligo species. Native staining TEM confirmed the presence of oligomeric elements in the LcrVoligo preparation (Fig. 3B), whereas any visible, clearly defined structures could be revealed neither in the Vuntreated fraction (monomers) nor in the fraction eluting in the void volume. Importantly, LcrVoligo structures were homogenous in size with external diameters of 8.3 ± 1.8 nm and internal diameters 3 ± 0.6 nm, somehow smaller than those observed with PcrV.
Intrinsic fluorescence and CD experiments were performed on the fractions of PcrV obtained after partial unfolding and refolding. The Voligo and Vmono fractions exhibited an intrinsic fluorescence maximum of emission at 337 nm, identical to the untreated sample. In the near (Fig. 4A) and far (Fig. 4B) UV regions, the CD spectra of the Voligo and Vmono fractions obtained after treatment were similar to the spectrum of the untreated monomeric sample, Vuntreated. The lower intensity of the signal observed with Voligo is most probably due to a higher level of light scattering caused by the oligomeric species. Nevertheless, these data strongly suggest that the global secondary and tertiary structures remain unchanged after oligomerization. Taken together, these results show that V antigens of Y. pestis and P. aeruginosa have the capacity to oligomerize into high-ordered ring-like structures whose estimated molecular mass is >130 kDa without gross modifications of tertiary structure.
FIGURE 4.
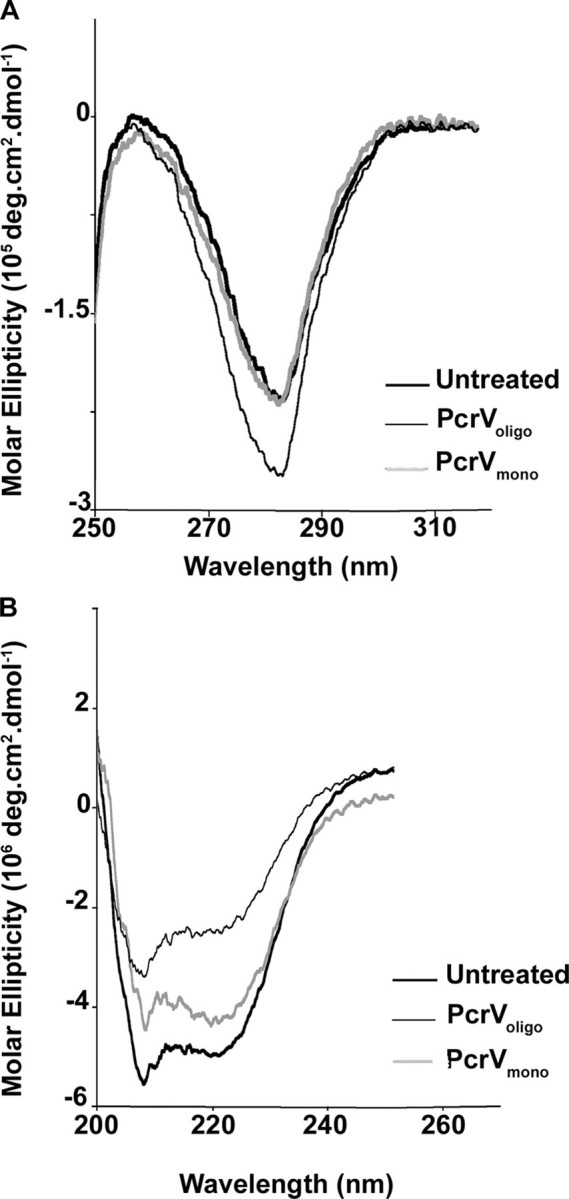
Oligomeric and monomeric forms of PcrV have similar global structural features. Structural features of PcrVuntreated (bold line), PcrVoligo (gray line), and PcrVmono (thin line) were examined by CD experiments. CD spectra of the different forms of PcrV were recorded in near-UV (A) and far-UV (B) to assess tertiary and secondary structures, respectively. Protein concentrations were 10 and 1 μm for near- and far-UV, respectively.
