

Abstract
Human telomerase reverse transcriptase (hTERT) underlies cancer cell immortalization, and the expression of hTERT is regulated strictly at the gene transcription. Here, we report that transcription factor Ets2 is required for hTERT gene expression and breast cancer cell proliferation. Silencing Ets2 induces a decrease of hTERT gene expression and increase in human breast cancer cell death. Reconstitution with recombinant hTERT rescues the apoptosis induced by Ets2 depression. In vitro and in vivo analyses show that Ets2 binds to the EtsA and EtsB DNA motifs on the hTERT gene promoter. Mutation of either Ets2 binding site reduces the hTERT promoter transcriptional activity. Moreover, Ets2 forms a complex with c-Myc as demonstrated by co-immunoprecipitation and glutathione S-transferase pulldown assays. Immunological depletion of Ets2, or mutation of the EtsA DNA motif, disables c-Myc binding to the E-box, whereas removal of c-Myc or mutation of the E-box also compromises Ets2 binding to EtsA. Thus, hTERT gene expression is maintained by a mechanism involving Ets2 interactions with the c-Myc transcription factor and the hTERT gene promoter, a protein-DNA complex critical for hTERT gene expression and breast cancer cell proliferation.
The Ets protein family consists of a large number of evolutionarily conserved gene transcription factors. Each Ets member has an 85-amino acid sequence called the Ets domain that adopts the winged helix-turn-helix DNA binding structure with high affinity for the core consensus sequence of GGAA/T in a variety of genes. Although many Ets proteins are transcription factors, some function as repressors, and others are shown to be both activators and repressors (1–4). Frequently, Ets1 and Ets2 are highly expressed in a number of human malignancies (1, 3, 5, 6). Overexpression of Ets1 and Ets2 stimulates cell proliferation, anchorage-independent growth, and tumorigenicity in nude mice (7, 8). Targeted disruption of a single allele of the Ets2 gene limits the growth of breast tumors in transgenic mice (9), whereas homozygote deletion is embryonic lethal (10). These findings suggest that Ets2 plays an important role in the development of breast cancer.
The molecular mechanisms of action of Ets2 in tumorigenesis remain largely unknown. Ets2 is phosphorylated on threonine 72 by ERK1/2 and the phosphorylated form accumulates in the cell nucleus (11). Stimulation of VEGFR2, ErbB2, or estrogen receptors results in activation of ERK1/25 kinases and phosphorylation of Ets2 in human breast cancer cells (12, 13). Several downstream target genes of Ets2 have been implicated as mediating Ets2-induced oncogenesis. These include Bcl-2, cyclin D1, and Bcl-XL in prostate cancer (14, 15). Recently, telomerase reverse transcriptase (hTERT) has been suggested to be a target gene of the Ets family (reviewed Ref. 16). However, controversy remains as to whether Ets2 is a transcription factor (17) or repressor (18, 19). Maida et al. showed that mutation of a putative Ets binding site proximal to the transcription site (–23TTCCTT–18) on the hTERT gene promoter inhibits, and overexpression of Ets2 stimulates, the recombinant hTERT gene promoter activity in vulvar cancer A431 cells (17). Xiao et al. (18, 19) showed that both Ets1 and Ets2 bind to the hTERT gene promoter and overexpression of either Ets1 or Ets2 inhibits telomerase activity, an effect independent of gene expression of c-Myc and Sp1 in human myeloid erythroleukemia K562 cells.
The regulation of hTERT gene transcription is an important step toward telomerase activation, maintenance of telomeres (ends of chromosomes), and continuous cell proliferation. This is particularly critical in cancer cells where telomeres are critically short in supporting constant cell proliferation. Previous studies show that c-myc is a primary transcription factor of the hTERT gene. Binding the E-box on the hTERT gene promoter, c-myc, stimulates hTERT gene promoter activity (16, 20). Myc regulates gene expression by binding cognate DNA sequences known as E-box (CACGTG). Although other transcriptional factors such as Ets1, Ets2, and Sp1 are also implicated as playing a role in regulating the hTERT gene, little is known of and how these transcription factors cooperate.
To establish the role of Ets1 and Ets2 in regulating telomerase activity, we chose to silence each of the Ets1 and Ets2 genes in breast cancer cells in which Ets2 is expressed at high levels (6), to observe their effects on hTERT gene expression and breast cancer cell proliferation. We found that knockdown of Ets1 has no effect on telomerase activity, but knockdown of Ets2 significantly disrupts hTERT gene expression and causes breast cancer cell death. Reconstitution with recombinant hTERT rescues the Ets2 knockdown-induced cell death. Mechanistic studies showed that Ets2 binds to the hTERT gene at two discrete Ets-binding sites of the gene promoter. Mutation of either site reduces the hTERT gene promoter activity. In addition, silencing Ets2 entrains a decreased gene expression of c-Myc, but reconstituted c-Myc in the Ets2 knockdown cells does not restore hTERT activity. Furthermore, Ets2 forms a complex with c-Myc regulating c-Myc binding to the E-box. Thus, the hTERT gene is stimulated with a cooperative interaction between Ets2 and c-Myc in their respective bindings to their individual binding sites on the hTERT gene promoter, which is critically required for telomerase activity and hTERT-dependent proliferation of human breast cancer cells.
EXPERIMENTAL PROCEDURES
Cell Culture—The human breast cancer MCF-7 cells were obtained from American Type Culture Collection (Rockville, MD) and grown in Dulbecco's modified Eagle's medium. Cell culture media were supplemented with 10% heat-inactivated fetal calf serum, penicillin (100 units/ml), and streptomycin (100 μg/ml) at 37 °C in a humidified 5% CO2 atmosphere. Cells were seeded in 24-well plastic plates, 6–10-cm dishes or eight-chamber glass slides (Nunc, Naperville, CT).
RNA Interference (RNAi) Constructs, Synthetic siRNAs, and Gene Silencing—For generation of Ets1- and Ets2-specific RNAi, specifically annealed oligonucleotides were cloned individually to the BamHI and BbsI sites immediately downstream of the initiating “G” of the U6 promoter. The annealed oligonucleotides used are listed below: Ets2a (5′-tttgtcttgtggatgatgttcttgaagcttgaagaacatcatccacaagactttttt-3′ and 5′-gatcaaaaaagtcttgtggatgatgttcttcaagcttcaagaacatcatccacaaga-3′), Ets2b (5′-tttgcacaggctttaattgtaaagaagcttgtttacaattaaagcctgtgctttttt-3′ and 5′-gatcaaaaaagcacaggctttaattgtaaacaagcttcaagcttgtttacaattaaagcctgtgc-3′), and Ets1 (5′-tttggcagaattcagtgaatcatcggaagcttgcgatgattcactgaattctgctttttt-3′ and 5′-gatcaaaaaagcagaattcagtgaatcatcgcaagcttccgatgattcactgaattctgc-3′). Correctly integrated clones were identified by HindIII digestion and verified by sequencing. The DNA fragment containing the U6 promoter and Ets2-specific hairpin was prepared by amplification with T7 and SP6 and gel purification. 500 ng of U6-Ets2RNAi DNA was co-transfected with 50 ng of a PGK-puromycin into MCF-7 cells using FuGENE 6 (Roche). After 24 h 25 μg/ml puromycin was added to the cultures and after 7 days of selection, cells were pooled and expanded for analysis. For transient gene silencing, synthetic small interference RNA (siRNA) targeting Ets1 and Ets2 were used. The siRNAs were purchased from Ambion.
Gene Expressions of Ets2, c-Myc, and hTERT—Wild type hTERT gene expression plasmid was prepared by subcloning hTERT full-length cDNA into the pcDNA3HA plasmid as described previously (21). The c-Myc gene expression plasmid was generated by inserting full-length c-Myc cDNA downstream of the EF-1α promoter. The gene expression plasmids were transfected into Ets2 knockdown clones (clone 2–15) using FuGENE 6, e.g. 20 μg of pEFBOS-c-Myc (or pEFBOS 1 FLAG) and a puromycin resistance clone. Individual clones were isolated and screened by PCR. Other Ets2 knockdown cell clones (clones 17–34) were also transfected in mass cell cultures. The transfection of hTERT was confirmed by telomerase activity assay and c-Myc expression was confirmed by Western blotting. The cell line and multiple clones displayed various levels of c-Myc protein; however, there was no difference between their growths (data not shown).
Real-time RT-PCR Analysis—Single-stranded cDNA was generated using 1 μg of total RNA as template and avian myeloblastosis virus reverse transcriptase (Promega), as per the manufacturer's instructions. A ⅕ dilution of the resulting cDNA was used for real-time RT-PCR of 45 cycles, using a Light Cycler (Roche, version 3.5 software). Fast start SYBR Green Master Mix (Roche) was used, with 10 pmol of each primer in 10-μl reactions. Primers specific for different genes are: hTERT (5′-CCACCTTGACAAAGTACAG-3′ and 5′-CGTCCAGACTCCGCTTCAT-3′) and human glyceraldehyde-3-phosphate dehydrogenase (5′-GAGAGACCCTCACTGCTG-3′ and 5′-GTAGGTATATGACAAGGTG-3′). Following gel electrophoresis on 2% agarose-TAE gel, the size and purity of each PCR product were analyzed. Each DNA sequence was subjected to BLAST analysis to confirm that the PCR represented the correct gene. All samples were subjected to PCR with glyceraldehyde-3-phosphate dehydrogenase as a control. To create a standard curve, five 10-fold dilutions of plasmid DNA were used as template (10–5–10–9 ng/μl). Results represent an average of three independent experiments.
