

Abstract
Whereas studies involving animal models of cardiovascular disease demonstrated that resveratrol is able to inhibit hypertrophic growth, the mechanisms involved have not been elucidated. Because studies in cells other than cardiomyocytes revealed that AMP-activated protein kinase (AMPK) and Akt are affected by resveratrol, we hypothesized that resveratrol prevents cardiac myocyte hypertrophy via these two kinase systems. Herein, we demonstrate that resveratrol reduces phenylephrine-induced protein synthesis and cell growth in rat cardiac myocytes via alterations of intracellular pathways involved in controlling protein synthesis (p70S6 kinase and eukaryotic elongation factor-2). Additionally, we demonstrate that resveratrol negatively regulates the calcineurin-nuclear factor of activated T cells pathway thus modifying a critical component of the transcriptional mechanism involved in pathological cardiac hypertrophy. Our data also indicate that these effects of resveratrol are mediated via AMPK activation and Akt inhibition, and in the case of AMPK, is dependent on the presence of the AMPK kinase, LKB1. Taken together, our data suggest that resveratrol exerts anti-hypertrophic effects by activating AMPK via LKB1 and inhibiting Akt, thus suppressing protein synthesis and gene transcription.
Pathological left ventricular hypertrophy is associated with coronary heart disease and all-cause mortality (1) and is a major clinical concern in cardiovascular medicine. At the cellular level, enlargement of the cardiac myocyte involves multiple events, including gene transcription and protein translation/synthesis, which are regulated by protein kinase signaling cascades. For gene transcription, the calcineurin-nuclear factor of activated T cells (NFAT)6 signaling pathway has been shown to play a major role in the development of pathological cardiac hypertrophy (2). Upon dephosphorylation by calcineurin, NFAT translocates to the nucleus of the cardiac myocyte where it mediates the transcription of numerous targets involved in hypertrophic growth (see Ref. 3 for review). In conjunction with alterations in gene expression, an important cellular process that must also occur during hypertrophy is enhanced protein synthesis. While the list of proteins involved in regulating protein synthesis in the cardiac myocyte is extensive, the p70S6 kinase (p70S6K) and eukaryotic elongation factor-2 (eEF2) play key roles in this process (4-8). Although the p70S6K, eEF2, and NFAT pathways are distinct in many ways, their regulation, in some instances, appears to intersect as all three of these proteins can be regulated by two upstream kinases, AMP-activated protein kinase (AMPK) and Akt.
Activation of Akt has been shown to promote cardiac hypertrophy via a number of pathways including the indirect activation of p70S6K (6) and the inhibition of glycogen synthase kinase-3β (GSK-3β) (9). Whereas the pro-hypertrophic effects of Akt activation in the heart are well-established (10-12), much less is known about the role of AMPK in the hypertrophic process. AMPK is a key regulator of energy homeostasis, which stimulates ATP-generating pathways and inhibits ATP-consuming pathways in response to the depletion of ATP by cellular stresses (13). We have previously shown that pharmacological and genetic activation of AMPK in the cardiac myocyte can inhibit protein synthesis associated with cardiomyocyte hypertrophy (5, 14) and that this effect was mediated by the inhibition of the p70S6K and eEF2 pathways. The anti-hypertrophic effects of AMPK activation was subsequently confirmed in vivo, as well as extended to include suppression of the calcineurin-NFAT signaling pathway (15). As such, a small molecule that could activate AMPK and inhibit Akt to prevent protein synthesis and NFAT activation could be an ideal treatment strategy for pathological cardiac hypertrophy.
One molecule that may fit this criteria is the polyphenol, resveratrol (3,5,4′-trihydroxystilbene). Indeed, in vivo administration of resveratrol (Resv) can blunt concentric hypertrophy and diastolic impairment in rats subjected to abdominal aortic banding (16). However, despite this finding, it is not known if Resv mediates this anti-hypertrophic effect via changes in vascular function to reduce hemodynamic load (17) or whether Resv has a direct effect on the cardiac myocyte. Assuming the latter, the molecular mechanisms behind these changes are also currently unknown. Therefore, based on these previous findings, we hypothesized that Resv can activate AMPK and inhibit Akt in the cardiac myocyte, and that these changes result in the suppression of the p70S6K, eEF2, and NFAT signaling pathways leading to the subsequent inhibition of protein synthesis and the development of cardiac hypertrophy.
EXPERIMENTAL PROCEDURES
Animal Care—All experimental procedures involving animals were approved by the University of Alberta Animal Policy and Welfare Committee, which adheres to the principles for biomedical research involving animals developed by the Council for International Organizations of Medical Sciences (CIOMS), and complies with the Canadian Council on Animal Health Care guidelines.
Cell Culture and Treatment—Cardiac myocytes were isolated from the hearts of 1-3-day-old neonatal rat pups and cultured in Dulbecco's Modified Eagle's Medium/Ham's Nutrient Mixture F-12 (Dulbecco's modified Eagle's medium/F-12) containing 5% fetal bovine serum, 10% horse serum, 50 μg/ml gentamicin, and 1% penicillin-streptomycin for 16 h essentially as described (18). 24 h prior to the initiation of experiments, the medium was changed to serum-free Dulbecco's modified Eagle's medium/F-12 containing 50 μg/ml gentamicin supplemented with 1× insulin, transferrin, sodium selenite (ITS)+3 liquid media supplement. All subsequent treatments of the myocytes were performed in this serum-free medium for either 1 or 24 h. Cells were treated with either 1 μl of 50 mm Resv (Sigma) to a final concentration of 50 μm or with 1 μl of 96% ethanol as vehicle (control), in the absence or presence of 10 μm phenylephrine (PE) (Sigma) for 24 h. The final concentration of ethanol in both instances was 0.096%. In some experiments, calcineurin inhibitors (500 ng/ml (416 nm) of cyclosporin A (CsA) or 150 ng/ml (182 nm) of FK506 (Alexis Biochemicals)) were used in combination with PE and/or Resv. In some experiments, cells were treated with vehicle or 100 μm Resv for 1 h. To visualize changes in cell size, myocytes were plated on fibronectin-coated coverslips at a density of 6.7 × 105 cells in 35-mm dishes, and treated as above. Cells were fixed and visualized using mouse anti-α-actinin (Sigma) and Texas Red-conjugated donkey anti-mouse antibodies (Jackson Immuno Research) as described previously (19). All mouse embryonic fibroblasts (MEFs) were cultured in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum and 1% penicillin-streptomycin.
