

Abstract
In eukaryotes, S-adenosyl-l-homocysteine hydrolase (Sah1) offers a single way for degradation of S-adenosyl-l-homocysteine, a product and potent competitive inhibitor of S-adenosyl-l-methionine (AdoMet)-dependent methyltransferases. De novophosphatidylcholine(PC)synthesisrequiresthreeAdoMet-dependent methylation steps. Here we show that down-regulation of SAH1 expression in yeast leads to accumulation of S-adenosyl-l-homocysteine and decreased de novo PC synthesis in vivo. This decrease is accompanied by an increase in triacylglycerol (TG) levels, demonstrating that Sah1-regulated methylation has a major impact on cellular lipid homeostasis. TG accumulation is also observed in cho2 and opi3 mutants defective in methylation of phosphatidylethanolamine to PC, confirming that PC de novo synthesis and TG synthesis are metabolically coupled through the efficiency of the phospholipid methylation reaction. Indeed, because both types of lipids share phosphatidic acid as a precursor, we find in cells with down-regulated Sah1 activity major alterations in the expression of the INO1 gene as well as in the localization of Opi1, a negative regulatory factor of phospholipid synthesis, which binds and is retained in the endoplasmic reticulum membrane by phosphatidic acid in conjunction with VAMP/synaptobrevin-associated protein, Scs2. The addition of homocysteine, by the reversal of the Sah1-catalyzed reaction, also leads to TG accumulation in yeast, providing an attractive model for the role of homocysteine as a risk factor of atherosclerosis in humans.
S-Adenosyl-l-homocysteine hydrolase (Sah1,2 EC 3.3.1.1.) is one of the most highly conserved enzymes from bacteria to mammals (1, 2). It plays a key role in the regulation of transmethylation reactions in all eukaryotic organisms by catalyzing the degradation of S-adenosyl-l-homocysteine (AdoHcy), the potent product inhibitor of AdoMet-dependent methyltransferases (see Fig. 1) (3-5). Targets of AdoMet-dependent methyltransferases include a wide spectrum of cellular compounds, such as DNA (5, 6), mRNA (5), histones H3 and H4 (7, 8), and other proteins as well as smaller metabolites, including lipids, e.g. phosphatidylethanolamine (9). Thus, by regulating AdoHcy levels in the cell, Sah1 activity has pleiotropic effects on gene expression, signal transduction, and lipid biosynthesis. In addition, the Sah1-catalyzed reaction is reversible, adding to the regulatory complexity of trans-methylation reactions. Whereas in vitro the equilibrium of the Sah1 reaction is in the biosynthetic direction to form AdoHcy from adenosine and homocysteine (10), quick metabolization of adenosine to ATP and inosine and homocysteine to methionine and cysteine and subsequently, glutathione, favors the catabolic direction of the reaction in vivo (11). Therefore, in vivo accumulation of homocysteine and/or adenosine favors the bio-synthetic reaction, resulting in AdoHcy accumulation under these conditions (12, 13).
FIGURE 1.

Role of Sah1p in lipid metabolism. PI, phosphatidylinositol; PS, phosphatidylserine; EA, ethanolamine; Cho, choline; Glucose-6-P, glucose-6-phosphate.
Many pathological disease states have been related to altered Sah1 function. Hypomethylation of DNA and high homocysteine/AdoHcy levels were shown to be associated with the pathology of cardiovascular diseases in mammals (14-16). Mice deficient in methylene tetrahydrofolate reductase, necessary for homocysteine to methionine remethylation, exhibit hyperhomocysteinemia and decreased methylation capacity along with neuropathology and aortic lipid deposition (17). A defect in hepatic phosphatidylethanolamine (PE) to phosphatidylcholine (PC) methylation leads to liver steatosis in mice (18) and was also shown to be associated with diabetes in mice and rats (19, 20). Complete loss of Sah1 function in mammals is deleterious for growth and development; deletion of the SAH1 locus is embryonic lethal in mice (21). However, recently patients deficient in Sah1 and exhibiting only 3-20% of mean control Sah1 activity were identified that displayed severe myopathy and mental retardation (22-24). Conflicting results exist as to the essential function of Sah1 in yeast (see below).
In addition to AdoHcy catabolism, Sah1 plays an important role both in mammals and yeast in homocysteine production, which is required for cysteine and glutathione synthesis (25, 26). Whereas in mammalian cells homocysteine is produced exclusively by Sah1, yeast possesses an additional option to synthesize homocysteine through the sulfur assimilation pathway that allows it to utilize sulfate as a source of sulfur (Fig. 1) (27-29).
In yeast as in mammalian cells the major membrane phospholipid, PC, is synthesized either de novo by sequential methylation of PE by AdoMet-dependent methyltransferases (Cho2 and Opi3 in yeast) or via the Kennedy pathway from diacylglycerol (DG) and CDP-activated choline (9, 30, 31). Although in yeast PC de novo synthesis is the dominating pathway, in mammals only 30% of total hepatic PC is synthesized by PE methylation de novo (9). Nevertheless, phospholipid methylation in mammals appears to be a major AdoMet-consuming pathway, since hepatic PC de novo synthesis is responsible for about 50% of plasma homocysteine levels (32). Accordingly, the highest Sah1 activity levels were detected in liver (21).
Previously, we have shown that SAH1 transcription in yeast is regulated in response to the lipid precursors, inositol and choline, and is coordinated with genes involved in phospholipid biosynthesis (2). This transcriptional regulation requires two transcriptional activators Ino2/Ino4 as well as the repressor Opi1 and is dependent on the phospholipid metabolite, phosphatidic acid (PA), in the endoplasmic reticulum (33). Ino2/Ino4 bind as heterodimeric activation complex to UASINO sequences present in the 5′ regions of all phospholipid biosynthetic genes (34, 35). In the absence of lipid precursors, Opi1, which may interact with Ino2 in the nucleus to block transcription, is retained in the endoplasmic reticulum by binding to PA (33). The presence of inositol leads to rapid consumption of PA pools in the ER and subsequent translocation of Opi1 into the nucleus to repress Ino2/Ino4-dependent transcription (33, 36, 37). Defective PE to PC methylation in cho2 or opi3 mutants leads to PA accumulation and, accordingly, to derepression of phospholipid biosynthetic genes, in particular INO1, which encodes inositol 3-phosphate synthase, the most highly regulated gene in this regulatory circuit (38, 39).
Depletion of Sah1 activity in yeast results in massive accumulation of lipid droplets and triacylglycerols during logarithmic growth (2). Here we show for the first time that Sah1 activity controls PC synthesis in yeast and that a decreased PC de novo synthesis both in Sah1-depleted cells and in cho2 and opi3 mutants defective in PE to PC methylation is accompanied by an increase in triacylglycerol levels. Reduction of Sah1 activity leads to deregulated INO1 expression, comparable with that observed in cho2 and opi3 mutants. Furthermore, homocysteine supplementation leads to AdoHcy accumulation through the Sah1-catalyzed reaction in vivo and, subsequently, inhibition of PE methylation and TG accumulation. We also show that the sulfur assimilation pathway in yeast is essential for survival in the absence of Sah1. These data demonstrate that Sah1 is not only important for regulating AdoMet-dependent methyl transfer reactions. It is also essential for providing homocysteine as a precursor for cysteine and glutathione synthesis in the absence of sulfur assimilation, which is lacking in mammalian cells. Taken together, Sah1 activity has a major impact on cellular lipid homeostasis, and its deficiency results in dysregulated lipid metabolism, leading to an imbalance of phospholipid and triacylglycerol synthesis, with implications for mammalian lipid-associated disorders.