Gene Transfection and Luciferase Gene Reporter Assay—The Ets consensus sites (EtsA and -B) were mutated with the QuikChange site-directed mutagenesis kit (Stratagene) in the hTERT luciferase reporter (pTERT-luc330, gift from Silvia Bacchetti and Yu-sheng Cong). The mutagenesis primers used for EtsA were 5′-CCC AGG ACC GCG CTT CTC ACG TGG CGG AGG G-3′ and 5′-CCC TCC GCC ACG TGA GAA GCG CGG TCC TGG G-3, and for EtsB were 5′-CAG CCC CTC CCC TTC TTT TCC GCG GCC CCG-3′ and 5′-CGG GGC CGC GGA AAA GAA GGG GAG GGG CTG-3′ (underlined were putative Ets binding sites and italic were the mutations). All the expression plasmids, wild type and mutants, were verified by DNA sequencing before use. All plasmids used in the transfection assay were prepared with the endotoxin-free plasmid Maxi-kit (Qiagen) and resuspended with endotoxin-free 0.1× Tris/EDTA buffer to a concentration of 1 μg/μl. MCF-7 cells (2 × 105) were placed in 1 ml of medium in 24-well tissue culture plates and incubated overnight. Next day, the wild type or mutant hTERT promoter reporter gene vector (1 μg) along with 0.2 μgof α-galactosidase expression vector were transfected into MCF-7 cells with FuGENE 6 (Roche). In 24 h of transfection, the cells were harvested, washed in PBS, and lysed in cell lysis buffer (Promega). Luciferase activity was measured with luciferase substrate purchased from Promega according to the procedures of the Luciferase Assay System (Promega), and readout in opaque 96-well plates using a plate reading luminometer. β-Galactosidase was used to normalize transfection efficiency. Experiments were performed in triplicate.
Electrophoretic Mobility Shift Assay—MCF-7 cell nuclear extract was incubated with 10,000 cpm of a 22-bp DNA oligonucleotide containing the EtsA and -B consensus sequence (wild type EtsA, 5′-GACCGCGCTTCCCACGTGGC-3′, mutant EtsA, 5′-GACCGCGCTTCTCACGTGGC-3′; wild type EtsB, 5′-CCCTCCCCTTCCTTTCCGCG-3′, and mutant EtsB, 5′-CCCTCCCCTTCTTTTCCGCG-3′). Probes were labeled with [γ-32P]dATP (Amersham Biosciences). Incubations were performed at room temperature for 30 min in the presence of 2 μg of poly(dI-dC) as nonspecific competitor and 10 mm Tris-HCl, pH 7.5, containing 100 mm NaCl, 1 mm EDTA, 5 mm dithiothreitol, 4% glycerol, and 100 μg/ml nuclease-free bovine serum albumin. For competition studies, unlabeled wild-type or mutant Ets oligonucleotides were added to the binding reaction 30 min before the addition of the radiolabeled probe. Specific Ets1 or Ets2 antibody (0.5 μl) was incubated with nuclear extracts for 20 min on ice before the binding reaction. All incubation mixtures were subjected to electrophoresis on native 5% (w/v) polyacrylamide gels, which were subsequently dried and subjected to autoradiography.
Chromatin Immunoprecipitation (ChIP) Assays—ChIP assays were performed using the ChIP Assay Kit (Upstate, Lake Placid, NY) according to the manufacturer's protocol. Briefly, MCF-7 cells (1 × 107) were fixed with formaldehyde (final concentration, 1%) for 10 min at room temperature. After washing with phosphate-buffered saline, cells were pelleted and resuspended in SDS lysis buffer (1% SDS, 10 mm EDTA, 50 mm Tris-HCl, pH 8.1, 1 mm dithiothreitol, 1 mm phenylmethylsulfonyl fluoride). The lysates were sonicated to generate the DNA fragments, diluted with dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mm EDTA, 16.7 mm Tris-HCl, pH 8.1, 167 mm NaCl), and pre-cleared by incubation with a salmon sperm DNA, protein A,agarose 50% slurry for 60 min at 4 °C. The supernatant was incubated with anti-Ets1, anti-Ets2, anti-acetylhistone H3 antibodies or IgG at 4 °C overnight. Immunocomplexes were collected with a salmon sperm DNA, protein A, agarose, 50% slurry, heated at 65 °C to reverse cross-linkage, and treated with 40 μg/ml proteinase K at 45 °C for 60 min. DNA was recovered by phenol/chloroform/ethanol precipitation and used as a template for PCR to amplify the proximal region containing the Ets site of the hTERT promoter or the distal region void of the Ets site as control. The primer pairs for the proximal region were 5′-GGC CGG GCT CCC AGT GGA TTC-3′ and 5′-CAG CGG GGA GCG CGC GGC ATC G-3′, and the primer pairs for the distal region were 5′-GGCAGGCACGAGTGATTTTA-3′ and 5′-CTGAGGCACGAGAATTGCTT-3. Primer pairs for PTHrP control were 5′-TGCCTCGAGCGTGTGAACA-3′ and 5-TCCCATAGCAATGTCTAATTAATCTGG-3. The PCR conditions were 30 s at 94 °C, 30 s at 58 °C, and 30 s at 72 °C for 35 cycles. The PCR products were resolved on a 7% polyacrylamide gel and stained with SYBR Gold (Molecular Probes, Eugene, OR).
GST Fusion Proteins—GST, GST-Ets1, and GST-Ets2 fusion proteins were produced in BL21 Escherichia coli under induction by isopropyl 1-thio-β-d-galactopyranoside at room temperature. Proteins were purified by affinity absorption using glutathione-Sepharose 4B beads. The recombinant GST, GST-Ets1, and GST-Ets2 proteins on the glutathione beads were incubated with nuclear protein extracts or total cell lysates of breast cancer MCF-7 cells at 4 °C for 1 h followed by extensive washing. Proteins on the beads were resolved on 8% SDS-PAGE and visualized by immunoblotting with anti-myc antibody and anti-GST antibodies.
Immunoprecipitation and Western Blotting Analysis—For immunoprecipitation, cells were lysed with triple detergent lysis buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 0.02% sodium azide, 0.1% SDS, 100 μg/ml phenylmethylsulfonyl fluoride, 1 μg/ml aprotinin, 1% Nonidet P-40, 0.5% sodium deoxycholate). Cell lysates (∼1 mg) were incubated with antibodies as indicated in individual experiments. Antibody complexes were captured using protein A-agarose beads (GE Healthcare) and washed three times with PBS. The beads carrying the immune complex were boiled in SDS sample buffer and analyzed by Western blotting. For total nuclear protein extraction, 1 × 106 cells were collected in cold PBS and resuspended in RIPA buffer (150 mm sodium chloride, 50 mm Tris-HCl, pH 7.4, 1 mm EDTA, 1 mm phenylmethylsulfonyl fluoride, 1% Triton X-100, 1% sodium deoxycholic acid, 0.1% SDS, 5 μg/ml aprotinin, 5 μg/ml leupeptin). Protein concentration was determined using a Bio-Rad protein assay. Proteins were separated by 10% SDS-PAGE and transferred onto nitrocellulose membrane by electroblotting. The nitrocellulose membranes were blocked at 4 °C overnight in 10% (w/v) nonfat milk (0.2% fetal calf serum, 0.05% Tween 20 in 1× PBS) and incubated at room temperature for 1 h with antibodies (Santa Cruz Biotechnology), followed by appropriate horseradish peroxidase-conjugated secondary antibody (diluted 1:1000, DAKO Australia). SuperSignal West PICO (Calbiochem) chemiluminescent substrate kit was used to detect and visualize protein antigens after exposure to BioMax autoradiographic film (Kodak). Alternatively x-ray film or Odyssey was used for autoradiography. The autoradiograph films were scanned and the bands quantified on a PhosphorImager (Fujifilm FLA-2000, Berthold).
Immunofluorescence—MCF-7 cells (1 × 105) were seeded in Chamber slides (Lab-Tek II, Nalge Nunc International) containing Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated fetal calf serum. Cells at 60–80% confluence were washed with PBS, starved by addition of serum-free Dulbecco's modified Eagle's medium for 24 h, followed by fixation with 4% paraformaldehyde in PBS for 10 min and blocking with CAS blocking solution (Zymed Laboratories Inc.) at room temperature for 30 min. The cells were incubated with the Ets2 polyclonal (Santa Cruz) or hTERT primary antibody at room temperature for 1 h, washed three times with PBS, 0.01% Triton X-100 for 5 min each, and incubated at room temperature for 1 h with fluorescein isothiocyanate-conjugated anti-rabbit secondary antibodies (Molecular Probes). For staining of the nuclei, cells were stained with 50 ng/ml Hoechst (Sigma) at room temperature for 10 min. Slides were washed with PBS, Triton X-100, mounted in anti-fade medium (Bio-Rad), and analyzed by fluorescence microscopy (Leica Instruments).
