

Abstract
Deletion of the distal region of chromosome 1 frequently occurs in a variety of human cancers, including aggressive neuroblastoma. Previously, we have identified a 500-kb homozygously deleted region at chromosome 1p36.2 harboring at least six genes in a neuroblastoma-derived cell line NB1/C201. Among them, only KIF1Bβ, a member of the kinesin superfamily proteins, induced apoptotic cell death. These results prompted us to address whether KIF1Bβ could be a tumor suppressor gene mapped to chromosome 1p36 in neuroblastoma. Hemizygous deletion of KIF1Bβ in primary neuroblastomas was significantly correlated with advanced stages (p = 0.0013) and MYCN amplification (p < 0.001), whereas the mutation rate of the KIF1Bβ gene was infrequent. Although KIF1Bβ allelic loss was significantly associated with a decrease in KIF1Bβ mRNA levels, its promoter region was not hypermethylated. Additionally, expression of KIF1Bβ was markedly down-regulated in advanced stages of tumors (p < 0.001). Enforced expression of KIF1Bβ resulted in an induction of apoptotic cell death in association with an increase in the number of cells entered into the G2/M phase of the cell cycle, whereas its knockdown by either short interfering RNA or by a genetic suppressor element led to an accelerated cell proliferation or enhanced tumor formation in nude mice, respectively. Furthermore, we demonstrated that the rod region unique to KIF1Bβ is critical for the induction of apoptotic cell death in a p53-independent manner. Thus, KIF1Bβ may act as a haploinsufficient tumor suppressor, and its allelic loss may be involved in the pathogenesis of neuroblastoma and other cancers.
Neuroblastoma is one of the most common malignant solid tumors occurring in infancy and childhood and accounts for 10% of all pediatric cancers (1). Neuroblastomas are derived from sympathetic neuroblasts with various clinical outcomes from spontaneous regression because of neuronal differentiation and/or apoptotic cell death to malignant progression. Extensive cytogenetic and molecular genetic studies identified that genetic abnormalities such as loss of short arm of chromosome 1 (1p), amplification of MYCN, and 17q gain are frequently observed and often associated with poor clinical outcome (2, 3). The actual prevalence of 1p deletion in neuroblastoma is ∼35% (4–9). The deleted regions were extensively mapped to identify the candidate tumor suppressor gene(s) that has been deleted out from this region (10–17). A chromosomal locus 1p36 is frequently deleted in aggressive neuroblastoma, pheochromocytoma, colon, liver, brain, breast, and other cancers (18, 19). Transfer of 1p chromosome segments into neuroblastoma-derived cell line NGP.1A.TR1 resulted in a significant suppression of their tumor formation (20). Furthermore, previous studies indicated that there is no single site of deletion on the distal part of 1p36, but there are at least three discrete regions that are commonly deleted in neuroblastoma, indicating that they may harbor potential tumor suppressor gene(s) (8).
Tumor suppressor genes, one of the main classes of cancer-associated genes, encode inhibitors of cell proliferation and/or activators of apoptotic cell death and are involved in a variety of molecular mechanisms behind cell growth suppression (21). Tumor suppressor genes frequently mutated in other malignancies do not appear to play a major role in the generation of neuroblastoma, indicating that development of this type of tumor employs one or more previously unidentified genetic pathways. To date, a majority of candidate tumor suppressor genes has been identified by mapping the minimal deleted region and searching for the intact homologous region of mutated genes. This experimental strategy fails when the second allele is silenced by promoter hypermethylation or the targeted gene is haploinsufficient for tumor suppression, a situation where loss of one allele confers a selective advantage for tumor growth. Several examples of such haploinsufficiency for tumor suppression have been demonstrated in the case of p27KIP1, p53, and PTEN (22, 23).
We and other investigators have previously identified a 500-kb homozygous deletion at 1p36.2 harboring at least six genes, PEX14, UFD2a, KIF1B, CORT, DFF45, and PGD, in a neuroblastoma-derived cell line NB1/C201 (12, 15, 24). In this study, we have demonstrated that only KIF1Bβ, a member of the kinesin 3 family genes (25), might be a tumor suppressor gene mapped to chromosome 1p36 in neuroblastoma. Kinesins are microtubule-dependent intracellular motors involved in the transport of organelles, vesicles, protein complexes, and RNA to specific destinations (26, 27). KIF1B encodes two alternatively spliced isoforms, including KIF1Bα and KIF1Bβ, and both form homodimers and transport mitochondria and synaptic vesicle precursors, respectively (28). The NH2-terminal motor domain of KIF1Bα is identical to KIF1Bβ, whereas COOH-terminal tails share no structural homology. A point mutation in the ATP-binding site within the motor domain of KIF1Bβ has been closely linked to Charcot-Marie Tooth disease type 2A (29).
In this study, we cloned a full-length KIF1Bβ cDNA, generated recombinant adenovirus encoding KIF1Bβ, and examined its biological role in neuroblastoma and other cell lines. We systematically analyzed KIF1Bβ for LOH,2 mutation, and promoter methylation. Our genetic and functional analyses clearly showed that KIF1Bβ is a tumor suppressor, although not a classic one. KIF1Bβ might act as a haploinsufficient tumor suppressor, and its down-regulation might potentially contribute to tumorigenesis of cancers, including neuroblastoma.
EXPERIMENTAL PROCEDURES
Cell Lines and Tumor Samples—Human neuroblastoma (NB) cell lines such as SH-SY5Y, NB1, and SK-N-BE were grown in RPMI 1640 medium supplemented with heat-inactivated fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin. NMuMG, COS7, HEK293, and HeLa cells were grown in Dulbecco's modified Eagle's medium containing 10% heat-inactivated fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin. Tumor DNA and RNA samples were obtained from our Neuroblastoma Resource Bank. Informed consent was obtained at each hospital.
GSE-mediated Tumor Formation in Nude Mice—GSE assay was performed as described previously (30). In brief, a cDNA fragment (nucleotide number 2658–3115 of GenBank™ accession number AB017133) corresponding to the unique region of KIF1Bβ was amplified by PCR-based strategy and subcloned into the HpaI site of the pLXSN vector in an antisense orientation to give pLXSN-antisense KIF1Bβ. NMuMG mammary gland cells (1 × 106 cells) infected with pLXSN or with pLXSN-antisense KIF1Bβ were inoculated subcutaneously into the femoral region of nude mice. In the experiments using live animals, we strictly followed the Chiba Cancer Center Research Institute guidelines and protocols for handling live animals.
