

Abstract
The anaphase-promoting complex (APC) regulates cell division in eukaryotes by targeting specific proteins for destruction. APC substrates generally contain one or more short degron sequences that help mediate their recognition and poly-ubiquitination by the APC. The most common and well characterized degrons are the destruction box (D box) and the KEN box. The budding yeast Acm1 protein, an inhibitor of Cdh1-activated APC (APCCdh1) also contains several conserved D and KEN boxes, and here we report that two of these located in the central region of Acm1 constitute a pseudosubstrate sequence required for APCCdh1 inhibition. Acm1 interacted with and inhibited substrate binding to the WD40 repeat domain of Cdh1. Combined mutation of the central D and KEN boxes strongly reduced both binding to the Cdh1 WD40 domain and APCCdh1 inhibition. Despite this, the double mutant, but not wild-type Acm1, was poly-ubiquitinated by APCCdh1 in vitro. Thus, unlike substrates in which D and KEN boxes promote ubiquitination, these same elements in the central region of Acm1 prevent ubiquitination. We propose that this unique property of the Acm1 degron sequences results from an unusually high affinity interaction with the substrate receptor site on the WD40 domain of Cdh1 that may serve both to promote APC inhibition and protect Acm1 from destruction.
The anaphase-promoting complex (APC),4 is a highly conserved multisubunit ubiquitin ligase and an important regulator of eukaryotic cell division (1). It targets key cell cycle proteins for proteolysis via the ubiquitin pathway (2), including securin, an inhibitor of chromosome segregation, and the S and M phase cyclin subunits of cyclin-dependent kinase (CDK). Securin proteolysis triggers the initiation of anaphase once all sister chromatids have been properly bi-oriented on the mitotic spindle during metaphase (3–5). Cyclin proteolysis leads to inactivation of CDK, which is necessary for cells to exit from mitosis (6–8). In addition, APC controls the destruction of numerous other proteins such as Aurora A kinase, Polo-like kinases, the APC co-activator Cdc20, the Skp2 F-box protein, various spindle components, and replication factors such as Cdc6, Dbf4, and geminin (1). Several of these substrates were recently reported to be overexpressed in a wide range of malignant cancers (9), highlighting the critical role APC plays in maintaining genomic stability and proper regulation of the cell division cycle.
Although many substrates of APC have been identified, the mechanism by which they are specifically recognized is still poorly understood. Substrate recognition was originally proposed to be mediated by the Cdc20 and Cdh1 proteins, two related WD40 repeat domain proteins that are essential for APC activity at different times during the cell cycle (10, 11). The identification of direct interactions between these co-activators and several substrates (12–16) led to a model for APC activation in which Cdc20 and Cdh1 acted as substrate-recruiting factors, much like the F-box proteins of the SCF ubiquitin ligase (17). However, more recent work suggests that APC and co-activator together contribute to substrate recognition (18–23), and evidence emerged that APC itself can interact directly with substrates (24, 25). Unfortunately, the exact mechanism by which Cdc20 and Cdh1 activate APC remains unclear.
Because substrate selectivity by APC in vivo is highly specific, one might expect substrates to share common sequence motifs required for their recognition. To a certain extent this is true. The destruction box (D box) was originally identified as a conserved 9-amino acid motif in sea urchin cyclin B (26). Mutations in the D box (consensus RXXLXXXXN) in cyclin B prevented its ubiquitination and proteolysis. Functional RXXLD boxes are found in the majority of APC substrates, but not all. Additional short amino acid sequences have subsequently been identified that can direct APC-dependent ubiquitination and proteolysis either in the absence of, or in conjunction with, D boxes. The most common and well characterized of these additional degrons is the KEN box, originally identified in human Cdc20 (27), but now found in many APC substrates. Additional degrons include the A box found in Xenopus Aurora A kinase (28, 29), the CRY box found in mammalian Cdc20 (30), a GxEN sequence in the Xenopus chromokinesin XKid (31), and an LXEXXXN sequence in budding yeast Spo13 (32). The KEN box was thought to be a specific signal for recognition by Cdh1 (27). This theory has been supported by several studies (33–35), but exceptions have also been found (12, 36, 37). Moreover, the existence of the other, apparently unrelated, degrons has further clouded the understanding of substrate recognition by APC. Also, many proteins that are clearly not APC substrates contain sequences that match the D box, KEN box, and other APC degrons. Thus, sequence context surrounding the degrons, and possibly other factors, are clearly important contributors to substrate recognition as well.
A recently identified mechanism for regulation of APC activity is pseudosubstrate inhibition. Pseudosubstrate inhibition is best characterized in kinases where intramolecular amino acid motifs mimicking a natural substrate sit in the kinase active site to suppress activity in the absence of an activating signal (38). The vertebrate Emi1 protein was recently proposed to be a pseudosubstrate inhibitor of the APC (39). Emi1 competitively inhibits substrate binding to the APC and co-activators (39, 40). This activity is dependent on a D box in Emi1 suggesting that Emi1 interacts with APC similar to a true substrate. But why is it not ubiquitinated like a substrate and targeted for proteolysis? Miller et al. (39) found that the zinc binding domain of Emi1 was essential for APC inhibition and removal of this region allowed Emi1 to be ubiquitinated and destroyed in a D box-dependent manner. Thus, the Emi1 D box promotes high affinity binding to the substrate recognition site on the coactivator-APC complex while the zinc binding region blocks the catalytic activity of APC. The yeast Mad3 protein, an essential component of the spindle checkpoint that inhibits APCCdc20 activity, was also proposed to act by a pseudosubstrate mechanism (36). This conclusion was based on the observation that mutations in two conserved KEN boxes and a D box in Mad3 compromise its interaction with Cdc20, its ability to block substrate binding to Cdc20, and its ability to function in the spindle checkpoint. Another recent report obtained similar results but also provided evidence that Mad3 is a substrate of APCCdh1 (37), raising the intriguing possibility that it inhibits one form of APC (APCCdc20) and is a substrate of the other (APCCdh1). Pseudosubstrate inhibitors will likely be useful models for further defining the determinants of APC substrate recognition.
Recently, the budding yeast Acm1 protein was identified as a stable binding partner of Cdh1 and an inhibitor of APCCdh1 activity (41, 42) that contributes to spindle pole body separation (43). Acm1 contains conserved D box and KEN box sequences, suggesting that it also could be a pseudosubstrate inhibitor similar to Emi1 and Mad3. We have addressed that hypothesis here by studying the effects of D and KEN box mutations in Acm1. We conclude that Acm1 is indeed a pseudosubstrate inhibitor of APCCdh1. Interestingly, we observed a unique property of a D and KEN box pair in the central region of Acm1 required for high affinity Cdh1 binding and APCCdh1 inhibition. Contrary to their typical functions in APC substrates, these sequences prevent APCCdh1-catalyzed ubiquitination of Acm1. This could be important to promote the inhibitory function of Acm1 by protecting it from destruction. We discuss the implications for our understanding of substrate recognition and mechanisms of APC activation.