Telomerase Activity Assay Analysis—Atelomeric repeat amplification protocol was employed to determine telomerase activity essentially as described previously (22). Briefly, cells treated with different reagents were washed and lysed by detaching and passing the cells though a 26½-gauge needle attached to a 1-ml syringe in pre-chilled telomeric repeat amplification protocol lysis buffer (0.5% CHAPS, 10 mm Tris-HCl, pH 7.5, 1 mm MgCl2, 63 mm KCl, 0.05% Tween 20, 1 mm EDTA, 10% glycerol, 5 mm β-mercaptoethanol and mixture protease inhibitors). The nuclei were isolated by centrifugation and protein content determined. Equal amounts of nuclear telomerase extracts (0.4 μg) were incubated with telomeric DNA substrate and dNTP, and de novo synthesized telomeric DNA, with or without phenol and chloroform extraction, was amplified by PCR using specific telomeric DNA primers in the presence of [α-32P]ATP (Amersham Biosciences) and Taq DNA polymerase. The resultant 32P-labeled telomeric DNA ladders were resolved by polyacrylamide slab gel electrophoresis followed by autoradiography. To monitor nonspecific PCR effects, additional primers were included: NT (ATCGCTTCTCGGCCTTTT) and TSNT (AATCCGTCGAGCAGAGTTAAAAGGCCGAGAAGCGAT). Negative controls that were treated with either RNase A or alkaline phosphatase to inactivate telomerase were included in each experiment.
Apoptosis and the Cell Cycle Analysis—Apoptosis and the cell cycle analysis were performed as described (23). Briefly, both adherent and floating cells were fixed with 80% cold ethanol, pelleted by centrifugation, stained with propidium iodide and analyzed using an EPICS 752 flow cytometer. Cell debris, doublets, and fixation artifacts were gated out, and G0-G1, S, G2-M, and apoptotic populations recorded on a logarithmic scale. Apoptotic cells were identified as the lower fluorescence peak (“sub-G1 peak” on DNA frequency histograms) due to their reduced DNA content or detected with Apoptaq TUNEL kit (Oncor Inc., Gaithersburg, MD). Alternatively, apoptotic cells were stained with Annexin V-FLUOS conjugate (Roche Diagnostics) and propidium iodide (Sigma) at room temperature for 15 min in an incubation buffer that facilitates binding per the manufacturer's instructions. Analyzed for Annexin V and propidium iodide staining in FL-2 and FL-3 channels, respectively, the percentages of stained cells were determined using a FACSCalibur flow cytometer (BD Biosciences). An acquisition gate was set to include ∼20,000 of the centrally located cells for each sample acquisition using linear forward scatter versus linear side scatter. This acquisition strategy resulted in ∼40,000 ungated events being included for each sample analysis. Dot-plot integration was determinate from the background fluorescence using unstained cells. This integration cursor placement remained unchanged when stained cells were analyzed.
RESULTS
Requirement of Ets2 for Telomerase Activity and hTERT Gene Expression in Breast Cancer Cells—To investigate the role of Ets1 and Ets2 in regulating telomerase, we selectively silenced each of the Ets1 and Ets2 genes using small hairpin RNAs (shRNA) or synthetic siRNA targeting Ets1 and Ets2 mRNA specifically. MCF-7 cells were transfected stably with Ets1 shRNA construct, Ets2 shRNA constructs, or transiently with siRNA for 48 h. Whereas silencing Ets1 had no significant effect on telomerase activity, silencing Ets2 induced a marked inhibition of telomerase activity to 30–40% of the controls (Fig. 1A). Immunofluorescence staining of the telomerase catalytic subunit hTERT showed significant reduction of hTERT in the nucleus of Ets2-silenced cells (Fig. 1B). Consistent with the reduced telomerase activity and hTERT immunoreactivity, hTERT gene expression was decreased by about 75% in Ets2-silenced cells compared with controls expressing GFP or Ets1 shRNA (Fig. 1C). Western blotting confirmed a significant reduction of the Ets1 and Ets2 immunoreactivities induced by Ets1 and Ets2 RNAi, respectively (Fig. 1D). Thus, silencing the Ets2 gene triggered an inhibition of hTERT gene expression and telomerase activity in breast cancer MCF-7 cells.
FIGURE 1.
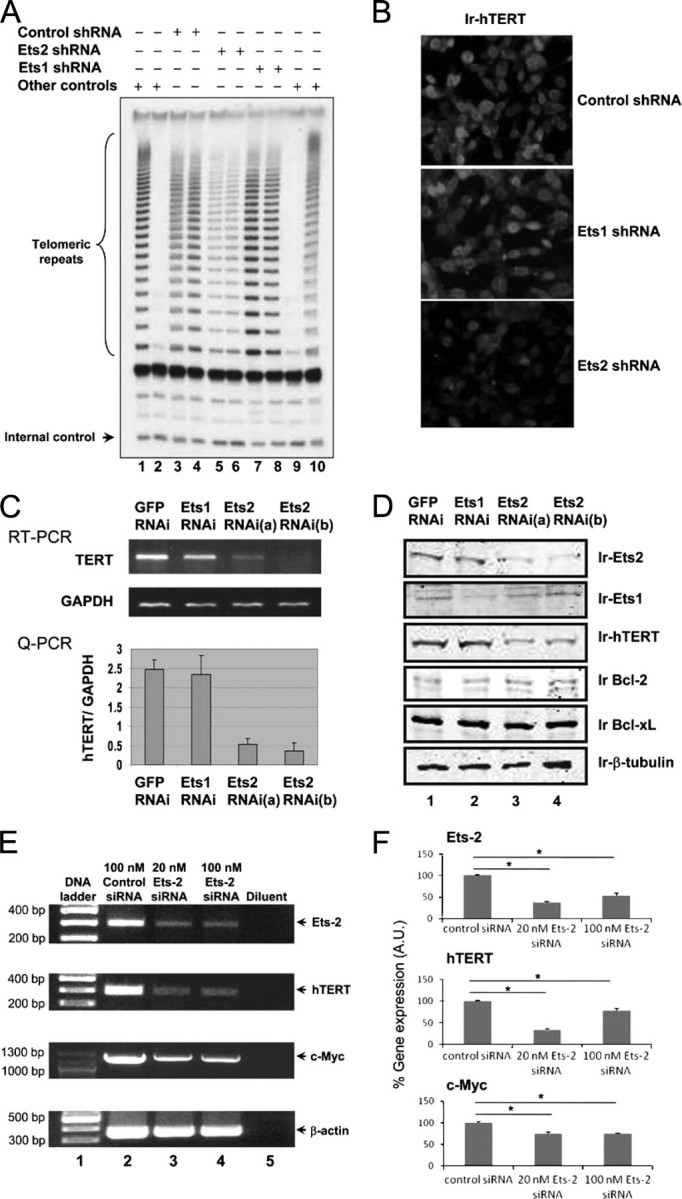
Silencing Ets2 but not Ets1 decreases gene expression of hTERT and telomerase activity in human breast cancer MCF-7 cells. A, effects of silencing Ets1 or Ets2 on telomerase activity in MCF-7 cells. Telomerase activity was determined by measuring de novo synthesis of telomeric DNA as described under “Experimental Procedures.” Cells were transfected in duplicate by Ets1 and Ets2 shRNA as indicated. Lanes 1 and 10 were telomerase-positive controls and lanes 2 and 9 were telomerase-negative controls. Internal control for amplification and DNA loading is indicated. B, immunofluorescence staining of hTERT in MCF-7 cells transfected with different shRNA as indicated. The microimages were recorded by immunofluorescent microscopy following incubation of the fixed cells with anti-hTERT antibodies and Cy3-coupled second antibodies. C, effects of silencing Ets2 and Ets1 on the gene expressions of hTERT. MCF-7 cells were transfected with the shRNA expression plasmids against Ets1 or Ets2 as indicated. Two Ets2 shRNA expression plasmids were used, and the GFP shRNA was used as control. In 48 h of siRNA plasmid transfection, total RNA was isolated from the MCF-7 cells and subjected to quantitative RT-PCR using specific primers of the hTERT gene. The quantitative PCR data are mean ± S.D. of three determinations. D, effects of shRNA expression constructs on gene expression determined by Western blotting. Cell lysates obtained from cells transfected with different shRNA for 48 h were probed with specific antibodies as indicated. Immunoreactive (Ir) Ets2, Ets1, hTERT, Bcl-2, Bcl-xl, and β-tubulin are indicated. E and F, RT-PCR determination of hTERT gene expression in cells transiently transfected with synthetic siRNA. Cells were transfected with the siRNA indicated for 48 h. Data are mean ± S.D. from three independent experiments. Asterisk indicates a significant difference with p < 0.05.
