

Abstract
Alginate is a family of linear copolymers of (1→4)-linked β-d-mannuronic acid and its C-5 epimer α-l-guluronic acid. The polymer is first produced as polymannuronic acid and the guluronic acid residues are then introduced at the polymer level by mannuronan C-5-epimerases. The structure of the catalytic A-module of the Azotobacter vinelandii mannuronan C-5-epimerase AlgE4 has been determined by x-ray crystallography at 2.1-Å resolution. AlgE4A folds into a right-handed parallel β-helix structure originally found in pectate lyase C and subsequently in several polysaccharide lyases and hydrolases. The β-helix is composed of four parallel β-sheets, comprising 12 complete turns, and has an amphipathic α-helix near the N terminus. The catalytic site is positioned in a positively charged cleft formed by loops extending from the surface encompassing Asp152, an amino acid previously shown to be important for the reaction. Site-directed mutagenesis further implicates Tyr149, His154, and Asp178 as being essential for activity. Tyr149 probably acts as the proton acceptor, whereas His154 is the proton donor in the epimerization reaction.
Alginate is a family of linear copolymers of (1→4)-linked β-d -mannuronic acid (M)5 and its C-5 epimer α-l-guluronic acid (G) produced by brown algae and some bacteria belonging to the Pseudomonas and Azotobacter genera (1–4). The M- and G-moieties are distributed irregularly as blocks of homopolymeric regions (M- or G-blocks) interspersed with blocks of alternating structure (MG-blocks). The relative amounts and sequence distributions of M and G vary considerably between alginates from different sources, and this variation in sequence and composition imparts different properties to alginates. G-blocks are rather stiff, and form strong, brittle gels with divalent cations such as Ca2+, whereas M-blocks are less rigid and do not form cationic gels. MG-blocks are the most flexible and have been shown to bind Ca2+ and form junction zones (5).
In Azotobacter vinelandii, alginate constitutes a major part of the coat surrounding cells differentiated into resting stage cysts (6, 7), and alginate negative strains do not encyst (8). Nevertheless, the polymer is also produced during vegetative growth, and under such conditions it may play a role in protecting the bacterium from oxidative stress (9).
The alginate polymer is produced as polymannuronic acid, and the guluronic acid residues are introduced at the polymer level by the action of mannuronan C-5-epimerases (10). All known alginate-producing bacteria possess a periplasmic C-5-epimerase, AlgG (11), but A. vinelandii also secretes a family of seven extracellular, Ca2+-dependent mannuronan C-5-epimerases, algE1–7 (12, 13). These enzymes are highly homologous and consist of one or two catalytic A-modules and one to seven regulatory R-modules, of which at least one is needed for full activity (14). Each of the extracellular A. vinelandii enzymes produces a distinctive M/G-pattern (14); AlgE2 and AlgE5 make relatively short G-blocks, whereas AlgE6 makes longer G-blocks (13, 15). AlgE4 almost exclusively produces alternating MG sequences (16).
AlgE4 is the smallest of the extracellular epimerases with a molecular mass of 57.7 kDa (553 amino acids). It contains one A-module, which is active on its own, comprising the 385 N-terminal amino acids, and one R-module of 142 residues, connected by a 7-residue linker. At the C terminus it has a small “S motif” of 19 residues of unknown function (12). Atomic force microscopy studies have revealed that the AlgE4 A-module binds more strongly to the alginate chain than the complete enzyme, indicating that the R-module is involved in the regulation of the enzyme-substrate binding strength (17). Nevertheless, the R-module on its own is also able to bind to the alginate chain (18).
AlgE4 has its optimum activity around pH 6.5–7.0 in the presence of 1–3 mm Ca2+ and at temperatures close to 37 °C (16). It is highly sensitive to alkaline pH, being inactive at pH > 8. It binds a minimum of 6 sugar residues in the catalytic site, and for short chains the third residue from the non-reducing end is the first to be epimerized (19). For longer chains the reaction proceeds toward the reducing end in a non-random, processive manner (20). On average, the enzyme epimerizes 10 units ((MG)10) in each reaction before leaving the chain (19).
An alignment of all known mannuronan C-5-epimerase sequences from bacteria and algae showed that they share an YG(F/I)DPH(D/E) motif (residues 149–155 in AlgE4) (21–23). Asp152 of this motif, which is also conserved in some pectate lyases, has been shown to be important for activity in AlgE7 (21). The reaction mechanism of C-5-epimerases has been proposed to be similar to the lyase reaction (24), involving three steps. First, the target uronic acid charge is neutralized, after which the proton at C-5 is abstracted. An enolate anion intermediate is formed, which is stabilized by resonance. In the lyase case, the enolate anion leads to β-elimination of the 4-O-glycosidic bond, whereas in the epimerase case β-elimination is prevented by donation of a proton to the opposite face of the sugar ring. Indeed in the bifunctional epimerase/lyase AlgE7 mutation of Asp152 eliminated both activities, suggesting that they occur at the same site (21). Here we report the three-dimensional structure of the AlgE4 A-module, which is responsible for generating MG-alginates, and discuss the location of the active site and a possible catalytic mechanism.
EXPERIMENTAL PROCEDURES
Growth of Bacteria—The bacterial strains and plasmids used in this study are listed in Table 1. Unless stated otherwise, bacteria were maintained at 37 °C in L broth (10 g of tryptone, 5 g of yeast extract, and 5 g of NaCl per liter) or on L agar (L broth supplemented with 15 g of agar per liter). Protein was expressed in 3× L broth (30 g of tryptone, 15 g of yeast extract, and 5 g of NaCl per liter). Media were supplemented with 200 μg/ml ampicillin or 12.5 μg/ml tetracycline when appropriate.
TABLE 1.