EXPERIMENTAL PROCEDURES
Strains, Plasmids, and Yeast Methods—Standard media and growth conditions were used for all yeast experiments.
All yeast strains and plasmids used in this study are listed in Table 1. Strain YKA294 was constructed by PCR-mediated replacement of the ACM1 coding sequence in YKA291 (42) with the KanMX4 cassette using standard procedures. Plasmids pHLP127 and pHLP128 expressing 3FLAG-tagged Cdh1 truncations 1–249 and 241–566, respectively, were constructed by amplifying the desired sequence by PCR and replacing the intact CDH1 sequence from pHLP130 (42) using NotI and XhoI restriction sites. pHLP273 was constructed by subcloning the XbaI-XhoI fragment from pHLP128 into p413ADH. Mutation of Acm1 degron residues (Arg and Leu of the D boxes and Lys, Glu, and Asn of the KEN box) to alanine were generated by site-directed mutagenesis using the QuikChange kit (Stratagene) and either pHLP117 or pHLP109 (42) as template. All mutations and all plasmids constructed using PCR were confirmed by DNA sequencing. Other plasmids and yeast strains have been described previously (see Table 1 for references).
TABLE 1.
Yeast strains and plasmids
|
Strain |
Background and relevant genotype |
Source |
||
| YKA150 | W303 MATa bar1::URA3 | (45) | ||
| YKA155 | W303 MATa bar1::URA3 CDC27-3FLAG:KanMX4 | (45) | ||
| YKA226 | BY4741 MATa 3HA-ACM1 | (42) | ||
| YKA247 | W303 MATa bar1::URA3 acm1::KanMX4 | (42) | ||
| YKA254 | W303 MATa acm1::KanMX4 | (49) | ||
| YKA257 | BY4741 MATa bar1::hisG 3HA-HSL1 acm1::KanMX4 | (42) | ||
|
YKA294 |
BY4741 MATa bar1::hisG 3FLAG-CDH1 acm1::KanMX4 |
This study |
||
|
Plasmid
namea |
Marker |
Promoter |
Expressed protein |
|
| pHLP109 | LEU2 | GAL1 | HA-Acm1 | |
| pHLP110 | LEU2 | GAL1 | HA-Acm1-5A | |
| pHLP117 | LEU2 | ACM1 | 3HA-Acm1 | |
| pHLP121 | LEU2 | GAL1 | HA-Acm1-db1 | |
| pHLP122 | LEU2 | GAL1 | HA-Acm1-db3 | |
| pHLP123 | LEU2 | GAL1 | HA-Acm1-ken | |
| pHLP126 | LEU2 | GAL1 | HA-Acm1-db1/db3/ken | |
| pHLP127 | LEU2 | ADH | 3FLAG-Cdh1-(1-249) | |
| pHLP128 | LEU2 | ADH | 3FLAG-Cdh1-(241-566) | |
| pHLP132 | LEU2 | ACM1 | 3HA-Acm1-db1 | |
| pHLP133 | LEU2 | ACM1 | 3HA-Acm1-db3 | |
| pHLP134 | LEU2 | ACM1 | 3HA-Acm1-ken | |
| pHLP140 | LEU2 | ACM1 | 3HA-Acm1-db1/db3 | |
| pHLP148 | LEU2 | ACM1 | 3HA-Acm1-db1/ken | |
| pHLP149 | LEU2 | ACM1 | 3HA-Acm1-db3/ken | |
| pHLP151 | LEU2 | ACM1 | 3HA-Acm1-db1/db3/ken | |
| pHLP158 | LEU2 | ACM1 | 3HA-Acm1-db2 | |
| pHLP159 | LEU2 | ACM1 | 3HA-Acm1-db1/db2 | |
| pHLP231 | TRP1 | GAL1 | 3FLAG-Cdh1 | |
| pHLP273 | HIS3 | ADH | 3FLAG-Cdh1-(241-566) | |
| pHLP274 | LEU2 | GAL1 | HA-Acm1-db3/ken | |
All plasmids carry a CEN/ARS origin for the low copy number.
Plasmids for in vitro transcription and translation to generate substrates for the ubiquitination assay were constructed by amplifying genes by PCR from yeast genomic DNA or available plasmid constructs and inserting the products into the NcoI and XhoI sites in pET28a. For HSL1 and FIN1, we used previously described gene fragments encoding the truncated amino acid sequences 667–872 and 1–152, respectively, which are sufficient for recognition by APCCdh1 (12, 44). ACM1 truncations were based on secondary structure predictions. Clb2 was synthesized from pRSET-CLB2 (18).
Protein Purification—To isolate APC, Cdc27–3FLAG was immunoaffinity-purified from YKA155 essentially as described (45) and stored in small aliquots at –80 °C immediately after elution from anti-FLAG antibody resin. 3FLAG-Cdh1 was purified as described (46) except phosphatase inhibitors were omitted and, following elution from anti-FLAG resin, was dialyzed into storage buffer (25 mm HEPES, pH 7.8, 100 mm NaCl, 50% glycerol) for 6 h. Aliquots were stored at –80 °C, and a working aliquot was kept at –20 °C, which maintained activity for at least a couple months. Ubc4-His6 and wild-type and mutant His6-Acm1 proteins were overexpressed in Escherichia coli and purified by Ni2+-affinity chromatography using 1-ml HisTrap columns and anÁKTA fast-protein liquid chromatography system (GE Healthcare), dialyzed into storage buffer overnight, and stored in aliquots at –80 °C. Working aliquots were kept at –20 °C. All protein concentrations were estimated by densitometric analysis of Coomassie Blue-stained polyacrylamide gels using a bovine serum albumin standard curve.
Ubiquitination Assay—l-[35S]Methionine (GE Healthcare or MP Biomedicals) was used to label APC substrates in TnT Quick Coupled Transcription/Translation reactions (Promega, Madison, WI) according to the product instructions. Ubiquitination reactions were based largely on assays from the Morgan and Barford groups (47, 48) and contained 200 nm yeast Uba1 (Boston Biochem), 10 μm Ubc4-His6, ∼100 nm 3FLAG-Cdh1, ∼2 nm APC, 1 mg/ml recombinant human ubiquitin (Boston Biochem), 20 μg/ml ubiquitin aldehyde (Boston Biochem), 25 μm MG-132 (Peptides International), and 3–4 μl of the appropriate TnT-generated 35S-labeled substrate all in 20 mm HEPES, pH 7.8, 5 mm ATP, 10 mm MgCl2, and 0.5 mm dithiothreitol. The Uba1, Ubc4-His6, and ubiquitin were preincubated together in the presence of Mg2+-ATP first for 10 min at 23 °C to generate ubiquitin-charged E2 prior to addition of the remaining components. Reactions (20 μl of total volume) were incubated at 23 °C for 30 min and stopped by boiling in SDS loading buffer. Products were resolved by SDS-PAGE, and dried gels were subjected to PhosphorImager analysis using a Molecular Dynamics Typhoon 8600 imaging system and ImageQuant software.