Construction of Expression Plasmids and Recombinant Adenovirus—KIF1Bβ splicing variants I, III, and IV fused to the FLAG epitope at their NH2 termini were amplified by PCR using cDNA prepared from CHP134 cells as a template and subcloned into pcDNA3.1 (Invitrogen). KIF1Bβ-GFP deletion constructs were produced by PCR-based amplification. The recombinant adenovirus was constructed as described previously (31). Briefly, KIF1Bβ cDNA was subcloned into pHMCMV6 adeno-shuttle vector. The shuttle vector was then digested with I-Ceu I and PI-Sce I and inserted into the identical restriction sites of the adenovirus expression vector pAdHM4. All of the recombinant vectors were verified by DNA sequencing. Recombinant adenoviruses were produced by transfecting the PacI-digested expression constructs into HEK293 cells. An expression vector encoding GFP was used to monitor the efficiency of infection.
Mutation Analysis—For the detection of KIF1Bβ mutations, we designed primer sets covering the motor domain and 2 kb of the 5′-upstream region of KIF1Bβ. After PCR-based amplification, PCR products were separated by 5% nondenaturing polyacrylamide gels. After electrophoresis, PCR products were gel-purified and subcloned into pGEM-T Easy Vector (Promega), and their DNA sequences were determined by an automated DNA sequencer (Applied Biosystems).
Flow Cytometry—Cells were fixed in ice-cold 70% ethanol, treated with 50 mm sodium citrate, 100 μg/ml RNase A, 50 μg/ml propidium iodide and subjected to FACS analysis (BD Biosciences) according to the manufacturer's instructions.
Construction of KIF1Bβ siRNA Expression Vector—An siRNA expression vector termed pMuniH1, in which the cytomegalovirus promoter of pcDNA 3.1 was replaced with the H1 promoter, was generated. Sense and antisense oligonucleotides for KIF1Bβ (nucleotide number 371–389 of GenBank™ accession number AB017183) were joined by a 9-base loop, annealed, and subcloned into pMuniH1.
Luciferase Reporter Assay—The genomic fragments corresponding nucleotide positions –887/+106, –630/+106, and –294/+106 of the KIF1Bβ gene were amplified from human placenta genomic DNA and cloned into pGL3-Basic luciferase reporter plasmid (Promega) to give pGL3(–887/+106), pGL3(–630/+106), and pGL3(–294/+106). For luciferase assay, SK-N-BE cells were transfected with pRL-TK (Promega) encoding Renilla luciferase cDNA and the indicated luciferase reporter constructs. Forty eight hours after transfection, firefly and Renilla luciferase activities were measured by dual-luciferase reporter assay system (Promega), and firefly luciferase activity was normalized to Renilla luciferase activity.
Methylation-specific PCR—The methylation status of the promoter region of KIF1Bβ was assessed by methylation-specific PCR as described previously (32).
Cell Cycle Analysis—Cells were fixed in 3.7% formaldehyde and permeabilized with 0.2% Triton X-100 and DNA was stained with 0.1 μg/ml of 4′,6′-diamidino-2-phenylindole. Cellular DNA content was analyzed by laser scanning cytometry (LSC2 System, Olympus).
Array-CGH Analysis—Array CGH analysis of 112 sporadic primary neuroblastomas using a chip carrying 2,464 bacterial artificial chromosome clones was conducted as described previously (33). All array-CGH data are available at NCBI Gene Expression Omnibus (GEO, www.ncbi.nlm.nih.gov) with accession number GSE 5784.
Caspase Assay—Caspase activity was measured by using caspase-3/7 assay system (Promega) according to the manufacturer's instructions.
Statistics—The Student's t test was used as a statistical method. Statistical significance was declared if the p value was <0.05.
RESULTS
Identification of KIF1Bβ as a Candidate Tumor Suppressor Mapped to Chromosome 1p36.2—To search for a candidate tumor suppressor gene(s), we first transferred each of the above-mentioned six genes into NB1 and nontransformed NMuMG mouse epithelial cells (30), and we found that only KIF1Bβ induces growth suppression in a dose-dependent manner (Fig. 1A). In contrast, our preliminary observations indicated that its alternative splicing variant KIF1Bα lacking a COOH-terminal rod region has marginal effects on cell growth in NB1 cells (data not shown). In support of these results, siRNA-mediated knockdown of KIF1Bβ in HeLa cells without 1p loss markedly enhanced their cell growth (Fig. 1B). In addition, enforced expression of KIF1Bβ induced growth retardation in p53-deficient H1299 cells and HeLa cells in which p53 is inactivated because of the presence of E6-AP (data not shown).
FIGURE 1.

KIF1Bβ has a growth-suppressive activity in vitro. A, NB1 (left panel) and NMuMG (right panel) cells were infected with recombinant adenovirus encoding LacZ or KIF1Bβ at the indicated multiplicity of infection (MOI). At the indicated time points after infection, the number of viable cells was measured. B, HeLa cells stably expressing control siRNA-2 or siRNA-5 against KIF1Bβ were established, and the expression levels of the endogenous KIF1Bβ were examined by RT-PCR (left panel). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control. Number of viable cells was measured at the indicated time points (right panel).
Overexpression of KIF1Bβ in Neuroblastomas-induced Apoptotic Cell Death—During PCR-based screening of human KIF1Bβ cDNA from human neuroblastoma cell lines, we identified at least four splicing variants that lacked exons 14 and/or 15 (supplemental Fig. S1). Similar splicing variants have also been observed in mice and rats (34). We successfully generated recombinant adenoviruses for human KIF1Bβ-I, -III, and -IV variants (Fig. 2A). Enforced expression of these splicing variants promoted apoptotic cell death in both SH-SY5Y (without 1p loss) and NB1 neuroblastoma cell lines as determined by MTT and FACS analyses (Fig. 2A and supplemental Fig. S2). Consistent with these results, colony formation assay showed that all these KIF1Bβ splicing variants strongly reduced the number of drug-resistant colonies in SH-SY5Y and NB1 neuroblastoma cells (Fig. 2B). These findings suggest that multiple KIF1Bβ splicing isoforms possess tumor suppressor activity. Intriguingly, the expression pattern of KIF1Bβ splicing variants was varied among various human tissues (supplemental Fig. S3).