Bacterial strains and plasmids
| Strains or plasmids | Relevant characteristicsa | Mutagenic oligonucleotideb | Refs. |
|---|---|---|---|
| E. coli | |||
| DH5α | 25 | ||
|
ER2566
|
Encodes T7 RNA polymerase
|
New England Biolabs
|
|
| Plasmids | |||
| pTYB4 | IMPACT-CN fusion vector containing a C-terminal chitin binding Tag | New England BioLabs | |
| pUC7Tc | pUC7 containing the tetAR genes from RK2, Apr, Tcr | 53 | |
| pBL5 | Derivative of pTrc99A encoding algE4 from A. vinelandii, Apr | 51 | |
| pTB22 | Derivative of pTYB4 in which a NcoI-XmaI fragment of pBL5 containing algE4 was inserted into the corresponding sites of the vector | This study | |
| pTB26 | Derivative of pTB22 in which the tetAR genes from a pUC7Tc BamHI-fragment was blunt-end ligated into the unique Eco47III site of the vector Tcr | This study | |
| pTB61 | algE4 H154F (removing BssSI) | CGGCTTCGACCCCttCGAGCAGACC | This study |
| pTB62 | algE4 Y149F (inserting BspEI) | CGAGATGTCCGGaTtCGGCTTC | This study |
| pTB63 | algE4 Y149H (inserting BspEI) | CGAGATGTCCGGacACGGCTTC | This study |
| pTB64 | algE4 K117E (removing SalI) | ACCAGCGGCgAaGTtGACGGCTGGT | This study |
| pTB65 | algE4 K117R (inserting NotI) | AACACCAGCGGCcgcGTCGACGGCTGGTTC | This study |
| pTB67 | algE4 F122Y (inserting RsaI) | CGACGGCTGGTaCAACGGCTATATC | This study |
| pTB69 | algE4 D173V (removing BglI) | CAACGGCCTCGtgGGaTTCGTCGCCGAC | This study |
| pTB70 | algE4 R195L (inserting BspMI) | GCCAACGACCtgCACGGCTTCAAC | This study |
| pTB71 | algE4 D152E (inserting BstBI) | ACGGCTTCGAaCCCCACGAG | This study |
| pTB72 | algE4 D152N (removing BssSI) | CTACGGCTTCaACCCCCAtGAGCAGACC | This study |
| pTB73 | algE4 H154Y (removing BssSI) | CTTCGACCCCtACGAGCAGACC | This study |
| pTB74 | algE4 R195K (removing BtgI) | CGCCAACGACaagCACGGCTTC | This study |
| pTB75 | algE4 D173N (inserting BsaI) | GACAACGGtCTCaACGGCTTCGTC | This study |
| pTB76 | algE4 D173E (inserting XhoI) | CAACGGCCTCGAgGGCTTC | This study |
| pTB78 | algE4 P153A (inserting BspHI) | CGGCTTCGACgCtCAtGAGCAGACC | This study |
| pTB79 | algE4 R249A (inserting Eco57I) | GACAACGCCgctGAAGGCGTGCTG | This study |
| pTB80 | algE4 F122V (inserting HpaI) | CGACGGCTGGgTtAACGGCTATATC | This study |
| pTB82 | algE4 D178E (removing SexAI) | CGGCTTCGTCGCCGAaTAtCTGGTC | This study |
| pTB83 | algE4 D178N (inserting NruI) | GACGGCTTCGTCGCgaAtTACCTG | This study |
| pTB84 | algE4 Q225E (removing BsgI, inserting BsrBI) | CCTGGTGGTGgAGCGGGGTCTG | This study |
| pTB87 | algE4 D152N, D173E; derivative of pTB72 (inserting XhoI) | CAACGGCCTCGAgGGCTTC | This study |
| pTB89 | algE4 D173E, P153A; derivative of pTB76 (inserting BspHI) | CGGCTTCGACgCtCAtGAGCAGACC | This study |
| pTB92 | algE4 Q225N (removing BsgI) | CCTGGTGGTGaAtCGGGGTCTGG | This study |
| pTB93 | algE4 K255R (inserting BspEI) | GTGCTGCTCcgGATGACCAGCGACATCAC | This study |
| pTB147 | algE4 E155A (removing BssSI) | GACCCCCACGcGCAGACCATCAACC | This study |
| pTB153 | algE4 Y149A (inserting NgoMIV) | CGAGATGTCCGGCgcCGGCTTCGACCC | This study |
| pTB154 | algE4 D152A (inserting BseRI) | GGCTACGGCTTCGctCCtCACGAGCAGACC | This study |
| pTB155 | algE4 H154A (removing BssSI, inserting PvuII) | GGCTACGGCTTCGACCCagctGAGCAGACCATC | This study |
| pTB156 | algE4 D173A (inserting NaeI) | CAACGGCCTCGcCGGCTTCGTCGC | This study |
| pTB157 | algE4 D178A (removing SexAI) | CTTCGTCGCCGcaTAtCTGGTCGACAG | This study |
| pTB158 | algE4 D173V, R195L; derivative of pTB69 (inserting BspMI) | GCCAACGACCtgCACGGCTTCAAC | This study |
| pHE186 | algE4 H154R (removing BssSI) | GCTTCGACCCaCgCGAGCAGACCATCAAC | This study |
| pHE187 | algE4 H154K (removing BssSI) | CGGCTTCGACCCtaAgGAGCAGACCATC | This study |
| pTB185 | algE4 H196A (inserting NaeI) | CTACGCCAACGACCGCgcCGGCTTCAAC | This study |
| pTB186 | algE4 H196F (inserting PvuI) | CTACGCCAACGAtCGCttCGGCTTCAAC | This study |
| pTB187 | algE4 H196Y (inserting PvrI) | CTACGCCAACGAtCGCtACGGCTTCAAC | This study |
| pTB188 | algE4 K117A (removing SalI) | CAACACCAGCGGCgcGGTtGACGGCTGGTTC | This study |
| pTB190 | algE4 Q156A (removing BssSI) | CTTCGACCCCCACGAagcGACCATCAACCTG | This study |
| pTB191 | algE4 E155A, Q156A (removing BssSI) | CTTCGACCCCCACGcGgcGACCATCAACCTG | This study |
| pTB192 | algE4 Q225A, K255A; derivative of pTB193 (inserting NruI) | GAAGGCGTGCTGCTCgcGATGACCAGCG | This study |
| pTB193 | algE4 Q225A (inserting SmaI) | GCCTGGTGGTGgccCGGGGTCTGGAGG | This study |
| pTB194 | algE4 K255A (removing NruI) | GAAGGCGTGCTGCTCgcGATGACCAGCG | This study |
The abbreviations used are: Apr, ampicillin resistance; Tcr, tetracycline resistance. Plasmids pTB61-194 and pHE186-187 are identical to pBL5 with the exception of the introduced mutation in algE4.
These are the forward primers; the reverse primers were complementary to them. Changed bases are shown in lower case. In pTB87, pTB89, pTB158, and pTB192, pBL5 was not used as the template.
Recombinant DNA Technology—Standard recombinant DNA procedures were done according to Sambrook and Russell (25), whereas transformations were carried out according to the RbCl transformation protocol (New England BioLabs). Plasmids were isolated using the Wizard® Plus SV minipreps DNA Purification System (Promega). All gene manipulations were done in Escherichia coli DH5α, and the pTB26 plasmid was later transferred to E. coli ER2566 (New England BioLabs). DNA sequencing was performed using the BigDye® Terminator version 1.1 Cycle Sequencing Kit (Applied Biosystems).
Production and Purification of Mannuronan Oligomers—Partial acid hydrolysis of mannuronan was performed essentially as described by Campa et al. (19). Briefly, after pre-hydrolysis at pH 5.6, the sample was incubated for 6 h at 95°C and pH 3.5, and then neutralized with NaOH. The resulting oligomannuronic acid hydrolysis mixture was fractionated on three serially connected columns of Superdex 30 preparative grade (HiLoad 2.6 × 60 cm, GE Healthcare) at a flow rate of 0.8 ml/min with 0.1 m NH4Ac (pH 6.9) at room temperature. Fractions of 5 ml were collected from three successive column runs and pooled. The pooled samples were stored at 4 °C prior to three cycles of freeze-drying to remove all traces of NH4Ac and to provide solid, purified oligomers of mannuronic acid.
Protein Expression and Purification—The A-module of AlgE4 was recombinantly expressed from pTB26, a derivative of the IMPACT-CN expression vector pTYB4 (New England BioLabs) with the sequence encoding the first 378 amino acids of AlgE4 inserted (Table 1). AlgE4A was expressed in E. coli ER2566 at 15 °C and purified in a single affinity chromatography step, using the IMPACT-CN Protein Fusion and Purification System (New England BioLabs), as described in the manual.