Co-immunoprecipitation—Co-IP assays were performed exactly as described (42, 49). Where indicated, NaCl concentration was adjusted from 100 mm to 400 mm. For immunoblotting, anti-HA 12CA5 (Roche Applied Science) was used at 1:1,000 and anti-FLAG M2 (Sigma) at 1:10,000 dilutions. Immunoblots were developed with ECL plus reagents (GE Healthcare).
Protein Stability and Cell Cycle Profiles—Protein stability was measured by galactose promoter shutoff assays exactly as described (49). Cell cycle expression profiles were determined by α-factor block and release and immunoblotting as described (42). In Vivo APC Inhibition Assay—Inhibition of APCCdh1 was measured in vivo as described previously (42).
RESULTS
Acm1 Is a General Inhibitor of APCCdh1 Activity Independent of 14-3-3 Binding and Phosphorylation—Acm1 purified from yeast was previously shown to inhibit the ubiquitination of Clb2 by APCCdh1 in vitro (41). We first wanted to know if Acm1 is a general inhibitor of APCCdh1 or is specific for Clb2, and also to determine if CDK phosphorylation and 14-3-3 protein binding, two known regulatory mechanisms controlling Acm1 stability (41, 42, 49), were important for inhibitory function. This information was critical to establishing an appropriate in vitro assay to study the mechanism of APCCdh1 inhibition by Acm1. To do this, we tested the ability of recombinant His6-Acm1 purified from E. coli to inhibit APCCdh1-catalyzed ubiquitination of the well characterized substrates Hsl1667–872 (12), Fin11–152 (44), and Pds1 (3), in addition to Clb2 (Fig. 1, A and B). Acm1 effectively inhibited ubiquitination of all four substrates and showed a similar concentration dependence for each. We conclude that Acm1 is a general inhibitor of APCCdh1 activity.
FIGURE 1.

Acm1 is a general inhibitor of APCCdh1, independent of CDK phosphorylation and 14-3-3 protein binding. A, APCCdh1-catalyzed ubiquitination of [35S]methionine-labeled substrates Clb2, Hsl1667–872, and Fin11–152 was assayed in the absence or presence of 125 nm recombinant His6-Acm1 purified from E. coli as described under “Experimental Procedures.” Reaction products (ubiquitin conjugates) indicated by the bracket are detected based on reduced mobility during SDS-PAGE. B, inhibition of APCCdh1-catalyzed ubiquitination of Clb2, Hsl1667–872, and Pds1 was measured as in A as a function of recombinant His6-Acm1 concentration. NC is a negative control lacking APC. Reaction products are labeled “Ubiq. Conj.” C, 10-fold serial dilutions of strain YKA247 expressing the indicated proteins from the GAL1 promoter on centromeric plasmids were spotted on rich media plates containing either glucose or galactose as the carbon source and grown for several days at 30 °C.
The results in Fig. 1 using recombinant His6-Acm1 also strongly suggest that CDK phosphorylation and 14-3-3 binding are not required for APC inhibition. To confirm this, we tested the ability of an Acm1 mutant lacking CDK phosphorylation sites, Acm1–5A (49), to inhibit APCCdh1 in vivo. Overexpression of Cdh1 is lethal due to constitutive APC activity that prevents proper cyclin accumulation and mitotic entry (10, 11). We previously showed that co-overexpression of Acm1 suppresses this lethality (42), establishing an assay to monitor APCCdh1 inhibition in vivo. The Acm1–5A mutant cannot be phosphorylated by CDK and is defective in 14-3-3 protein binding (49), yet when overexpressed in cells containing a lethal Cdh1 level, it fully restores viability like wild-type Acm1 (Fig. 1C). Thus, Acm1 does not require phosphorylation or binding to the 14-3-3 proteins Bmh1 and Bmh2 to act as an inhibitor of APCCdh1.
Conserved D Box and KEN Box Sequences in Acm1 Are Required for High Affinity Binding to the Substrate Receptor Site on the Cdh1 WD40 Domain—Alignment of Saccharomyces cerevisiae Acm1 with orthologs from other Saccharomyces species and other budding yeasts of the order Saccharomycetales revealed conserved sequence motifs common to APC substrates (Fig. 2). These include a D box near the N terminus (D box 1) and a D box (D box 3) and KEN box in the central region. An additional D box in the central region (D box 2) is not conserved. We speculated that the conserved degron-like sequences might be important for APC inhibition and that Acm1 might act as a pseudosubstrate inhibitor like Emi1 and Mad3 (36, 39). To test this, we made D box mutations (RXXLto AXXA) and KEN box mutations (KEN to AAA) alone and in various combinations. First, we examined the ability of mutant proteins expressed with an N-terminal 3HA tag from the ACM1 promoter to bind endogenous 3FLAG-Cdh1. Mutation of D box 1 (db1) and D box 2 (db2) had no effect on Cdh1 binding using a co-IP assay (Fig. 3A). Mutation of D-box 3 (db3) or the KEN-box (ken) reduced binding to Cdh1 and combining the two mutations (db3/ken) eliminated binding to Cdh1 in this assay. Therefore, these two motifs are required for the stable interaction between Acm1 and Cdh1 detected by co-IP.
FIGURE 2.

Acm1 contains substrate-like degron sequences that have been conserved during evolution. A, sections of Acm1 containing conserved D box (RXXL) and KEN box motifs from 6 Saccharomyces species aligned with ClustalW are shown. Consensus residues are highlighted in gray. Note that D box 2 is not conserved. B, a similar alignment of Acm1 orthologs from more distantly related budding yeasts with S. cerevisiae Acm1, illustrating conservation of D box 1, D box 3, and the KEN box. In both panels the asterisk indicates an invariant residue, “:” is a conservative substitution, and “.” is a semi-conservative substitution.
FIGURE 3.