EXPERIMENTAL PROCEDURES
Strains and Culture Conditions—The strains used in this study, except for the AID strain, are derived from the BX back-ground and are listed in Table 1. The tetO7-SAH1 strain, expressing SAH1 under control of the heterologous tetO7 promoter, was obtained from Open Biosystems. Precultures were grown in YPD complete media containing 10 g/liter yeast extract (Difco), 20 g/liter peptone (Difco), and 20 g/liter glucose (Merck) or in minimal SD media containing 6.7 g/liter yeast nitrogen base without amino acids (Difco), 20 g/liter glucose (Merck) and supplemented with amino acids and bases as described (2). Synthetic, inositol- and choline-free media that also lacked threonine (-I/-C/-threo) contained 10% glucose (40). Where indicated, methionine (20 μg/ml), cysteine (0.1 mm), inositol (80 μm), choline (1 mm), and homocysteine (1 mm) were added. Deuterated homocysteine (d4-Hcy, CDN Isotopes, Quebec, Canada) was added to the -I/-C/-threo medium at 1 mm. For repression of SAH1 expression under control of the tetO7 promoter, doxycycline was added at concentrations ranging from 0 to 50 μg/ml, as indicated. Media were solidified by the addition of 20 g/liter agar.
TABLE 1.
Strains used in this study
| Strain | Genotype | Source |
|---|---|---|
| YNM1 | MATahis3Δ1 met25Δ0 leu2Δ0 ura3Δ0 lys2Δ0 | Laboratory strain |
| YNM8 | MATahis3Δ1 leu2Δ0 ura3Δ0 lys2Δ0 | This study |
| YNM9 | MATahis3-1 leu2-0 pSAH1::kanR-tet07-TATA URA3::CMV-tTA | This study |
| YNM2 | MATahis3-1 leu2-0 met25-0 pSAH1::kanR-tet07-TATA URA3::CMV-tTA | Open Biosystems |
| YNM3 | MATahis3Δ1 leu2Δ0 met25Δ0 ura3Δ0 cho2::kanMX4 | Euroscarf |
| YNM4 | MATahis3Δ1 leu2Δ0 met25Δ0 ura3Δ0 opi3::kanMX4 | Euroscarf |
| YNM5 | MATahis3Δ1 met25Δ0 leu2Δ0 ura3Δ0 lys2Δ0 met6::kanMX4 str4::kanMX4 | This study |
| YNM6 | MATα his3Δ1 leu2Δ0 ura3Δ0 lys2Δ0 met6::kanMX4 str4::kanMX4 | This study |
| YNM7 | MATa/α; his3Δ1/his3Δ1 leu2Δ0/leu2Δ0 lys2Δ0/LYS2 MET25/met25Δ0 ura3Δ0/ura3Δ0 sah 1::kanMX4/SAH1 | Euroscarf |
| YNM10 | MATα his3Δ1 leu2Δ0 ura3Δ0 lys2Δ0 pUG36[Opil-GFP] | This study |
| YNM11 | MATα his3Δ1 leu2Δ0 ura3Δ0 lys2Δ0 sah 1::kanMX4 pUG36[Opil-GFP] | This study |
| AID | MATa/α ade1/ade1 ino1-13/ino1-13 | S.A. Henry |
Cloning of the MET25 Gene and Repair of the met25 Mutation—The MET25 wild type gene was amplified from BY4742 genomic DNA by PCR using 5′-ACTAATTAAGTTAGTCAAGGCGCCA-3′ and 5′-TCATTACGCACACTCATGGTTTTT-3′ primers and Tag DNA polymerase (Fermentas) following standard procedures. The obtained 2.7-kilobase MET25 fragment was transformed into yeast strains YNM1 and YNM2 (both met25) by the method of Gietz (41), replacing the met25 locus with the MET25 wild type gene. Correct integration at the MET25 locus and methionine prototrophy of the resultant transformants was confirmed by colony PCR and growth tests on media plates lacking methionine, respectively.
AdoHcy Quantification—AdoHcy was extracted by the method of Gellekink (42) with minor modifications. Yeast cells were grown overnight in 5 ml of YPD at 30 °C with shaking, inoculated in 100 ml of YPD medium to A600 = 0.1, and grown for additional 72 h. Cells were washed with sterile water and inoculated to A600 = 0.025 in -I/-C/-threo medium with or without supplements as indicated. Over a period of 25 h samples were collected, stored at -70 °C, or processed immediately. For AdoHcy extraction 100 A600 units of yeast cells were harvested, resuspended in 5 ml of 0.091 m acetic acid, and broken with glass beads in a Mercken-schlager homogenizer (Braun Bio-tech International) under CO2 cooling. The supernatant was separated from glass beads by centrifugation. 15 nmol of d3-AdoMet (CDN Isotopes) were added as internal standard to every sample before solid-phase extraction (SPE) on Bond Elut PBA columns (Varian Inc.) (42). AdoHcy was separated by HPLC (Rheos 2000 pump, Flux Instruments, Basel, Switzerland) using an ACE®3 C18 column (100 × 2.1 mm, Advanced Chromatography Technologies, Aberdeen, UK). The gradient started from 96% solvent A (0.1% formic acid in water) and increased to 100% solvent B (acetonitrile, 0.1% formic acid) over 9 min at a flow rate of 100 μl/min. AdoHcy concentration was measured on an LCQ Duo Ion Trap mass spectrometer (Thermo, San Jose, CA) using MS/MS-Scans for m/z 385 (AdoHcy), 389 (d4-AdoHcy) and 402 (d3-AdoMet) and with a TSQ-7000 Triple Quadrupole mass spectrometer performing the multiple reaction monitoring (MRM) scans for m/z 385→136 (AdoHcy), m/z 389→136 (d4-AdoHcy), and m/z 402→136 (d3-AdoMet). Calibration curves of AdoMet and AdoHcy (Sigma Aldrich) in the range of 1 to 50 μm were used for quantification.
Lipid Analysis—Yeast lipids were extracted by the Folch method (43). Briefly, 50 A600 units of yeast cells grown under identical conditions as used for AdoHcy extraction (see above) were harvested and broken with glass beads in a Mercken-schlager homogenizer (Braun Biotech International) under CO2 cooling. Lipids were extracted using chloroform/methanol 2:1 (v/v). Samples were applied onto silica gel TLC plates (Merck) with an automated sampler (Camag Automatic TLC Sampler 4). Total lipids were separated using light petroleum/diethyl ether/acetic acid 32:8:0.4 (v/v/v). Triacylglycerols and total phospholipids were visualized by carbonization and quantified by densitometric scanning as described (44) using triolein as the standard.
Phospholipid composition was analyzed by normal-phase HPLC using a YMC-Pack™ Diol column (250 × 4.6 mm, 5 μm) on an Agilent 1100 HPLC system (Agilent Technologies, Waldbronn, GER) equipped with an evaporative light scattering detector (PL-ELS 1000, Polymer Labs, Amherst, MA). The gradient was based on a previously reported method (45) and changed from acetone to acetone/methanol (46.2/53.8 v/v) in the presence of hexane (7 volume %), acetic acid (1.9%), and triethylamine (1.6%) over 32 min at a flow rate of 0.4 ml/min. Quantification of phospholipids was achieved by external standardization using lipid standards from Avanti Polar Lipids Inc. (Alabaster, AL).
RNA Isolation and Real Time PCR—An overnight culture grown in minimal medium was inoculated to A600 = 0.05 in -I/-C/-threo synthetic medium with or without doxycycline supplementation and grown for 6 h at 30°C under shaking. RNA was prepared using SV Total RNA Isolation System (Promega). 1 μg of total RNA was converted into cDNA using Superscript™ reverse transcriptase (Invitrogen). Expression of SAH1 (primers 5′-CCGAAGTTACCTGGTCCTCTTTGA-3′ and 5′-ACCGGAAGCGGCAATAGC-3′ and TagMan probe 5′-CGACTCAAGATCATGCCGCCGC-3′) and INO1 genes (primers 5′-GGAATGACGTTTATGCTCCTTTTAA-3′ and 5′-GTCCCAACCAGAGACGACAAA-3′ and TagMan probe 5′-CTGTTGCCCATGGTTAGCCCAAACG-3′) was measured by real time PCR using Universal PCR Master Mix (Applied Biosystems) and normalized to ACT1 (primers 5′-CGCCTTGGACTTCGAACAAG-3′ and 5′-GACCATCTGGAAGTTCGTAGGATT-3′ and TagMan probe 5′-TGCAAACCGCTGCTCAATCTTCTTCAAT-3′). Calculation of relative expression was according to the suppliers' protocol.