FIGURE 2.

Enforced expression of KIF1Bβ induces growth suppression in neuroblastoma-derived cell lines. A, MTT assay. Neuroblastoma-derived SH-SY5Y and NB1 cells were infected with the indicated recombinant adenoviruses, including empty adenovirus (pAdNull) at a 200 multiplicity of infection (MOI)(filled boxes) or left untreated (open boxes). Twenty four hours after infection, infected SH-SY5Y and NB1 cells were seeded at a density of 1 × 103 cells/96-well plates and allowed to attach. Ninety six hours after infection, 10 μl of MTT solution was added to each well and incubated for 3 h at 37°C (left panel). Right panels show the expression of the indicated splicing variants of KIF1Bβ as examined by immunoblotting (IB) with anti-FLAG antibody. B, colony formation assay. SH-SY5Y and NB1 cells were transfected with an empty plasmid or with the indicated expression plasmids. Forty eight hours after transfection, cells were transferred into fresh medium containing 500 μg/ml of G418 and incubated for 2 weeks. After selection with G418, G418-resistant viable colonies were stained with Giemsa solution, and number of colonies was scored.
Knockdown of KIF1Bβ Expression Accelerates Growth of NMuMG Cells and Tumor Formation in Nude Mice—We then asked whether genetic disruption of KIF1Bβ gene could be critical for tumorigenesis. For this purpose, we employed a genetic suppressor element (GSE) strategy (30). A mouse genomic DNA corresponding to a KIF1Bβ cDNA fragment (nucleotide number 2658–3115 of GenBank™ accession number AB017133) encoding the unique region of KIF1Bβ was subcloned into the retrovirus pLXSN vector in an antisense orientation to give pLXSN-KIF1Bβ-AS. NMuMG cells, immortalized mouse mammary gland cells, stably infected with pLXSN-KIF1Bβ-AS, showed more than 80% reduction in endogenous KIF1Bβ expression (Fig. 3A) and formed significantly larger colonies than empty vector-infected control cells in soft agar medium (Fig. 3B). In addition, all eight mice subcutaneously transplanted with NMuMG cells stably infected with pLXSN-KIF1Bβ-AS displayed remarkable tumor growth (Fig. 3C). On the other hand, only two of eight mice transplanted with the empty vector-infected cells formed tumors, which were smaller in both cases (note log scale in Fig. 3C). The tumors formed by cells lacking KIF1Bβ expression were histologically diagnosed as poorly differentiated invasive ductal carcinoma and frequently metastasized to the lung (Fig. 4). Thus, it is likely that KIF1Bβ exerts tumor-suppressive function in vivo.
FIGURE 3.

Tumor formation in vivo. A, NMuMG cells were infected with an empty retrovirus vector (pLXSN) or with pLXSN bearing mouse antisense KIF1Bβ (pLXSN-KIF1Bβ-AS). Genomic integration of the antisense KIF1Bβ was examined by PCR (upper panel). Lower panel shows the expression levels of KIF1Bβ as examined by RT-PCR. Arrows indicate the positions of PCR products corresponding to KIF1Bβ and GAPDH. B, NMuMG cells (5 × 104 cells) infected with pLXSN or pLXSN-KIF1Bβ-AS were suspended in 3 ml of 0.4% low melting agarose dissolved in culture medium, plated onto agarose bed consisting of 0.8% low-melting agarose, and incubated at 37 °C for 5 weeks. C, tumor formation in nude mice. NMuMG cells (1 × 106 cells) infected with the indicated retroviruses were injected subcutaneously and tumor volumes were estimated weekly (lower panel). Upper panels show tumors generated in nude mice.
FIGURE 4.
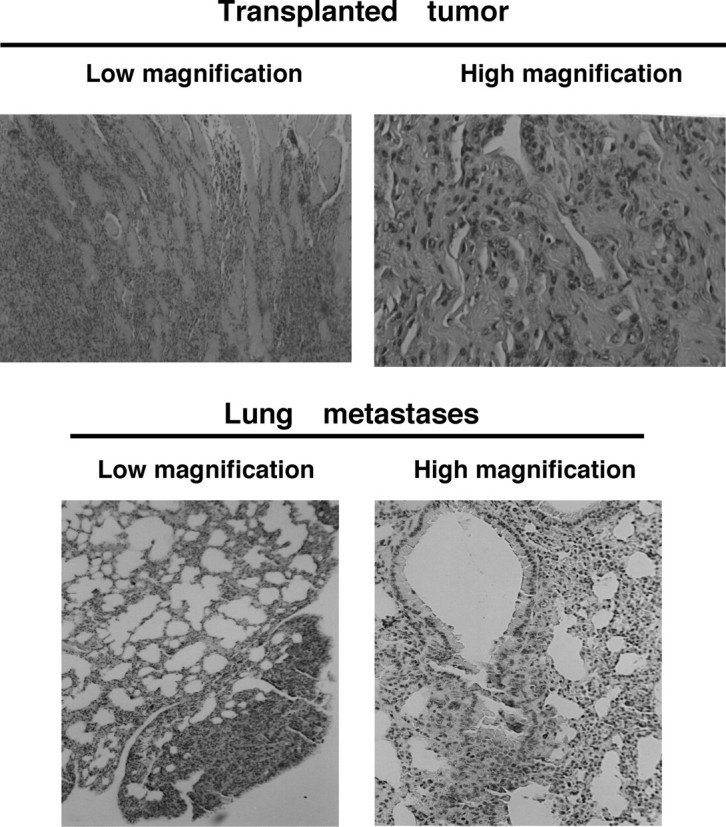
Histology of tumors generated in nude mice. Representative photographs of tumors (upper panels) and lung metastasis (lower panels) are shown. For histological analyses, tumor tissues were removed from animals and immediately fixed in 10% formaldehyde and embedded in paraffin, and 3-μm sections were stained with hematoxylin and eosin.