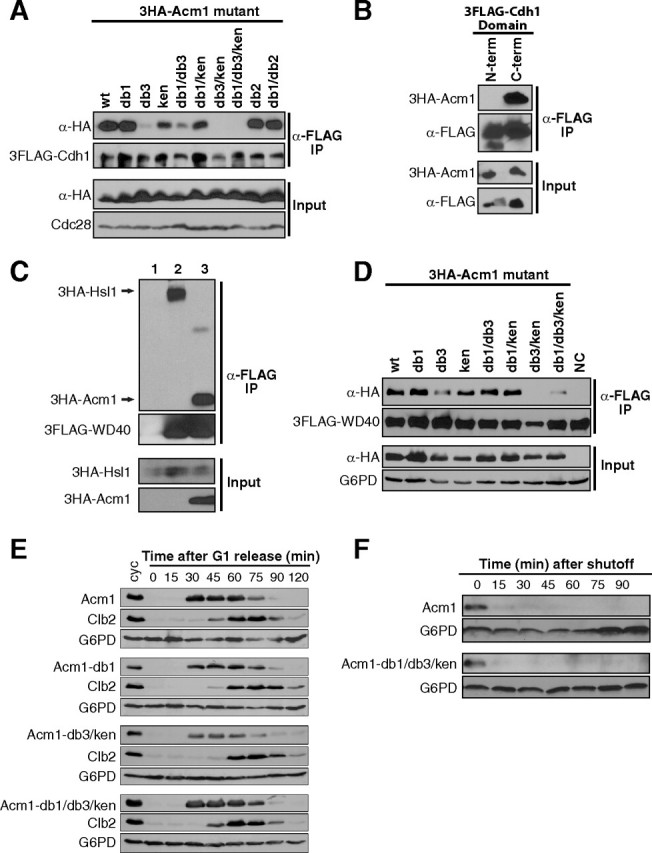
Central D box and KEN box sequences in Acm1 are required for high affinity binding to the Cdh1 WD40 domain. A, yeast strain YKA294 expressing endogenous 3FLAG-Cdh1 and containing centromeric plasmids expressing wild-type or the indicated mutant 3HA-Acm1 proteins from the ACM1 promoter were grown to mid-log phase. An anti-FLAG IP was performed from cell extracts and co-purification of 3HA-tagged protein monitored by anti-HA immunoblotting. Cdc28 is a loading control. B, the same procedure as in panel A using strain YKA226 expressing endogenous 3HA-Acm1 and containing a centromeric plasmid expressing 3FLAG-tagged N-terminal (amino acids 1–249) or C-terminal (amino acids 241–566) domains of Cdh1 expressed from the ADH promoter. C, yeast strain YKA257 expressing endogenous 3HA-Hsl1 in an acm1Δ background was transformed with empty control plasmids (lane 1), a centromeric plasmid expressing 3FLAG-Cdh1WD40 from the ADH promoter (lane 2), or the same 3FLAG-Cdh1WD40 plasmid plus a centromeric plasmid expressing 3HA-Acm1 from the GAL1 promoter. Cells were grown to mid-log phase in raffinose-containing medium, then 2% galactose added and cells harvested after 2 h and subjected to α-FLAG co-IP. D, the same experiment described in panel A, except strain YKA247 contained centromeric plasmids expressing 3FLAG-Cdh1WD40 (amino acids 241–566 only) from the ADH promoter and the indicated 3HA-Acm1 variant from the ACM1 promoter. Also, NaCl concentration in the co-IP buffer was increased from 100 mm to 400 mm. G6PD is a loading control. NC is a control lacking a 3HA-tagged Acm1 protein. E, synchronous cultures of strain YKA254 harboring centromeric plasmids expressing wild-type or the indicated mutant 3HA-Acm1 protein from the ACM1 promoter were obtained by G1α-factor arrest and then released into fresh medium. Samples taken at the indicated time points were analyzed by anti-HA, anti-Clb2, and anti-G6PD (loading control) immunoblotting, and α-factor was added back at 60 min to re-arrest cells in the subsequent G1. cyc, asynchronous cycling cells. F, the stability of HA-Acm1 and the HA-Acm1-db1/db3/ken mutant were assessed by promoter shutoff in α-factor-arrested G1 YKA150 cells. After arrest, expression was induced with galactose for 2 h and quenched by addition of glucose and cycloheximide (time 0). The level of each protein at the indicated time points was monitored by anti-HA immunoblotting. G6PD is a loading control.
Previous evidence suggested that Acm1 could inhibit the interaction of certain substrates with Cdh1 (41, 42). Because the WD40 domain of Cdh1 is thought to contain a D-box receptor that contributes to substrate binding (20), and because Acm1 interaction with Cdh1 is dependent on a D box and KEN box, we tested whether Acm1 specifically interacts with and inhibits substrate binding to the Cdh1 WD40 domain. First, we independently expressed either the N-terminal regulatory or C-terminal WD40 domain of Cdh1 as a 3FLAG fusion and monitored association with endogenous 3HA-Acm1 by co-IP. The boundary between the domains was chosen based on a stable naturally occurring proteolytic fragment of Cdh1 that contains the entire WD40 region (data not shown). Acm1 interacted very strongly with the Cdh1 WD40 domain but showed no interaction with the N-terminal domain (Fig. 3B). The 14-3-3 proteins Bmh1 and Bmh2 were also present in the WD40 complex, as expected (not shown).
The APC substrate Hsl1 forms a stable, direct interaction with Cdh1 that is dependent on a D box and KEN box and required for its proteolysis (12). We found that 3HA-Hsl1 also associated stably with the 3FLAG-Cdh1WD40 domain, consistent with the notion that the WD40 domain contains the substrate degron receptor(s) on Cdh1 (Fig. 3C, lane 2). Overexpressed 3HA-Acm1 displaced 3HA-Hsl1 from 3FLAG-Cdh1WD40 (Fig. 3C, lane 3), demonstrating that Acm1 and Hsl1 binding to the Cdh1 WD40 domain are mutually exclusive. The most likely explanation is high affinity competitive binding of Acm1 to the substrate receptor site.
To confirm that the association between Acm1 and Cdh1WD40 was also dependent on the central degron sequences we repeated the co-IPs from Fig. 3A using 3FLAG-Cdh1WD40 instead of full-length 3FLAG-Cdh1. Surprisingly, all Acm1 mutants associated with Cdh1WD40 to a similar extent (not shown). This suggested that additional sequences within Acm1 contribute to stable WD40 binding. The discrepancy is likely explained by the overexpression of 3FLAG-Cdh1WD40 from the ADH promoter, whereas full-length 3FLAG-Cdh1 used in Fig. 3A was expressed from its natural genomic locus. We therefore created more stringent co-IP conditions by increasing the salt concentration to 400 mm, matching that used in the original identification of the Acm1-Cdh1 complex (42). Under these conditions the results were similar to those observed with full-length Cdh1 (Fig. 3D). The db3 and ken mutations additively disrupted the Acm1-WD40 interaction. Collectively these results are consistent with a model for APC inhibition in which substrate-like degron sequences in Acm1 (in conjunction with additional unidentified sequence) allow it to stably occupy the substrate receptor site on the Cdh1 WD40 domain and competitively inhibit association of true substrates. This suggests that Acm1 acts as a pseudosubstrate inhibitor of APCCdh1.