Role of the C-terminal α12 Helix in Oligomerization—The crystal structure of LcrV shows that the C-terminal part folds into a 37-residue long α-helix (α12), which is arranged into an intramolecular coiled-coil together with α7, forming a unique hydrophobic “zipper” motif (Fig. 5 and Ref. 36). Description of LcrV interaction with LcrG (42) in bacterial cytoplasm leads to the hypothesis that the protein may also adopt an “open” conformation leaving helices 7 and 12 available for intermolecular interactions (36, 43).
FIGURE 5.
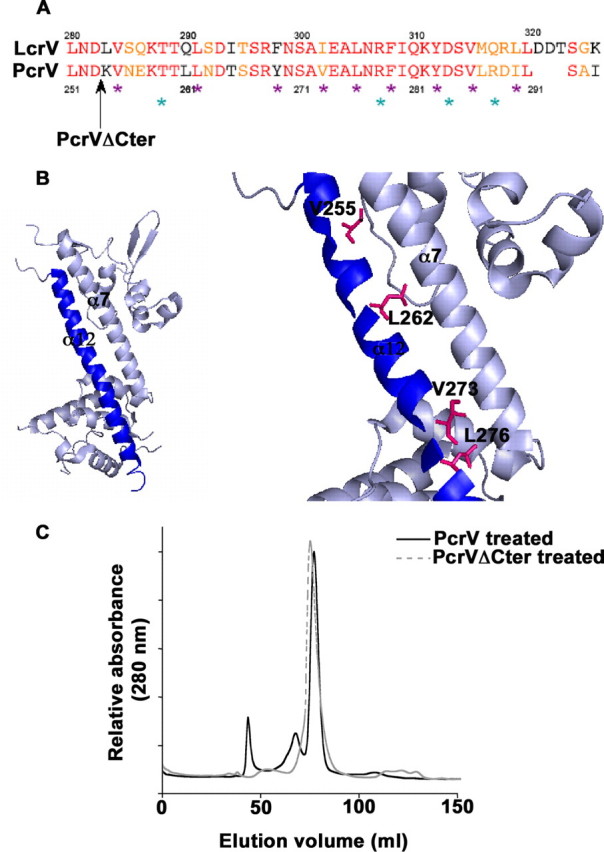
The conserved C-terminal α12 helix plays a key role in the protein functionality. A, alignment of LcrV and PcrV C-terminal α12 helix sequences. Identical residues and similar residues are shown in red and orange, respectively. The black arrow shows the end of the protein sequence of the PcrVΔCter mutant. Mutated residues are outlined by violet stars for hydrophobic residues (Val255, Leu262, Tyr269, Val273, Leu276, Phe279, Tyr283, Val286, and Ile290) and cyan stars for hydrophilic residues (Thr259, Arg278, Asp284, and Arg288). B, schematic of the LcrV structure (PDB 1R6F (36)). The C-terminal α12 helix is represented in blue. Zoom on the area of interest: mutated residues affecting Pseudomonas cytotoxicity (Val255, Leu262, Leu276, and Val273) are shown by stick representation. C, capacity of oligomerization of PcrVΔCter was assessed by pH treatment followed by size exclusion chromatography. Chromatogram of PcrVΔCter is overlaid with the chromatogram of pH-treated wild-type PcrV. PcrVΔCter is inefficient in forming the high molecular weight oligomeric state of PcrV.
To investigate the role of the PcrV α12 C-terminal region in the oligomerization process, we produced truncated PcrV (VΔCter), lacking the last 41 amino acids (Fig. 5A), and assayed its ability to multimerize. The VΔCter is a stable, correctly folded protein, as determined by CD analysis (data not shown) and behaves as a monomer in solution as verified by SEC and native mass spectrometry (molecular mass 27,683.44 Da; predicted 27,723 Da). However, when submitted to the pH treatment described above, PcrVΔCter did not give rise to any additional peak in the SEC experiment, strongly suggesting that it is unable to oligomerize (Fig. 5C). This result implies that the last 41 amino acids, being part of the α12 C-terminal helix, are required for multimer formation.