[3H]Phenylalanine Incorporation—Myocytes were treated with [3H]phenylalanine (Amersham Biosciences) (1 μCi/ml) at the time of drug treatment, incubated for 24 h, and harvested for [3H]phenylalanine incorporated as previously described (5).
Immunoblot Analysis—Cardiac myocytes or MEFs were harvested and subjected to immunoblot analysis as described (5) using a variety of phosphoprotein and protein-specific antibodies as indicated.
Mouse Embryonic Fibroblasts—Mouse embryonic fibroblasts (MEFs) isolated from wild-type (WT) and AMPKα1/α2-null genotype mice were generated as we have previously described (20). WT and Akt1/2-null MEFs were generously provided by Dr. N. Hay, University of Illinois. MEFs isolated from WT and LKB1-null genotype mice were a gift of Dr. R. Depinho and Dr. N. Bardeesy, Harvard Medical School. All experiments involving MEFs were performed in the presence of serum.
Luciferase Activity and Calcineurin Activity Measurements—Myocytes were cultured as described above and infected with Ad.GFP or Ad.NFAT-Luc-Promoter adenovirus (Seven Hills Bioreagents) at the multiplicity of infection (MOI) of 10. Cells were treated 24 h post-infection with vehicle or 50 μm Resv in the absence or presence of 10 μm PE for 24 h. In some experiments, 500 ng/ml of CsA or 150 ng/ml of FK506 were used in combination with PE and/or Resv. For MEFs, a MOI of 50 was used for each adenovirus. Then 48 h post-infection, cells were treated with 100 μm Resv or vehicle for 1 h. In some instances, cells were harvested with the reporter lysis buffer supplied in the luciferase assay system kit (Promega). For the calcineurin activity measurements, myocytes were harvested with the lysis buffer plus protease inhibitor supplied in the calcineurin cellular assay kit (BIOMOL). Samples were assessed as per the manufacturer's instructions. Calcineurin activity was also determined in an in vitro dephosphorylation assay using a γ-32P-labeled RII-peptide (corresponding to the regulatory domain of protein kinase A; the RII peptide, DLDVPIPGRFDRRVSVAAE) as previously described (21). Briefly, 20 μl of lysate was added to 15 μm γ-32P-labeled RII-peptide, and the reaction was carried out for 15 min at 30 °C in the presence of 1.5 μm okadaic acid with a total reaction volume of 60 μl. Following phosphatase reaction, the mixture was applied to an AG-50W0X8 cation exchange resin to separate out the γ-32P-labeled RII-peptide. Calcineurin activity was calculated based on the pmol of released phosphate/min/mg protein.
Statistical Analysis—All data are presented as means ± S.E. of the mean (S.E.). Statistical analysis for comparison of two groups was performed with Student's unpaired two-tailed t test or two-tailed Mann-Whitney test as appropriate. For comparison of three groups, one way analysis of variance (ANOVA) followed by the Bonferroni multiple comparisons test was used for the determination of statistical analysis. In some instances, the Kruskal-Wallis test followed by Dunn's multiple comparisons test was used. Student's one sample t test was utilized in cases where the control was set as 1. A value of p < 0.05 was considered significant.
RESULTS
Resveratrol Inhibits Phenylephrine-induced Cellular Hypertrophy and Protein Synthesis—Neonatal rat cardiac myocytes treated with 10 μm PE for 24 h demonstrated a dramatic increase in cell size as well as a significant 1.89-fold increase in protein synthesis over control-treated cells (Fig. 1, A and B), which was reduced by simultaneous treatment of 50 μm Resv. To investigate the molecular signaling mechanisms that may contribute to the Resv-induced reduction in hypertrophic growth, cell lysates from cardiac myocytes treated with PE in the absence or presence of Resv were subjected to immunoblot analysis using anti-phospho-p70S6K (T389) and anti-phospho-p70S6K (T421/S424) antibodies. While p70S6K undergoes multisite phosphorylation, phosphorylation at these sites are closely related to an increase in its activity (22, 23). Although PE increased the phosphorylation status of p70S6K significantly compared with control, Resv was able to decrease p70S6K phosphorylation to basal levels (Fig. 1, C and D). Furthermore, immunoblot analysis of eEF2 using anti-phospho-eEF2 (T56) antibody as a marker of its activity (where phosphorylation indicates inactivation, Refs. 5, 24) demonstrated that Resv significantly increased eEF2 phosphorylation in cells in the absence or presence of PE (Fig. 1E).
FIGURE 1.