Opi Test—The test for Opi phenotype was performed as described previously with minor modifications (46, 47). Briefly, strains were grown overnight in minimal medium, and 5-μl aliquots of A600 = 1 dilutions each were spotted onto -I/-C/-threo plates with or without doxycycline supplementation as indicated. The plates were incubated at 30 °C for 24 h and sprayed with a suspension of an inositol auxotrophic tester strain (AID). Secretion of inositol resulting in a growth ring of the tester strain around the spotted cultures was scored after further incubation for 24-48 h at 30 °C.
Microscopic Analysis of Opi1-GFP Localization—Wild type and sah1 mutant cells expressing C-terminal GFP-tagged Opi1 from plasmid pUG36 (48) were grown overnight in minimal SD media lacking uracil. Before microscopy, cells were inoculated into fresh -I/-C/-ura media containing 2 μg/ml methionine and 1 mm cysteine (see above) and sequentially re-inoculated in 3 cycles (8-12 h each) to obtain a logarithmically growing cell population with homogeneous Opi1-GFP expression. GFP fluorescence was analyzed on a Leica TCS4d confocal microscope with 488-nm excitation and 525/50-nm band pass filter detection. Quantitative image analysis was performed using Image J software (rsb.info.nih.gov/ij). Briefly, GFP fluorescence intensity profiles were calculated along a linear axis through the cell, normalized, and plotted (see also Fig. 6C). Intensity maxima (±S.D.), corresponding to the nuclear rim/ER, were correlated to intensity values inside and outside the nucleus, from at least 20 cells/nuclei in each preparation.
FIGURE 6.

Sah1 activity interferes with regulation of phospholipid synthesis. A, down-regulation of SAH1 results in overproduction and secretion of inositol (opi phenotype) in strain YNM2 (tet07SAH1 met25) grown in the presence of doxycycline. Wild type YNM1 (met25) and strains YNM3 (cho2 met25), YNM4 (opi3 met25), and YNM2 (tet07SAH met25) were grown overnight in minimal medium, and each 5 μl of A600 = 1 dilutions were spotted onto inositol- and choline-free plates in the absence and presence of doxycycline. On the following day an inositol auxotrophic AID strain was sprayed onto the plates, and the plates were incubated for another day at 30 °C and kept for 2 days at 4 °C before scanning. DC, doxycycline. B, INO1 expression in strain YNM2 (tet07SAH1 met25). RNA was prepared from wild type YNM1 (met25) and YNM2 (tet07SAH1 met25) cultures grown in -I/-C/-threo (black bars) and +I/-C/-threo (80 μm inositol, white bars) with and without doxycycline supplementation as indicated and converted to cDNA. INO1 expression was measured by real-time PCR using INO1-specific TagMan probe and normalized to ACT1. The data are the representative results from two independent experiments. C, subcellular and subnuclear localization of the transcriptional repressor Opi1. Wild type YNM10 (columns a and b) and sah1 mutant YNM11 cells (columns c and d) expressing Opi1-GFP from plasmid pUG36 (48) were grown in inositol- and choline-free medium supplemented with 2 μg/ml methionine and 1 mm cysteine in the absence (columns a and c) and presence of 10 μm inositol (columns b and d). Arrows indicate the appearance of bright Opi1-GFP foci at the nuclear envelope; these foci are actually below the diffraction limit but appear as larger structures due to their bright fluorescence. Top row, GFP fluorescence; middle row, transmission images; bottom row, representative histograms (cross-sections, dashed lines in fluorescence micrographs) of Opi1-GFP fluorescence across the nuclei of the cells and quantitative assessment of fluorescence intensities (arbitrary units ± S. D.) of the boxed areas, stretching the nuclear rim or the lumen of the nucleus, and cytosolic background. At least 20 cells were measured in each experiment using Image J software. Bar = 5 μm.
RESULTS
Sah1 Is Essential in Yeast in the Absence of Sulfur Assimilation—Previous studies have yielded ambiguous results as to an essential function of Sah1 in yeast. To address the reported discrepancies and to generate a conditional system that allowed systematic studies of the impact of Sah1 on lipid metabolism, we made use of a strain expressing SAH1 under control of the doxycycline-regulatable tetO7 promoter (Fig. 2A). As shown in Fig. 2B, expression of SAH1 in strain YNM2 (tet07SAH1 met25), as determined by real time PCR, was elevated compared with wild type during growth without doxycycline supplementation, consistent with previous results showing that the tetO7 promoter is rather strong in the absence of repressor. However, SAH1 expression was strongly dependent on the doxycycline concentration and was completely abolished in the presence of 2 μg/ml doxycycline in the medium. This concentration was also sufficient to fully inhibit growth of strain YNM2 (Fig. 2C). The addition of cysteine or homocysteine only marginally restored growth of this strain in the presence of 2 μg/ml doxycycline; however, it indicated to us that the essential function of Sah1 may be related to methionine or cysteine homeostasis.
FIGURE 2.

Characterization of a conditional, doxycycline-regulatable allele of SAH1. A, schematic representation of the chromosomal tetO7-SAH1 construct of strain YNM2. DC, doxycycline. B, SAH1 expression in strain YNM2 (tet07SAH1 met25) as a function of doxycycline concentration, measured by real-time PCR using SAH1-specific TagMan probe, and normalized to ACT1. Strains were grown in -I/-C/-threo media for 6 h in the absence and presence of doxycycline. C, growth of wild type YNM1 (met25) and YNM2 (tet07SAH1 met25) strains on synthetic defined inositol- and choline-free media plates (-I/-C/-threo). 2 μg/ml doxycycline, 20 μg/ml methionine, 1 mm homocysteine, or 0.1 mm cysteine were added as indicated. 5-μl dilutions of A600 = 1, 0.1, 0.01, and 0.001 (black triangles) were spotted onto the plates. Images were taken after 2 days of growth. n.s., no supplementation. D, tetrad analysis of diploid strain YNM7, heterozygous for sah1/SAH1 and met25/MET25 loci on YPD complete media plates. Symbols on the right mark the various genotypes for nine complete tetrads. The sah1 mutation gives rise to medium size colonies, and the met25 sah1 double mutation yields very small colonies; met25 mutants grow like wild type on YDP plates that contain methionine. Growth of four progeny strains after tetratype segregation of sah1 and met25 mutations was tested on synthetic minimal plates containing 1 mm methionine and supplemented with doxycycline, homocysteine, and cysteine, as indicated. Homocysteine or cysteine supplementation slightly improved growth of the sah1 met25 double deletion strain. 5-μl dilutions of A600 = 1, 0.1, 0.01, 0.001 (black triangles) were spotted onto the plates. Images were taken after 2 days of growth.
Indeed, MATa strains of the Euroscarf collection (and strains from the tetO7 promoter replacement collection) contain a mutation in the MET25 gene, encoding O-acetylhomoserine sulfhydrylase, which renders the sulfur assimilation pathway inactive (49). Because in yeast sulfur assimilation is an integral part of homocysteine and methionine homeostasis, we have repaired the met25 deletion in strain YNM2 by replacing the mutated allele with the MET25 wild type gene. Indeed, the addition of doxycycline to the medium did not affect the growth of strain YNM9 (tet07SAH1 MET25), demonstrating that Sah1 is only essential in the absence of Met25. To analyze this further, we have subjected strain YNM7 heterozygous for sah1 and met25 mutations (sah1/SAH1 met25/MET25) to tetrad analysis (Fig. 2D). On methionine-containing plates, the resulting progeny developed either big (MET25 or met25), medium size (sah1 MET25), or very small (sah1 met25) colonies. Supplementation of either homocysteine or cysteine in combination with methionine, but not methionine alone, supported slow growth of sah1 or sah1 met25 deletion strains (Fig. 2D). Taken together, these data demonstrate that (i) Sah1 is not an essential enzyme and is only indispensable for the yeast cell in the absence of sulfur assimilation and (ii) elimination of Sah1 function results in strongly reduced growth that cannot be completely rescued by amino acid supplementation, as a consequence of lacking AdoHcy catabolism. Because sulfur assimilation is absent in mammals, we have used strain YNM2 that lacks MET25 and, thus, more closely resembles the mammalian metabolism for studies on Sah1 function.