Secondary Structure Determination—Circular dichroism studies of AlgE4A were carried out with a protein concentration of 5 μm in 100 μm HEPES (pH 6.9), at 293 K, and 5 mm CaCl2 on a JASCO J-175 spectropolarimeter. The resulting spectrum was the average of 12 scans recorded at a rate of 50 nm/min; it was analyzed by the program K2d (26).
Crystallization—Crystallization conditions were difficult to find; over 500 different conditions were tested before the first protein crystals appeared. AlgE4A crystals were obtained after two months from hanging drop experiments with 1.6–1.7 m sodium malonate (pH 7–8) as precipitant at room temperature, using drops of 3 μl of protein (11 mg/ml) and 3 μl of reservoir solution (27). The lozenge-shaped crystals had a typical size of 0.3 × 0.2 × 0.1 mm3.
X-ray Data Collection—Before data collection, crystals were soaked for 1 min in a cryoprotectant solution containing 1.7 m malonate and 20% glycerol, directly followed by flash cooling. Data were collected in house at 100 K on a DIP2030H image plate detector (Bruker-AXS, Delft, The Netherlands) using Cu-Kα radiation from a Bruker-AXS FR591 rotating-anode generator equipped with Franks mirrors. Intensity data were processed using DENZO and SCALEPACK (28). A highly redundant native data set to 2.1-Å resolution was obtained. The crystals belong to space group P21212 (Table 2). With two monomers of 39.8 kDa in the asymmetric unit, the VM is 3.0 Å3/Da (29), and the calculated solvent content is 59%. Preparation of heavy atom derivatives was done by conventional soaking experiments (30) with 1–5 mm of the heavy atom compound. For substrate binding studies crystals were soaked for 18 h in a saturated solution of M4 (an oligomer of four (1→4)-linked β-d -mannuronic acid residues) in 50% (w/v) polyethylene glycol 3350, and 10 mm CaCl2 at pH 6.5. The M4 dataset was collected at Bruker-AXS, Delft, using Cu-Kα radiation from a Bruker-AXS Microstar generator with Montel200 multilayer graded optics, and a Proteum-R diffractometer system with a 3-axis goniostat, at a temperature of 100 K.
TABLE 2.
Data collection and phasing statistics
| Crystal/derivative | Native | EMP (C2H5HgPO4) | PtCl4 | M4 |
|---|---|---|---|---|
| Cell (Å) | 174.0, 121.3, 44.7 | 174.0, 121.2, 44.6 | 174.0, 121.3, 44.7 | 169.6, 121.8, 45.1 |
| Resolution range (Å) | 50.0-2.1 (2.18-2.1) | 50.0-2.8 (2.9-2.8) | 50.0-3.2 (3.31-3.2) | 43.6-2.7 |
| No. of unique reflections | 51,691 (4,598) | 19,544 (1,886) | 14,451 (1,368) | 26,719 |
| Completeness (%) | 92.3 (83.6) | 81.2 (80.7) | 88.5 (87.4) | 99.2 (93.8) |
| Overall I/σ (I) | 12.4 (1.8) | 11.6 (3.5) | 11.8 (5.3) | 10.7 (1.9) |
| Rsym (%) | 9.8 (63.8) | 9.6 (30.6) | 13.0 (29.7) | 10.0 (56.1) |
| No. of sites | 2 | 1 | - |
Structure Determination—The structure of AlgE4A was determined by multiple isomorphous replacement with two heavy atom derivatives (Table 2). Heavy atom positions were located with the program SOLVE (31), which was also used for refining the heavy atom parameters and for calculating phases. Density modification and phase extension to 2.1 Å was done with RESOLVE (31), which was also used to automatically build fragments of the main chain. After RESOLVE, the overall figure of merit was 0.45. At this stage, a 2.1-Å electron density map was calculated and visually inspected with the program O (32). The map was judged not to be interpretable; no electron density was visible for residues that had not been built by RESOLVE. Moreover, the direction of the main chain could not be deduced and no side chain density was visible. Therefore, in an iterative procedure, phases were improved with the ARP/wARP procedure (33) combined with model building by hand. When 55% of the main chain had been built in this way, the phases could not be further improved. By assuming that the two heavy atoms from the EMP derivative occupy the same positions in the protein, and by overlaying parts of the already built main chain, the non-crystallographic symmetry operators could be determined. This allowed non-crystallographic symmetry averaging during refinement with CNS (34), and the gradual completion of the model building. The structure refinement converged at 2.1Åtoan Rfree value of 23.5% and a conventional Rfactor of 19.5%. The final model consists of 750 amino acids, residues 2–375 from molecule A and residues 2–377 from molecule B, 520 water molecules, 1 glycerol molecule, and 2 calcium ions. Assuming that the B-values of these calcium ions are similar to those of the neighboring protein atoms the occupancies of the calcium ions are close to 0.5 (Table 3). The stereochemistry of the final structure was evaluated using the program PROCHECK (35) (Table 3). For the M4 experiment the native structure was used as the starting model. Initial 2Fo – Fc and Fo – Fc maps clearly showed density for an M3 trisaccharide product as well as for calcium and chloride ions. The atomic coordinates of the final models and structure factors have been deposited in the Protein Data Bank (entries 2PYG for the native structure and 2PYH for the M4 structure).
TABLE 3.
Refinement statistics
| Native | M4 | |
|---|---|---|
| Resolution (Å) | 50.0-2.1 | 43.6-2.7 |
| No. of reflections in working set | 48,999 | 25,216 |
| No. of reflections in test set | 2,624 | 1,336 |
| Rwork (%) | 19.4 | 23.3 |
|
Rfree (%)
|
23.5
|
27.9
|
| Root mean square deviations from ideal | ||
| Bond length (Å) | 0.006 | 0.007 |
|
Bond angles (deg)
|
1.29
|
1.30
|
| Ramachandran plot | ||
| Non-glycine residues in most favored regions (%) | 84.3 | |
| Non-glycine residues in additional allowed regions (%) | 14.8 | |
|
Non-glycine residues in generously allowed regions (%)
|
1.0
|
|
| Number of non-hydrogen atoms (average B values (A)2) total/main chain/side chain | ||
| Residues in chain A | 2-375 | 1-376 |
| Protein molecule A | 2,782 (33.5/33.0/34.0) | 2,799 (49.8/49.6/50.0) |
| Residues in chain B | 2-377 | 1-377 |
| Protein molecule B | 2,798 (31.0/32.5/31.7) | 2,806 (47.5/47.2/47.8) |
| Water | 520 (42.0) | 61 (36.6) |
| Calcium ions | 2 (33.8) | 7 (72.5) |
| Glycerol | 1 (60.6) | |
| Mannuronic acid | 3 (107.0) |
Construction and Characterization of AlgE4 Mutants—Mutants of AlgE4 were constructed by the QuikChange™ Site-directed Mutagenesis Kit (Stratagene) according to the manufacturer's instructions using the primers shown in Table 1. AlgE4 mutant proteins (plasmids pTB61–pTB65; pTB67; pTB69–pTB76; pTB78–pTB80; pTB82–pTB84; pTB87; pTB89; pTB92–pTB93; pTB147; pTB153–pTB158; pTB185–pTB188; pTB190–pTB194; pHE186–pHE187) and the wild type AlgE4 (plasmid pBL5) were expressed in E. coli DH5α at 37 °C, partially purified by fast protein liquid chromatography on a HiTrap-Q column (GE Healthcare), and their catalytic activities measured in an isotope assay essentially as previously described (14), using two purification buffers and a disruption buffer containing 20 mm MOPS (pH 6.9) supplemented with 2 mm CaCl2, 2 mm CaCl2, 1 m NaCl, and 4 mm CaCl2, respectively. Protein concentrations were estimated by the Bio-Rad protein assay (36).