Oligomerization Incompetent Mutants Are Unable to Insert Functional Translocon into Host Cell Membranes—To test the impact of the α12 C-terminal deletion in vivo, we engineered P. aeruginosa ΔPcrV mutant strains expressing either wild-type PcrV or PcrVΔCter, and tested them in cytotoxicity assays on macrophages. As expected, a P. aeruginosa strain carrying VΔCter was unable to provoke macrophage cell death, although the protein was synthesized and secreted at the wild-type level (Fig. 6, A and B). The crystal structure of LcrV shows that one part of the α12 helix (residues 305 to 326) is perfectly amphipathic, with the hydrophobic side of the helix being composed of residues Ala301, Ile302, Ala304, Leu305, Phe308, Ile309, Val315, Met316, Leu319, and Leu320, which are conserved in PcrV (Fig. 5A).
FIGURE 6.

PcrVL276D and V255D/L262D mutants are impaired in their post-secretory role in vivo, which correlates with inefficient oligomerization. A, loss of T3SS activity was assessed by infection of macrophages. Lactate dehydrogenase release of infected cells was measured at 3 h post-infection. P. aeruginosa strains PcrVΔCter, PcrVL276D, and PcrVV255D/L262D are noncytotoxic. B, global functionality of the type III machinery was checked by Western blotting on secreted proteins PopB and PcrV following in vitro induction of the system. C, translocon insertion capacity of two noncytotoxic PcrV mutants. Hemoglobin release was measured at 1 h post-infection. The presence of PcrV, PopB, and PopD in membrane fractions of RBC was revealed by immunodetection. Mutants are nonhemolytic and less efficient in inserting translocon proteins in cell membranes. Negative control was obtained from noninfected cells (N.I.). D, capacity of oligomerization of PcrVL276D and PcrVV255D/L262D was assessed by pH treatment followed by SEC. Chromatograms of PcrV mutants are overlaid with the chromatogram of pH-treated wild-type PcrV. Mutants are less efficient in forming the high molecular weight oligomeric forms of PcrV.
To pin down the role of the α12 helix, several individual amino acid substitutions were introduced in PcrV and modified proteins were tested for their capacity to complement the ΔPcrV mutant. We first targeted the amphipathic part of the helix and substituted hydrophilic (Arg278, Lys282, and Arg288) and hydrophobic (Leu276, Phe279, Tyr283, Val286, and Ile290) residues into alanine (Fig. 5A). With the exception of Leu276, all mutants exhibited a wild-type phenotype with respect to secretion and cytotoxicity. Remarkably, the changes of Leu276 to either Ala or Asp resulted in a strong decrease in cytotoxicity, although the secretion in vitro was not affected (Fig. 6, A and B). Interestingly, Leu276 (which corresponds to Leu305 in LcrV) is the last residue of the α12 helix engaged in intramolecular coiled-coil formation, being positioned just before the “kink” in the helix (Fig. 5 and Ref. 36). Therefore, additional mutations were engineered on residues positioned lengthwise to α12 and being fully engaged within the “zipper” motif (Fig. 5A). Single mutations V255D, T259D, L262D, and V273D have a pronounced effect on bacterial cytotoxicity, whereas the double mutation V255D/L262D completely abolished cytotoxicity toward macrophages (Fig. 6A). All mutants secreted wild-type levels of PcrV and PopB (Fig. 6B) indicating that the secretion apparatus was intact. In summary, the post-secretory, biological function of PcrV is tightly associated with the C-terminal helix of the protein, involving notably hydrophobic residues positioned within the α12 helix and engaged in intramolecular coiled-coil formation.