To explore the specificity and mechanisms of hTERT gene repression induced by Ets2 gene silencing, we examined Bcl-2 and Bcl-xL that were shown to be downstream target genes of Ets2 (14). No significant change of the Bcl-2 or Bcl-xL gene expression was observed by Western blotting using specific antibodies in the same experiment where decreased gene expressions of Ets2 and hTERT were observed (Fig. 1D). However, we found that the proto-oncogene c-Myc was reduced significantly (Fig. 1E). The decrease of c-Myc was about 25% of controls, versus ∼70% decrease of the hTERT gene expression (Fig. 1F). The data suggest that hTERT and c-Myc are downstream target genes of Ets2, and that the hTERT gene is regulated by Ets2 directly and indirectly via c-Myc in human breast cancer cells.
Ets2 Gene Silencing, hTERT Gene Repression, and Breast Cancer Cell Death—Because little is known of the cellular consequence of Ets regulation of telomerase activity, we determined the effect of gene silencing of Ets2 on breast cancer cell death by measuring Annexin V and propidium iodide staining-positive cells using fluorescence-activated cell sorting (FACS) analysis. As shown in Fig. 2A, 48 h post-transfection of MCF-7 cells with different shRNA plasmids, there was a significant inhibition of cell proliferation in cell cultures transfected with Ets2 shRNA, whereas control cells underwent exponential cell population doublings. The reduction of cell numbers in the Ets2-transfected cells was by about 70–75% of the controls transfected with GFP shRNA (Fig. 2A). Significant floating cells were noted under the microscope and staining of the attached cells with crystal violet blue showed a significant cell loss (not shown). DNA damage in the apoptotic cells was confirmed by staining with ApopTaq in the Ets2, but not Ets1, silenced breast cancer cells (Fig. 2B). FACS analysis showed a significant increase in Annexin V and propidium iodide staining double-positive apoptotic cells in the cell culture transfected with Ets2 shRNA, when compared with that in control cells or cells transfected with Ets1 siRNA (Fig. 2C).
FIGURE 2.

Silencing Ets2 induces telomerase inhibition-dependent breast cancer cell death. A, effects of silencing Ets1 and Ets2 on breast cancer cell proliferation. MCF-7 cells at 1 × 105 cells per well in 24-well plates were transfected with four different shRNA expression plasmids individually as indicated and cell numbers were monitored on each day of the transfection for 3 days. The results were mean ± S.D. of three determinations of cell numbers from three experiments. B, staining of apoptotic cells in MCF-7 cell cultures. Cells were transfected for 48 h with different shRNA expression plasmids as indicated and labeled with ApopTaq for DNA breaks. Nuclear total DNA was stained with Hoechst. Micrographs were from fluorescence microscopy at ×10 magnification. C, effect of silencing Ets1 or Ets2 by specific siRNAs on apoptosis. Cells were stained with Annexin V and propidium iodide, and analyzed by FACS. Annexin V and propidium iodide double positive cells in the upper right panel of B were expressed as a percentage of total cells. D, re-establishment of hTERT gene expression in Ets2-silenced cells. Cells with Ets2 gene silencing constructs were transfected with HA-hTERT for 48 h. Cell lysates were probed with specific antibodies for Ets2 and hTERT with β-tubulin as control. E, effects of hTERT overexpression and Ets2 transduction on apoptosis induced by silencing Ets2. MCF-7 cells stably transfected with the different shRNA expression plasmids as indicated were transduced with hTERT gene expression plasmids, Ets2 fusion protein, or GST control for 24 h. Cells were stained with propidium iodide followed by FACS. The percentages of cells with a hypodiploid (sub-2N) DNA content indicative of apoptosis are shown. F, effects of Ets2 siRNA on apoptosis in cultured cells expressing hTERT. Cells with or without recombinant GFP-hTERT or GFP only were transfected with Ets2 siRNA for 48 h followed by FACS. Data are representatives of three experiments.
Thus, silencing Ets2 is paralleled by the fall of hTERT gene expression (Fig. 1) and the rise of apoptosis in cultured human breast cancer cells. To determine whether Ets2 silencing-induced cell death is mediated by the inhibition of telomerase activity, we overexpressed recombinant hTERT by transfecting the Ets2 knockdown breast cancer cells with the CMV-FLAG-hTERT gene expression plasmid and determined if the constitutive expression of hTERT rescues Ets2 silencing-induced cell death. Forty-eight h following the gene expression of hTERT, reconstituted telomerase activity was established (not shown) and recombinant hTERT protein was confirmed in the Ets2-silenced cells (Fig. 2D). Examination of apoptosis under different conditions showed that overexpression of hTERT markedly reduced the cell death in the cell cultures where Ets2 was knocked down (Fig. 2, E and F). The Ets2 shRNA-induced cell death was ∼46% without hTERT gene expression but ∼13% with the established hTERT overexpression (Fig. 2E), and the Ets2 siRNA-induced cell death was 17 versus 11% in the control or in the presence of recombinant hTERT (Fig. 2F). Thus, overexpression of hTERT reduced levels of apoptosis induced by Ets2 shRNA and rescued Ets2 siRNA-induced apoptosis, suggesting that telomerase inhibition plays a significant part in the increased apoptosis induced by silencing the Ets2 gene.
To confirm the importance of Ets2 gene expression in regulating the hTERT gene for breast cancer cell survival and proliferation, we determined the effects of silencing the Ets2 gene on hTERT gene expression, telomerase activity, and apoptotic cell death in the human breast cancer PMC42 and cervical cancer HeLa cell cultures. As shown in Fig. 3A, whereas silencing hTERT with hTERT siRNA induced an increased PMC42 cell death, silencing Ets2 caused no increase in HeLa cell death. Moreover, both Ets2 siRNA and hTERT siRNA induced increased cell death in PMC42 cell cultures (Fig. 3B). Consistently, Ets2 gene silencing was accompanied by a significant down-regulation of hTERT gene expression and telomerase activity in PMC42 cells (Fig. 3, C and D) but not in HeLa cells (data not shown). To investigate the apoptotic effect selectively on breast cancer cells of Ets2 silencing (Fig. 3E), we examined if Ets2 silencing-induced cell death in PMC42 cell cultures is also dependent on hTERT gene inhibition. PMC42 cells were co-transfected with the Ets2 siRNA and hTERT gene expression plasmid. In the presence of hTERT gene expression as shown in Fig. 3, C and D, Ets2 siRNA did not induce a significant increase in PMC42 cell death (Fig. 3F). The data suggest that Ets2 is critical in telomerase activity and cell proliferation of human breast cancer PMC42 cells with specificity and that silencing Ets2 induces telomerase inhibition-dependent breast cancer cell death.
FIGURE 3.
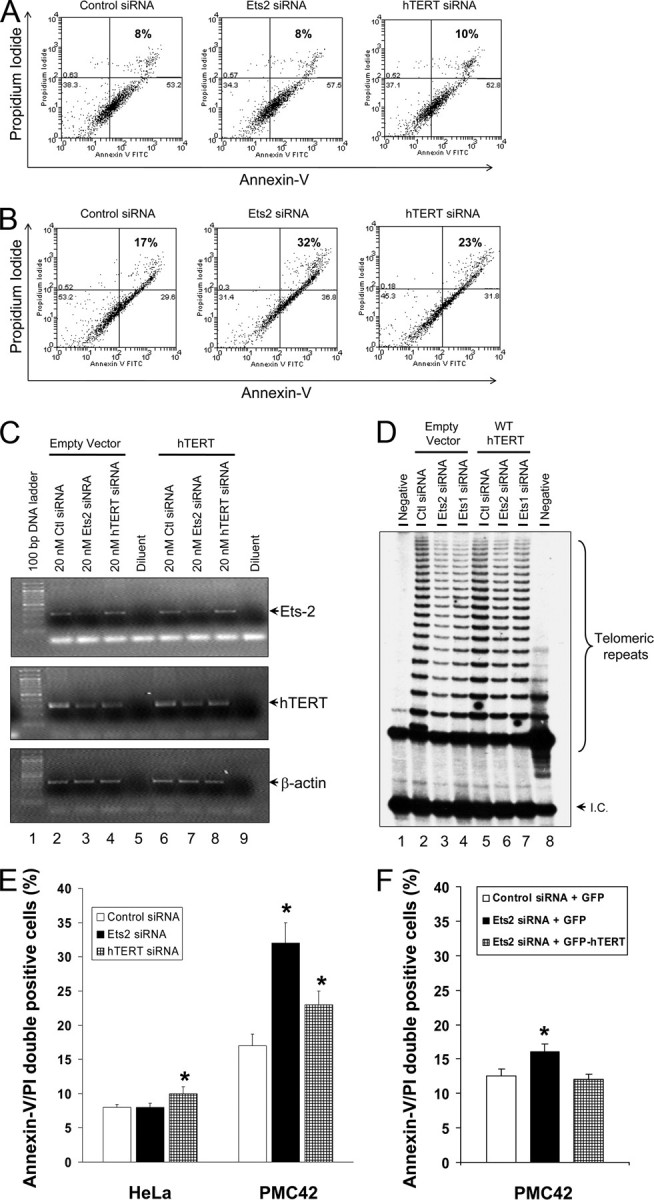
Silencing Ets2 but not Ets1 decreases gene expression of hTERT and telomerase activity, and increases apoptosis in human breast cancer PMC42 cells. A, effect of silencing Ets2 and hTERT gene expression by specific siRNAs on cervical cancer HeLa cell death. Cells were transfected with specific siRNA for 48 h followed by staining with Annexin V and propidium iodide for FACS analysis. B, effect of silencing Ets2 and hTERT gene expression by specific siRNAs on PMC42 breast cancer cell death. Cells were transfected with specific siRNA for 48 h followed by staining with Annexin V and propidium iodide for FACS analysis. C, gene expression analysis of Ets2 and hTERT in PMC42 cells transfected with Ets2 or hTERT siRNA plus or minus hTERT gene expression plasmid. Cells were co-transfected for 48 h as indicated and analyzed for mRNA by RT-PCR with specific primers. D, telomerase activity assayed by telomeric repeat amplification protocol as described under “Experimental Procedures” in cellular lysates from the same experiments as in panel C. E, effects of silencing Ets2 on HeLa and PMC42 cell death. Experiments were carried as in panels A and B. Data are mean ± S.D. from three similar experiments. F, effect of hTERT gene expression on Ets2 siRNA-induced PMC42 cell death. Cells were co-transfected with Ets2 siRNA and hTERT gene expression plasmid for 48 h followed by FACS analysis of apoptosis. Data are mean ± S.D. of three similar experiments. Asterisks indicate significant differences in comparison with control with p < 0.01.