Sah1 Activity Controls AdoHcy Content in Yeast—Sah1 is the only enzyme responsible for degrading AdoHcy in yeast and other eukaryotes. Thus, we have analyzed the impact of modulated Sah1 activity on AdoHcy levels in strain YNM2 grown in the absence and presence of doxycycline. As expected and shown in Fig. 3, depletion of Sah1 in YNM2 cells in the presence of 2 μg/ml doxycycline resulted in 10-fold accumulation of AdoHcy compared with wild type. Interestingly, AdoHcy levels in cells grown in the absence of doxycycline were also about 2-fold higher than in wild type, which may be explained by a highly activated methylation metabolism, in accordance with elevated phospholipid methylation under these conditions (see below).
FIGURE 3.

Relative AdoHcy levels in YNM2 (tet07SAH1 met25) strain overexpressing or down-regulating SAH1 compared with wild type YNM1 (met25). Cultures were pregrown in YPD, inoculated at A600 = 0.025 in fresh inositol- and choline-free medium without threonine, and supplemented with methionine (20 μg/ml) and doxycycline (0 and 2 μg/ml). The probes were taken to the time points indicated (see “Experimental Procedures”). Here early log phase, 15 h, is shown. AdoHcy is extracted via SPE and analyzed by HPLC-ESI-MS/MS (C-18 column, water-acetonitrile gradient, TSQ 7000 Triple-Quad MS (42)). See also Fig. 8A.
Down-regulation of SAH1 Leads to TG Accumulation and Inhibition of PE to PC Methylation and Interferes with Regulation of Phospholipid Synthesis—We have previously observed that modulation of SAH1 expression by virtue of the GAL1 promoter affects lipid homeostasis (2). As shown in Fig. 4, A and B, down-regulation of SAH1 in strain YNM2 by the addition of doxycycline results in accumulation of lipid droplets during early logarithmic growth, a phase that is typically characterized by low TG content in wild type cells (50). Neutral lipid composition of wild type and strain YNM2 grown in the presence and absence of doxycycline was quantified as described under “Experimental Procedures.” Both wild type YNM1 (met25) and mutant YNM2 (tet07SAH1 met25) strains were inoculated at very low cell density of A600 = 0.025 to allow efficient repression of the tetO7 promoter by doxycycline, and lipid composition was analyzed in early log phase (15 h). As shown in Fig. 4C (white bars), the triacylglycerol content increased 3-fold over wild type and more than 2.5-fold over the control in early log phase cells treated with doxycycline. TG content was elevated throughout the entire growth curve in YNM2 cells treated with doxycycline (25 h; data not shown).
FIGURE 4.
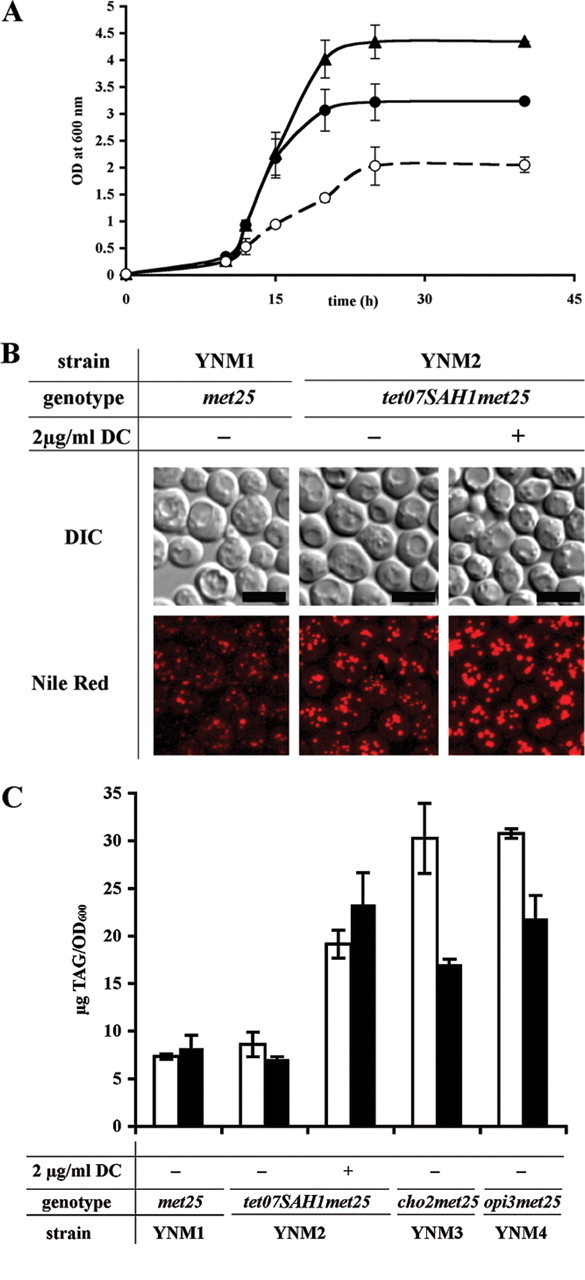
Impact of defective phospholipid methylation on TG and lipid droplet content during early log phase (15 h). A, growth of wild type YNM1 (met25)(filled triangles, solid line), YNM2 (tet07SAH1 met25) without doxycycline supplementation (filled circles, solid line), and YNM2 with 2 μg/ml doxycycline supplementation (open circles, dashed line) in synthetic defined -I/-C/-threo media. Inoculation was at (OD600) = 0.025. B, Nile red staining of lipid droplets in YNM1 (met25) and YNM2 (tet07SAH1 met25) cells grown with or without doxycycline (DC) supplementation. Nile red staining was performed as described (62). Bar = 5 μm. C, TG (triacylglycerol, TAG) accumulation in wild type YNM1 (met25), YNM2 (tet07SAH1 met25) with and without doxycycline supplementation, YNM3 (cho2 met25) and YNM4 (opi3 met25) defective in PC de novo synthesis in the absence and presence of 1 mm choline. Cultures were pregrown in YPD and transferred to fresh inositol- and choline-free media supplemented with or without 2 μg/ml doxycycline and 1 mm choline (white bars, without choline; black bars, with choline). At the indicated time points, lipids were extracted and analyzed by TLC as described under “Experimental Procedures.” Data are from at least three independent experiments. Data from early log phase (15 h) are shown.
We hypothesized that this increase in TG mass was due to reduced PE to PC methylation under conditions of Sah1 deficiency, leading to accumulation and preferred conversion of lipid precursors, PA and DG, into triacylglycerol. Thus, we predicted that cho2 and opi3 mutants, which are deficient in PE and PL methyltransferases, respectively, should have a similar impact on TG homeostasis. Indeed, as shown in Fig. 4C (white bars), TG levels were elevated 4-fold in cho2 and opi3 mutants, respectively, similarly to strain YNM2 grown in the presence of doxycycline. These data demonstrate that a defect in PE to PC methylation rather than an indirect effect on other AdoMet-dependent methyltransferases is responsible for TG accumulation under conditions of reduced Sah1 activity. The addition of choline, which enters PC synthesis via the Kennedy pathway and, thus, bypasses de novo methylation, alleviated TG accumulation by only 30% (Fig. 4C, black bars) in strains with defective de novo synthesis, suggesting limited availability of DG for phospholipid synthesis.