Resveratrol blunts phenylephrine-induced cardiac myocyte hypertrophy and protein synthesis via the p70S6K, eEF2, and calcineurin-NFAT signaling pathways. A, qualitative representation of the cell size of neonatal rat cardiac myocytes treated with ethanol (Control) or 50 μm resveratrol (Resv), in the absence or presence of 10 μm phenylephrine (PE) for 24 h. Changes in cell size were visualized following fixation on coverslips using mouse anti-α-actinin and Texas Red-conjugated donkey anti-mouse antibodies. B, measurement of protein synthesis using [3H]phenylalanine incorporation with counts expressed in DPM (disintegrations per minute). Values are means ± S.E. with each experiment performed in duplicate (n = 4). *, p < 0.01 versus Control; #, p < 0.05 versus PE; ^, p < 0.05 versus Resv; assessed by ANOVA and Bonferroni multiple comparisons test. C-E, representative immunoblot and densitometry of cellular extracts from cardiac myocytes treated as described above. Values are expressed as means ± S.E. and analyzed with the Kruskal-Wallis test followed by Dunn's multiple comparisons test. C, cell lysates blotted with anti-phospho-p70S6K (T389) and anti-actin antibodies; *, p < 0.05 versus Control; #, p < 0.01 versus PE (n = 9-10). D, cell lysates blotted with anti-phospho-p70S6K (T421/S424) and anti-actin antibodies; *, p < 0.05 versus Control; #, p < 0.05 versus PE (n = 5-6). E, cell lysates blotted with anti-phospho-eEF2 (T56) and anti-eEF2 antibodies; *, p < 0.05 versus Control; #, p < 0.01 versus PE (n = 8-9). F, cellular extracts of cardiac myocytes infected with Ad.NFAT-Luc-Promoter adenovirus and treated as described above were assessed for NFAT-dependent transcription, measured as luciferase activity expressed in relative light units. Values are means ± S.E. with each sample assessed in duplicate (n = 5); **, p < 0.001 versus Control; *, p < 0.05 versus Control; #, p < 0.001 versus PE, analyzed by ANOVA and Bonferroni multiple comparisons test. G and H, myocytes infected with Ad.NFAT-Luc-Promoter adenovirus were treated with 500 ng/ml (416 nm) of CsA or 150 ng/ml (182 nm) of 506 with or without 50 μm Resv, in the presence of 10 μm PE for 24 h. G, NFAT-dependent transcription measured from cellular extracts is presented as a percentage (%) compared with PE treatment, where values are means ± S.E. with each sample assessed in duplicate (n = 4); *, p < 0.001 versus Resv; #, p < 0.001 versus CsA; ^, p < 0.001 versus FK506, analyzed by ANOVA and Bonferroni multiple comparisons test. H, calcineurin activity measured from cellular extracts is presented as a percentage (%) compared with PE treatment, where values are means ± S.E. with each sample assessed in duplicate (n = 4-5), analyzed by ANOVA.
Resveratrol Inhibits NFAT-dependent Transcription as well as Calcineurin Activity—To further explore the possible mechanisms of action for the anti-hypertrophic effects of Resv, NFAT-dependent transcriptional activity was measured using a NFAT-luciferase reporter transgene (Ad.NFAT-Luc-Promoter) (3). Rat cardiac myocytes with adenoviral-mediated delivery of a luciferase reporter gene driven by the α-myosin heavy chain promoter containing multiple NFAT binding sites were treated with PE in the absence or presence of Resv. Myocytes treated with Resv displayed a significant and dramatic reduction in NFAT-dependent transcription compared with vehicle controls (Fig. 1F). To investigate whether the effect of Resv on NFAT transcriptional activity was mediated via modification of calcineurin activity, rat cardiac myocytes treated with PE were co-treated with Resv and/or known calcineurin inhibitors, CsA and FK506, at doses known to significantly reduce calcineurin activity in these cells (500 ng/ml (416 nm) and 150 ng/ml (182 nm), respectively (19)). The data show that at these concentrations, Resv was significantly more effective at inhibiting PE-induced NFAT transcription than CsA and FK506 (Fig. 1G), and that there were no additive effects when co-treated with these calcineurin inhibitors (Fig. 1H). Interestingly, Resv was able to inhibit calcineurin to similar levels as that observed with CsA and FK506 using both the calcineurin cellular assay kit (Fig. 1H) as well as using the γ-32P-labeled RII-peptide in vitro dephosphorylation assay (data not shown). Together, these data suggest that an additional signaling mechanism is involved which results in a more pronounced reduction of NFAT-dependent transcription (Fig. 1G) beyond that observed via inhibition of calcineurin alone.
Chronic Resveratrol Treatment Increases Phosphorylation of AMPK but Not Akt—Consistent with previous studies showing that AMPK activation can inhibit cardiac hypertrophy via the p70S6K (5, 14, 15), eEF2 (5, 15), and NFAT pathways (15), the level of phosphorylated AMPK was significantly increased in Resv-treated cardiac myocytes in the absence or presence of PE (Fig. 2A). In accordance with increased phosphorylation of AMPK in Resvtreated cells, the level of phosphorylated acetyl-CoA carboxylase (ACC), which is also used as an intracellular marker of AMPK activation, was significantly increased (Fig. 2B). As both AMPK and Akt have been shown to be involved in regulating cardiac myocyte growth (5, 10-12, 25), we also investigated the involvement of Akt as a possible mediator of the effects of Resv in PE-induced cardiac myocyte hypertrophy. Interestingly, we did not observe any changes in the phosphorylation status of Akt or its downstream target GSK-3β, in the presence of Resv compared with any other groups (Fig. 2, C and D) despite a previous report suggesting that Akt is inhibited by Resv in vascular smooth muscle cells (26). However, cardiac myocytes treated with 100 μm Resv for 1 h and then rinsed and incubated in medium in the absence of Resv for 24 h, showed no changes in either the phosphorylation of AMPK or Akt (not shown), yet still displayed alterations in the phosphorylation of p70S6K and eEF2 (Fig. 2, E-G). These findings suggest that Akt activity may have been inhibited acutely by Resv, which had a longer lasting effect on the phosphorylation status of p70S6K and eEF2 despite Akt activity being normalized by 24 h. Based on these data, we investigated the effects of acute treatment of Resv in the cardiac myocyte.