Ets2 Binding and Stimulating the hTERT Gene Promoter—With the vital connection between Ets2 transcription factor, the hTERT gene, and breast cell proliferation, we further examined the action of Ets2 on the hTERT gene interface of Ets2 binding sites. Inspection of the hTERT gene as well as the genes coding for rat TERT and mouse TERT revealed two consensus DNA elements (the EtsA and EtsB) that are conserved in the proximal core promoters of the TERT genes potentially for Ets2 binding (Fig. 4A). For the human gene of TERT, both EtsA (–243CCTT–246) and EtsB (–96CCTT–99) appear to localize within the unmethylated transcriptional active regulatory regions (24). To determine whether the putative Ets sites at –243 and –96 bp are each required for Ets2 binding, we created single nucleotide mutations for the aCTT sequence (instead of the wild type CCTT motif) in the 300-bp hTERT proximal core promoter upstream of a luciferase reporter gene (Fig. 4A). The mutant luciferase reporter gene expression constructs were then introduced into MCF-7 cells by transient transfection, with wild type and empty plasmids as parallel controls. Significantly, mutation of either EtsA or EtsB in the hTERT gene promoter resulted in a marked reduction of the hTERT gene promoter activity. The mutant promoters showed gene transcriptional activity of only ∼30% of the controls, an inhibition that was comparable with a positive control with mutation in the E-box (Fig. 4B). Thus, our data confirmed that EtsB is important for hTERT gene transcription as reported previously (17) and revealed that the EtsA site adjacent to an E-box is also indispensable for the hTERT gene promoter activity.
FIGURE 4.

Effect of hTERT promoter mutation on Ets2 binding to the hTERT gene promoter. A, schematic of the proximal hTERT gene promoter highlighting the positions of the EtsA and EtsB binding sites in relation to the E-box. Shown is also an alignment of the EtsA and E-box sites between human and rodent TERT promoters. B, effects of the gene mutations on the hTERT gene promoter activity. The putative Ets binding sites on the hTERT gene promoter were mutated as indicated in the recombinant hTERT gene promoter upstream of a luciferase reporter gene by site-directed mutagenesis. MCF-7 cells were transfected with the hTERT gene promoter wild type, or mutants carrying a mutation at the E-box, EtsA, or EtsB sites as indicated for 24 h. Cell luciferase activity was determined relative to a β-galactosidase control. The c-Myc and two Ets binding site (EtsA and -B) are underlined and mutated nucleotides are bold. C and D, effects of silencing Ets transcription factors on the hTERT gene promoter activity. MCF-7 cells were transfected for 24 h with the gene expression plasmids coding the individual shRNAs as indicated (C), or the Ets1, Ets2, PEA3, or ELF5 transcription factors as indicated (D). E and F, direct binding of Ets2 to the hTERT gene promoter EtsA (E) and EtsB (F) sites in vitro. Nuclear extracts of MCF-7 cells were incubated with [γ-32P]dATP-labeled hTERT promoter nucleotide probes containing wild type EtsA and E-box elements, or the EtsA mutation, in the presence or absence of Ets2 antibodies or 5-fold excess amounts of unlabeled probe competitors. The wild type and mutant forms of the EtsB probes were similarly incubated with the nuclear extracts. Binding of Ets2 to the hTERT promoter DNA was resolved by non-denatured acrylamide gel electrophoresis. G, Ets2 binding to the hTERT gene promoter in vivo by ChIP. MCF-7 cell lysates were incubated with different antibodies and the immune complexes were analyzed for hTERT gene DNA analysis by PCR using specific primers for the proximal (top panel) and distal (middle) panel of the hTERT gene promoter DNA. Acetylhistone H3 antibodies were used as positive control. The data are from one of three typical experiments. H, Ets2 binding to the PTHrP gene promoter serving as positive controls. Indicated antibodies were incubated with MCF-7 cell lysates and the immune complexes were analyzed for PTHrP gene promoter DNA by PCR.
With the EtsA and -B sites potentially required for Ets2 binding, we next co-transfected MCF-7 cells with Ets2 shRNA and the hTERT promoter luciferase reporter gene, and determined if silencing Ets2 results in hTERT gene promoter inhibition. We found that Ets2 shRNA caused a significant decrease of the hTERT gene promoter activity, but Ets1 shRNA had no effect on the hTERT promoter activity (Fig. 4C). To attest a specific causal effect of Ets2 to the hTERT gene, we transduced recombinant Ets2 into the MCF-7 cells where the endogenous Ets2 gene has been silenced, asking if the exogenous recombinant Ets2 stimulates the hTERT promoter activity. Indeed, we found that reconstitution of Ets2 led to an improved hTERT promoter activity, with activity returned to 70% of the controls following the Ets2 protein transduction (Fig. 4C). Thus, Ets2 alone appeared to be sufficient to up-regulate the hTERT gene promoter activity, supporting a direct action of Ets2 on the hTERT gene. To determine whether the hTERT promoter is stimulated by overexpression of Ets2, we transduced the recombinant Ets2 protein or Ets2 gene expression plasmids in MCF-7 cells without silencing the Ets2 gene and found that neither recombinant Ets2 protein (not shown) nor Ets2 gene overexpression plasmids altered the hTERT gene promoter activity (Fig. 4D). Overexpression of Ets1, PEA3, or ELF5 in the MCF-7 cells resulted in no significant change in the hTERT gene promoter activity either (Fig. 4D). These data suggest that in the MCF-7 cells, the hTERT gene promoter is regulated to a maximal level by endogenous Ets2 with no additive or synergistic effect from PEA3 (17, 25) or competitive inhibition from Ets1 (18).
To establish Ets2 binding to the canonical EtsA and EtsB elements on the hTERT gene promoter, we carried out the electrophoretic mobility shift assay using the sequences flanking EtsA and EtsB sites separately, and the ChIP analysis using specific antibodies. Incubation of the MCF-7 cell nuclear lysates with the radiolabeled EtsA or EtsB hTERT promoter DNA probes produced markedly shifted complexes of significant binding activity (Fig. 4, E and F, lane 1 in each panel). The shifted complex was identified to contain and depend on Ets2 using specific antibodies; inclusion of Ets2 antibodies in the binding reactions produced a displacement of Ets2 binding, showing an Ets2-dependent formation of the complex (Fig. 4F, compare lanes 1 and 2). Inclusion of a 5-fold molar excess of unlabeled DNA effectively competitively inhibited the binding activity to the radiolabeled probes (Fig. 4, E and F, compare lane 3 with 1 in each panel). In contrast, inclusion of a 5-fold molar excess of unlabeled DNA that carried a mutation did not compete with Ets2 to displace binding to the radiolabeled probe (Fig. 4, E and F, compare the last lane with lane 1 in each panel). These data demonstrated a direct binding of Ets2 to the hTERT promoter DNA in vitro.