Interestingly, none of the mutations we created in degron sequences affected the cell cycle expression profile of Acm1 in synchronized cultures (Fig. 3E). All Acm1 mutants were absent from G1 cells, appeared at the onset of S phase, and disappeared rapidly in mitosis, similar to wild-type Acm1. The mutations also had no significant impact on the stability of Acm1 in GAL promoter shutoff assays in G1-arrested cells (Fig. 3F). Typically, mutation of D-box and KEN-box sequences in APC substrates results in significant stabilization in vivo, particularly in G1. We previously provided evidence supporting the existence of an APC-independent proteolytic mechanism for Acm1 in late mitosis and G1 (49). The lack of effects of degron mutations on Acm1 stability is consistent with these previous observations and further supports pseudosubstrate (rather than true substrate) functions for the conserved central D box 3 and KEN box.
D Box and KEN Box Mutations in Acm1 Prevent APCCdh1 Inhibition in Vivo and in Vitro—If Acm1 inhibits APCCdh1 activity by blocking association of substrates with the Cdh1 WD40 domain, then the db3/ken double mutation that compromises binding to Cdh1 should also compromise APCCdh1 inhibition. We tested this both in vivo and in vitro. The Acm1-db1 and Acm1-ken mutants suppressed Cdh1-induced growth arrest like wild-type Acm1 and the Acm1-db3 mutant exhibited only a slight decrease in suppression (Fig. 4A), suggesting little to no loss of APCCdh1 inhibition. In contrast, the Acm1-db3/ken double mutant and the Acm1-db1/db3/ken triple mutant exhibited dramatically reduced suppression of the Cdh1-induced arrest. The residual suppression observed with these two mutants may result from the contribution of other regions in Acm1 to Cdh1 binding revealed in our co-IP studies with the WD40 domain. Based on this experiment, though, the third D box and KEN box are clearly critical for function of Acm1 as an APCCdh1 inhibitor in vivo, again consistent with a pseudosubstrate mechanism.
FIGURE 4.

The central D box and KEN box in Acm1 are required for full inhibition of APCCdh1 activity. A, 10-fold serial dilutions of strain YKA247 expressing the indicated proteins from the GAL1 promoter on centromeric plasmids were spotted on rich media plates containing either glucose or galactose and grown for several days at 30 °C. The α-HA immunoblot on the right compares the level of galactose-induced overexpression of each of the Acm1 variants. B, APC-catalyzed ubiquitination of Clb2 was assayed in the absence or presence of 500 nm wild-type (wt) recombinant His6-Acm1 or the indicated Acm1 mutants. Relative activity was obtained from the total ubiquitin conjugate signal (bracket). C, same as B except Hsl1667–872 was used as the substrate. D, the same quantities of the indicated recombinant His6-Acm1 proteins used in B and C were compared by anti-His6 immunoblot to demonstrate equivalent concentrations. E, inhibition of APC-catalyzed ubiquitination of Clb2 was measured as a function of inhibitor concentration for wild-type (wt) His6-Acm1 and the db3/ken and db1 mutants. F, identical to E except Hsl1667–872 was used as the substrate.
We next compared the ability of Acm1 and various degron mutants to inhibit APCCdh1 activity in the in vitro ubiquitination assay. At an inhibitor concentration of 500 nm Acm1 almost completely inhibited ubiquitination of Clb2 and Hsl1667–872, whereas the Acm1-db3/ken and Acm1-db1/db3/ken mutants showed little to no inhibition (Fig. 4, B and C). We also evaluated inhibition across a range of Acm1 concentrations and included Acm1-db1 to test whether D box 1 contributes to APCCdh1 inhibition in vitro (Fig. 4, E and F). Acm1 and Acm1-db1 inhibited ubiquitination of Clb2 and Hsl1667–872 with similar concentration dependence, confirming that D box 1 is not required for Acm1 inhibitory function. Acm1-db3/ken, in contrast, was a relatively poor inhibitor, suggesting that the decrease in binding to Cdh1 compromised its ability to inhibit APC activity. However, Acm1-db3/ken appeared to retain a low level of APCCdh1 inhibition, consistent with the in vivo inhibition assay.
Acm1 Can Be Mono-ubiquitinated by APCCdh1 in Vitro—Although Acm1 is a potent inhibitor of APCCdh1, when we tested it as a substrate of APCCdh1 in vitro we found it could be fairly efficiently ubiquitinated (Fig. 5A, first two lanes). To narrow down the region within Acm1 responsible for the observed ubiquitination we tested several truncated forms of Acm1 lacking sequence from the N and/or C termini. Acm1 fragments lacking the first 42 amino acids were poor substrates, whereas fragments containing the intact N terminus were all ubiquitinated to a similar extent (Fig. 5A). Although the percentage of substrate converted to product was substantial in these experiments, product formation appeared limited primarily to mono-ubiquitin conjugates (based on the size of the distinct product bands). To confirm this we compared product formation in the presence of either ubiquitin or methyl-ubiquitin (which supports conjugation to substrate lysines but blocks poly-ubiquitin chain formation) using Acm1, Clb2, or Hsl1667–872 as substrate. Whereas the size of ubiquitin conjugates formed on Clb2 and Hsl1667–872 was dramatically reduced by substitution of methyl-ubiquitin as expected, the pattern of ubiquitin conjugation to Acm1 was unchanged (Fig. 5B). In our in vitro assay, Acm1 can be efficiently mono-ubiquitinated by APCCdh1 but is not a good substrate for poly-ubiquitination.
FIGURE 5.

The central D box and KEN box restrict poly-ubiquitination of Acm1 by APCCdh1. A, full-length Acm1 (amino acids 1–209) and N- and C-terminal truncated forms were synthesized and 35S-labeled by in vitro coupled transcription/translation and used as substrates in an APCCdh1 ubiquitination assay. B, products of APCCdh1-catalyzed reactions using ubiquitin and methylated ubiquitin (me-Ubiquitin) were compared for the 35S-labeled substrates Acm1, Clb2, and Hsl1667–872. Arrows indicate the unmodified substrates. C, ubiquitination of Acm1 and Acm1-db1 by APCCdh1 were compared. The dominant mono-ubiquitin product (mono-ub) is indicated. The percentage of substrate converted to mono-ubiquitin conjugate was determined with Image-QuaNT. D, products of APCCdh1-catalyzed reactions using ubiquitin and methyl-ubiquitin were compared for the 35S-labeled substrates wild-type (wt) Acm1 and the Acm1-db3/ken mutant. E, ubiquitination of wild-type Acm1, Acm1-db3/ken, and Acm1-db1/db3/ken by APCCdh1 were compared. For Acm1-db3/ken, the dependence of poly-ubiquitination on yeast Ubc4 and Cdh1 are also shown. F, ubiquitination of full-length Acm1-(1–209) and the wild-type and db3/ken forms of an Acm1 fragment encompassing residues 42–177 by APCCdh1 was compared. Arrows point to unmodified substrate. Note the complete conversion of the Acm142–177 db3/ken substrate to poly-ubiquitin conjugates.