It was shown previously that AdoHcy is an in vitro competitive inhibitor for both Cho2 and Opi3 methyltransferases with respect to AdoMet (51). Thus, we further addressed the impact of Sah1 on lipid metabolism by more detailed lipid analyses. Phospholipid composition of strain YNM2 grown in inositol- and choline-free (white bars) or choline-supplemented (black bars) media in the absence and presence of doxycycline was analyzed. In the presence of 2 μg/ml doxycycline the PC content of strain YNM2 was reduced by almost 60%, similarly to cho2 mutant cells (Fig. 5, A and B) if choline was absent. PC was below detection limit in the opi3 mutant, which instead accumulated the monomethylated PE intermediate (52). PC levels were increased in the YNM2 strain grown in the absence of doxycycline compared with wild type, in agreement with about 2-fold elevated SAH1 expression (Figs. 5B and 2B) under these conditions. This observation further supports in vitro data (51) that phospholipid methylation is very sensitive to AdoHcy accumulation and may be stimulated by its enhanced removal, also in vivo.
FIGURE 5.

Sah1p controls PC de novo synthesis in yeast. A, analysis of phospholipids by HPLC; chromatograms of lipid extracts from wild type YNM1 (met25) and strains YNM3 (cho2 met25), YNM4 (opi3 met25), and YNM2 (tet07SAH1 met25), grown with and without doxycycline (DC) supplementation (2 μg/ml), as indicated. NL, neutral lipid; PMME, phosphatidyl monomethyl ethanolamine. B, PC content in the lipid extracts of wild type YNM1 (met25), YNM3 (cho2 met25), YNM4 (opi3 met25), and YNM2 (tet07SAH1 met25) strains grown with and without doxycycline and choline supplementation (white bars, without choline; black bars, with choline) as indicated. Data from early log phase (15h) are shown. mAU, milliabsorbance units.
Consistent with a major defect in phospholipid metabolism, we observed that strains with reduced Sah1 activity also displayed defective regulation of INO1, resulting in overproduction and secretion of inositol into the growth medium; this Opi (overproducer of inositol) phenotype (46, 47) is characteristic for cho2 and opi3 mutants deficient in phospholipid metabolism (38). As shown in Fig. 6A, a set of isogenic strains YNM1 (“wild type” control; met25), YNM3 (cho2 met25), YNM4 (opi3 met25), and YNM2 (tet07SAH1 met25) was tested for inositol overproduction in the presence and absence of doxycycline. Indeed, down-regulation of SAH1 in strain YNM2 grown in the presence of doxycycline led to an Opi phenotype similarly to cho2 and opi3 mutants. To quantitate the impact of defective methylation on INO1 expression, we have analyzed the INO1 mRNA steady state levels in strain YNM2 grown in the presence and absence of doxycycline. As shown in Fig. 6B, down-regulation of SAH1 resulted in 16-fold overexpression of the INO1 gene compared with wild type cells. Interestingly, INO1 expression was not fully repressed under these conditions by the addition of 80 μm inositol, which otherwise strongly represses INO1 transcription in wild type (39), consistent with a significant regulatory function of Sah1 activity in phospholipid metabolism.
Sah1 Deficiency Precludes Translocation of Opi1-GFP into the Nucleus in the Presence of Inositol—Opi1 is a transcriptional repressor of phospholipid biosynthetic genes and acts as a sensor of PA in the endoplasmic reticulum; upon PA depletion in the ER membrane, Opi1 translocates into the lumen of the nucleus to repress Ino2/Ino4-dependent gene expression (33). Because our hypothesis predicts accumulation of PA under conditions of Sah1 deficiency, we have assessed PA levels in the ER membrane by analyzing Opi1-GFP localization by fluorescence microscopy and quantitative image analysis in wild type (YNM10) and the sah1 mutant (YNM11) strains. In the absence of inositol in the growth media, Opi1-GFP localized to the endoplasmic reticulum and nuclear envelope in both wild type and sah1 mutant (Fig. 6C, panels a and c), consistent with the de-repressed levels of INO1 expression in these strains (Fig. 6B). Remarkably, Opi1-GFP levels under these derepressed conditions were lower in the lumen of the nuclei of the sah1 mutant compared with wild type, which is also reflected in the elevated level of INO1 expression in this strain (Fig. 6B) and which further supports the validity of the microscopic assay. Within 10 min after the addition of 10 μm inositol, Opi1-GFP translocated into the nucleus in wild type cells (Fig. 6C, panel b) but remained largely excluded from the nuclear lumen in the sah1 mutant (Fig. 6C, panel d) and was rather sequestered to foci at the nuclear rim, outside the nucleus. The subcellular distribution of Opi1-GFP in wild type and sah1 mutant cells in the absence or presence of 10 μm inositol was assessed by quantitative image analysis. These data show that translocation of the transcriptional repressor Opi1 into the lumen of the nucleus upon inositol supplementation is strongly reduced in the sah1 mutant. The reduced flux through the de novo phospholipid biosynthetic pathway may, thus, lead to the accumulation of phosphatidic acid in the ER, with consequences for the transcriptional regulation of lipid synthesis, in particular the PA/Scs2-dependent localization of the transcriptional repressor Opi1 (33), and expression of the INO1 gene. Because PA also serves as a precursor for TG, the potential rise of PA levels as a consequence of defective phospholipid synthesis in methylation-deficient cells may be alleviated by increasing TG synthesis.
Homocysteine Supplementation Induces AdoHcy Synthesis through the Sah1 Reaction—Homocysteine generated by Sah1 is a key intermediate in sulfur and methylation metabolism. To study the impact of an imbalanced homocysteine metabolism, we have constructed a mutant strain YNM5 (met25 met6 str4), which is deficient in homocysteine metabolism both through trans-sulfuration and trans-methylation pathways (Fig. 1). This mutant accumulates homocysteine generated by the Sah1 reaction in vivo lacking cystathionine β-synthase and methionine synthase required for its conversion to cysteine and methionine and is, thus, a cysteine and methionine auxotroph. As shown in Fig. 7A, this strain becomes highly sensitive to homocysteine, presumably due to its conversion to AdoHcy by Sah1. Mutant strain YNM6 (met6 str4), which also lacks cystathionine β-synthase and methionine synthase but is capable of sulfur assimilation that leads to additional homocysteine synthesis, grew very poorly even without homocysteine supplementation. The addition of 1 mm homocysteine completely abolished growth of this mutant. These data show that elevated homocysteine levels become toxic to the cells, most likely through reversal of the Sah1 reaction that leads to elevated AdoHcy levels.
FIGURE 7.

AdoHcy accumulates in the presence of homocysteine. A, growth of wild type YNM1(met25), and YNM5 (met25 met6 str4), and YNM6 (met6 str4) mutant strains on inositol- and choline-free plates in the presence and absence of 1 mm homocysteine. 5-μl dilutions of A600 = 1, 0.1, 0.01, and 0.001 (black triangles) were spotted onto the plates. Images were taken after 2 days of growth. n.s., no supplementation. B, accumulation of d4-AdoHcy by YNM5 (met25 met6 str4) mutant cells grown in the presence of d4-Hcy. YNM5 (met25 met6 str4) mutant cells were pregrown in YPD medium and inoculated at A600 = 0.025 in fresh inositol- and choline-free medium containing 1 mmd4-Hcy, and after 15 h of growth (early log phase), AdoHcy and AdoMet were extracted and analyzed by HPLC-ESI-MS/MS (C-18 column, water-acetonitrile gradient, TSQ 7000 Triple-Quad MS (42)) as described under “Experimental Procedures.” AdoHcy, d4-AdoHcy, AdoMet, and d3-AdoMet were traced in the chromatograms by virtue of their characteristic m/z values, as indicated in the figure. C, relative AdoHcy content in the YNM5 (met25 met6 str4) mutant strain in the presence and absence of homocysteine. YNM5 mutant cells were pregrown in YPD medium and inoculated at A600 = 0.025 in fresh inositol- and choline-free medium with or without 1 mm homocysteine supplementation. At the indicated time points AdoHcy was extracted and analyzed by HPLC-ESI-MS/MS as described under “Experimental Procedures.” Data from early log phase (15 h) are shown.