FIGURE 2.

Chronic resveratrol treatment increases AMPK and ACC phosphorylation without changing Akt and GSK-3β phosphorylation, while acute treatment has lasting effects on p70S6K and eEF2 phosphorylation levels. A-D, representative immunoblot and densitometry of cellular extracts from neonatal rat cardiac myocytes treated with ethanol (Control) or 50 μm resveratrol (Resv), in the absence or presence of 10 μm phenylephrine (PE) for 24 h. Values are expressed as means ± S.E. and analyzed with the Kruskal-Wallis test followed by Dunn's multiple comparisons test. A, cell lysates blotted with anti-phospho-α-AMPK (T172) and anti-α-AMPK antibodies; *, p < 0.01 versus Control; #, p < 0.05 versus PE (n = 9-10). B, cell lysates blotted with anti-phospho-ACC (S79) antibody and peroxidase-labeled streptavidin, which detects both isoforms of ACC (upper and lower bands) and were quantified together; *, p < 0.05 versus Control; #, p < 0.05 versus PE (n = 5-7). C, cell lysates blotted with anti-phospho-Akt (S473) and anti-Akt antibodies (n = 9). D, cell lysates blotted with anti-phospho-GSK-3β (S9) and anti-GSK-3β antibodies (n = 9). E-G, representative immunoblot of cellular extracts from neonatal rat cardiac myocytes treated with ethanol (Control) or 100 μm Resv for 1 h, followed by 24 h of incubation with serum-free medium (n = 4). E, cell lysates blotted with anti-phospho-p70S6K (T389) and anti-actin antibodies, (F) anti-phospho-p70S6K (T421/S424) and anti-actin antibodies, and (G) anti-phospho-eEF2 (T56) and anti-eEF2 antibodies.
Acute Resveratrol Treatment Increases AMPK Phosphorylation and Decreases Akt Phosphorylation and Has More Dramatic Effects on the Phosphorylation of p70S6K and eEF2—Rat cardiac myocytes treated with 100 μm Resv for 1 h demonstrated a striking and significant decrease in the phosphorylation of p70S6K at both phosphorylation sites as well as a significant increase in eEF2 phosphorylation (Fig. 3, A-C). Consistent with these enhanced effects of p70S6K and eEF2 phosphorylation, AMPK and ACC phosphorylation levels were significantly increased with 1 h Resv treatment (Fig. 3, D and E). In addition, phosphorylation levels of Akt and GSK-3β were significantly reduced 1 h following Resv treatment (Fig. 3, F and G). Together, these data show that Resv is able to significantly alter AMPK and Akt activity despite the short treatment duration in the cardiac myocyte. Although this short treatment time with Resv precluded us from observing changes in cell size or measuring rates of protein synthesis, these dramatic effects on major regulators of protein synthesis are likely involved in the substantial inhibition of protein synthesis and cardiac myocyte cell growth observed with longer Resv treatment.
FIGURE 3.

Acute treatment with resveratrol has dramatic effects on the p70S6K and eEF2 signaling pathways, while affecting both AMPK and Akt signaling pathways. Representative immunoblot and densitometry of cellular extracts from neonatal rat cardiac myocytes treated with ethanol (Control) or 100 μm resveratrol (Resv) for 1 h. Values are expressed as means ± S.E. and analyzed with Student's one sample t test as control was set to 1. A, cell lysates blotted with anti-phospho-p70S6K (T389) and anti-actin antibodies, *, p < 0.0001 versus Control (n = 4); (B) anti-phospho-p70S6K (T421/S424) and anti-actin antibodies, *, p < 0.001 versus Control (n = 4); (C) anti-phospho-eEF2 (T56) and anti-eEF2 antibodies, *, p < 0.05 versus Control (n = 9); (D) anti-phospho-α-AMPK (T172) and anti-α-AMPK antibodies, *, p < 0.05 versus Control (n = 6); (E) antiphospho-ACC (S79) antibody and peroxidase-labeled streptavidin, with both ACC bands being quantified together, *, p < 0.05 versus Control (n = 7); (F) anti-phospho-Akt (S473) and anti-Akt antibodies, *, p < 0.001 versus Control (n = 4); and (G) anti-phospho-GSK-3β (S9) and anti-GSK-3β antibodies, *, p < 0.01 versus Control (n = 4).
The Effects of Resveratrol on AMPK Are Mediated via LKB1—In the heart, a major upstream kinase involved in regulating AMPK activity is LKB1 (27). To investigate whether Resv activates AMPK via LKB1, we utilized WT and LKB1-null MEFs treated with 100 μm Resv for 1 h. Consistent with what was observed in the cardiac myocyte, Resv treatment increased the phosphorylation status of AMPK in WT MEFs (Fig. 4A). However, Resv was unable to activate AMPK in LKB1-null MEFs, suggesting that the effect of Resv on AMPK is mediated via LKB1 (Fig. 4A). In addition, the absence of LKB1 had no affect on the ability of Resv to inhibit Akt (Fig. 4A), providing evidence that LKB1 is not necessary for Resv to inhibit Akt.
FIGURE 4.