RESULTS
Structure of the Mannuronan C-5-epimerase AlgE4 A-module—The crystal structure of the A-module of AlgE4 (AlgE4A) was determined at 2.1-Å resolution by the multiple isomorphous replacement method using two heavy atom derivatives (Table 2). There are two molecules, A and B, in the asymmetric unit. Residues 2–375 from molecule A and residues 2–377 from molecule B could be built into the electron density. Superimposition of the two molecules reveals that they are very similar with 373 Cα atoms overlaying with an root mean square deviation of only 0.20 Å.
The AlgE4A molecule folds into a right-handed parallel β-helix structure comprising 12 complete turns with overall dimensions of 67 × 37 × 36 Å (Fig. 1). The number of amino acid residues per turn varies from 20 to 40. The β-helix turns form four parallel β-sheets, named PB1, PB2a, PB2b, and PB3, consistent with the naming of the β-sheets in polygalacturonase II (37). Not being part of the β-helix, residues 307–309 and 316–318 make up a small two-stranded antiparallel β-sheet, whereas residues 19–32 form an amphipathic α-helix that caps the N-terminal end of the β-helix. The C-terminal end of the β-helix is covered by the C-terminal end of the second molecule in the asymmetric unit. This predominantly β-helical structure agrees with circular dichroism studies (Fig. 2), which suggest an α-helix content of 5%, a β-strand content of 47%, and a coil content of 48%, whereas, according to the DSSP algorithm (38), the crystal structure contains 3.5% α-helix, 46.5% β-strand, and 50% coiled coil.
FIGURE 1.
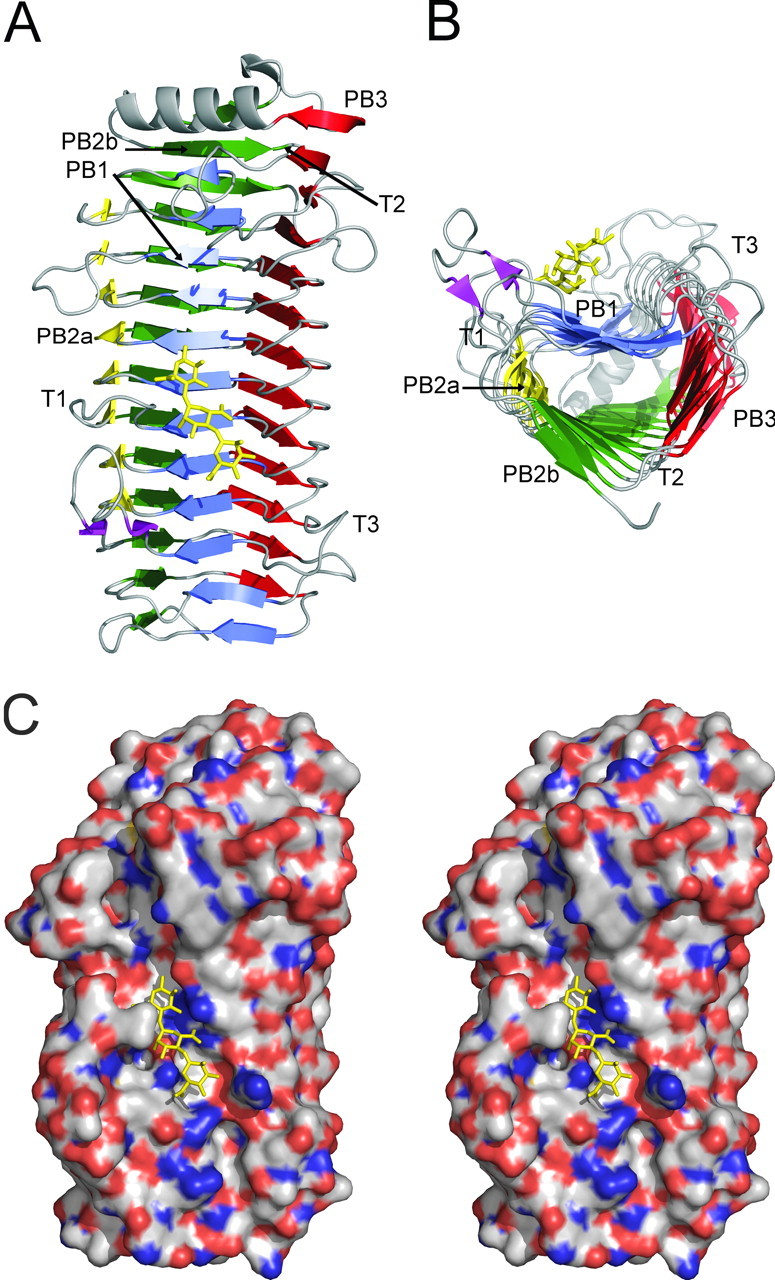
Structure of AlgE4A. A mannuronan trisaccharide (M3) bound in the active cleft is shown in stick representation. A, the secondary structure elements forming the β-helix fold. The N-terminal α-helix is shown at the top. The 4 sheets forming the helix are colored blue (PB1), yellow (PB2a), green (PB2b), and red (PB3). The T1–T3 turns are also indicated. B, the same structure as in A, seen from the C-terminal end. C, stereo view of the electrostatic potential surface. Positive potential is shown in blue and negative potential in red. The figure was produced using PyMol (52).
FIGURE 2.

Circular dichroism spectrum of 5 μm AlgE4A in 100 μm HEPES (pH 6.9), 20 °C, and 5 mm CaCl2. The spectrum of AlgE4A indicates high β-sheet structure content.
Turns connect the β-strands in adjacent β-sheets. The T1 turns between PB1 and PB2 (a or b) vary in length from 1 to 19 amino acids, whereas the T2 turns between PB2 (a or b) and PB3 are short, consisting of one amino acid residue, except in the first three turns, which comprise 8, 2, and 7 residues, respectively. Between PB3 and PB1, the T3 turns vary from 4 to 12 amino acid residues. The T1 and T3 turns are longer toward the N- and C-terminal parts of the β-helix, respectively, and these extensions form a cleft across the center of the molecule, of which the bottom is made up by PB1. In other β-helix proteins the active site is located in the cleft between the long T1 and T3 loops (37, 39), and also in AlgE4A the catalytically important residue Asp152 (21) is situated in this cleft.