To test whether those amino acids are also important for the oligomerization process in vitro, the L276D and V255D/L262D mutations were engineered in the PcrV-overexpression plasmid, and the mutated proteins were purified and submitted to pH-induced unfolding and refolding. Refolded PcrVL276D and PcrVV255D/L262D were analyzed by SEC and the elution profiles were compared with wild-type protein. After pH treatment, mutated PcrV proteins mostly behave as monomers, displaying only a small shoulder corresponding to multimeric forms in SEC analysis. The amount of the oligomeric species compared with monomers was found to be 4.1 and 4.9% for PcrVL276D and PcrVV255D/L262D, respectively (compared with the 30% oligomers obtained with the wild-type protein and 9.7% obtained with PcrVΔCter), showing that the mutants have a reduced capacity to form oligomers (Fig. 6D).
As the function of PcrV on the tip of the type III secretory needle is thought to guide the insertion of Pop translocators within host cell membranes, we checked whether P. aeruginosa strains carrying PcrVΔCter and PcrVL276D are able to insert PopB and PopD. RBCs have been shown to be an adequate model to appreciate the incorporation of the Pop/Yop translocon within host membranes, as hemolysis occurs upon the formation of the pore within membranes, an event dependent on functional PcrV (12, 24, 31). After RBCs were infected with the indicated strains, hemolysis was checked and membranes were purified on sucrose gradients as described previously (22). The wild-type strain and ΔV/Vwt lysed RBCs after a 1-h infection, whereas all PcrV mutants displayed a CHAΔPcrV phenotype. Immunodetection of pore proteins showed that PopB is inserted within membranes of RBCs infected by all strains, whereas PopD was not present in mutant strains CHAΔV/VΔCter and CHAΔV/VL276D (Fig. 6C).
Taken together, mutations in or deletions of the C-terminal α12 helix of PcrV do not affect secretion of the Pop proteins, but prevent the formation of the functional Pop pore within the host membranes. Therefore, the absence of the correct PcrV structure abolishes the translocon function and, in consequence, bacterial cytotoxicity.
DISCUSSION
PcrV and LcrV are an integrative part of the T3S machinery that allows the passage of bacterial toxic proteins into the host eukaryotic cell in two continuous steps: secretion and translocation. Scanning transmission electron microscopy images of native Yersinia and Shigella needles showed that those proteins are organized into distinct structures on the distal part of the T3S needle (19, 21, 34, 41).
In this work, we demonstrated the ability of PcrV and LcrV to multimerize into doughnut-like structures composed of at least four subunits, as revealed by native mass spectrometry. The oligomerization step required the partial unfolding of the monomer, which was achieved in vitro by controlled denaturation/renaturation using variations of pH. This treatment was chosen to artificially mimic what happens in vivo. Indeed, it may reflect the conformational changes imposed to all T3S substrates, including V proteins, which are thought to travel through the 3-nm wide secretion channel in a partially unfolded state (44). Unfolding of the molecules prior to secretion is thought to be accomplished in vivo by a T3S-specific ATPase, located at the base of the secretion apparatus (45, 46). In the Salmonella T3SS, the InvC ATPase directly binds to the chaperone-substrate complex and induces chaperone release and unfolding of the substrate in an ATP-dependent manner (39). This partially unfolded state concerns only the tertiary structure with conservation of the secondary structures (47). The pH treatment performed on PcrV efficiently allowed us to create such a conformation, e.g. molten globule state.
The oligomeric state of the tip complexes was proposed to be either tetrameric or pentameric, based either on modeling data (7) or by analyzing mass-per-length measurements performed on native needles (41). More recently, genetic and biochemical studies on Shigella needles proposed that the tip complex is a heteropentamer harboring one additional IpaB molecule that is the homologue of the PopB/YopB translocator (48). In our study, the broad peak obtained on SEC and the observations by TEM indicated that a population of different oligomers was obtained upon pH treatment. Native mass spectrometry revealed masses corresponding to tetramers, trimers, and dimers. Because oligomers are submitted to harsh conditions during the native mass spectrometry experiment, especially toward hydrophobic interactions, it is possible that even if higher oligomeric species are formed, they may have been destroyed during the analysis.