The effect of resveratrol on AMPK phosphorylation is mediated by LKB1 without cross-talk between AMPK and Akt, and its effect on NFAT-dependent transcription is mainly dependent on AMPK not Akt. A-C, representative immunoblots of cellular extracts from mouse embryonic fibroblasts (MEFs) treated with ethanol (Control) or 100 μm resveratrol (Resv) for 1 h, blotted with anti-phospho-α-AMPK (T172), anti-α-AMPK, anti-phospho-Akt (S473), and anti-Akt antibodies. A, wild-type (WT) and LKB1-null MEFs; B, WT and AMPKα1/α2-null MEFs; and C, WT and Akt1/2-null MEFs were utilized (n = 3). D and E, AMPKα1/α2-null and Akt1/2-null MEFs with their respective WT MEFs were infected with Ad.NFAT-Luc-Promoter adenovirus and then treated with ethanol (Control) or 100 μm Resv for 1 h. NFAT-dependent transcription measured from cellular extracts is presented as a percentage (%) compared with control-treated, where values are means ± S.E. D,*, p < 0.01 versus WT control-treated; #, p < 0.05 versus AMPKα1/α2-null control-treated (n = 4-6), assessed by two-tailed Mann-Whitney test. E,*, p < 0.001 versus WT control-treated; #, p < 0.001 versus Akt1/2-null control-treated (n = 3), assessed by Student's unpaired two-tailed t test.
Cross-talk between AMPK and Akt Is Not Involved in Mediating the Effects of Resveratrol—Because we (18, 28) and others (29-32) have demonstrated cross-talk between AMPK and Akt, we determined whether the effect of Resv on either of these two kinases was mediated by the other. In agreement with our previous findings, WT MEFs from both AMPKα1/α2-null littermates and from Akt1/2-null littermates demonstrated increased phosphorylation of AMPK following a 1-h treatment with Resv (Fig. 4, B and C). Furthermore, the absence of Akt1/2 had no effect on the ability of Resv to activate AMPK (Fig. 4C), and the absence of AMPKα1/α2 had no effect on the ability of Resv to inhibit Akt (Fig. 4B). Together, these data suggest that Resv exerts independent effects on AMPK and Akt, without subsequent cross-talk between the two kinases. As both AMPK (15, 33) and Akt (34-36) have been previously shown to regulate NFAT activation in the cardiac myocyte, we utilized our NFAT-luciferase reporter construct in WT, AMPKα1/α2-null, and Akt1/2-null MEFs. In both WT MEF populations, Resv significantly reduced NFAT-dependent transcription to ∼20% of that observed in vehicle-treated MEFs (Fig. 4, D and E), a value which is similar to that observed in cardiac myocytes. However, Resv failed to inhibit most of the NFAT activity in the nucleus in AMPKα1/α2-null MEFs (Fig. 4D), whereas Resv treatment inhibited NFAT-dependent transcription in the Akt1/2-null MEFs to a similar extent as in WT MEFs (Fig. 4E). These data indicate that the ability of Resv to inhibit NFAT transcriptional activity is dependent on AMPK and not Akt.
DISCUSSION
Whereas previous in vivo studies in rats demonstrated that Resv is effective in preventing pressure overload-induced cardiac hypertrophy (16, 17), the mechanisms involved in the anti-hypertrophic effect have not been clearly defined. Our data show that Resv is able to diminish the activity of both p70S6K and eEF2 (Fig. 1, C-E), suggesting that the anti-hypertrophic effects of Resv may be mediated by the inhibition of protein synthesis. In addition, our data show that Resv had a profound inhibitory effect on NFAT-dependent transcription in the cardiac myocyte, further suggesting that Resv can prevent hypertrophic growth by blocking NFAT-mediated regulation of gene transcription known to be involved in pathological cardiac hypertrophy (2). Moreover, Resv appears to exert its effect on NFAT in part by the reduction of the activity of calcineurin, an upstream regulator of NFAT, as well as an additional as yet unidentified signaling mechanism to achieve this dramatic inhibition of PE-induced NFAT activation (Fig. 1G). The ability of Resv to significantly block NFAT transcriptional activity via calcineurin and this yet unknown pathway could result in further suppression of the hypertrophic response at the transcription level, thereby strengthening the anti-hypertrophic effects of Resv. As such, Resv appears to prevent hypertrophic growth by inhibiting pathways involved in both protein synthesis and gene transcription.
Because we have previously shown that pharmacological activation of AMPK by AICAR results in suppression of cardiac myocyte hypertrophy via inhibition of the p70S6K and eEF2 signaling pathways (5), we investigated the possibility that Resv exerts its anti-hypertrophic effects via AMPK activation. In agreement with this, we show that AMPK is activated in the cardiac myocyte during both acute and chronic treatment with Resv (Figs. 2A and 3D) and that this activation not only affects the p70S6K and eEF2 signaling pathways (Figs. 1, C-E and 3, A-C), but also the calcineurin-NFAT pathway (Fig. 1F) to promote the inhibition of cardiac hypertrophy. In addition, because a major upstream kinase involved in regulating AMPK activity in the heart is LKB1 (27), we examined whether AMPK activation by Resv was mediated via LKB1. Using LKB1-null MEFs we show that the presence of LKB1 is necessary for Resv to activate AMPK and that the nuclear localization of NFAT is regulated by the LKB1/AMPK signaling axis (Fig. 4, A and D). Although we did not measure AMP:ATP levels in Resv-treated cells, given that LKB1 appears to mediate the effects of Resv and that AMP does not activate LKB1 in a purified in vitro system (37), it is likely that changes in AMP:ATP ratios are not involved. This is entirely consistent with what has been found in Neuro2a cells where Dasgupta et al. (38) reported no change in AMP:ATP ratio in Resv-treated cells yet a significant activation of AMPK. Whether Resv has a direct effect on LKB1 or whether it is mediated via another effector molecule is currently unknown. Indeed, as Resv is one of the most potent SIRT1 activators found in nature and AMPK activation was also noted in the livers of Resv-fed mice (39), it is possible that one of the effectors responsible for Resv-stimulated AMPK activation via LKB1 is SIRT1. Indeed, evidence for this has now been provided in hepatocytes (40). However, although we cannot rule out the possibility of SIRT1 regulating LKB1 activity, the effect of Resv on AMPK in neurons was shown to be independent of SIRT1 activation (38). Moreover, we have previously shown that pharmacological activators of AMPK that are distinct from Resv also inhibit protein synthesis and cardiac hypertrophy (5), providing further support that SIRT1 may not be involved in this signaling pathway.