A calcium ion with a pentagonal-bipyramidal coordination is bound in the hairpin loop between strands β7 and β8 (Fig. 3), with side chains of Ser91, Thr97, and Asp133 and the main chain carbonyl oxygen atoms of Glu95 and Gly124 coordinating the ion. No other calcium ions were present in the native structure, but after soaking crystals in 10 mm CaCl2 five additional calcium binding sites were identified at the surface close to Glu376–Ile357, Glu229, Thr114–Arg111, Asp112–Glu145–Asp169, and Asp308–Thr310. In addition, a chloride ion is bound to Arg90 under these conditions. A glycerol molecule from the cryoprotectant is present in the active site in molecule A, close to His154, Asp178, and Tyr149. Glycerol hydroxyl groups are known to often mimic binding of the oxygen atoms of a saccharide substrate (40). One cis-peptide is present between Glu155 and Gln156. In proteins in which non-proline cis-peptide bonds have been identified, they often occur at or near functionally important sites and are likely involved in catalysis (41).
FIGURE 3.

Stereo view of amino acids surrounding the substrate binding site. A bound mannuronan trisaccharide (M3) molecule is shown in stick representation. Glu155 and Gln156 form a cis-peptide. The image was constructed in PyMOL (52).
Location of the Active Site—To investigate the interaction of AlgE4A with substrate, crystals were soaked in a solution of M4, a tetrasaccharide of four (1→4)-linked β-d -mannuronic acid residues. After overnight soaking electron density was only observed for a trisaccharide (M3), bound in subsites –3, –2, and –1 (subsite nomenclature according to Ref. 42, the epimerization reaction takes place in subsite +1). The binding of the trisaccharide does not influence the overall structure of the enzyme; the room mean square deviation of all Cα atoms is only 0.27 Å between native and trisaccharide-bound AlgE4A. The bound trisaccharide adopts a linear conformation and has its non-reducing end in subsite –3 toward the C-terminal end of the protein (Fig. 3; see Table 4 for a summary of the hydrogen bonding interactions between the bound trisaccharide and AlgE4A). A shorter soaking time of 4 h revealed some extra electron density in the +1 subsite, close to Asp152, a residue previously implicated in catalysis (21), suggesting that during the overnight soaking the substrate had degraded. However, density in the +1 subsite was not clear enough to allow reliable modeling of a tetrasaccharide.
TABLE 4.
Interactions between bound sugar atoms and AlgE4A
| Sugar atom | Protein atom | Distance |
|---|---|---|
| Å | ||
| M-3 O-5 | Lys255 NZ | 3.7 |
| M-3 O-6A | Lys255 NZ | 4.0 |
| M-3 O-6B | Lys255 NZ | 3.2 |
| M-2 O-2 | Gln225 OE1 | 2.5 |
| M-2 O-3 | Lys255 NZ | 3.8 |
| M-2 O-5 | Gln225 NE2 | 3.2 |
| M-2 O-6A | Arg195 NH2 | 3.5 |
| His196 NE2 | 2.9 | |
| Gln225 NE2 | 3.7 | |
| M-2 O-6B | Arg195 NH1 | 4.0 |
| M-1 O-1 | Tyr149 OH | 3.0 |
| M-1 O-2 | Tyr149 OH | 2.5 |
| M-1 O-3 | Asn199 ND2 | 3.0 |
| M-1 O-3 | Arg195 NH2 | 2.6 |
| M-1 O-6B | Leu228 N | 2.9 |
To assess the importance of other amino acids for catalysis and binding of the alginate substrate, numerous mutants of AlgE4 were constructed (Fig. 4), based on its alignment with AlgE1–7 and their essentially inactive homologue AlgY (13). Four residues, Tyr149, Asp152, His154, and Asp178, all located in subsite +1, were found to be absolutely essential for AlgE4 activity. The first three of these amino acids are conserved in all known mannuronan C-5-epimerases, whereas Asp178 seems to be conserved in the A. vinelandii enzymes only. Tyr149, His154, and Asp178 point with their side chains into the +1 subsite (Fig. 3), and are likely to interact with a sugar bound at this subsite. Even conservative replacements of these three residues result in essentially inactive enzymes, strongly suggesting a role in catalysis. Replacement of Asp152 by a Glu makes the enzyme practically inactive and only 1% of the activity remains when it is replaced by an Asn (Fig. 4). Thus, both the charge and the size of this residue are important.
FIGURE 4.

Characterization of AlgE4 mutants. Relative activities are shown compared with the wild type activity (100%). Enzymes showing less than 0.05% activity are considered inactive. Only enzymes with decreased activities to less than 60% are considered as being significantly different from the native enzyme.
In the active site, Pro153 and the cis-peptide between Glu155 and Gln156 appear as important elements for providing an exact geometry around the catalytic site. Indeed replacement of Pro153 with Ala resulted in a 10-fold drop in activity. An even more dramatic effect was seen when replacing Glu155 with Ala, leading to a near 1000-fold drop in activity. This residue is unlikely to interact directly with the substrate but the substitution probably leads to the disruption of the cis peptide. In contrast, the replacement of Gln156 with Ala leads to a 10-fold activity drop, whereas replacements by other amino acids (results not shown) result in no detectable activity changes, implying that Gln156 is neither essential for the cis peptide nor for the catalysis, however, in combination with E155A, Q156A leads to an essentially inactive enzyme.
Residues Involved in Substrate Binding—Exchange of Arg195 with Lys reduced the activity of AlgE4 ∼30-fold, whereas its replacement by Leu made the enzyme essentially inactive (0.2% activity) (Fig. 4). Arg195 binds the alginate chain in subsite –1 (Table 4). Its side chain orientation is stabilized by hydrogen bonding interactions with the carboxylate group of Asp173 (2.8 and 3.1 Å). The Asp173 side chain has additional hydrogen bonds with the peptide nitrogen atom of His196 (3.2 Å), and with the N-δ2 atom of Asn199 (3.0 Å). Substitution of Asp173 with the somewhat larger Glu apparently leaves these interactions mostly intact, because this mutant protein displays only a slight drop in activity, and has limited additive effect in combination with D152N or P153A. In contrast, the D173N mutant has lost most of its activity, presumably due to an impaired interaction with Arg195. Thus Asp173 appears to be important for maintaining a productive conformation of Arg195 for substrate binding, consistent with the activity data for D173A, D173V, R195L, and the corresponding double mutant (Fig. 4). Indeed, the Asp173–Arg195 pair is absolutely conserved in A. vinelandii AlgE1–7. Substitutions of His196 by Ala, or Tyr, reduced the AlgE4 activity 20–30-fold, whereas its replacement by Phe resulted in a 100-fold activity drop. The N-ε2 atom of His196 interacts with the M-6OA in subsite –2, implying an important role of His196 in substrate binding.