To provide in vivo proof of a reversal of the Sah1 reaction, we have supplemented mutant strain YNM5 (met25 met6 str4) with 1 mm deuterated homocysteine, d4-Hcy. Indeed, 5 pmol/A600 of deuterated d4-AdoHcy were detectable after 15 h of incubation with d4-Hcy in cell extracts of strain YNM5 (Fig. 7B). Furthermore, AdoHcy levels increased 10-fold upon homocysteine supplementation in this strain (Fig. 7C). These data demonstrate that the Sah1-catalyzed reaction is reversible in vivo and that elevated levels of homocysteine generate increased amounts of AdoHcy in the absence of alternative pathways for homocysteine metabolization.
Homocysteine Supplementation Results in TG Accumulation in Yeast—Reduced Sah1 activity results in increased AdoHcy content, defective PE to PC methylation, and accumulation of TG. Because homocysteine supplementation also leads to an increased AdoHcy content in the presence of active Sah1, we have analyzed lipid composition of YNM5 (met25 met6 str4) strain grown in the absence or presence of homocysteine. As stated above, this strain lacks enzymes involved in homocysteine metabolism through trans-sulfuration and trans-methylation pathways but is able to utilize homocysteine for AdoHcy production through the Sah1-catalyzed reaction. As expected, strain YNM5 displayed 50% increased TG content compared with wild type, even in the absence of homocysteine supplementation (Fig. 8A). The presence of 1 mm homocysteine led to TG accumulation both in wild type and YNM5 strains, comparable with cho2 and opi3 mutant strains defective in de novo PC synthesis (Fig. 4C). The elevated PC content of the mutant, compared with wild type, is most likely due to the slower growth and altered cell size of the triple mutant. Nevertheless, PC levels dropped both in wild type and strain YNM5 in the presence of homocysteine (Fig. 8B).
FIGURE 8.

Impact of homocysteine supplementation on lipid homeostasis in yeast. A, TG (triacylglycerol, TAG) accumulation in the presence of homocysteine. Wild type YNM1 (met25) and YNM5 (met25 met6 str4) mutant cells were pregrown in YPD medium and inoculated at A600 = 0.025 in fresh inositol- and choline-free medium with or without 1 mm homocysteine supplementation. At the indicated time points lipids were extracted and analyzed by TLC as described under “Experimental Procedures.” Data from early log phase (15 h) are shown. B, PC content of wild type YNM1(met25) and YNM5 (met25 met6 str4) strains grown with and without homocysteine supplementation, as indicated. Data from early log phase (15 h) are shown.
In summary, elevation of intracellular homocysteine levels by homocysteine supplementation stimulates AdoHcy synthesis by the Sah1-catalyzed reaction. AdoHcy accumulation, which can also be induced by inactivating Sah1, causes inhibition of PE to PC methylation. The reduced flux of the diacylglycerol backbone into phospholipids leads to the accumulation of phosphatidic acid in the ER, with consequences for the transcriptional regulation of lipid synthesis. In addition, excess PA is detoxified by the generation of triacylglycerols, leading to lipid droplet accumulation, with potential pathological consequences.
DISCUSSION
Understanding the regulatory cross-talk between metabolic pathways is a major challenge in biomedical research. This is particularly true for membrane lipid synthesis, which connects to multiple essential cellular processes, such as secretion, signaling, and proliferation. Synthesis of membrane phospholipids and storage triacylglycerols has to be coordinated with nutrient availability and cell cycle control. Both types of glycerolipids share the same precursors, phosphatidic acid and dia-cylglycerol; however, the regulatory mechanisms governing lipid precursor partitioning toward lipids required for proliferation or storage are largely unknown.
We have previously identified a novel and unexpected regulatory link between Sah1, key enzyme of methylation metabolism, and lipid metabolism (2). Sah1 plays a dual role by degrading AdoHcy, which is a byproduct and strong competitive inhibitor of AdoMet-dependent methylation reactions and provides homocysteine, which may enter the trans-methylation pathway for methionine regeneration or the trans-sulfuration pathway for cysteine and glutathione synthesis. SAH1 expression is transcriptionally coordinated with phospholipid biosynthesis, and Sah1-defective mutants are characterized by increased triacylglycerol synthesis and lipid droplet proliferation (2). Supporting the notion that Sah1 offers a single way for AdoHcy catabolism, Sah1 deficiency results in AdoHcy accumulation both in human patients deficient in Sah1 as well as in yeast sah1 mutants (22-24, 53). An impact of Sah1 function on AdoMet-dependent methylation is further suggested by low levels of plasma PC as well as elevated guanidinoacetate in human patients deficient in Sah1 (22-24).
Sah1 is one of the most highly conserved biosynthetic enzymes, with more than 70% identity between human and yeast orthologs (1). This high level of sequence conservation is astonishing and highlights the important cellular function of the enzyme. Indeed, Sah1 is essential in mammals (21); still, conflicting data exists as to its essential function in yeast (2, 27, 53, 54). Here we show that Sah1 in yeast plays important roles by regulating AdoMet-dependent methylation of phospholipids. It is also essential for homocysteine and cysteine production in the absence of the sulfur assimilation pathway. Thus, sah1 null mutants of yeast are only viable upon homocysteine or cysteine supplementation or when the sulfur assimilation pathway remains active. Mammals, in contrast to yeast, lack the sulfur assimilation pathway, and are thus dependent on Sah1 for homocysteine synthesis.
In this study we present evidence that S-adenosyl-l-homocysteine hydrolase is a major regulator of lipid metabolism in yeast. By regulating cellular levels of AdoHcy, Sah1 controls PE to PC de novo methylation. Similarly to mutants defective in PE to PC methylation, cho2 and opi3, lack of Sah1 activity was expected to have a major impact on the level of phosphatidic acid. PA plays an important role in the transcriptional regulation of phospholipid synthesis by retaining, in conjunction with the type II integral membrane protein Scs2 (VAP; VAMP/synaptobrevin-associated protein), the transcriptional repressor Opi1 outside the nucleus and away from the transcription activation complex that is required for UASINO-dependent gene expression (33). Down-regulation of SAH1 indeed leads to an increased transcription of the most strongly UASINO-dependent gene, INO1, both in the absence and presence of inositol, suggesting elevated PA levels in the nuclear envelope/ER. Indeed, Opi1-GFP localization differed in the sah1 mutant and was not responsive to the presence of 10 μm inositol, which induced nuclear translocation of Opi1-GFP in the wild type.
Because PA is a precursor for both phospholipids and triacylglycerols, we hypothesized that impaired conversion to phospholipids, e.g. under conditions of Sah1p depletion as well as in methylation-defective mutants, results in preferred utilization of PA for TG synthesis. Indeed, depletion of Sah1 had a similar effect on TG accumulation as mutations in phospholipid methyltransferases, Cho2 and Opi3. These data under-score the importance of the Sah1-catalyzed reaction in the regulation of lipid synthesis, with potential implications for lipid pathologies. Our data are in accordance with the observation that defective phospholipid methylation leads to accumulation of triacylglycerols in mouse liver (27). Also, patients deficient in Sah1 exhibit in particular, elevated AdoHcy levels, low levels of plasma PC, and subcutaneous and hepatic fat accumulation (22-24).