LOH of KIF1Bβ Locus Is Frequently Observed in Primary Advanced Neuroblastomas—We next sought to search for LOH at chromosome 1p36 in 112 sporadic neuroblastomas using array-based comparative genomic hybridization (array-CGH). Similar to previous reports, the smallest region of overlap at the distal region of chromosome 1p identified in 37 primary neuroblastomas with 1p loss was between 1p36.22 and 1pter and included KIF1B, CHD5, TP73, and SKI (supplemental Fig. S4). Thirty two percent (30/95) of neuroblastomas examined had lost one KIF1Bβ allele as determined by quantitative real time genomic PCR (Table 1). KIF1Bβ was hemizygously deleted in 18% of early neuroblastomas (stages 1 and 2, n = 51), in 55% of advanced neuroblastomas (stages 3 and 4, n = 38) (p = 0.0013), in 13% of primary neuroblastomas with a single copy of MYCN (n = 70), and in 84% of MYCN-amplified primary neuroblastomas (n = 25) (p < 0.001). No homozygous deletion was detected in the primary tumors examined.
TABLE 1.
Frequency of LOH of the KIF1B gene
LOH was examined by both array-CGH and quantitative real time PCR using genomic DNA obtained from primary neuroblastomas (tumor cells component, >70%). The cutoff value of the LOH score was 0.8 in the latter.
|
Category |
n |
KIF1B LOH |
|
|---|---|---|---|
| LOH (+) | % | ||
| Stage | |||
| 1 | 36 | 6 | 17 |
| 2 | 15 | 3 | 20 |
| 3 | 7 | 3 | 43 |
| 4 | 31 | 18 | 58 |
|
4s
|
6
|
0
|
0
|
| MYCN | |||
| Single copy | 70 | 9 | 13 |
| Amplification | 25 | 21 | 84 |
| Total | 95 | 30 | 32 |
Decreased Expression of KIF1Bβ Is Associated with Monoallelic Loss of the Gene in Primary Neuroblastomas—We examined expression levels of KIF1Bβ mRNA in 102 primary neuroblastomas by using both semi-quantitative and quantitative real time PCRs. As shown in Fig. 5, A and B, expression levels of KIF1Bβ mRNA were significantly higher in tumors at favorable stages (1, 2, and 4s, 1.654 ± 0.257, mean ± S.E., n = 60) than in those at advanced stages (3 and 4, 0.503 ± 0.180, n = 42, p < 0.001). To address whether its expression levels could be correlated with number of alleles at the KIF1Bβ gene locus, we examined primary tumors with a diploid karyotype. As shown in the lower panel of Fig. 5B, tumors with monoallelic loss of KIF1Bβ gene locus expressed significantly lower levels of KIF1Bβ mRNA (0.126 ± 0.092, n = 13) as compared with those with two KIF1Bβ alleles (0.364 ± 0.035, n = 16, p = 0.019). These results suggest that KIF1Bβ is a haploinsufficient tumor suppressor gene in high risk neuroblastomas.
FIGURE 5.

Expression levels of KIF1Bβ in primary neuroblastomas. A, semi-quantitative RT-PCR analysis. Total RNA was prepared from favorable (n = 16; stages 1 and 2, MYCN single copy) and unfavorable (n = 16; stages 3 and 4, MYCN amplified) neuroblastomas and subjected to semi-quantitative RT-PCR to examine the expression levels of KIF1Bβ. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control. B, quantitative real time PCR. Expression levels of KIF1Bβ were standardized using the corresponding glyceraldehyde-3-phosphate dehydrogenase value of each neuroblastoma sample. The relative expression levels of KIF1Bβ in favorable (stages 1, 2, and 4s) and advanced (stages 3 and 4) neuroblastomas are shown (upper panel). Lower panel shows a significant correlation between mono-allelic loss of KIF1Bβ gene and its lower expression levels.
No Promoter Methylation and Rare Mutations Are Observed in Neuroblastoma Cell Lines and Primary Neuroblastomas—Our initial mutation searches of KIF1Bβ gene were focused on its motor domain and the proximal (∼2 kb) promoter region in 21 primary neuroblastomas with 1p36 LOH and in 17 neuroblastoma cell lines. As shown in Table 2, we identified only a silent mutation GCC-GCG (at codon 95) in two primary tumors, a 2-bp (CC) deletion (at –113/–114) and G-A base change (at –336) in the KIF1Bβ promoter region in three primary tumors and four neuroblastoma cell lines. Because these aberrations were also found in the control samples, it is likely that these base changes reflect single nucleotide polymorphisms of the Japanese population.
TABLE 2.
Mutation analyses of KIF1Bβ gene
| S. no. | Case no. | Exon 4-6 | KIF exon 15 | KIF promotor F12 | KIF promotor F11 | F/UFa |
|---|---|---|---|---|---|---|
| 1 | NB-1 | NA | ||||
| 2 | NB-2 | G→A | Δ2 bp (-113-4) | G→A (-366) | NA | |
| 3 | NB-3 | UF | ||||
| 4 | NB-4 | G→A | UF | |||
| 5 | NB-5 | / | NA | |||
| 6 | NB-6 | GCC/GCG (95) | / | Δ2 bp (-113-4) | G→A (-366) | F |
| 7 | NB-7 | / | UF | |||
| 8 | NB-8 | / | UF | |||
| 9 | NB-9 | / | F | |||
| 10 | NB-10 | / | F | |||
| 11 | NB-11 | / | UF | |||
| 12 | NB-12 | / | UF | |||
| 13 | NB-13 | GCC/GCG (95) | / | Δ2 bp (-113-4) | G→A (-366) | UF |
| 14 | NB-14 | / | NA | |||
| 15 | NB-15 | / | NA | |||
| 16 | NB-16 | / | NA | |||
| 17 | NB-17 | / | NA | |||
| 18 | NB-18 | / | NA | |||
| 19 | NB-19 | / | UF | |||
| 20 | NB-20 | / | NA | |||
| 21 | NB-21 | / | F | |||
| 22 | NB-GAMB | G>A | Δ2 bp (-113-4) | G→A (-366) | Cell line | |
| 23 | NB-GOTO/P3 | Cell line | ||||
| 24 | NB-KAN | G>A | Δ2 bp (-113-4) | G→A (-366) | Cell line | |
| 25 | NB-LHN | G>A | Δ2 bp (-113-4) | G→A (-366) | Cell line | |
| 26 | NB-NB9 | Cell line | ||||
| 27 | NB-NB69 | / | Cell line | |||
| 28 | NB-NBLS | / | Cell line | |||
| 29 | NB-NBTu-1 | / | Cell line | |||
| 30 | NB-NLF | / | Cell line | |||
| 31 | NB-NMB | / | Δ2 bp (-113-4) | G→A (-366) | Cell line | |
| 32 | NB-OAN | / | Cell line | |||
| 33 | NB-SK-N-AS | / | Cell line | |||
| 34 | NB-SK-N-BE | / | Cell line | |||
| 35 | NB-SK-N-SH | / | Cell line | |||
| 36 | NB-SH-SY5Y | / | Cell line | |||
| 37 | NB-CHP134 | / | Cell line | |||
| 38 | NB-TGW | GCC/GCG (95) | / | Cell line |
For Shimada classification, F indicates favorable histology; UF indicates unfavorable histology, and NA indicates not analyzed.