To determine if the highly conserved D box 1 was responsible for the mono-ubiquitination observed in Fig. 5A we compared APCCdh1-catalyzed ubiquitin conjugation to wild-type Acm1 and the Acm1-db1 mutant (Fig. 5C). The percentage of substrate converted to mono-ubiquitin conjugate was greatly reduced in the absence of D box 1, demonstrating that this sequence is a functional APC degron in vitro even though it is dispensable for pseudosubstrate inhibition of APC.
D Box and KEN Box Mutations Allow Acm1 to be Poly-ubiquitinated by APCCdh1 in Vitro—Residual mono-ubiquitination was still observed on Acm1-db1, which lacks an intact D box 1. We expected this residual ubiquitination to be dependent on D box 3 and the KEN box in the central region of Acm1 that are critical for high affinity binding to the substrate receptor site on Cdh1 and APCCdh1 inhibition. Surprisingly, the Acm1-db3/ken mutant appeared to be poly-ubiquitinated by APCCdh1 based on increased size of ubiquitin conjugates and their sensitivity to methyl-ubiquitin, similar to Clb2 and Hsl1667–872 (Fig. 5D). The poly-ubiquitination of Acm1-db3/ken was highly specific, because it required the yeast E2 enzyme Ubc4, yeast Cdh1, and yeast APC (Fig. 5E). Moreover, it was largely independent of D box 1, because the triple degron mutant Acm1-db1/db3/ken supported similar poly-ubiquitination (Fig. 5E) and an Acm1 fragment (residues 42–177) lacking the N terminus and harboring the db3/ken double mutation was an even more efficient substrate, being completely converted to high molecular weight poly-ubiquitin conjugates (Fig. 5F). These results are striking, because they suggest the central D box and KEN box in Acm1, instead of promoting ubiquitination like degrons in known APC substrates, actually restrict Acm1 ubiquitination by APCCdh1. The results also imply the existence of additional sequence(s) within Acm1 capable of supporting its recognition by APCCdh1, consistent with the weak inhibitory function of Acm1-db3/ken in vivo (Fig. 4A) and the retention of some binding affinity for the Cdh1 WD40 domain observed in the low salt co-IP experiments (not shown).
DISCUSSION
From this study we conclude that Acm1 is a general pseudosubstrate inhibitor of APCCdh1 and that inhibition is independent of regulatory CDK phosphorylation and 14-3-3 protein binding. Acm1 inhibits the ubiquitination of several well characterized APCCdh1 substrates in vitro. Our results suggest that inhibition of APC activity occurs via the stable binding of Acm1 to the WD40 domain of Cdh1, which competitively blocks substrate binding. The interaction between Acm1 and the Cdh1 WD40 domain and inhibition of APC activity are both dependent on a conserved D box and KEN box in the central region of Acm1. Acm1 mutants lacking these degron sequences, despite compromised APC inhibitory activity, are poly-ubiquitinated like substrates by APCCdh1, demonstrating that Acm1 contains the necessary structural elements for association with APC as a substrate (including acceptor lysines in appropriate positions). Acm1 therefore joins Emi1 and Mad3 as a family of proteins that negatively regulate APC activity by a pseudosubstrate mechanism. While our manuscript was under review, two additional reports appeared describing similar roles for Acm1 D and KEN boxes in pseudosubstrate inhibition of APCCdh1 (50, 51).
Pseudosubstrate inhibition appears to be a commonly evolved mechanism for regulating APC activity. A related mode of APC inhibition was also found to be biologically relevant recently. Substrates can inhibit the proteolysis of other substrates through simple competition for APC binding. Most notably, the Schizosaccharomyces pombe mes1 protein, previously identified as a meiotic inhibitor of APC (52), also turns out to be a true APC substrate (53). Its presence during meiosis may be sufficient to inhibit the complete destruction of cyclin B simply by occupying APC binding sites long enough. It remains to be seen if there is a post-translational regulatory switch that converts mes1 from an inhibitor into a substrate. Also, mouse securin has been shown to inhibit cyclin B proteolysis to regulate mitotic entry by a substrate competition mechanism (54). These observations suggest that affinity of substrates for APC may help dictate the timing of their ultimate destruction, in addition to a previous proposal suggesting that processivity dictates the order of substrate degradation by APC (55). The residual inhibition of APCCdh1 by the Acm1-db3/ken mutant in our studies may represent a form of substrate competition.
There is an important distinction between the pseudosubstrate mechanisms of Emi1 and Acm1. Emi1 contains a C-terminal zinc binding domain that is essential for APC inhibition (39). A separate D box in Emi1 is required for recognition by APC. Thus, binding and inhibition are provided by two distinct regions of Emi1. Loss of the zinc binding domain converts Emi1 from an inhibitor into a D box-dependent substrate. Acm1, on the other hand, does not contain a zinc binding domain. And in contrast to Emi1, mutation of degron sequences themselves converted Acm1 from an inhibitor into a substrate, at least in vitro. This is a very surprising result, because in all cases reported so far, mutations in degron sequences either have no effect or reduce ubiquitination and proteolysis of substrates. But in Acm1, mutation of the conserved central D and KEN boxes resulted in greatly increased poly-ubiquitination.
The poly-ubiquitination of Acm1-db3/ken and the residual APCCdh1 inhibition and WD40 domain binding exhibited by Acm1-db3/ken reveal that other sequences in addition to D box 3 and the KEN box must exist within Acm1 and contribute to the interaction with Cdh1. The highly conserved D box 1 would seem a likely candidate but mutation of this sequence had no significant effect on the poly-ubiquitination of Acm1-db3/ken even though it did contribute to the mono-ubiquitination of wild-type Acm1. Moreover, a truncated form of the Acm1-db3/ken mutant lacking the N-terminal 42 amino acids that contain D box 1 was an even more efficient substrate than the full-length db3/ken mutant. This implicates sequences near the N and/or C termini as contributors to APC inhibition. So far these sequences remain unidentified.