Elevated plasma homocysteine levels are considered a risk factor for many pathological conditions, including cardiovascular diseases (55). However, in vivo studies showed that increased Hcy is associated with elevated plasma AdoHcy levels (56) and that AdoHcy rather than Hcy contributes to cardiovascular disease (16, 57). Accordingly, AdoHcy but not homocysteine itself is toxic to yeast cells compromised in homocysteine metabolism through the trans-sulfuration pathway (58). Because cardiovascular disease is well correlated with global DNA hypomethylation in patients (16), it was suggested that AdoHcy-induced hypomethylation represents an alternative mechanism for the pathogenesis of diseases caused by hyper-homocysteinemia (59). On the other hand, diet-induced hyper-homocysteinemia in mice leads to cholesterol and triacylglycerol accumulation in liver (60). We show that homocysteine supplementation of yeast leads to TG accumulation via an Sah1-dependent mechanism both in wild type and a met25 met6 str4 mutant, which is deficient in homocysteine catabolism. According to this model, homocysteine is converted by Sah1 to AdoHcy, which attenuates AdoMet-dependent methyltransferase activities and leads to an inhibition of phospholipid methylation, accumulation of phosphatidic acid in the ER, and its preferred conversion into triacylglycerols. Supporting a direct involvement of Sah1 in the pathology of lipid accumulation in mammals, supplementation with betaine, which aids in remethylation of homocysteine, attenuates alcoholic steatosis by restoring PE to PC methylation in liver (61), presumably through normalizing hepatocellular AdoMet/AdoHcy ratios. Altogether, our findings uncover an unexpected regulatory link between phospholipid methylation and triacylglycerol accumulation and demonstrate a novel molecular mechanism for homocysteine as a risk factor for lipid pathologies.
This work was supported by the Austrian Science Fund FWF, Project P18094-B14 (to O. T.) and Project SFB Lipotox (F3005) (to S. D. K.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: Sah1, S-adenosyl-l-homocysteine hydrolase; AdoMet, S-adenosyl-l-methionine; Hcy, homocysteine; AdoHcy, S-adenosyl-l-homocysteine; PC, phosphatidylcholine; PE, phosphatidylethanol-amine; TG, triacylglycerol; Cho, choline; INO1, inositol-3-phosphate synthase; Cho2, phosphatidylethanolamine methyltransferase; Opi3, phospholipid methyltransferase; MET25, O-acetylhomoserine sulfhydrylase; STR4, cystathionine-β-synthase; ER, endoplasmic reticulum; YPD, yeast extract/peptone/dextrose; SPE, solid-phase extraction; HPLC, high performance liquid chromatography; MS, mass spectroscopy; GFP, green fluorescent protein; ESI, electrospray ionization.
References
- 1.Mushegian, A. R., Garey, J. R., Martin, J., and Liu, L. X. (1998) Genome Res. 8 590-598 [DOI] [PubMed] [Google Scholar]
- 2.Tehlivets, O., Hasslacher, M., and Kohlwein, S. D. (2004) FEBS Lett. 577501 -506 [DOI] [PubMed] [Google Scholar]
- 3.Wolfe, M. S., and Borchardt, R. T. (1991) J. Med. Chem. 341521 -1530 [DOI] [PubMed] [Google Scholar]
- 4.Lee, H., Kim, J. H., Chae, Y. J., Ogawa, H., Lee, M. H., and Gerton, G. L. (1998) Biol. Reprod. 581437 -1444 [DOI] [PubMed] [Google Scholar]
- 5.Chiang, P. K., Gordon, R. K., Tal, J., Zeng, G. C., Doctor, B. P., Pardhasaradhi, K., and McCann, P. P. (1996) FASEB J. 10471 -480 [PubMed] [Google Scholar]
- 6.Lindsay, H., and Adams, R. L. (1996) Biochem. J. 320473 -478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Strahl, B. D., Grant, P. A., Briggs, S. D., Sun, Z. W., Bone, J. R., Caldwell, J. A., Mollah, S., Cook, R. G., Shabanowitz, J., Hunt, D. F., and Allis, C. D. (2002) Mol. Cell. Biol. 221298 -1306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davis, C. D., and Ross, S. A. (2007) Nutr. Rev. 6588 -94 [DOI] [PubMed] [Google Scholar]
- 9.Vance, D. E. (2006) FEBS Lett. 5805430 -5435 [DOI] [PubMed] [Google Scholar]
- 10.De La Haba, G., and Cantoni, G. L. (1959) J. Biol. Chem. 234603 -608 [PubMed] [Google Scholar]
- 11.Hoffman, D. R., Marion, D. W., Cornatzer, W. E., and Duerre, J. A. (1980) J. Biol. Chem. 25510822 -10827 [PubMed] [Google Scholar]
- 12.Isa, Y., Mishima, T., Tsuge, H., and Hayakawa, T. (2006) J. Nutr. Sci. Vitaminol. (Tokyo) 52 479-482 [DOI] [PubMed] [Google Scholar]
- 13.Moffatt, B. A., Stevens, Y. Y., Allen, M. S., Snider, J. D., Pereira, L. A., Todorova, M. I., Summers, P. S., Weretilnyk, E. A., Martin-McCaffrey, L., and Wagner, C. (2002) Plant Physiol. 128812 -821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zaina, S., Lindholm, M. W., and Lund, G. (2005) J. Nutr. 1355 -8 [DOI] [PubMed] [Google Scholar]
- 15.Dayal, S., Bottiglieri, T., Arning, E., Maeda, N., Malinow, M. R., Sigmund, C. D., Heistad, D. D., Faraci, F. M., and Lentz, S. R. (2001) Circ. Res. 881203 -1209 [DOI] [PubMed] [Google Scholar]
- 16.Castro, R., Rivera, I., Struys, E. A., Jansen, E. E., Ravasco, P., Camilo, M. E., Blom, H. J., Jakobs, C., and Tavares de Almeida, I. (2003) Clin. Chem. 491292 -1296 [DOI] [PubMed] [Google Scholar]
- 17.Chen, Z., Karaplis, A. C., Ackerman, S. L., Pogribny, I. P., Melnyk, S., Lussier-Cacan, S., Chen, M. F., Pai, A., John, S. W., Smith, R. S., Bottiglieri, T., Bagley, P., Selhub, J., Rudnicki, M. A., James, S. J., and Rozen, R. (2001) Hum. Mol. Genet. 10 433-443 [DOI] [PubMed] [Google Scholar]
- 18.Li, Z., Agellon, L. B., Allen, T. M., Umeda, M., Jewell, L., Mason, A., and Vance, D. E. (2006) Cell Metab. 3 321-331 [DOI] [PubMed] [Google Scholar]
- 19.Pan, H. J., Agate, D. S., King, B. L., Wu, M. K., Roderick, S. L., Leiter, E. H., and Cohen, D. E. (2006) FEBS Lett. 5805953 -5958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hartz, C. S., Nieman, K. M., Jacobs, R. L., Vance, D. E., and Schalinske, K. L. (2006) J. Nutr. 1363005 -3009 [DOI] [PubMed] [Google Scholar]