Being positioned at the junction between the secretory needle and the translocon, the LcrV/PcrV protein family plays a decisive role in proper insertion of translocators (PopB and PopD in Pseudomonas) into the host plasma membrane during infection (11, 12, 22). The estimated sizes of the doughnut-like structures observed by negative-staining TEM (internal diameter of 3-4 nm and external diameter of 8-10 nm) are similar to the size of the homo- or hetero-oligomers of PopB and PopD obtained previously in the presence of liposomes (14), suggesting that the oligomeric structure of V may serve as an assembly platform for the translocators just before the membrane insertion. Furthermore, the evidence of loss of cytotoxicity toward macrophages and the inefficient hemolysis capability exhibited by the PcrV mutants defective in oligomerization strongly support the idea that the insertion of a functional translocon into host cell membranes requires the formation of a stable complex of oligomeric PcrV/LcrV on the needle extremity. The direct interactions between the V proteins and translocators are currently being investigated, and may require proper multimeric organization of all components.
We demonstrated that the last 41 amino acids at the C terminus of PcrV are essential for oligomerization of the protein in vitro and its function in bacterial cytotoxicity. The produced truncated protein is stable and properly folded. In P. aeruginosa, the truncation does not perturb the secretion of either PcrVΔCter itself, or of other T3SS-dependent proteins (PopB, PopD, or ExoS), indicating that only the post-secretory role of PcrV is affected. The C terminus potentially folds into a long α helix, α12, which together with α7, could form an unique intramolecular coiled-coil (36). Remarkably, the mutants engineered so that they perturb the formation of the coiled-coil structure were found to be noncytotoxic in cellular models of infection while not influencing the secretion of ExoS, PopB, PopD, and PcrV itself. In addition, in vitro oligomerization of the mutant proteins could not be induced, as evaluated by SEC. Of note, the heptad repeat LX3-LX2-LX3L of LcrV within the α7 helix was found to be involved in specific interactions with the N terminus of its intracellular partner LcrG, strongly suggesting that the V molecule may alter between a “closed,” monomeric shape and an open form, prone to interact with its partners. In addition, as already noted by Derewenda et al. (36) the C-terminal helix is preceded by a flexible loop that may be pulled apart leaving α12 and α7 helices free for other types of interactions.
Interestingly, PcrV and LcrV are fully functionally exchangeable only if the cognate translocators are co-expressed (22, 49, 50). Based on this observation, functional and scanning transmission electron microscopy analyses of LcrV/PcrV hybrids in which N- and C-terminal domains were exchanged and then expressed in Yersinia, suggested that the globular N-terminal domain of the protein is required for proper insertion of cognate translocators into host membranes (41). Surprisingly, using the same hybrids in P. aeruginosa,5 we were not able to make the same conclusion; whereas the LcrVNter-PcrV hybrid was able to complement a ΔpcrV mutant in Pseudomonas, the PcrVNter-LcrV hybrid was not, which would be expected if the participation of only N-terminal domains in insertion of translocators is indispensable. Based on these observations, we suggest that the orientation and function of globular domains may be more complicated than proposed in the original model, with both globular domains (Fig. 5B) being critical for correct multimerization of the protein into the functional tip. Using CD analysis and tryptophan fluorescence, we showed that the global helical structure does not change when passing from monomers to oligomers. This observation, and the probability of PcrV to exhibit closed and open conformations via the flexible loop, allows us to speculate that the oligomerization may occur via a so called “domain swapping” mechanism (51, 52). Association between adjacent V subunits may occur by intermolecular exchange of α7 and α12 helices, which must be available when the protein comes out from the secretion channel.