To further extend our mutation searches, we have examined the presence or absence of KIF1Bβ mutations within its whole coding region in 100 primary neuroblastoma tissues. Finally, we found out the missense mutations (N737S) in six independent cases. However, their functional significances remained unclear.
Methylation of CpG islands in the promoters has been considered to be another well recognized molecular mechanism behind the inactivation of the tumor suppressor gene. To determine whether the methylation of CpG island could contribute to the inactivation of KIF1Bβ, the region spanning exon 1 and 5′-upstream sequences (nucleotide number –877 to +106) of KIF1Bβ was cloned and analyzed for promoter activity by luciferase reporter assay. As shown in Fig. 6, A and B, KIF1Bβ promoter region existed at nucleotide position between –630 and –294. We then identified KIF1Bβ CpG islands within the promoter region and investigated whether these CpGs could be methylated in primary neuroblastomas as well as cell lines. The methylation-specific PCR analysis demonstrated that all of the CpG clusters are unmethylated, suggesting that KIF1Bβ is not inactivated by methylation (Fig. 6C).
FIGURE 6.

Identification of the promoter region of human KIF1Bβ gene and its methylation status. A, nucleotide sequence of 5′-upstream region and a part of exon 1 of human KIF1Bβ gene. +1 indicates the first nucleotide of exon 1. CpG island is indicated by underline. B, luciferase reporter assay. Neuroblastoma-derived SK-N-BE cells were transiently co-transfected with the constant amount of pRL-TK encoding Renilla luciferase cDNA and the indicated luciferase reporter constructs. Forty eight hours after transfection, cells were lysed, and their luciferase activities were measured. C, methylation-specific PCR. Methylation status of the promoter region of KIF1Bβ gene in primary neuroblastomas was examined by methylation-specific PCR. M, methylated; UM, unmethylated; C, control.
The COOH-terminal Region between FHA and Pleckstrin Homology Domains of KIF1Bβ Is Responsible to Induce Apoptotic Cell Death—To map a critical domain(s) of KIF1Bβ responsible for its tumor-suppressive function, we generated NH2-terminal deletion mutants of KIF1Bβ splicing isoform IV lacking motor domain (KIF1Bβ-IV-Del 1-GFP), motor and FHA domains (KIF1Bβ-IV-Del 2-GFP), coiled-coil domain (KIF1Bβ-IV-Del 3-GFP), and FHA and coiled-coil domains (KIF1Bβ-IV-Del 4-GFP) fused with enhanced green fluorescent protein at their COOH termini (Fig. 7A). COS7 cells were transfected with wild-type or with KIF1Bβ-IV-Del-GFP fusion constructs and followed by live confocal laser scanning microscopy. Forty eight hours after transfection, KIF1Bβ-GFP-positive cells began to lose their normal cell morphology. Seventy two hours after transfection, most of the cells underwent apoptotic cell death and detached from the cell culture dish (Fig. 7, B and D). Expression of splicing variants-I, -III, and -IV along with deletion mutants of splicing variant-IV lacking the motor domain (with or without FHA domain) also induced apoptotic cell death. In contrast, expression of KIF1Bβ mutants lacking the COOH-terminal rod domain did not promote apoptotic cell death (Fig. 7, A and B). Under our experimental conditions, enforced expression of KIF1Bβ variant-IV resulted in a significant increase in the caspase activities (Fig. 7C), suggesting that KIF1Bβ-mediated apoptotic cell death might be regulated in a caspase-dependent manner. Our further analysis using other deletion mutants revealed that the 807 amino acids death-inducing region is located between FHA and pleckstrin homology domains (data not shown).
FIGURE 7.

The coiled-coil region is required for KIF1Bβ-mediated apoptotic cell death. A, schematic representation of GFP-tagged KIF1Bβ deletion mutants and summary of their ability to induce apoptotic cell death. B, COS7 cells were transiently transfected with the indicated expression plasmids. Forty eight hours after transfection, morphologies of GFP-positive cells were examined by a confocal laser scanning microscope. C, caspase activity. HeLa cells were transiently transfected with the indicated expression plasmids. Forty eight hours after transfection, cell lysates were prepared, and their caspase activities were measured. Statistically significant differences are indicated by asterisks (p < 0.05). D, time course experiments. The indicated expression plasmids were transiently introduced into COS7 cells. Forty eight hours after transfection, changes of morphology of GFP-positive cells were monitored for 12 h.
To determine whether the kinesin activity of KIF1Bβ could be necessary for its tumor-suppressive function, we introduced a Q98L mutation within a consensus ATP-binding site of KIF1Bβ-IV splicing variant (Fig. 8A). This mutation disrupts the motor function of KIF1Bβ (29). In addition, a KIF1Bβ-IV splicing variant carrying two point mutations (Q560A and D568A) within its highly conserved amino acid residues of FHA domain, which may be critical for binding to Ser/Thr-phosphorylated motifs of the interacting proteins, was also generated. In addition to these two mutants, we also generated an additional mutant bearing Q98L, Q560A, and D568A. These three mutants, however, retained an ability to induce apoptotic cell death, suggesting that KIF1Bβ-mediated apoptotic cell death does not require its ability to transport cargo using its motor domain (Fig. 8B).