To determine in vivo binding between Ets2 and the hTERT gene promoter, we performed ChIP using specific antibodies against either Ets1 or Ets2, with anti-acetylhistone H3 as positive control and IgG as negative control. Precipitation of Ets1 with specific antibodies resulted in no co-precipitation of the hTERT gene promoter DNA (Fig. 4G, top panel, lane 2), but Ets2 antibodies co-precipitated the hTERT gene promoter DNA as resolved by PCR using specific primers (Fig. 4G, top panel, lane 3). Analysis for a distal region of the hTERT gene promoter DNA devoid of the Ets site was also performed in the Ets2 precipitates, and the result showed no detection of the region (–2916 to –2763) ((Fig. 4G, middle panel), suggesting that Ets2 precipitated the proximal region of the hTERT gene promoter specifically. For another positive control, we determined a known binding of Ets2 to the parathyroid hormone-related protein (PTHrP) gene promoter (26). As shown in Fig. 4H, the binding of Ets2 to the hTERT gene promoter was comparable with Ets2 binding the PTHrP gene promoter DNA when compared against the common control of acetylhistone H3 interaction with DNA (Fig. 4, G, lane 3 versus H, lanes 4 and 5). Thus, our data revealed that Ets2 binds to EtsA and EtsB sites in vitro and the hTERT gene promoter directly in vivo in human breast cancer cells.
Ets2 Regulating c-Myc Interaction with the hTERT Promoter—Because knockdown Ets2 is associated with decreased c-Myc gene expression (Fig. 1, E and F) and the EtsA juxtaposes immediately to a c-Myc binding E-box (–237GTGCAC–242) (Fig. 4A), we investigated a potentially direct interaction between Ets2 and c-Myc pathways in regulating the hTERT gene. As shown in Fig. 5A, expression of c-Myc did not return the gene promoter activity of hTERT inhibited by silencing Ets2, suggesting that c-Myc regulation of the hTERT gene requires Ets2. To determine a plausible cooperative binding of Ets2 and c-Myc to their respective EtsA and E-box binding sites (–237GTGCACCCTT–246, underlined is the E-box and italic is EtsA), we mutated the EtsA and E-box together or individually, and examined the binding of Ets2 and c-Myc to the hTERT gene promoter DNA. As shown in Fig. 5B, the presence of c-Myc antibodies not only abolished c-Myc binding to the E-box but also reduced Ets2 binding to EtsA site (lanes 3 and 4). Mutation of the E-box abolished the binding of c-Myc and also compromised the binding Ets2 to the hTERT promoter DNA (lane 6). Thus, these data suggest that both Ets2 and c-Myc are important in their respective bindings to the EtsA and E-box (–237GTGCACCCTT–246, underlined is the E-box and italic is EtsA) in the hTERT gene promoter. This conclusion was supported by the data that reconstitution of c-Myc did not restore Ets2 knockdown-induced decrease of the hTERT gene promoter activity (Fig. 5A) showing that c-Myc alone failed to stimulate the hTERT gene promoter activity in the absence of Ets2.
FIGURE 5.

Functional interdependence between Ets2 and c-Myc bindings to the hTERT gene promoter. A, expression of c-Myc failed to stimulate the hTERT gene promoter activity in cells expressing Ets2 shRNA. Cells with or without Ets2 silencing and c-Myc gene expression were transfected with the hTERT promoter luciferase reporter gene with or without mutation at the E-box or both E-box and EtsA sites. The hTERT gene promoter activity was determined by measuring luciferase activity. B, effects of mutations of the E-box on the binding of Ets2 to EtsA and c-Myc to E-box. Nuclear extracts of MCF-7 cells were incubated with [γ-32P]dATP-labeled hTERT promoter nucleotide probes containing wild type or mutated sequences singly or in combination, as indicated. Ets2 and c-Myc bindings are indicated. The single and double asterisks indicate 1:10 and 1:5 dilution of c-Myc antibodies. C and D, effects of immunological depletion of Ets2 and c-Myc on the respective bindings of Ets2 to EtsA and c-Myc to E-box. Nuclear extracts of MCF-7 cells were incubated with [γ-32P]dATP-labeled hTERT promoter nucleotide probes containing E-box and EtsA sequences (C) or the Sp1 site (D) in the presence or absence of different antibodies in control cells and Ets2 silenced cells as indicated. The bindings to the hTERT promoter sequences in the presence of different antibodies were resolved in non-denatured acrylamide gels. E, establishment of recombinant c-Myc gene expression in Ets2-silenced cells. Cells expressing the Ets2 shRNA were transfected with c-Myc gene expression plasmid for 24 h, and c-Myc gene expression was determined by Western blotting. F, effect of c-Myc gene expression on Ets2 gene silencing-induced inhibition of c-Myc binding to the hTERT gene promoter. Three groups of cells expressing normal Ets2 (lanes 1–4), low Ets2 (lanes 5–8), or low Ets2 plus high c-Myc (lanes 9–12) were subjected to ChIP using different antibodies as indicated. The hTERT gene promoter bound by Ets2 and c-Myc was determined by PCR. Results are representatives of three experiments. IB, immunoblot.
To investigate a potentially tripartite binding between the hTERT promoter sequence (–237GTGCACCCTT–246, underlined is the E-box and italic is EtsA), c-Myc and Ets2, we further analyzed the effects of removal of Ets2 or c-Myc on the bindings of c-Myc and Ets2, respectively, to the hTERT promoter sequence. As shown in Fig. 5C, in the presence of Ets2 antibodies, the binding of c-Myc to the hTERT promoter DNA was inhibited (lane 2 versus lanes 1 and 5), an effect similar to that of c-Myc antibodies (lane 4 versus lane 2) and dissimilar to that of Ets1 antibodies (lanes 4 versus lane 3). The data that immunologically chelating Ets2 impaired c-Myc binding to the hTERT promoter DNA suggest Ets2 participation in regulating c-Myc binding to the E-box. Silencing Ets2 also inhibited c-Myc binding activity to the hTERT promoter DNA (Fig. 5C, lanes 6–8). Similar to the data shown in Fig. 5B, the c-Myc antibodies once again inhibited not only c-Myc binding but Ets2 binding to the hTERT promoter DNA (Fig. 5C, lane 4), suggesting that c-Myc is involved in Ets2 interaction with the EtsA site. As specificity controls, normal IgG produced no effect on the binding of either Ets2 or c-Myc (Fig. 5C, lane 5), and as shown in Fig. 5D, Ets2 and c-Myc antibodies had no effect on binding to the Sp1 sites of the hTERT promoter DNA oligonucleotide probe in the MCF-7 cells with or without Ets2 gene silencing.
To determine the effect of Ets2 on c-Myc binding to the hTERT gene promoter in vivo, we silenced the Ets2 gene, reconstituted c-Myc gene expression, and determined the bindings of Ets2 and c-Myc to the hTERT gene promoter. As shown in Fig. 5E, silencing Ets2 once again entrained c-Myc repression. However, silencing of Ets2 not only abolished Ets2 binding but also inhibited c-Myc binding to the hTERT promoter (Fig. 5F, lanes 7 and 8 versus lanes 3 and 4). Re-establishment of c-Myc did not recover c-Myc binding in the absence of Ets2 binding to the hTERT promoter (Fig. 5F, lanes 11 and 12 versus lanes 3 and 4). These data together suggest a cooperative interaction of Ets2 and c-Myc with the hTERT gene promoter DNA, with an interdependent action and mutual regulation between Ets2 and c-Myc in their bindings to the respective EtsA and E-box sites in the hTERT promoter.
Ets2 Complex with c-Myc—With the direct sequence connection between the EtsA and E-box DNA elements, and the cooperative bindings of Ets2 and c-Myc to EtsA and E-box, respectively, we investigated if Ets2 and c-Myc form a complex by a co-immunoprecipitation approach. Incubation of MCF-7 cell lysates with specific c-Myc antibodies to immunoprecipitate c-Myc resulted in co-precipitated immunoreactive Ets2 (Fig. 6A). Incubation of the cell lysates with Ets2 specific antibodies also resulted in a complex containing immunoreactive c-Myc (Fig. 6B). Confirming a specific interaction of c-Myc with Ets2, experiments showed that c-Myc was absent in the immunoprecipitates obtained using specific antibodies against either Ets1 or ELF5. To confirm the binding specificity of Ets2 and its ability to pull down c-Myc, we expressed and purified Ets1 and Ets2 GST fusion proteins and carried out a GST fusion protein pulldown assay. Incubations of GST-Ets1 or GST-Ets2 with breast cancer MCF-7 cell lysates or nuclear fraction showed that GST-Ets2 precipitated c-Myc from both total and nuclear lysates (Fig. 6C). In marked contrast, GST pulled down no c-Myc, and GST-Ets1 precipitated discernable c-Myc immunoreactivity that was significantly much less than that precipitated with GST-Ets2 from cellular lysates (Fig. 6C, lane 3 versus lane 4) and the nuclear fraction (Fig. 6C, lane 7 versus lane 8). Note, a significantly higher band was detected from GST-Ets2 as well as GST-Ets1 pulldown pellets with unknown identity, which requires further investigation. Thus, our data showed for the first time that the two transcription factors, Ets2 and c-Myc, can form a complex in the context of regulating hTERT gene transcription in breast cancer cells.
FIGURE 6.