Gln225 interacts with the alginate chain in subsite –2. Exchange of this residue with Ala, Asn, or Glu reduced the activity about 10–20-fold, demonstrating its importance for substrate binding. The replacement of Lys255 by an Arg decreased the activity only slightly, but the activity decreased considerably when Lys255 was exchanged for an Ala. Lys255 is located near subsite –3 and may interact, either directly or via a water molecule, with the carboxylate of the mannuronic acid residue at subsite –3 (Table 4). The importance of these residues for substrate binding is also evident from the nearly total loss of activity for the double mutant Q225A, K255A. Mutants replacing Arg249 in subsite –4, on the other hand, keep most of the activity, implying that this residue is not important for substrate binding. Replacement of Lys117 in the proximity of subsites +1 and +2 into an Arg or Ala reduced the AlgE4 activity 4–6-fold. When it was changed to Glu (the amino acid found in AlgE7) the activity dropped to 3%. The positive charge is thus important at this site. Finally, Phe122 is located close to subsite +2 and probably interacts with the substrate in this site. Replacing Phe122 with Tyr resulted in only a minor drop in activity, whereas the replacement with Val led to a virtually inactive enzyme. The absence of structural data on the substrate binding mode in subsite +2 unfortunately precludes an explanation of these data.
DISCUSSION
AlgE4 is the first structurally characterized β-helix protein that catalyzes a C-5-epimerization reaction. Most β-helix proteins are hydrolases and lyases involved in the degradation of acidic polysaccharides. A Dali structural alignment search (43) revealed nine β-helix polysaccharide hydrolases and lyases with three-dimensional structures highly similar to AlgE4A (with Z-scores over 20). However, these proteins do not share any sequence homology that could suggest a common evolutionary origin, and their proposed catalytic residues are not conserved in AlgE4A.
In view of the proposed similarities in the catalytic mechanisms of alginate lyases and mannuronan C-5-epimerases, a comparison with the active sites of alginate lyases was made. So far, five alginate lyase crystal structures are available from 3 different polysaccharide lyase (PL) families (see Ref. 44). The alginate lyase A1-III from Sphingomonas species A1 (A1-III) is a PL-5 enzyme. Its structure (PDB 1HV6 (45)) is predominantly α-helical with a deep tunnel-like cleft, in which a mannuronic acid trimer can bind in an extended conformation (42). The other four crystal structures are the PL-7 alginate lyases PA1167 from Pseudomonas aeruginosa (PDB 1VAV (46)), ALY-1 from Corynebacterium sp. (PDB 1UAI (47)), A1-II′ from Sphingomonas sp. A1 (PDB 2CWS (48)), and a PL-18 alginate lyase from Alteromonas sp. 272 (PDB 1J1T). Although these enzymes consist, like AlgE4A, almost entirely of β-strands, their structures are quite different from that of AlgE4A. Instead of the parallel β-helix architecture of AlgE4 they display a jellyroll β-sandwich topology with antiparallel β-strands.
Osawa et al. (47) noted a striking agreement in spatial arrangement of four amino acid residues in the center of the substrate binding clefts of ALY-1 (PL-7) and A1-III (PL-5). These residues were Gln/Asn, His, Arg, and Tyr. They were proposed to be the catalytic residues, with His functioning as the base in the reaction mechanism. The same spatial arrangement is also found in the other three lyases, and mutational studies of A1-II′ have indicated that the Tyr residue is particularly crucial for the catalytic reaction (48).
AlgE4A displays the same spatial arrangement of Asp152, His154, Lys117, and Tyr149 in its active site (Fig. 5), and the mannuronic trisaccharide binds in a very similar orientation and position to that of the bound trisaccharide in A1-III. Although four active site amino acid residues seem to be positioned similarly, other important residues (particularly Asp178) and the environment at subsites –2 and –3 are very different.
FIGURE 5.

Superimposition of the active site residues of ALY-1 in green (His119, Gln117, Arg72, and Tyr195), A1-III (His192, Asn191, Arg239, Tyr246, and trisaccharide) in lilac, and AlgE4A (His154, Asp152, Lys117, and Tyr149 and trisaccharide) in CPK colors. The image was constructed in PyMOL (52).
The catalytic mechanism of mannuronan C-5-epimerases has previously been proposed to proceed in three steps: the abstraction of H-5, an inversion of the hexopyranose ring, and the donation of a proton to the opposite side of the pyranose ring (24). Tyr149, Asp152, His154, and Asp178 are all positioned in the vicinity of the catalytic subsite (+1); moreover the replacement of these residues with other amino acids generally resulted in essentially inactive enzymes. To analyze the possible role of these residues in catalysis, a mannuronic acid residue in the 4C1 chair conformation was modeled into subsite +1, by extending the experimentally observed bound M3 trisaccharide chain with an extra M-unit (Fig. 6). The constraints imposed by the active site architecture resulted in only a limited number of possible positions for the +1 mannuronic acid residue without causing steric clashes. The allowed conformations are all very similar, and in all of them the +1 residue is oriented with its C-5 hydrogen atom pointing toward the Tyr149 hydroxyl group. Tyr149 is thus in a position fully compatible with a role as a general base (AA-2 in the model of Gacesa (24)), that abstracts the C-5-bound proton from the +1 M-residue. At the other face of the +1 sugar His154 is located, in an excellent position to act as a general acid (AA-3 (24)) that donates a proton to the C-5 atom of the +1 sugar, flipped into the 1C4 chair conformation (Fig. 7).
FIGURE 6.

2Fo – Fc electron density of the native active site residues generated from a simulated-annealing composite omit map contoured at 1.5 σ. The M residue in subsite –1 from the M3 structure is superposed on the native structure, the M residue in the +1 site has been obtained by computer modeling. The figure was produced using PyMol (52).
FIGURE 7.

Proposed catalytic mechanism of AlgE4. A and B, the alginate polymer enters the catalytic site. B and C, the carboxylate moiety of the mannuronic acid in subsite +1 is protonated, enabling it to form a hydrogen bond with Asp152 (and/or 178), which stabilizes the substrate-enzyme complex. C, upon deprotonation of Tyr149 (via Arg195) the alkoxide ion group extracts H-5 from the re-face of the mannuronic acid in subsite +1. C and D, a double bond is formed, which makes the conformation of the +1 mannuronic acid partially planar. D, the protonated His154 performs a nucleophilic attack on the C-5 atom of the +1 sugar from the si-face with the concomitant flip of the +1 sugar ring into the 1C4 chair conformation of guluronic acid. D and E, the carboxylic acid moiety on sugar +1 is deprotonated. E and F, the epimerized sugar leaves the active site and His154 is protonated again. F, the epimerase is ready to perform a new reaction.
The Tyr149 hydroxyl group is at hydrogen bonding distance to the Arg195 side chain (held in position by Asp173), which provides a proton relay path to the solvent. This proposed role of Tyr149 resembles the proton abstraction step in chondroitin AC lyase, which also uses a tyrosine residue to abstract a proton, in this case from the C-5 atom of a glucuronic acid (49, 50). In addition, the reaction catalyzed by this latter enzyme proceeds via a similar putative enolic transition state as in the mannuronan C-5-epimerases and alginate lyases (42, 48). AlgE4 differs from the chondroitin AC lyase, though, in that the glycosidic bond between the –1 and +1 sugars remains intact, and Tyr149 does not donate its proton to the glycosidic oxygen. The essential role of His154 as a proton donor in M → G catalysis explains the lack of activity of AlgE4 at pH > 8 (16), at which pH the His154 side chain presumably is deprotonated.