FIGURE 8.

Motor and FHA domains are not required for KIF1Bβ-mediated growth suppression. A, schematic drawing of mutant forms of KIF1Bβ. Point mutations (Q98L, Q560A, and D568A) were introduced into KIF1Bβ by using the QuickChange XL site-directed mutagenesis kit (Stratagene) following the manufacturer's recommendations. B, colony formation assay. SH-SY5Y cells were transfected with an empty vector or with the indicated expression vectors. Forty eight hours after transfection, cells were transferred into fresh medium containing 500 μg/ml of G418. Two weeks after selection, G418-resistant colonies were fixed and stained with Giemsa solution, and number of drug-resistant colonies was scored.
DISCUSSION
In this study, we have shown that the KIF1Bβ gene is hemizygously deleted especially in aggressive primary neuroblastoma tumors, and its mutation is infrequent. The expression of KIF1Bβ was kept at quite a low level in aggressive neuroblastoma subsets, even though no methylation of its promoter region was observed. One of the well known haploinsufficient tumor suppressors is the cyclin-dependent kinase inhibitor p27KIP1 (35). The heterozygous mice of p27KIP1 developed tumors when mice were treated with tumor-promoting agents, and tumors retained the normal p27KIP1 allele. Additionally, hemizygous loss of p27KIP1 and/or reduced expression level of p27KIP1 conferred poor prognosis in human cancers (36). Taken together, our present results suggest that, like p27KIP1, KIF1Bβ is a haploinsufficient tumor suppressor gene of neuroblastoma, and its function to induce apoptotic cell death is regulated in a p53-independent manner. Although homozygous deletion or mutations of KIF1Bβ were rarely detectable in this study, several losses of function mutations in the coding region of KIF1Bβ gene in a large number of primary neuroblastomas, pheochromocytomas, and medulloblastomas have now been identified.3 Homozygous deletion of KIF1B in mice resulted in death just after birth because of apnea. However, heterozygous mice are viable with a phenotype resembling Charcot-Marie-Tooth disease type 2A (29). To date, no information has been available in the literature regarding spontaneous tumor formation in KIF1B-heterozygous mice. It is possible that these mice have not been followed long enough or that loss of one KIF1B allele is not sufficient for tumor formation and requires cooperating mutations for spontaneous tumor formation. Since there is functional disruption of wild-type p53 because of its mislocalization, haploinsufficiency of the KIF1Bβ gene might contribute to tumorigenesis of aggressive neuroblastomas with 1p LOH and MYCN amplification (37). KIF1Bβ might also be involved in tumorigenesis in combination with other contiguous 1p36.3 genes such as p73 (38) and CHD5 (39).
Finally, we have identified four different splicing variants of KIF1Bβ. However, colony formation assay revealed that all of the splicing variants almost equally suppress cell growth, indicating that its tumor-suppressive function may not be dependent on alternative splicing events. The deletion construct termed Del 2-GFP encoding amino acid residues 637–1576 induced apoptotic cell death similar to wild-type KIF1Bβ. Therefore, this region containing two predicted coiled-coils (amino acid residues 668–737 and 841–863) alone is sufficient for pro-apoptotic function of KIF1Bβ. The coiled-coil motifs are amphipathic oligomerization motifs. The tumor suppressor par-4 with a potential coiled-coil structure induced apoptotic cell death in prostate cancer cell lines (40, 41). Moreover, a putative coiled-coil domain of potential tumor suppressor protein, Prohibitin, has been shown to be sufficient to repress E2F1-mediated transcription and induction of apoptotic cell death (42).
In neuroblastoma, polyploidy is very common, which is often associated with a better prognosis. The precise molecular mechanisms underlying this phenomenon still remain unclear. Recently, defects in mitotic spindle check point gene products such as MAD1, MAD2, BUB1, BUB3, and BUBR1 have been implicated in the generation of polyploidy (43). Intriguingly, attached cells expressing GFP-tagged KIF1Bβ splicing variants exhibited a perturbation of G2/M progression and multinucleation (supplemental Fig. S5). The precise molecular mechanisms by which KIF1Bβ could promote these cellular abnormalities and apoptotic cell death are currently unknown. On the other hand, down-regulation of KIF1Bβ resulted in augmented cell proliferation in vitro and tumor formation in vivo, indicating that KIF1Bβ might have a critical role in the regulation of mitosis like other mitotic kinesins (44). It is conceivable that KIF1Bβ might act in a dominant inhibitory manner to sequester fundamental cytoplasmic factors that are required for proper cell cycle progression. In this connection, we are undertaking to identify the KIF1Bβ-binding partner(s), which might clarify the molecular mechanisms behind growth suppression and/or apoptotic cell death mediated by KIF1Bβ.
The nerve growth factor (NGF) dependence of tumor cells through the TrkA-p75NTR receptor complex plays a critical role in the regulation of the spontaneous regression and differentiation in neuroblastoma (45). NGF depletion-induced apoptotic cell death is blocked in aggressive neuroblastoma (46). The findings showing that expression of KIF1Bβ also increases during apoptotic cell death triggered by NGF depletion in PC12 cells3 strengthen the significance of the tumor suppressor function of KIF1Bβ in primary neuroblastomas and pheochromocytoma. Indeed, some kinesin family proteins are involved in the regulation of apoptotic cell death in developing neurons (47). In conclusion, our present results unveiled that KIF1Bβ, mapped to chromosome 1p36.2, is the candidate tumor suppressor gene of the kinesin family functioning in a manner of haploinsufficiency.
Supplementary Material
Acknowledgments
We thank Hajime Kageyama for help in FACS analysis and DNA sequencing; Yuki Nakamura, Natsue Kitabayashi, and Ayaka Nobusato for their excellent technical assistance; Drs. Keizo Takenaga, Kou Miyazaki, Hisashi Tokita, Nobumoto Tomioka, Yoko Nakamura, and Daihachiro Tomotsune for helpful discussions; and Drs. E. Thavathiru and Margaret Das for critical reading of the manuscript.