Ets2 and c-Myc interact in vitro and in cultured human breast cancer cells. A, co-immunoprecipitation of Ets2 with c-Myc. MCF-7 cell lysates were incubated with normal IgG or anti-c-Myc antibody for 60 min at room temperature before capturing the c-Myc immune complex using protein A-coated Sepharose beads. Precipitated proteins were determined by immunoblotting (IB) using the antibodies as indicated. B, co-immunoprecipitation (IP) of c-Myc with Ets2. Immunoprecipitation was conducted using anti-Ets2 and other Ets antibodies as indicated in reverse order to that shown in A. Co-immunoprecipitation of c-Myc with Ets2 was determined by immunoblotting. Data are representatives of one of three similar experiments. C, in vitro binding between Ets2 and c-Myc by GST pulldown assay. GST, GST-Ets1, and GST-Ets2 purified on glutathione beads were incubated with the nuclear fraction or total cell lysates of MCF-7 cells, which was followed by centrifugation, stringent wash, and subsequent protein analysis in the precipitates by Western blotting. Immunoreactivity of c-Myc and GST proteins was revealed using specific anti-myc and anti-GST antibodies. Results were from one of three similar experimental results. D, quantification of c-Myc binding to GST fusion proteins by densitometry. Results are mean ± S.D. of detected c-Myc normalized by GST proteins as ratios from three similar experiments. Asterisks indicate significant differences in comparison with GST-Ets1 or GST only with p < 0.001.
DISCUSSION
The regulation of hTERT gene expression is crucial for cancer cell immortality by a variety of factors (20, 27, 28). In the present study, we present three novel findings in Ets2 regulating the hTERT gene and breast cancer cell proliferation: first, we show that Ets2 is required not only for hTERT gene transcription and telomerase activity, but also for continuous proliferation of human breast cancer cells. In addition, we define two non-canonical Ets2-binding sites in the proximal region of the hTERT promoter that are both required to mediate the effect of Ets2 on hTERT gene transcription. Furthermore, we show that Ets2 binds to c-Myc in a collaborative regulation of the hTERT gene expression in breast cancer cells. These findings provide insight into the mechanisms of maintaining hTERT gene expression and cancer cell proliferation at the molecular levels involving Ets2 and its interplay with c-Myc.
Knockdown of Ets2, but not that of Ets1, induces significant repression of the hTERT gene in association with growth arrest of breast cancer cell proliferation and increased apoptosis. The depression of the hTERT gene is unlikely to be secondary to a major cellular event such as cell death, but appears to be specific, as Ets1 (closest analog of Ets2) does not produce a comparable effect on the hTERT gene, no change in Bcl-2 or Bcl-xL is observed, and the Ets2 silencing-induced cell death is reversible by hTERT. The specific effect of Ets2 retraction on the gene repression of hTERT in breast cancer cells reflects an important mechanism for the maintenance or up-regulation of hTERT gene transcription by Ets2. Thus, our findings do not support the paradox of Ets2 negative regulation of the hTERT gene by Ets2 overexpression (18, 19), although we cannot exclude the possibility that Ets2 operates at the hTERT gene by partnering with another protein when Ets2 is overexpressed in a cell type-specific manner.
Consistent with an inhibition of telomerase activity induced by Ets2 gene silencing, we show that the cellular functional consequence of Ets2 regulation of the hTERT gene expression and telomerase activity is to underpin continuous cell proliferation. This is demonstrated by the data that Ets2 knockdown-induced cell death is dependent on hTERT gene repression, and prevented by overexpression of recombinant hTERT, consistent with a cell proliferative function of hTERT (29, 30). Although our data do not exclude other mechanisms than telomerase activation in Ets2 regulation of breast cancer cell proliferation, hTERT repression and telomerase inhibition appear to play a significant role in Ets2 knockdown-induced cell death. Consistent with previous studies that inhibition of telomerase induces rapid cell death (21, 31), inhibition of telomerase activity may render telomeres unstable to stress stimuli, predisposing cancer cells to undergo cell aging and apoptosis. Our findings may provide an explanation for the mechanisms of Ets2-associated prostate cancer cell oncogenesis (15) and Ets2 down-regulation-associated prostate cancer cell growth inhibition and apoptosis (14).
Investigating the link between Ets2 and the hTERT gene promoter, and characterizing the interface of their molecular interactions, we have found two novel features in Ets2 regulating the transcriptional activity of the hTERT gene. First, Ets2 interacts with two sites of the hTERT gene promoter in a bipartite manner in vitro and in vivo, as evidenced by the gel shift assay, chromatin immunoprecipitation, and site-directed mutagenesis. We show that both non-canonical EtsA and EtsB elements are required, although they are 144 bp apart in the proximal region of the hTERT promoter (Fig. 4). Mutation of either EtsA or EtsB site disables the hTERT gene transcriptional activity. Given that Ets2 protein potentially forms a homodimer with the unfolded N-terminal α helix (HI-1) swapped between two Ets2 monomers when bound to DNA (32), it is compelling to speculate that Ets2 binds to the two sites of EtsA and EtsB simultaneously in a dimer form, so as to bring the two DNA binding elements in close proximity to facilitate formation of a transcriptosome on the hTERT gene.
The second feature at the interface of molecular interaction between Ets2 and the hTERT gene promoter is the involvement of c-Myc. The interaction of Ets2 with EtsA of the hTERT gene promoter appears to be regulated by the binding of c-Myc to the E-box that is immediately juxtaposed to the 3′ EtsA element of the hTERT proximal core promoter. In the presence of c-Myc antibodies, not only is the c-Myc binding inhibited, but Ets2 binding is also compromised, to the hTERT gene promoter DNA sequences. Mutation of the E-box also inhibits Ets2 binding. Supporting a reciprocal cooperation between EtsA and E-box, mutation of EtsA compromises E-box binding by c-Myc. In addition, we show that the converse is also likely to be true in the regulation of c-Myc binding to the E-box by Ets2 binding to the EtsA neighboring to E-box. Ets2 binding to the EtsA site appears to be a precondition to the binding of c-Myc. Consequently, knocking down Ets2 also inhibits the binding of c-Myc to the hTERT gene promoter. Overexpression of c-Myc does not improve the reduced hTERT gene promoter activity induced by Ets2 gene silencing. These data suggest that c-Myc binding to the E-box adjacent to EtsA depends on the local structural conformational change that is brought about by Ets2 binding to the EtsA. Thus, Ets2 and c-Myc collaborate in their optimal bindings to EtsA and E-box, respectively, on the hTERT gene promoter. Collaboration between EtsA and E-box may be also regulated by epigenetic mechanisms to allow Ets2/c-Myc complex to dock onto these binding sites of the hTERT promoter.
With the findings of mutual regulations between Ets2 and c-Myc in their interactions with the hTERT gene promoter, we hypothesized that Ets2 physically interacts with c-Myc, and thus carried out co-immunoprecipitation experiments. In support of the hypothesis, our data show for the first time that Ets2 indeed binds c-Myc, as evidenced in the co-immunoprecipitated complex and in vitro binding assay. Thus, a regulatory complex of hTERT gene transcription may be present containing the hTERT promoter DNA, c-Myc and Ets2. This possibility is supported by the data: 1) that the contiguous EtsA and E-box sites are both required for the intact bindings of Ets2 and c-Myc on the hTERT gene promoter; 2) that c-Myc alone without Ets2 fails to stimulate hTERT gene expression; 3) that both Ets2 and c-Myc are required in their binding to EtsA and E-box, respectively; and 4) that Ets2 and c-Myc bind to each other in their immune complexes. It is possible that Ets2 binds c-Myc to form a complex bridging the adjacent DNA-binding sites. Mutation of either EtsA or EtsB, or deficiency of either Ets2 or c-Myc, may cause inactivation of hTERT gene transcription leading to inhibition of telomerase activity.
Consistent with our findings on Ets2 binding to the EtsA site playing an indispensable role in mediating Ets2 up-regulation of the hTERT gene and c-Myc binding on the hTERT promoter, the EtsA element has recently been shown to be a significant single nucleotide polymorphism in telomerase activity and telomere length of non-small cell lung cancer (33). Sequencing a 716-bp DNA segment upstream of the hTERT transcription starting code in 66 non-small cell lung cancers reveals a polymorphic substitution of thymine –245 to cytosine in the EtsA motif (–246TTCC–243) of the hTERT gene. The single nucleotide polymorphism occurs at both alleles of the hTERT gene in about 10% of the patients, and at a single allele in about 53% of the patients (33). Consistent with EtsA mediation of Ets2 regulation of telomerase activity, the –245Thr → 245Cys polymorphism correlates with low levels of telomerase activity and short telomeres compared with wild type alleles (33). Thus, the EtsA element at the location of –246 to –243 is a key structure of the hTERT gene in mediating Ets2 positive regulation of telomerase activity in cancer (33). Further studies are required to determine EtsA single nucleotide polymorphism in breast cancer patients and potential regulation of hTERT gene expression by Ets2 in normal cell development.