Shigella IpaD and BipB from Burkholderia pseudomallei (the two V homologues) possess, as do PcrV and LcrV, an internal coiled-coil, and in addition a four-helix bundle at the N terminus that, when removed from the protein, induces the formation of multimers as revealed by SEC (53). It was suggested that this part of the protein is a kind of chaperone, which flips away and promotes the exposure of the intramolecular coiled-coil only at the top of the needle. We found that native secreted V molecules, recovered from bacterial culture supernatants folds directly into monomers (not shown), when not interacting with the needle. It seems thus that in vivo, only needle-attached V proteins assemble into oligomer-forming structures that are visible by microscopy, suggesting a crucial role of local monomer concentration and needle interaction. However, how this interaction occurs and promotes oligomerization still needs to be investigated.
Active and passive immunization by LcrV and PcrV provides animals with a high level of protection against infections by Y. pestis and P. aeruginosa, respectively (15-18). Protective anti-PcrV Mab166 recognizes the region from amino acids 144 to 257 (16), partially overlapping the α7 helix involved in coiled-coil formation. The same antibody is capable of affecting the insertion of translocators in red blood cells membranes (22). Creation of oligomeric V species in vitro will allow the production of other specific antibodies with the expected higher level of protection than the existing one. Determination of the exact structure of V oligomers formed on the needle extremity may lead to new development of therapies against Pseudomonas- and Yersinia-caused infections.
Supplementary Material
Acknowledgments
We thank Karin Pernet-Gallay (Grenoble Institut des Neurosciences) for forming G.C. with transmission electron microscopy and sample staining and David Lascoux (Institut de Biologie Structurale, Grenoble) for taking over the native mass spectrometry experiments. We thank Vincent Forge (LCBM, CEA, Grenoble) for precious advice on fluorescence and circular dichroism experiments and Andréa Dessen (Institut de Biologie Structurale, Grenoble) for helpful discussions and critical reading of the manuscript.
This work was supported in part by Research Contract 06.70.151. 00.470.75.96 from Délégation Générale pour l'Armement (DGA) and the French Cystic Fibrosis Association Vaincre la Mucoviscidose. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables S1 and S2.
Footnotes
The abbreviations used are: T3SS, type III secretion system; GST, glutathione S-transferase; RBC, red blood cell; SEC, size exclusion chromatography; TEM, transmission electron microscopy.
C. Gébus, unpublished data.
References
- 1.Cornelis, G. R. (2006) Nat. Rev. Microbiol. 4 811-825 [DOI] [PubMed] [Google Scholar]
- 2.Galan, J. E., and Wolf-Watz, H. (2006) Nature 444 567-573 [DOI] [PubMed] [Google Scholar]
- 3.Blocker, A., Komoriya, K., and Aizawa, S. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 3027-3030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoiczyk, E., and Blobel, G. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 4669-4674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Journet, L., Agrain, C., Broz, P., and Cornelis, G. R. (2003) Science 302 1757-1760 [DOI] [PubMed] [Google Scholar]