This work was supported by the Ministry of Education, Culture, Sports, Science and Technology of Japan and the Ministry of Health, Labor and Welfare of Japan. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1-S5.
Footnotes
The abbreviations used are: LOH, loss of heterogeneity; CGH, comparative genomic hybridization; FHA, forkhead-associated; GSE, genetic suppressor element; KIF, kinesin superfamily protein; NB, neuroblastoma; NGF, nerve growth factor; FACS, fluorescence-activated cell sorter; siRNA, short interfering RNA; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; RT, reverse transcription; GFP, green fluorescent protein.
S. Schlisio and W. G. Kaelin, Jr., personal communication.
References
- 1.Westemann, F., and Schwab, M. (2002) Cancer Lett. 184 127–147 [DOI] [PubMed] [Google Scholar]
- 2.Brodeur, G. M., Seeger, R. C., Schwab, M., Varmus, H. E., and Bishop, J. M. (1984) Science 224 1121–1124 [DOI] [PubMed] [Google Scholar]
- 3.Caron, H. (1995) Med. Pediatr. Oncol. 24 215–221 [DOI] [PubMed] [Google Scholar]
- 4.Gehring, M., Berthold, R., Edler, L., Schwab, M., and Amler, L. C. (1995) Cancer Res. 55 5366–5369 [PubMed] [Google Scholar]
- 5.White, P. S., Maris, J. M., Beltinger, C., Sulman, E., Marshall, H. N., Fujimori, M., Kaufman, B. A., Biegel, J. A., Allen, C., Hilliard, C., Valentine, M. B., Look, A. T., Enomoto, H., Sakiyama, S., and Brodeur, G. M. (1995) Proc. Natl. Acad. Sci. U. S. A. 92 5520–5524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martinsson, T., Sjoberg, R. M., Hedborg, F., and Kongner, P. (1995) Cancer Res. 55 5681–5686 [PubMed] [Google Scholar]
- 7.White, P. S., Thompson, P. M., Seifried, B. A., Sulman, E. P., Jensen, S. J., Guo, C., Maris, J. M., Hogarty, M. D., Allen, C., Biegel, J. A., Matise, T. C., Gregory, S. G., Reynolds, C. P., and Brodeur, G. M. (2001) Med. Pediatr. Oncol. 36 37–41 [DOI] [PubMed] [Google Scholar]
- 8.Brodeur, G. M. (2003) Nat. Rev. Cancer 3 203–216 [DOI] [PubMed] [Google Scholar]
- 9.White, P. S., Thompson, P. M., Gotoh, T., Okawa, E. R., Igarashi, J., Kok, M., Winter, C., Gregory, S. G., Hogarty, M. D., Maris, J. M., and Brodeur, G. M. (2005) Oncogene 24 2684–2694 [DOI] [PubMed] [Google Scholar]
- 10.Hogarty, M. D., Liu, X., Guo, C., Thompson, P. M., Weiss, M. J., White, P. S., Sulman, E. P., Brodeur, G. M., and Maris, J. M. (2000) Med. Pediatr. Oncol. 6 512–515 [DOI] [PubMed] [Google Scholar]
- 11.Nakagawara, A., Ohira, M., Kageyama, H., Mihara, M., Furuta, S., Machida, T., Takayasu, H., Islam, A., Nakamura, Y., Takahashi, M., Shishikura, T., Kaneko, Y., Toyoda, A., Hattori, M., Sakaki, Y., Ohki, M., Horii, A., Soeda, E., Inazawa, J., Seki, N., Kuma, H., Nozawa, I., and Sakiyama, S. (2000) Med. Pediatr. Oncol. 35 516–521 [DOI] [PubMed] [Google Scholar]
- 12.Ohira, M., Kageyama, H., Mihara, M., Furuta, S., Machida, T., Shishikura, T., Takayasu, H., Islam, A., Nakamura, Y., Takahashi, M., Tomioka, N., Sakiyama, S., Kaneko, Y., Toyoda, A., Hattori, M., Sakaki, Y., Ohki, M., Horii, A., Soeda, E., Inazawa, J., Seki, N., Kuma, H., Nozawa, I., and Nakagawara, A. (2000) Oncogene 19 4302–4307 [DOI] [PubMed] [Google Scholar]
- 13.Bauer, A., Savelveva, L., Claas, A., Praml, C., Berthold, F., and Schwab, M. (2001) Genes Chromosomes Cancer 31 228–239 [DOI] [PubMed] [Google Scholar]
- 14.Caron, H., Spieker, N., Godfried, M., Veenstra, M., van Sluis, P., de Kraker, J., Voûte, P., and Versteeg, R. (2001) Genes Chromosomes Cancer 30 168–174 [DOI] [PubMed] [Google Scholar]
- 15.Chen, Y. Z., Soeda, E., Yang, H. W., Takita, J., Chai, L., Horii, A., Inazawa, J., Ohki, M., and Hayashi, Y. (2001) Genes Chromosomes Cancer 31 326–332 [DOI] [PubMed] [Google Scholar]
- 16.Ejeskar, K., Sjoberg, R. M., Abel, F., Kogner, P., Ambros, P. F., and Martinsson, T. (2001) Med. Pediatr. Oncol. 36 61–66 [DOI] [PubMed] [Google Scholar]
- 17.Mosse, Y. P., Greshock, J., Margolin, A., Naylor, T., Cole, K., Khazi, D., Hii, G., Winter, C., Shahzad, S., Asziz, M. U., Biegel, J. A., Weber, B. L., and Maris, J. M. (2005) Genes Chromosomes Cancer 43 390–403 [DOI] [PubMed] [Google Scholar]
- 18.Schwab, M., Praml, C., and Amler, L. C. (1996) Genes Chromosomes Cancer 16 211–229 [DOI] [PubMed] [Google Scholar]
- 19.Schwab, M., Westermann, F., Hero, B., and Berthold, F. (2003) Lancet 4 472–480 [DOI] [PubMed] [Google Scholar]
- 20.Bader, S. A., Fasching, C., Brodeur, G. M., and Stanbridge, E. J. (1991) Cell Growth & Differ. 5 245–255 [PubMed] [Google Scholar]