D box 1 is very strictly conserved in Acm1 orthologs, and it was surprising that it had no effect on Cdh1 binding and APCCdh1 inhibition. Interestingly, a recent report identified D box 1 as a functional degron for targeting by APCCdc20 in early anaphase (50). Our previous study found Acm1 proteolysis in late anaphase and G1 to be independent of APC, however this additional mode of regulation in early anaphase (which we possibly missed due to occlusion of the N-terminal D box 1 by an epitope tag) provides an explanation for the strict conservation of D box 1 and suggests that the multiple degron motifs in Acm1 allow it to be recognized as a substrate by one form of the APC and act as a potent inhibitor of the other form. Consistent with this, Enquist-Newman et al. (50) found that Acm1 was unable to inhibit APCCdc20 activity. The presence of distinct degrons within a single protein that are recognized by both forms of the APC makes Acm1 a unique and useful model for better understanding the determinants for substrate recognition.
What makes the central degron motifs of Acm1 such effective inhibitors of APCCdh1? The answer is currently unclear, but our results highlight the important role that sequence context must play in modulating degron function. We speculate that APC inhibition results from the unusually stable interaction between Acm1 and the Cdh1 WD40 domain (mediated both by the central degron motifs and other sequences). Although evidence suggests a clear role for co-activators in substrate degron binding (19, 20) these interactions are difficult to detect in many cases, suggesting that they tend to be transient, or weak. In contrast, Acm1 forms a stoichiometric, highly stable complex with Cdh1 (42) that can prevent substrate binding, suggesting that the affinity of this interaction is much higher than Cdh1-substrate interactions. It has been proposed that the WD40 domain of Cdh1 directly contacts substrate D boxes (20). Our results are fully consistent with this model and suggest that stable interaction between substrate degrons and the co-activator proteins may be selected against, being mutually exclusive with processive ubiquitin ligation. In the case of Acm1, the high affinity interaction with Cdh1 appears to serve two purposes. It blocks recognition of APCCdh1 substrates while simultaneously protecting itself from APCCdh1-mediated proteolysis, resulting in highly stable and potent APCCdh1 inhibition.
Why would weak interactions between substrates and co-activators be preferred? One possibility is that processive assembly of a poly-ubiquitin chain on substrates requires disengagement of substrate from the co-activator after one ubiquitin ligation event to allow repositioning in the ligase active site prior to the next one. It is important to note here that degron binding sites on the core APC are thought to exist as well, and it has been proposed that cooperativity between degron recognition sites on the co-activators and core APC might be one mechanism contributing to functional substrate binding (18, 21, 24, 25). Recently, a novel motif called the TEK box present in both ubiquitin and several human APC substrates was identified that plays an important role in substrate binding and poly-ubiquitination (56). The authors proposed that a TEK box binding site on APC or the E2 UbcH10 first orients substrates in the active site for initial ubiquitin transfer and subsequently the TEK box of the conjugated ubiquitin displaces the substrate TEK box to allow poly-ubiquitin chain formation (56). Although there is no evidence yet for a TEK box-like mechanism in yeast, this model supports the idea that substrates are repositioned in the APC active site after the initial ubiquitin transfer to facilitate assembly of the large, bulky poly-ubiquitin chain. A highly stable substrate-co-activator interaction may not allow disengagement, and the processive ubiquitin transfer reaction would be blocked. This is consistent with our observation that wild-type Acm1 can be mono-ubiquitinated fairly efficiently in vitro but that poly-ubiquitin chain assembly does not occur unless affinity of the Cdh1-Acm1 interaction is greatly reduced by mutation of the central degron sequences. However, other mechanisms are possible as well. Testing this model and other possible models is clearly an interesting and important focus of future studies aimed at understanding substrate recognition and APC activation by the Cdc20 and Cdh1 co-activator proteins.
Acknowledgments
We thank Jessica Schoenherr and Hongzi Liang for technical contributions. We are grateful to Henrik Dohlman, Christoph Borchers, and Evgeniy Petrotchenko for guidance and helpful comments in the early stages of this work. We also thank David Morgan for communication of results prior to publication. This is journal paper 18369 from the Purdue University Agricultural Experiment Station.
This work was supported by an American Cancer Society Institutional Research Grant to the Purdue Cancer Center and by an American Heart Association Scientist Development Grant (to M. C. H.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviationsusedare: APC, anaphase-promotingcomplex; CDK, cyclin-dependent kinase; APCCdh1, Cdh1-dependent anaphase-promoting complex; APCCdc20, Cdc20-dependent anaphase-promoting complex; D box, destruction box; co-IP, co-immunoprecipitation; HA, hemagglutinin; G6PD, glucose-6-phosphate dehydrogenase; db1, db2, and db3, mutations in D box 1, 2, and 3.
References
- 1.Peters, J. M. (2002) Mol. Cell 9931 –943 [DOI] [PubMed] [Google Scholar]
- 2.Hershko, A., and Ciechanover, A. (1998) Annu. Rev. Biochem. 67425 –479 [DOI] [PubMed] [Google Scholar]
- 3.Cohen-Fix, O., Peters, J. M., Kirschner, M. W., and Koshland, D. (1996) Genes Dev. 103081 –3093 [DOI] [PubMed] [Google Scholar]
- 4.Funabiki, H., Yamano, H., Kumada, K., Nagao, K., Hunt, T., and Yanagida, M. (1996) Nature 381438 –441 [DOI] [PubMed] [Google Scholar]
- 5.Leismann, O., Herzig, A., Heidmann, S., and Lehner, C. F. (2000) Genes Dev. 142192 –2205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holloway, S. L., Glotzer, M., King, R. W., and Murray, A. W. (1993) Cell 731393 –1402 [DOI] [PubMed] [Google Scholar]