An essential feature of the binding mode of the +1 mannuronic acid is that its carboxylic acid moiety points into the interior of the enzyme, and is at hydrogen bonding distance from the carboxylic acid side chains of Asp152 and Asp178. It has been observed that for the activity of pectate/pectin lyases, which proceeds through a similar transition state as the mannuronan C-5 epimerases (24), an uncharged or methylated sugar carboxylate significantly enhances reactivity. The close interactions of the +1 carboxylic acid and the presumably negatively charged Asp152 (and/or Asp178) side chain ensures that the sugar carboxylate will be protonated (AA-1 substrate stabilization (24)), and thus will be in an uncharged state promoting reactivity.
An intriguing question is how AlgE4 prevents the lyase side reaction. In principle, Tyr149 could donate the abstracted proton of the glycosidic oxygen of the bond between the –1 and +1 uronic acid residue, similar to, e.g. chondroitin AC lyase. We propose that the interaction between Tyr149 and Arg195 may provide an effective pathway to remove the proton from the active site, thus reducing the lyase activity. In addition, the hydrogen bonding interaction between the O-3 hydroxyl group of the +1 sugar and the ring O-5 atom of the –1 sugar residue may help to prevent the two sugars moving apart, which will also diminish the lyase reaction.
Although calcium is needed for the activity of AlgE4, no Ca2+ ion is found near the active site of the enzyme, neither in the native state nor after calcium soaking. However, the Ca2+ ion that was identified in the loop between strands β7 and β8 could very well explain the calcium dependence. In the absence of Ca2+ at this position, the loop would probably adopt a different conformation and the active site cleft could be disrupted.
Various positively charged protein side chains (Arg90, Lys84, Lys117, Arg195, His196, and Lys255) were identified that could stabilize the negative charge of the carboxylate side chains of the substrate. Of these residues, Arg195 and His196 bind the carboxylate side chain of the sugar residue at subsite –2, whereas Lys255 binds the one at subsite –3. Arg90 binds a chloride ion suggesting that it is also capable of binding the carboxylate side chain of a sugar residue. All these positively charged residues are positioned on one side of the active site cleft. The AlgE4-R-module also has a positively charged cleft on the surface of its β-helix (18). Connecting the C-terminal residue of the catalytic A-module to the N terminus of the R-module by a peptide bond would create one long β-helix protein, in which a continuous long substrate binding groove runs over the surface of the full-length AlgE4 epimerase (18), thus rationalizing the contribution of the R-module to substrate binding.
AlgE4 is a processive enzyme, which binds a minimum of six residues in the active site (19). The structure of the A-module shows that the polymer, which has to slide through the active site, is enclosed by two “clamps.” The first clamp is between Gly53 and the Ca2+ binding loop with Tyr93 at the tip, and Arg90 at the bottom. The second clamp is between Arg195 and a loop with Leu228 at the tip, in the –2 subsite. These two clamps span a distance of about 20 Å, which can accommodate 5 sugar residues in a stretched conformation. The +1 site is at about 10 Å from either clamp. If the polymer chain has to slide through these positions it is also clear that, because of steric hindrance, O-2 and O-3 acetylated polymers cannot be epimerized, which explains why AlgE4A has no activity on such substrates. The importance of the second clamp is supported by hybrid enzyme studies in which residues 215 to 262 from AlgE4A, a processive enzyme, were transferred to AlgE2, a non-processive enzyme, converting it into a presumably processive enzyme (51).
It is still unclear how enzymes with the same catalytic residues distinguish between the formation of MG/GG-blocks and cleavage of the alginate chain. This reaction specificity must come from the environment of the active site, limiting which molecules effectively bind and also which reaction will take place. By comparing AlgE4 to the highly G-block forming enzyme AlgE6 or the bifunctional AlgE7 it is apparent that only subtle differences result in very different activities. Now that the structure of AlgE4 is known, it is possible to form and test hypotheses as to which amino acids are responsible for the various properties. The answers to these questions will also create a deeper understanding of the reaction mechanism of these enzymes. Given the striking similarities of the active sites of the studied epimerases and lyases, the results presented here may also help in understanding substrate binding, and hence the substrate specificities of the different enzymes involved in alginate cleavage or epimerization.
Acknowledgments
We thank Drs. A Coetzee and B. Schierbeek for collection of the M4 data at Bruker-AXS and T. Pijning for critical reading of the manuscript.
This work was supported by a grant from the Norwegian Research Council. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The atomic coordinates and structure factors (codes 2PYG and 2PYH) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
Footnotes
The abbreviations used are: M, β-d -mannuronic acid; MG-blocks, stretches of contiguous alternating structure ((MG)n); G, α-l -guluronic acid; G-blocks, stretches of contiguous G-residues; EMP, ethyl-mercury-phosphate; PL, polysaccharide lyase; MOPS, 3-(N-morpholino)propanesulfonic acid.
References
- 1.Fischer, F. G., and Dörfel, H. (1955) Hoppe-Seyler's Z. Physiol. Chem. 302 186–203 [PubMed] [Google Scholar]
- 2.Drummond, D. W., Hirst, E. L., and Percival, E. (1962) J. Chem. Soc. pp. 1208–1216
- 3.Linker, A., and Jones, R. S. (1964) Nature 204 187–188 [DOI] [PubMed] [Google Scholar]
- 4.Gorin, P. A. J., and Spencer, J. F. T. (1966) Can. J. Chem. 44 993–998 [Google Scholar]
- 5.Donati, I., Holtan, S., Mørch, Y. A., Borgogna, M., Dentini, M., and Skjåk-Bræk, G. (2005) Biomacromolecules 6 1031–1040 [DOI] [PubMed] [Google Scholar]