In addition to the effects of Resv on the LKB1/AMPK pathway, our data show that Resv can also inhibit Akt activation. Although Resv has been shown to inhibit Akt phosphorylation and reduce p70S6K phosphorylation and protein synthesis stimulated by angiotensin II in rat aortic smooth muscle cells (26), its effect on the Akt/p70S6K pathway in the cardiac myocyte was heretofore unknown. Interestingly, we did not observe any changes in the phosphorylation status of Akt or GSK-3β, the downstream target of Akt, in the neonatal rat cardiac myocytes treated with Resv for 24 h (Fig. 2, C and D) yet observed a profound decrease in Akt and GSK-3β phosphorylation after 1 h of treatment (Fig. 3, F and G). Consistent with a decrease in Akt activity, we observed a striking and significant decrease in the phosphorylation of p70S6K and a significant increase in eEF2 phosphorylation (Fig. 3, A-C), suggesting dramatic inhibitory effects on pathways involved in regulating protein synthesis. However, Resv treatment inhibited NFAT-dependent transcription even in Akt1/2-null MEFs (Fig. 4E), suggesting that Akt signaling is not responsible for the inhibitory effects of Resv on NFAT transcriptional activity. That said, the Akt1/2-null MEFs still contain minor amounts of Akt3 activity (Fig. 4C), which may be sufficient to maintain Akt signaling. Although this is a possibility, we believe it is unlikely, because Resv is still able to increase phosphorylation of AMPK to the same extent as WT MEFs despite the dramatic decrease in Akt activity in the Akt1/2-null MEFs. This supports the notion that the effects of Resv on AMPK are independent from Akt. In contrast, Resv failed to inhibit most of the NFAT-dependent transcription in the nucleus of AMPKα1/α2-null MEFs (Fig. 4D), indicating that the ability of Resv to inhibit NFAT transcriptional activity is dependent on AMPK. While the majority of the effect of Resv on the nuclear localization of NFAT appears to be dependent on AMPK, there also appears to be at least one other AMPK-independent pathway that alters NFAT transcriptional activity in response to Resv treatment. Though it is tempting to speculate that the Resv-induced inhibition of Akt and increased activation of GSK-3β (Fig. 3, F and G) may also promote the nuclear export of NFAT, our finding that NFAT-dependent transcription was inhibited by Resv in Akt1/2-null MEFs to the same extent as in WT MEFs (Fig. 4E) does not support this concept. Furthermore, although we conclude that Resv can inhibit NFAT-dependent transcription in the MEFs, given the short duration of treatment with Resv in these cells, we cannot rule out the possibility that synthesis of the luciferase protein is altered by the p70S6K and eEF2 pathways independent from NFAT transcriptional activity.
In summary, our data show that Resv can activate AMPK and inhibit Akt signaling in the cardiac myocyte, and that these Resv-mediated effects can inhibit cardiac myocyte hypertrophy (Fig. 5). Our data also show that the anti-hypertrophic effects of Resv can be attributed to decreased protein synthesis via inhibition of the p70S6K and eEF2 signaling pathways, and reduced NFAT translocation to the nucleus via calcineurin activity suppression. Additionally, we provide evidence that LKB1 is necessary for Resv to activate AMPK, suggesting that LKB1 and not AMPK may be a target of Resv. Taken together, our findings further support the growing body of evidence suggesting that AMPK activation has therapeutic potential for inhibiting hypertrophic growth and shed considerable light on the mechanisms involved in the anti-hypertrophic effects of Resv.
FIGURE 5.
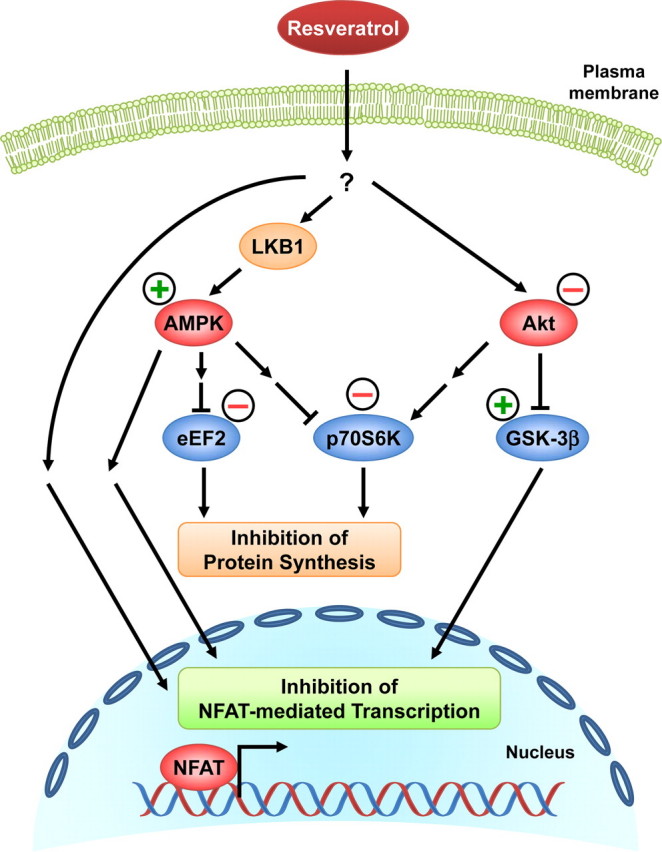
Proposed mechanisms of the anti-hypertrophic effects of resveratrol. Resveratrol, either directly or indirectly, activates AMPK via LKB1 and inhibits Akt. This results in a reduction of p70S6K and eEF2 activities and the subsequent inhibition of protein synthesis. The reduced Akt activity relieves the suppression of GSK-3β activity, which may also contribute to the inhibition of NFAT-mediated transcription. AMPK activation by resveratrol appears to mediate a major component of the diminished NFAT transcriptional activity in the nucleus via inhibition of calcineurin and yet to be identified mechanism(s). Resveratrol treatment may also decrease NFAT-mediated transcription via pathways independently of AMPK and Akt. Together, all of these resveratrol-mediated effects contribute to the inhibition of cardiac myocyte hypertrophy.