- 21.Sherr, C. J. (2004) Cell 116 235–246 [DOI] [PubMed] [Google Scholar]
- 22.Cook, W. D., and McCaw, B. J. (2000) Oncogene 19 3434–3438 [DOI] [PubMed] [Google Scholar]
- 23.Fodde, R., and Smits, R. (2002) Science 298 761–763 [DOI] [PubMed] [Google Scholar]
- 24.Nagai, M., Ichimiya, S., Ozaki, T., Seki, N., Mihara, M., Furuta, S., Ohira, M., Tomioka, N., Nomura, N., Sakiyama, S., Kubo, O., Takakura, K., Hori, T., and Nakagawara, A. (2000) Int. J. Oncol. 16 907–916 [DOI] [PubMed] [Google Scholar]
- 25.Lawrence, C. J., Dawe, R. K., Christie, K. R., Cleveland, D. W., Dawson, S. C., Endow, S. A., Goldstein, L. S., Goodson, H. V., Hirokawa, N., Howard, J., Malmberg, R. L., McIntosh, J. R., Miki, H., Mitchison, T. J., Okada, Y., Reddy, A. S., Saxton, W. M., Schliwa, M., Scholey, J. M., Vale, R. D., Walczak, C. E., and Wordeman, L. (2004) J. Cell Biol. 167 19–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hirokawa, N. (1998) Science 279 519–526 [DOI] [PubMed] [Google Scholar]
- 27.Goldstein, L. S., and Philp, A. V. (1999) Annu. Rev. Cell Dev. Biol. 15 141–183 [DOI] [PubMed] [Google Scholar]
- 28.Nangaku, M., Sato-Yoshitake, R., Okada, Y., Noda, Y., Takemura, R., Yamazaki, H., and Hirokawa, N. (1994) Cell 79 1209–1220 [DOI] [PubMed] [Google Scholar]
- 29.Zhao, C., Takita, J., Tanaka, Y., Setou, M., Nakagawa, T., Takeda, S., Yang, H. W., Terada, S., Nakata, T., Takei, Y., Saito, M., Tsuji, S., Hayashi, Y., and Hirokawa, N. (2001) Cell 105 587–597 [DOI] [PubMed] [Google Scholar]
- 30.Garkavtsev, I., Kazarov, A., Gudkov, A., and Riabowol, K. (1996) Nat. Genet. 14 415–420 [DOI] [PubMed] [Google Scholar]
- 31.Mizuguchi, H., and Kay, M. A. (1998) Hum. Gene Ther. 9 2577–2583 [DOI] [PubMed] [Google Scholar]
- 32.Herman, J. G., Graff, J. R., Myohansen, S., Nelkin, B. D., and Baylin, S. B. (1996) Proc. Natl. Acad. Sci. U. S. A. 93 9821–9826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tomioka, N., Oba, S., Ohira, M., Misra, A., Fridlyand, J., Ishii, S., Nakamura, Y., Isogai, E., Hirata, T., Yoshida, Y., Todo, S., Kaneko, Y., Albertson, D. G., Pinkel, D., Feuerstein, B. G., and Nakagawara, A. (2008) Oncogene 27 441–449 [DOI] [PubMed] [Google Scholar]
- 34.Gong, T. W., Winnicki, R. S., Kohrman, D. C., and Lomax, M. I. (1999) Gene (Amst.) 239 117–127 [DOI] [PubMed] [Google Scholar]
- 35.Fero, M. L., Randel, E., Gurley, K. E., Roberts, J. M., and Kemp, C. J. (1998) Nature 396 177–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blain, S. W., Scher, H. I., Cordon-Cardo, C., and Koff, A. (2003) Cancer Cell 3 111–115 [DOI] [PubMed] [Google Scholar]
- 37.Moll, U. M., LaQuaglia, M., Benard, J., and Riou, G. (1995) Proc. Natl. Acad. Sci. U. S. A. 92 4407–4411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ichimiya, S., Nimura, Y., Kageyama, H., Takada, N., Sunahara, M., Shishikura, T., Nakamura, Y., Sakiyama, S., Seki, N., Ohira, M., Kaneko, Y., McKeon, F., Caput, D., and Nakagawara, A. (2001) Med. Pediatr. Oncol. 36 42–44 [DOI] [PubMed] [Google Scholar]
- 39.Bagchi, A., Papazoglu, C., Wu, Y., Capurso, D., Brodt, M., Francis, D., Bredel, M., Vogel, H., and Mills, A. A. (2007) Cell 128 459–475 [DOI] [PubMed] [Google Scholar]
- 40.Sells, S. F., Han, S. S., Muthukkumar, S., Maddiwar, N., Johnstone, R., Boghaert, E., Gillis, D., Liu, G., Nair, P., Monnig, S., Collini, P., Mattson, M. P., Sukhatme, V. P., Zimmer, S. G., Wood, D. P., Jr., McRoberts, J. W., Shi, Y., and Rangnekar, V. M. (1997) Mol. Cell. Biol. 17 3823–3832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dutta, K., Engler, F. A., Cotton, L., Alexandrov, A., Bedi, G. S., Colquhoun, J., and Pascal, S. M. (2003) Protein Sci. 12 257–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Joshi, B., Ko, D., Ordonez-Ercan, D., and Chellappan, D. (2003) Biochem. Biophys. Res. Commun. 312 459–466 [DOI] [PubMed] [Google Scholar]
- 43.Bharadwaj, R., and Yu, H. (2004) Oncogene 23 2016–2027 [DOI] [PubMed] [Google Scholar]
- 44.Endow, S. A. (1999) Eur. J. Biochem. 262 12–18 [DOI] [PubMed] [Google Scholar]
- 45.Nakagawara, A. (1998) Med. Pediatr. Oncol. 31 113–115 [DOI] [PubMed] [Google Scholar]
- 46.Nakagawara, A., Arima-Nakagawara, M., Scavarda, N. J., Azar, C. G., Cantor, A. B., and Brodeur, G. M. (1993) N. Engl. J. Med. 328 847–854 [DOI] [PubMed] [Google Scholar]
- 47.Midorikawa, R., Takei, Y., and Hirokawa, N. (2006) Cell 125 371–383 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.