- 21.Miller, M. W., Duhl, D. M., Winkes, B. M., Arredondo-Vega, F., Saxon, P. J., Wolff, G. L., Epstein, C. J., Hershfield, M. S., and Barsh, G. S. (1994) EMBO J. 131806 -1816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buist, N. R., Glenn, B., Vugrek, O., Wagner, C., Stabler, S., Allen, R. H., Pogribny, I., Schulze, A., Zeisel, S. H., Baric, I., and Mudd, S. H. (2006) J. Inherited Metab. Dis. 29 538-545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baric, I., Fumic, K., Glenn, B., Cuk, M., Schulze, A., Finkelstein, J. D., James, S. J., Mejaski-Bosnjak, V., Pazanin, L., Pogribny, I. P., Rados, M., Sarnavka, V., Scukanec-Spoljar, M., Allen, R. H., Stabler, S., Uzelac, L., Vugrek, O., Wagner, C., Zeisel, S., and Mudd, S. H. (2004) Proc. Natl. Acad. Sci. U. S. A. 1014234 -4239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baric, I., Cuk, M., Fumic, K., Vugrek, O., Allen, R. H., Glenn, B., Maradin, M., Pazanin, L., Pogribny, I., Rados, M., Sarnavka, V., Schulze, A., Stabler, S., Wagner, C., Zeisel, S. H., and Mudd, S. H. (2005) J. Inherited Metab. Dis. 28 885-902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Finkelstein, J. D. (2006) J. Nutr. 136 Suppl. 6,1750 -1754 [Google Scholar]
- 26.Mendoza-Cozatl, D., Loza-Tavera, H., Hernandez-Navarro, A., and Moreno-Sanchez, R. (2005) FEMS Microbiol. Rev. 29653 -671 [DOI] [PubMed] [Google Scholar]
- 27.Hansen, J., and Johannesen, P. F. (2000) Mol. Gen. Genet. 263535 -542 [DOI] [PubMed] [Google Scholar]
- 28.Breton, A., and Surdin-Kerjan, Y. (1977) J. Bacteriol. 132224 -232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thomas, D., and Surdin-Kerjan, Y. (1997) Microbiol. Mol. Biol. Rev. 61 503-532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vance, J. E., and Vance, D. E. (2004) Biochem. Cell Biol. 82113 -128 [DOI] [PubMed] [Google Scholar]
- 31.de Kroon, A. I. (2007) Biochim. Biophys. Acta 1771343 -352 [DOI] [PubMed] [Google Scholar]
- 32.Stead, L. M., Brosnan, J. T., Brosnan, M. E., Vance, D. E., and Jacobs, R. L. (2006) Am. J. Clin. Nutr. 83 5-10 [DOI] [PubMed] [Google Scholar]
- 33.Loewen, C. J., Gaspar, M. L., Jesch, S. A., Delon, C., Ktistakis, N. T., Henry, S. A., and Levine, T. P. (2004) Science 3041644 -1647 [DOI] [PubMed] [Google Scholar]
- 34.Chen, M., Hancock, L. C., and Lopes, J. M. (2007) Biochim. Biophys. Acta 1771310 -321 [DOI] [PubMed] [Google Scholar]
- 35.Bachhawat, N., Ouyang, Q., and Henry, S. A. (1995) J. Biol. Chem. 27025087 -25095 [DOI] [PubMed] [Google Scholar]
- 36.Gaspar, M. L., Aregullin, M. A., Jesch, S. A., Nunez, L. R., Villa-Garcia, M., and Henry, S. A. (2007) Biochim. Biophys. Acta 1771241 -254 [DOI] [PubMed] [Google Scholar]
- 37.Jesch, S. A., Liu, P., Zhao, X., Wells, M. T., and Henry, S. A. (2006) J. Biol. Chem. 28124070 -24083 [DOI] [PubMed] [Google Scholar]
- 38.Henry, S. A., and Patton-Vogt, J. L. (1998) Prog. Nucleic Acid Res. Mol. Biol. 61 133-179 [DOI] [PubMed] [Google Scholar]
- 39.Carman, G. M., and Henry, S. A. (2007) J. Biol. Chem. 28237293 -37297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jesch, S. A., Zhao, X., Wells, M. T., and Henry, S. A. (2005) J. Biol. Chem. 2809106 -9118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gietz, R. D., Schiestl, R. H., Willems, A. R., and Woods, R. A. (1995) Yeast 11 355-360 [DOI] [PubMed] [Google Scholar]
- 42.Gellekink, H., van Oppenraaij-Emmerzaal, D., van Rooij, A., Struys, E. A., den Heijer, M., and Blom, H. J. (2005) Clin. Chem. 511487 -1492 [DOI] [PubMed] [Google Scholar]
- 43.Folch, J., Lees, M., and Sloane Stanley, G. H. (1957) J. Biol. Chem. 226497 -509 [PubMed] [Google Scholar]
- 44.Schneiter, R., and Daum, G. (2006) Methods Mol. Biol. 31375 -84 [DOI] [PubMed] [Google Scholar]
- 45.Sas, B., Peys, E., and Helsen, M. (1999) J. Chromatogr. A 864179 -182 [DOI] [PubMed] [Google Scholar]
- 46.Greenberg, M. L., Goldwasser, P., and Henry, S. A. (1982) Mol. Gen. Genet. 186157 -163 [DOI] [PubMed] [Google Scholar]
- 47.McGee, T. P., Skinner, H. B., and Bankaitis, V. A. (1994) J. Bacteriol. 1766861 -6868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Niedenthal, R. K., Riles, L., Johnston, M., and Hegemann, J. H. (1996) Yeast 12 773-786 [DOI] [PubMed] [Google Scholar]
- 49.Thomas, D., Barbey, R., and Surdin-Kerjan, Y. (1990) J. Biol. Chem. 26515518 -15524 [PubMed] [Google Scholar]
- 50.Kurat, C. F., Natter, K., Petschnigg, J., Wolinski, H., Scheuringer, K., Scholz, H., Zimmermann, R., Leber, R., Zechner, R., and Kohlwein, S. D. (2006) J. Biol. Chem. 281491 -500 [DOI] [PubMed] [Google Scholar]
- 51.Gaynor, P. M., and Carman, G. M. (1990) Biochim. Biophys. Acta 1045156 -163 [DOI] [PubMed] [Google Scholar]
- 52.McGraw, P., and Henry, S. A. (1989) Genetics 122317 -330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mizunuma, M., Miyamura, K., Hirata, D., Yokoyama, H., and Miyakawa, T. (2004) Proc. Natl. Acad. Sci. U. S. A. 1016086 -6091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Winzeler, E. A., Shoemaker, D. D., Astromoff, A., Liang, H., Anderson, K., Andre, B., Bangham, R., Benito, R., Boeke, J. D., Bussey, H., Chu, A. M., Connelly, C., Davis, K., Dietrich, F., Dow, S. W., El Bakkoury, M., Foury, F., Friend, S. H., Gentalen, E., Giaever, G., Hegemann, J. H., Jones, T., Laub, M., Liao, H., Liebundguth, N., Lockhart, D. J., Lucau-Danila, A., Lussier, M., M'Rabet, N., Menard, P., Mittmann, M., Pai, C., Rebischung, C., Revuelta, J. L., Riles, L., Roberts, C. J., Ross-MacDonald, P., Scherens, B., Snyder, M., Sookhai-Mahadeo, S., Storms, R. K., Veronneau, S., Voet, M., Volckaert, G., Ward, T. R., Wysocki, R., Yen, G. S., Yu, K., Zimmermann, K., Philippsen, P., Johnston, M., and Davis, R. W. (1999) Science 285901 -906 [DOI] [PubMed] [Google Scholar]
- 55.Vollset, S. E., Refsum, H., Tverdal, A., Nygard, O., Nordrehaug, J. E., Tell, G. S., and Ueland, P. M. (2001) Am. J. Clin. Nutr. 74130 -136 [DOI] [PubMed] [Google Scholar]
- 56.Loehrer, F. M., Tschopl, M., Angst, C. P., Litynski, P., Jager, K., Fowler, B., and Haefeli, W. E. (2001) Atherosclerosis 154147 -154 [DOI] [PubMed] [Google Scholar]
- 57.Kerins, D. M., Koury, M. J., Capdevila, A., Rana, S., and Wagner, C. (2001) Am. J. Clin. Nutr. 74 723-729 [DOI] [PubMed] [Google Scholar]
- 58.Christopher, S. A., Melnyk, S., James, S. J., and Kruger, W. D. (2002) Mol. Genet. Metab. 75 335-343 [DOI] [PubMed] [Google Scholar]
- 59.Detich, N., Hamm, S., Just, G., Knox, J. D., and Szyf, M. (2003) J. Biol. Chem. 27820812 -20820 [DOI] [PubMed] [Google Scholar]
- 60.Werstuck, G. H., Lentz, S. R., Dayal, S., Hossain, G. S., Sood, S. K., Shi, Y. Y., Zhou, J., Maeda, N., Krisans, S. K., Malinow, M. R., and Austin, R. C. (2001) J. Clin. Investig. 1071263 -1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kharbanda, K. K. (2007) World J. Gastroenterol. 134947 -4954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wolinski, H., and Kohlwein, S. D. (2008) in Membrane Trafficking (Vancura, A., ed) Vol.457 , Humanna Press, Totowa, NJ