Acknowledgments
We thank Suzanne Kovacic for expert technical assistance.
This research was supported in part by grants from the Canadian Institutes of Health Research (CIHR) and the Heart and Stroke Foundation of Canada (HSFC). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: NFAT, nuclear factor of activated T cells; p70S6K, p70S6 kinase; eEF2, eukaryotic elongation factor-2; AMPK, AMP-activated protein kinase; GSK-3β, glycogen synthase kinase-3β; Resv, resveratrol; PE, phenylephrine; CsA, cyclosporin A; MEFs, mouse embryonic fibroblasts; WT, wild-type; MOI, multiplicity of infection; ACC, acetyl-CoA carboxylase; ANOVA, analysis of variance.
References
- 1.Benjamin, E. J., and Levy, D. (1999) Am. J. Med. Sci. 317 168-175 [DOI] [PubMed] [Google Scholar]
- 2.Wilkins, B. J., Dai, Y. S., Bueno, O. F., Parsons, S. A., Xu, J., Plank, D. M., Jones, F., Kimball, T. R., and Molkentin, J. D. (2004) Circ. Res. 94 110-118 [DOI] [PubMed] [Google Scholar]
- 3.Wilkins, B. J., and Molkentin, J. D. (2004) Biochem. Biophys. Res. Commun. 322 1178-1191 [DOI] [PubMed] [Google Scholar]
- 4.Horman, S., Beauloye, C., Vertommen, D., Vanoverschelde, J. L., Hue, L., and Rider, M. H. (2003) J. Biol. Chem. 278 41970-41976 [DOI] [PubMed] [Google Scholar]
- 5.Chan, A. Y., Soltys, C. L., Young, M. E., Proud, C. G., and Dyck, J. R. (2004) J. Biol. Chem. 279 32771-32779 [DOI] [PubMed] [Google Scholar]
- 6.Wang, L., Wang, X., and Proud, C. G. (2000) Am. J. Physiol. Heart Circ. Physiol. 278 H1056-H1068 [DOI] [PubMed] [Google Scholar]
- 7.Boluyt, M. O., Zheng, J. S., Younes, A., Long, X., O'Neill, L., Silverman, H., Lakatta, E. G., and Crow, M. T. (1997) Circ. Res. 81 176-186 [DOI] [PubMed] [Google Scholar]
- 8.Everett, A. D., Stoops, T. D., Nairn, A. C., and Brautigan, D. (2001) Am. J. Physiol. Heart Circ. Physiol. 281 H161-H167 [DOI] [PubMed] [Google Scholar]
- 9.Cross, D. A., Alessi, D. R., Cohen, P., Andjelkovich, M., and Hemmings, B. A. (1995) Nature 378 785-789 [DOI] [PubMed] [Google Scholar]
- 10.Shiojima, I., Yefremashvili, M., Luo, Z., Kureishi, Y., Takahashi, A., Tao, J., Rosenzweig, A., Kahn, C. R., Abel, E. D., and Walsh, K. (2002) J. Biol. Chem. 277 37670-37677 [DOI] [PubMed] [Google Scholar]
- 11.Shioi, T., McMullen, J. R., Kang, P. M., Douglas, P. S., Obata, T., Franke, T. F., Cantley, L. C., and Izumo, S. (2002) Mol. Cell. Biol. 22 2799-2809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shiojima, I., Sato, K., Izumiya, Y., Schiekofer, S., Ito, M., Liao, R., Colucci, W. S., and Walsh, K. (2005) J. Clin. Invest. 115 2108-2118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corton, J. M., Gillespie, J. G., and Hardie, D. G. (1994) Curr. Biol. 4 315-324 [DOI] [PubMed] [Google Scholar]
- 14.Noga, A. A., Soltys, C. L., Barr, A. J., Kovacic, S., Lopaschuk, G. D., and Dyck, J. R. (2007) Am. J. Physiol. Heart Circ. Physiol. 292 H1460-H1469 [DOI] [PubMed] [Google Scholar]
- 15.Li, H. L., Yin, R., Chen, D., Liu, D., Wang, D., Yang, Q., and Dong, Y. G. (2007) J. Cell. Biochem. 100 1086-1099 [DOI] [PubMed] [Google Scholar]
- 16.Juric, D., Wojciechowski, P., Das, D. K., and Netticadan, T. (2007) Am. J. Physiol. Heart Circ. Physiol. 292 H2138-H2143 [DOI] [PubMed] [Google Scholar]
- 17.Liu, Z., Song, Y., Zhang, X., Liu, Z., Zhang, W., Mao, W., Wang, W., Cui, W., Zhang, X., Jia, X., Li, N., Han, C., and Liu, C. (2005) Clin. Exp. Pharmacol. Physiol. 32 1049-1054 [DOI] [PubMed] [Google Scholar]
- 18.Kovacic, S., Soltys, C. L., Barr, A. J., Shiojima, I., Walsh, K., and Dyck, J. R. (2003) J. Biol. Chem. 278 39422-39427 [DOI] [PubMed] [Google Scholar]
- 19.Molkentin, J. D., Lu, J. R., Antos, C. L., Markham, B., Richardson, J., Robbins, J., Grant, S. R., and Olson, E. N. (1998) Cell 93 215-228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laderoute, K. R., Amin, K., Calaoagan, J. M., Knapp, M., Le, T., Orduna, J., Foretz, M., and Viollet, B. (2006) Mol. Cell Biol. 26 5336-5347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fruman, D. A., Mather, P. E., Burakoff, S. J., and Bierer, B. E. (1992) Eur. J. Immunol. 22 2513-2517 [DOI] [PubMed] [Google Scholar]