- 6.Lin, L. P., and Sadoff, H. L. (1969) J. Bacteriol. 100 480–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sadoff, H. L. (1975) Bacteriol. Rev. 39 516–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Campos, M. E., Martínez-Salazar, J. M., Lloret, L., Moreno, S., Núñez, C., Espín, G., and Soberón-Chávez, G. (1996) J. Bacteriol. 178 1793–1799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sabra, W., Zeng, A. P., Lünsdorf, H., and Deckwer, W. D. (2000) Appl. Env. Microbiol. 66 4037–4044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Valla, S., Li, J. P., Ertesvåg, H., Barbeyron, T., and Lindahl, U. (2001) Biochimie (Paris) 83 819–830 [DOI] [PubMed] [Google Scholar]
- 11.Rehm, B. H., Ertesvåg, H., and Valla, S. (1996) J. Bacteriol. 178 5884–5889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ertesvåg, H., Høidal, H. K., Hals, I. K., Rian, A., Doseth, B., and Valla, S. (1995) Mol. Microbiol. 16 719–731 [DOI] [PubMed] [Google Scholar]
- 13.Svanem, B. I. G., Skjåk-Bræk, G., Ertesvåg, H., and Valla, S. (1999) J. Bacteriol. 181 68–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ertesvåg, H., and Valla, S. (1999) J. Bacteriol. 181 3033–3038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramstad, M. V., Ellingsen, T. E., Josefsen, K. D., Høidal, H. K., Valla, S., Skjåk-Bræk, G., and Levine, D. W. (1999) Enzyme Microb. Technol. 24 636–646 [Google Scholar]
- 16.Høidal, H., Ertesvåg, H., Skjåk-Bræk, G., Stokke, B. T., and Valla, S. (1999) J. Biol. Chem. 274 12316–13322 [DOI] [PubMed] [Google Scholar]
- 17.Sletmoen, M., Skjåk-Bræk, G., and Stokke, B. T. (2004) Biomacromolecules 5 1288–1295 [DOI] [PubMed] [Google Scholar]
- 18.Aachmann, F. L., Svanem, B. I. G., Güntert, P., Petersen, S. B., Valla, S., and Wimmer, R. (2006) J. Biol. Chem. 281 7350–7356 [DOI] [PubMed] [Google Scholar]
- 19.Campa, C., Holtan, S., Nilsen, N., Bjerkan, T. M., Stokke, B. T., and Skjåk-Bræk, G. (2004) Biochem. J. 381 155–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hartmann, M., Holm, O. B., Johansen, G. A. B., Skjåk-Bræk, G., and Stokke, B. T. (2002) Biopolymers 63 77–88 [DOI] [PubMed] [Google Scholar]
- 21.Svanem, B. I. G., Strand, W. I., Ertesvåg, H., Skjåk-Bræk, G., Hartmann, M., Barbeyron, T., and Valla, S. (2001) J. Biol. Chem. 276 31542–31550 [DOI] [PubMed] [Google Scholar]
- 22.Nyvall, P., Corre, E., Boisset, C., Barbeyron, T., Rousvoal, S., Scornet, D., Kloareg, B., and Boyen, C. (2003) Plant Physiol. 133 726–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Douthit, S. A., Dlakic, M., Ohman, D. E., and Franklin, M. J. (2005) Bacteriol. J. 187 4573–4583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gacesa, P. (1987) FEBS Lett. 212 199–202 [Google Scholar]
- 25.Sambrook, J., and Russell, D. (2001) Molecular Cloning: A Laboratory Manual, Third Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY
- 26.Andrade, M. A., Chacón, P., Merelo, J. J., and Morán, F. (1993) Protein Eng. 6 383–390 [DOI] [PubMed] [Google Scholar]
- 27.McPherson, A. (2001) Protein Sci. 10 418–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Otwinowski, Z., and Minor, W. (1997) Methods Enzymol. 276 307–326 [DOI] [PubMed] [Google Scholar]
- 29.Matthews, B. W. (1968) J. Mol. Biol. 33 491–497 [DOI] [PubMed] [Google Scholar]
- 30.Bluhm, M. M., Bodo, G., Dintzis, H. M., and Kendrew, J. C. (1958) Proc. R. Soc. A 246 369–389 [Google Scholar]
- 31.Terwilliger, T. C. (2003) Methods Enzymol. 374 22–37 [DOI] [PubMed] [Google Scholar]
- 32.Jones, T. A., Zou, J. Y., Cowan, S. W., and Kjeldgaard, M. (1991) Acta Crystallogr. Sect. A 47 110–119 [DOI] [PubMed] [Google Scholar]
- 33.Morris, R. J., Perrakis, A., and Lamzin, V. S. (2003) Methods Enzymol. 374 229–244 [DOI] [PubMed] [Google Scholar]
- 34.Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T., and Warren, G. L. (1998) Acta Crystallogr. Sect. D Biol. Crystallogr. 54 905–921 [DOI] [PubMed] [Google Scholar]
- 35.Laskowski, R. A., MacArthur, M. W., Moss, D. S., and Thornton, J. M. (1993) J. Appl. Crystallogr. 26 283–291 [Google Scholar]
- 36.Bradford, M. M. (1976) Anal. Biochem. 72 248–254 [DOI] [PubMed] [Google Scholar]
- 37.van Santen, Y., Benen, J. A. E., Schröter, K. H., Kalk, K. H., Armand, S., Visser, J., and Dijkstra, B. W. (1999) J. Biol. Chem. 274 30474–30480 [DOI] [PubMed] [Google Scholar]
- 38.Kabsch, W., and Sander, C. (1983) Biopolymers 22 2577–2637 [DOI] [PubMed] [Google Scholar]
- 39.Jenkins, J., Shevchik, V. E., Hugouvieux-Cotte-Pattat, N., and Pickersgill, R. W. (2004) J. Biol. Chem. 279 9139–9145 [DOI] [PubMed] [Google Scholar]
- 40.van Pouderoyen, G., Snijder, H. J., Benen, J. A. E., and Dijkstra, B. W. (2003) FEBS Lett. 554 462–466 [DOI] [PubMed] [Google Scholar]
- 41.Jabs, A., Weiss, M. S., and Hilgenfeld, R. (1999) J. Mol. Biol. 286 291–304 [DOI] [PubMed] [Google Scholar]
- 42.Yoon, H. J., Hashimoto, W., Miyake, O., Murata, K., and Mikami, B. (2001) J. Mol. Biol. 307 9–16 [DOI] [PubMed] [Google Scholar]
- 43.Holm, L., and Sander, C. (1993) J. Mol. Biol. 233 123–138 [DOI] [PubMed] [Google Scholar]
- 44.Coutinho, P. M., and Henrissat, B. (1999) in Recent Advances in Carbohydrate Bioengineering (Gilbert, H. J., Davies, G., Henrissat, B., and Svensson, B., eds) pp. 3–12, The Royal Society of Chemistry, Cambridge
- 45.Yoon, H. J., Mikami, B., Hashimoto, W., and Murata, K. (1999) J. Mol. Biol. 290 505–514 [DOI] [PubMed] [Google Scholar]
- 46.Yamasaki, M., Moriwaki, S., Miyake, O., Hashimoto, W., Murata, K., and Mikami, B. (2004) J. Biol. Chem. 279 31863–31872 [DOI] [PubMed] [Google Scholar]
- 47.Osawa, T., Matsubara, Y., Muramatsu, T., Kimura, M., and Kakuta, Y. (2005) J. Mol. Biol. 345 1111–1118 [DOI] [PubMed] [Google Scholar]
- 48.Yamasaki, M., Ogura, K., Hashimoto, W., Mikami, B., and Murata, K. (2005) J. Mol. Biol. 352 11–21 [DOI] [PubMed] [Google Scholar]
- 49.Huang, W. J., Boju, L., Tkalec, L., Su, H. S., Yang, H. O., Gunay, N. S., Linhardt, R. J., Kim, Y. S., Matte, A., and Cygler, M. (2001) Biochemistry 40 2359–2372 [DOI] [PubMed] [Google Scholar]
- 50.Rye, C. S., Matte, A., Cygler, M., and Withers, S. G. (2006) ChemBioChem 7 631–637 [DOI] [PubMed] [Google Scholar]
- 51.Bjerkan, T. M., Lillehov, B. E., Strand, W. I., Skjåk-Bræk, G., Valla, S., and Ertesvåg, H. (2004) Biochem. J. 381 813–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.DeLano, W. L. (2002) The PyMOL Molecular Graphics System, DeLano Scientific, Palo Alto, CA
- 53.Blatny, J. M., Brautaset, T., Winther-Larsen, H. C., Karunakaran, P., and Valla, S. (1997) Plasmid 38 35–51 [DOI] [PubMed] [Google Scholar]