- 6.Marlovits, T. C., Kubori, T., Sukhan, A., Thomas, D. R., Galan, J. E., and Unger, V. M. (2004) Science 306 1040-1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deane, J. E., Roversi, P., Cordes, F. S., Johnson, S., Kenjale, R., Daniell, S., Booy, F., Picking, W. D., Picking, W. L., Blocker, A. J., and Lea, S. M. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 12529-12533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hakansson, S., Bergman, T., Vanooteghem, J. C., Cornelis, G., and Wolf-Watz, H. (1993) Infect. Immun. 61 71-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neyt, C., and Cornelis, G. R. (1999) Mol. Microbiol. 33 971-981 [DOI] [PubMed] [Google Scholar]
- 10.Sarker, M. R., Neyt, C., Stainier, I., and Cornelis, G. R. (1998) J. Bacteriol. 180 1207-1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blocker, A., Gounon, P., Larquet, E., Niebuhr, K., Cabiaux, V., Parsot, C., and Sansonetti, P. (1999) J. Cell Biol. 147 683-693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goure, J., Pastor, A., Faudry, E., Chabert, J., Dessen, A., and Attree, I. (2004) Infect. Immun. 72 4741-4750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Faudry, E., Vernier, G., Neumann, E., Forge, V., and Attree, I. (2006) Biochemistry 45 8117-8123 [DOI] [PubMed] [Google Scholar]
- 14.Schoehn, G., Di Guilmi, A. M., Lemaire, D., Attree, I., Weissenhorn, W., and Dessen, A. (2003) EMBO J. 22 4957-4967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bacon, G. A., and Burrows, T. W. (1956) Br. J. Exp. Pathol. 37 481-493 [PMC free article] [PubMed] [Google Scholar]
- 16.Frank, D. W., Vallis, A., Wiener-Kronish, J. P., Roy-Burman, A., Spack, E. G., Mullaney, B. P., Megdoud, M., Marks, J. D., Fritz, R., and Sawa, T. (2002) J. Infect. Dis. 186 64-73 [DOI] [PubMed] [Google Scholar]
- 17.Hill, J., Leary, S. E., Griffin, K. F., Williamson, E. D., and Titball, R. W. (1997) Infect. Immun. 65 4476-4482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anderson, G. W., Jr., Leary, S. E., Williamson, E. D., Titball, R. W., Welkos, S. L., Worsham, P. L., and Friedlander, A. M. (1996) Infect. Immun. 64 4580-4585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mueller, C. A., Broz, P., Muller, S. A., Ringler, P., Erne-Brand, F., Sorg, I., Kuhn, M., Engel, A., and Cornelis, G. R. (2005) Science 310 674-676 [DOI] [PubMed] [Google Scholar]
- 20.Espina, M., Olive, A. J., Kenjale, R., Moore, D. S., Ausar, S. F., Kaminski, R. W., Oaks, E. V., Middaugh, C. R., Picking, W. D., and Picking, W. L. (2006) Infect. Immun. 74 4391-4400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sani, M., Botteaux, A., Parsot, C., Sansonetti, P., Boekema, E. J., and Allaoui, A. (2007) Biochim. Biophys. Acta 1770 307-311 [DOI] [PubMed] [Google Scholar]
- 22.Goure, J., Broz, P., Attree, O., Cornelis, G. R., and Attree, I. (2005) J. Infect. Dis. 192 218-225 [DOI] [PubMed] [Google Scholar]
- 23.Lee, V. T., Tam, C., and Schneewind, O. (2000) J. Biol. Chem. 275 36869-36875 [DOI] [PubMed] [Google Scholar]
- 24.Marenne, M. N., Journet, L., Mota, L. J., and Cornelis, G. R. (2003) Microb. Pathog. 35 243-258 [DOI] [PubMed] [Google Scholar]
- 25.Holder, I. A., Neely, A. N., and Frank, D. W. (2001) Infect. Immun. 69 5908-5910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leary, S. E., Williamson, E. D., Griffin, K. F., Russell, P., Eley, S. M., and Titball, R. W. (1995) Infect. Immun. 63 2854-2858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weeks, S., Hill, J., Friedlander, A., and Welkos, S. (2002) Microb. Pathog. 32 227-237 [DOI] [PubMed] [Google Scholar]
- 28.Dacheux, D., Attree, I., Schneider, C., and Toussaint, B. (1999) Infect. Immun. 67 6164-6167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Toussaint, B., Delic-Attree, I., and Vignais, P. M. (1993) Biochem. Biophys. Res. Commun. 196 416-421 [DOI] [PubMed] [Google Scholar]