- 22.Dennis, P. B., Pullen, N., Kozma, S. C., and Thomas, G. (1996) Mol. Cell Biol. 16 6242-6251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pearson, R. B., Dennis, P. B., Han, J. W., Williamson, N. A., Kozma, S. C., Wettenhall, R. E., and Thomas, G. (1995) EMBO J. 14 5279-5287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang, L., and Proud, C. G. (2002) FEBS Lett. 531 285-289 [DOI] [PubMed] [Google Scholar]
- 25.Shibata, R., Ouchi, N., Ito, M., Kihara, S., Shiojima, I., Pimentel, D. R., Kumada, M., Sato, K., Schiekofer, S., Ohashi, K., Funahashi, T., Colucci, W. S., and Walsh, K. (2004) Nat. Med. 10 1384-1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haider, U. G., Sorescu, D., Griendling, K. K., Vollmar, A. M., and Dirsch, V. M. (2002) Mol. Pharmacol. 62 772-777 [DOI] [PubMed] [Google Scholar]
- 27.Sakamoto, K., Zarrinpashneh, E., Budas, G. R., Pouleur, A. C., Dutta, A., Prescott, A. R., Vanoverschelde, J. L., Ashworth, A., Jovanovic, A., Alessi, D. R., and Bertrand, L. (2006) Am. J. Physiol. Endocrinol. Metab. 290 E780-E788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Soltys, C. L., Kovacic, S., and Dyck, J. R. (2006) Am. J. Physiol. Heart Circ. Physiol. 290 H2472-H2479 [DOI] [PubMed] [Google Scholar]
- 29.Ouchi, N., Kobayashi, H., Kihara, S., Kumada, M., Sato, K., Inoue, T., Funahashi, T., and Walsh, K. (2004) J. Biol. Chem. 279 1304-1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zou, M. H., Kirkpatrick, S. S., Davis, B. J., Nelson, J. S., Wiles, W. G., Schlattner, U., Neumann, D., Brownlee, M., Freeman, M. B., and Goldman, M. H. (2004) J. Biol. Chem. 279 43940-43951 [DOI] [PubMed] [Google Scholar]
- 31.Bertrand, L., Ginion, A., Beauloye, C., Hebert, A. D., Guigas, B., Hue, L., and Vanoverschelde, J. L. (2006) Am. J. Physiol. Heart Circ. Physiol. 291 H239-H250 [DOI] [PubMed] [Google Scholar]
- 32.Horman, S., Vertommen, D., Heath, R., Neumann, D., Mouton, V., Woods, A., Schlattner, U., Wallimann, T., Carling, D., Hue, L., and Rider, M. H. (2006) J. Biol. Chem. 281 5335-5340 [DOI] [PubMed] [Google Scholar]
- 33.Folmes, K. D., Witters, L. A., Allard, M. F., Young, M. E., and Dyck, J. R. (2007) Am. J. Physiol. Heart Circ. Physiol. 293 H3456-H3464 [DOI] [PubMed] [Google Scholar]
- 34.Barac, Y. D., Zeevi-Levin, N., Yaniv, G., Reiter, I., Milman, F., Shilkrut, M., Coleman, R., Abassi, Z., and Binah, O. (2005) Cardiovasc. Res. 68 75-86 [DOI] [PubMed] [Google Scholar]
- 35.Ni, Y. G., Berenji, K., Wang, N., Oh, M., Sachan, N., Dey, A., Cheng, J., Lu, G., Morris, D. J., Castrillon, D. H., Gerard, R. D., Rothermel, B. A., and Hill, J. A. (2006) Circulation 114 1159-1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li, R., Zheng, W., Pi, R., Gao, J., Zhang, H., Wang, P., Le, K., and Liu, P. (2007) FEBS Lett. 581 3311-3316 [DOI] [PubMed] [Google Scholar]
- 37.Suter, M., Riek, U., Tuerk, R., Schlattner, U., Wallimann, T., and Neumann, D. (2006) J. Biol. Chem. 281 32207-32216 [DOI] [PubMed] [Google Scholar]
- 38.Dasgupta, B., and Milbrandt, J. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 7217-7222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baur, J. A., Pearson, K. J., Price, N. L., Jamieson, H. A., Lerin, C., Kalra, A., Prabhu, V. V., Allard, J. S., Lopez-Lluch, G., Lewis, K., Pistell, P. J., Poosala, S., Becker, K. G., Boss, O., Gwinn, D., Wang, M., Ramaswamy, S., Fishbein, K. W., Spencer, R. G., Lakatta, E. G., Le Couteur, D., Shaw, R. J., Navas, P., Puigserver, P., Ingram, D. K., de Cabo, R., and Sinclair, D. A. (2006) Nature 444 337-342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hou, X., Xu, S., Maitland-Toolan, K. A., Sato, K., Jiang, B., Ido, Y., Lan, F., Walsh, K., Wierzbicki, M., Verbeuren, T. J., Cohen, R. A., and Zang, M. (2008) J. Biol. Chem. [DOI] [PMC free article] [PubMed]