- 7.Sigrist, S., Jacobs, H., Stratmann, R., and Lehner, C. F. (1995) EMBO J. 144827 –4838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Surana, U., Amon, A., Dowzer, C., McGrew, J., Byers, B., and Nasmyth, K. (1993) EMBO J. 121969 –1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lehman, N. L., Tibshirani, R., Hsu, J. Y., Natkunam, Y., Harris, B. T., West, R. B., Masek, M. A., Montgomery, K., van de Rijn, M., and Jackson, P. K. (2007) Am. J. Pathol. 1701793 –1805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schwab, M., Lutum, A. S., and Seufert, W. (1997) Cell 90683 –693 [DOI] [PubMed] [Google Scholar]
- 11.Visintin, R., Prinz, S., and Amon, A. (1997) Science 278460 –463 [DOI] [PubMed] [Google Scholar]
- 12.Burton, J. L., and Solomon, M. J. (2001) Genes Dev. 152381 –2395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hilioti, Z., Chung, Y. S., Mochizuki, Y., Hardy, C. F., and Cohen-Fix, O. (2001) Curr. Biol. 111347 –1352 [DOI] [PubMed] [Google Scholar]
- 14.Pfleger, C. M., Lee, E., and Kirschner, M. W. (2001) Genes Dev. 152396 –2407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schwab, M., Neutzner, M., Mocker, D., and Seufert, W. (2001) EMBO J. 205165 –5175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sorensen, C. S., Lukas, C., Kramer, E. R., Peters, J. M., Bartek, J., and Lukas, J. (2001) Mol. Cell Biol. 213692 –3703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vodermaier, H. C. (2001) Curr. Biol. 11R834 –R837 [DOI] [PubMed] [Google Scholar]
- 18.Passmore, L. A., McCormack, E. A., Au, S. W., Paul, A., Willison, K. R., Harper, J. W., and Barford, D. (2003) EMBO J. 22786 –796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burton, J. L., Tsakraklides, V., and Solomon, M. J. (2005) Mol. Cell 18533 –542 [DOI] [PubMed] [Google Scholar]
- 20.Kraft, C., Vodermaier, H. C., Maurer-Stroh, S., Eisenhaber, F., and Peters, J. M. (2005) Mol. Cell 18543 –553 [DOI] [PubMed] [Google Scholar]
- 21.Carroll, C. W., Enquist-Newman, M., and Morgan, D. O. (2005) Curr. Biol. 1511 –18 [DOI] [PubMed] [Google Scholar]
- 22.Eytan, E., Moshe, Y., Braunstein, I., and Hershko, A. (2006) Proc. Natl. Acad. Sci. U. S. A. 1032081 –2086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Passmore, L. A., and Barford, D. (2005) EMBO Rep. 6873 –878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hayes, M. J., Kimata, Y., Wattam, S. L., Lindon, C., Mao, G., Yamano, H., and Fry, A. M. (2006) Nat. Cell Biol. 8607 –614 [DOI] [PubMed] [Google Scholar]
- 25.Yamano, H., Gannon, J., Mahbubani, H., and Hunt, T. (2004) Mol. Cell 13137 –147 [DOI] [PubMed] [Google Scholar]
- 26.Glotzer, M., Murray, A. W., and Kirschner, M. W. (1991) Nature 349132 –138 [DOI] [PubMed] [Google Scholar]
- 27.Pfleger, C. M., and Kirschner, M. W. (2000) Genes Dev. 14655 –665 [PMC free article] [PubMed] [Google Scholar]
- 28.Castro, A., Vigneron, S., Bernis, C., Labbe, J. C., Prigent, C., and Lorca, T. (2002) EMBO Rep. 31209 –1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Littlepage, L. E., and Ruderman, J. V. (2002) Genes Dev. 162274 –2285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reis, A., Levasseur, M., Chang, H. Y., Elliott, D. J., and Jones, K. T. (2006) EMBO Rep. 71040 –1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Castro, A., Vigneron, S., Bernis, C., Labbe, J.-C., and Lorca, T. (2003) Mol. Cell Biol. 234126 –4138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sullivan, M., and Morgan, D. O. (2007) J. Biol. Chem. 28219710 –19715 [DOI] [PubMed] [Google Scholar]
- 33.Hendrickson, C., Meyn, M. A., 3rd, Morabito, L., and Holloway, S. L. (2001) Curr. Biol. 111781 –1787 [DOI] [PubMed] [Google Scholar]
- 34.Hagting, A., Den Elzen, N., Vodermaier, H. C., Waizenegger, I. C., Peters, J. M., and Pines, J. (2002) J. Cell Biol. 1571125 –1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zur, A., and Brandeis, M. (2002) EMBO J. 214500 –4510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burton, J. L., and Solomon, M. J. (2007) Genes Dev. 21655 –667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.King, E. M., van der Sar, S. J., and Hardwick, K. G. (2007) PLoS ONE 2 e342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kemp, B. E., Pearson, R. B., House, C., Robinson, P. J., and Means, A. R. (1989) Cell. Signal. 1303 –311 [DOI] [PubMed] [Google Scholar]
- 39.Miller, J. J., Summers, M. K., Hansen, D. V., Nachury, M. V., Lehman, N. L., Loktev, A., and Jackson, P. K. (2006) Genes Dev. 202410 –2420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reimann, J. D., Gardner, B. E., Margottin-Goguet, F., and Jackson, P. K. (2001) Genes Dev. 153278 –3285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dial, J. M., Petrotchenko, E. V., and Borchers, C. H. (2007) J. Biol. Chem. 2825237 –5248 [DOI] [PubMed] [Google Scholar]
- 42.Martinez, J. S., Jeong, D. E., Choi, E., Billings, B. M., and Hall, M. C. (2006) Mol. Cell Biol. 269162 –9176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Crasta, K., Lim, H. H., Giddings, T. H., Jr., Winey, M., and Surana, U. (2008) Nat. Cell Biol. 10665 –675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Woodbury, E. L., and Morgan, D. O. (2007) Nat. Cell Biol. 9106 –112 [DOI] [PubMed] [Google Scholar]
- 45.Hall, M. C., Torres, M. P., Schroeder, G. K., and Borchers, C. H. (2003) J. Biol. Chem. 27816698 –16705 [DOI] [PubMed] [Google Scholar]
- 46.Hall, M. C., Warren, E. N., and Borchers, C. H. (2004) Cell Cycle 31278 –1284 [DOI] [PubMed] [Google Scholar]
- 47.Carroll, C. W., and Morgan, D. O. (2005) Methods Enzymol. 398219 –230 [DOI] [PubMed] [Google Scholar]
- 48.Passmore, L. A., Barford, D., and Wade Harper, J. (2005) Methods Enzymol. 398195 –219 [DOI] [PubMed] [Google Scholar]
- 49.Hall, M. C., Jeong, D. E., Henderson, J. T., Choi, E., Bremmer, S. C., Iliuk, A. B., and Charbonneau, H. (2008) J. Biol. Chem. 28310396 –10407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Enquist-Newman, M., Sullivan, M., and Morgan, D. O. (2008) Mol. Cell 30437 –446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ostapenko, D., Burton, J. L., Wang, R., and Solomon, M. J. (2008) Mol. Cell Biol., in press [DOI] [PMC free article] [PubMed]
- 52.Izawa, D., Goto, M., Yamashita, A., Yamano, H., and Yamamoto, M. (2005) Nature 434529 –533 [DOI] [PubMed] [Google Scholar]
- 53.Kimata, Y., Trickey, M., Izawa, D., Gannon, J., Yamamoto, M., and Yamano, H. (2008) Dev. Cell 14446 –454 [DOI] [PubMed] [Google Scholar]
- 54.Marangos, P., and Carroll, J. (2008) Nat. Cell Biol. 10445 –451 [DOI] [PubMed] [Google Scholar]
- 55.Rape, M., Reddy, S. K., and Kirschner, M. W. (2006) Cell 12489 –103 [DOI] [PubMed] [Google Scholar]
- 56.Jin, L., Williamson, A., Banerjee, S., Philipp, I., and Rape, M. (2008) Cell 133653 –665 [DOI] [PMC free article] [PubMed] [Google Scholar]