- 30.Chuanchuen, R., Narasaki, C. T., and Schweizer, H. P. (2002) BioTechniques 33 760, 762-763 [DOI] [PubMed] [Google Scholar]
- 31.Dacheux, D., Goure, J., Chabert, J., Usson, Y., and Attree, I. (2001) Mol. Microbiol. 40 76-85 [DOI] [PubMed] [Google Scholar]
- 32.Dacheux, D., Toussaint, B., Richard, M., Brochier, G., Croize, J., and Attree, I. (2000) Infect. Immun. 68 2916-2924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DeLano, W. L. (2002) PyMol, DeLano Scientific, Palo Alto, CA
- 34.Espina, M., Ausar, S. F., Middaugh, C. R., Baxter, M. A., Picking, W. D., and Picking, W. L. (2007) Protein Sci. 16 704-714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nanao, M., Ricard-Blum, S., Di Guilmi, A. M., Lemaire, D., Lascoux, D., Chabert, J., Attree, I., and Dessen, A. (2003) BMC Microbiol. 3 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Derewenda, U., Mateja, A., Devedjiev, Y., Routzahn, K. M., Evdokimov, A. G., Derewenda, Z. S., and Waugh, D. S. (2004) Structure 12 301-306 [DOI] [PubMed] [Google Scholar]
- 37.Hamad, M. A., and Nilles, M. L. (2007) J. Bacteriol. 189 6734-6739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lawton, D. G., Longstaff, C., Wallace, B. A., Hill, J., Leary, S. E., Titball, R. W., and Brown, K. A. (2002) J. Biol. Chem. 277 38714-38722 [DOI] [PubMed] [Google Scholar]
- 39.Akeda, Y., and Galan, J. E. (2005) Nature 437 911-915 [DOI] [PubMed] [Google Scholar]
- 40.Arai, M., and Kuwajima, K. (2000) Adv. Protein. Chem. 53 209-282 [DOI] [PubMed] [Google Scholar]
- 41.Broz, P., Mueller, C. A., Muller, S. A., Philippsen, A., Sorg, I., Engel, A., and Cornelis, G. R. (2007) Mol. Microbiol. 65 1311-1320 [DOI] [PubMed] [Google Scholar]
- 42.Nilles, M. L., Williams, A. W., Skrzypek, E., and Straley, S. C. (1997) J. Bacteriol. 179 1307-1316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nilles, M. L. (2004) Structure 12 357-358 [DOI] [PubMed] [Google Scholar]
- 44.Cordes, F. S., Komoriya, K., Larquet, E., Yang, S., Egelman, E. H., Blocker, A., and Lea, S. M. (2003) J. Biol. Chem. 278 17103-17107 [DOI] [PubMed] [Google Scholar]
- 45.Akeda, Y., and Galan, J. E. (2004) J. Bacteriol. 186 2402-2412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gauthier, A., and Finlay, B. B. (2003) J. Bacteriol. 185 6747-6755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stebbins, C. E., and Galan, J. E. (2003) Nat. Rev. Mol. Cell. Biol. 4 738-743 [DOI] [PubMed] [Google Scholar]
- 48.Veenendaal, A. K., Hodgkinson, J. L., Schwarzer, L., Stabat, D., Zenk, S. F., and Blocker, A. J. (2007) Mol. Microbiol. 63 1719-1730 [DOI] [PubMed] [Google Scholar]
- 49.Broms, J. E., Sundin, C., Francis, M. S., and Forsberg, A. (2003) J. Infect. Dis. 188 239-249 [DOI] [PubMed] [Google Scholar]
- 50.Frithz-Lindsten, E., Holmstrom, A., Jacobsson, L., Soltani, M., Olsson, J., Rosqvist, R., and Forsberg, A. (1998) Mol. Microbiol. 29 1155-1165 [DOI] [PubMed] [Google Scholar]
- 51.Bennett, M. J., Choe, S., and Eisenberg, D. (1994) Proc. Natl. Acad. Sci. U. S. A. 91 3127-3131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rousseau, F., Schymkowitz, J. W., and Itzhaki, L. S. (2003) Structure 11 243-251 [DOI] [PubMed] [Google Scholar]
- 53.Johnson, S., Roversi, P., Espina, M., Olive, A., Deane, J. E., Birket, S., Field, T., Picking, W. D., Blocker, A. J., Galyov, E. E., Picking, W. L., and Lea, S. M. (2007) J. Biol. Chem. 282 4035-4044 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.