Additional to a direct action of Ets2 on the hTERT gene promoter, our data that silencing Ets2 represses the c-Myc gene suggest that Ets2 regulation of the hTERT gene involves direct regulation of c-Myc action and indirect transcription-dependent regulation of c-Myc gene expression. This finding of Ets2 regulating the c-Myc gene is consistent with previous studies showing that a triple DNA-mediated down-regulation of Ets2 expression is associated with reduced gene expression of c-Myc (14) and that Ets2 dominant-negative mutation inhibits the CSF-1-induced c-Myc gene (34). By using the gene silencing approach, we confirm that Ets2 is required for c-Myc gene expression in breast cancer cells. This regulatory role of Ets2 is in contrast to a negative effect of Ets1, showing a specificity of Ets2 toward not only the hTERT gene but also the hTERT upstream regulator c-Myc gene. Ets2 is therefore likely a key transcription factor up-regulating the hTERT gene expression through actions on the hTERT gene and interactions with c-Myc pathway.
In summary, Ets2 transcription factor is required for hTERT gene expression and hTERT-dependent proliferation of human breast cancer cells. The transcriptional regulation of the hTERT gene by Ets2 involves Ets2 binding to EtsA and EtsB elements in the proximal core promoter region of the hTERT gene. Ets2 and c-Myc bind to each other and to their respective EtsA and E-box sites cooperatively. Silencing Ets2, or mutation of either of the Ets2 binding sites, is sufficient to switch off telomerase activity and induce cancer cell death. Thus, these findings provide significant insight into the molecular mechanisms of telomerase regulation at gene expression levels in breast cancer cells.
Acknowledgments
We thank Dr. Silvia Bacchetti for kind gifts of the hTERT-luc constructs.
This work was supported in part by research grants from the National Health and Medical Research Council of Australia, Australia Research Council, and Cancer Council of Victoria, Australia. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: ERK, extracellular signal-regulated kinase; TERT, telomerase reverse transcriptase; ChIP, chromatin immunoprecipitation; PTHrP, parathyroid hormone-related protein; RNAi, RNA interference; HA, hemagglutinin; siRNA, small interfering RNA; RT, reverse transcriptase; PBS, phosphate-buffered saline; GST, glutathione S-transferase; FACS, fluorescence-activated cell sorting; GFP, green fluorescent protein; CHAPS, 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid.
References
- 1.Seth, A., and Watson, D. K. (2005) Eur. J. Cancer 41 2462–2478 [DOI] [PubMed] [Google Scholar]
- 2.Mavrothalassitis, G., and Ghysdael, J. (2000) Oncogene 19 6524–6532 [DOI] [PubMed] [Google Scholar]
- 3.Myers, E., Hill, A. D., Kelly, G., McDermott, E. W., O'Higgins, N. J., Buggy, Y., and Young, L. S. (2005) Clin. Cancer Res. 11 2111–2122 [DOI] [PubMed] [Google Scholar]
- 4.Li, R., Pei, H., and Watson, D. K. (2000) Oncogene 19 6514–6523 [DOI] [PubMed] [Google Scholar]
- 5.Hsu, T., Trojanowska, M., and Watson, D. K. (2004) J. Cell Biochem. 91 896–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galang, C. K., Muller, W. J., Foos, G., Oshima, R. G., and Hauser, C. A. (2004) J. Biol. Chem. 279 11281–11292 [DOI] [PubMed] [Google Scholar]
- 7.Hahne, J. C., Okuducu, A. F., Kaminski, A., Florin, A., Soncin, F., and Wernert, N. (2005) Oncogene 24 5384–5388 [DOI] [PubMed] [Google Scholar]
- 8.Foos, G., and Hauser, C. A. (2000) Oncogene 19 5507–5516 [DOI] [PubMed] [Google Scholar]
- 9.Neznanov, N., Man, A. K., Yamamoto, H., Hauser, C. A., Cardiff, R. D., and Oshima, R. G. (1999) Cancer Res. 59 4242–4246 [PubMed] [Google Scholar]
- 10.Yamamoto, H., Flannery, M. L., Kupriyanov, S., Pearce, J., McKercher, S. R., Henkel, G. W., Maki, R. A., Werb, Z., and Oshima, R. G. (1998) Genes Dev. 12 1315–1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang, B. S., Hauser, C. A., Henkel, G., Colman, M. S., Van Beveren, C., Stacey, K. J., Hume, D. A., Maki, R. A., and Ostrowski, M. C. (1996) Mol. Cell. Biol. 16 538–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Svensson, S., Jirstrom, K., Ryden, L., Roos, G., Emdin, S., Ostrowski, M. C., and Landberg, G. (2005) Oncogene 24 4370–4379 [DOI] [PubMed] [Google Scholar]
- 13.Man, A. K., Young, L. J., Tynan, J. A., Lesperance, J., Egeblad, M., Werb, Z., Hauser, C. A., Muller, W. J., Cardiff, R. D., and Oshima, R. G. (2003) Mol. Cell. Biol. 23 8614–8625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carbone, G. M., Napoli, S., Valentini, A., Cavalli, F., Watson, D. K., and Catapano, C. V. (2004) Nucleic Acids Res. 32 4358–4367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sementchenko, V. I., Schweinfest, C. W., Papas, T. S., and Watson, D. K. (1998) Oncogene 17 2883–2888 [DOI] [PubMed] [Google Scholar]
- 16.Dwyer, J., Li, H., Xu, D., and Liu, J. P. (2007) Ann. N. Y. Acad. Sci. 1114 36–47 [DOI] [PubMed] [Google Scholar]
- 17.Maida, Y., Kyo, S., Kanaya, T., Wang, Z., Yatabe, N., Tanaka, M., Nakamura, M., Ohmichi, M., Gotoh, N., Murakami, S., and Inoue, M. (2002) Oncogene 21 4071–4079 [DOI] [PubMed] [Google Scholar]
- 18.Xiao, X., Athanasiou, M., Sidorov, I. A., Horikawa, I., Cremona, G., Blair, D., Barret, J. C., and Dimitrov, D. S. (2003) Exp. Mol. Pathol. 75 238–247 [DOI] [PubMed] [Google Scholar]
- 19.Xiao, X., Phogat, S. K., Sidorov, I. A., Yang, J., Horikawa, I., Prieto, D., Adelesberger, J., Lempicki, R., Barrett, J. C., and Dimitrov, D. S. (2002) Leukemia 16 1877–1880 [DOI] [PubMed] [Google Scholar]
- 20.Li, H., Xu, D., Li, J., Berndt, M. C., and Liu, J. P. (2006) J. Biol. Chem. 281 25588–25600 [DOI] [PubMed] [Google Scholar]
- 21.Cao, Y., Li, H., Deb, S., and Liu, J. P. (2002) Oncogene 21 3130–3138 [DOI] [PubMed] [Google Scholar]
- 22.Li, H., Zhao, L., Yang, Z., Funder, J. W., and Liu, J. P. (1998) J. Biol. Chem. 273 33436–33442 [DOI] [PubMed] [Google Scholar]
- 23.Gong, J., Traganos, F., and Darzynkiewicz, Z. (1994) Anal. Biochem. 218 314–319 [DOI] [PubMed] [Google Scholar]
- 24.Zinn, R. L., Pruitt, K., Eguchi, S., Baylin, S. B., and Herman, J. G. (2007) Cancer Res. 67 194–201 [DOI] [PubMed] [Google Scholar]
- 25.Takahashi, A., Higashino, F., Aoyagi, M., Yoshida, K., Itoh, M., Kyo, S., Ohno, T., Taira, T., Ariga, H., Nakajima, K., Hatta, M., Kobayashi, M., Sano, H., Kohgo, T., and Shindoh, M. (2003) Cancer Res. 63 8338–8344 [PubMed] [Google Scholar]
- 26.Lindemann, R. K., Braig, M., Hauser, C. A., Nordheim, A., and Dittmer, J. (2003) Biochem. J. 372 787–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu, J. P. (1999) FASEB J. 13 2091–2104 [DOI] [PubMed] [Google Scholar]
- 28.Bayne, S., Jones, M. E., Li, H., and Liu, J. P. (2007) Ann. N. Y. Acad. Sci. 1114 48–55 [DOI] [PubMed] [Google Scholar]
- 29.Rahman, R., Latonen, L., and Wiman, K. G. (2005) Oncogene 24 1320–1327 [DOI] [PubMed] [Google Scholar]
- 30.Massard, C., Zermati, Y., Pauleau, A. L., Larochette, N., Metivier, D., Sabatier, L., Kroemer, G., and Soria, J. C. (2006) Oncogene 25 4505–4514 [DOI] [PubMed] [Google Scholar]
- 31.Li, S., Crothers, J., Haqq, C. M., and Blackburn, E. H. (2005) J. Biol. Chem. 280 23709–23717 [DOI] [PubMed] [Google Scholar]
- 32.Lee, G. M., Donaldson, L. W., Pufall, M. A., Kang, H. S., Pot, I., Graves, B. J., and McIntosh, L. P. (2005) J. Biol. Chem. 280 7088–7099 [DOI] [PubMed] [Google Scholar]
- 33.Hsu, C. P., Hsu, N. Y., Lee, L. W., and Ko, J. L. (2006) Eur. J. Cancer 42 1466–1474 [DOI] [PubMed] [Google Scholar]
- 34.Sapi, E., Flick, M. B., Rodov, S., and Kacinski, B. M. (1998) Cancer Res. 58 1027–1033 [PubMed] [Google Scholar]