

Abstract
Xanthomonas campestris GumK (β-1,2-glucuronosyltransferase) is a 44-kDa membrane-associated protein that is involved in the biosynthesis of xanthan, an exopolysaccharide crucial for this bacterium's phytopathogenicity. Xanthan also has many important industrial applications. The GumK enzyme is the founding member of the glycosyltransferase family 70 of carbohydrate-active enzymes, which is composed of bacterial glycosyltransferases involved in exopolysaccharide synthesis. No x-ray structures have been reported for this family. To better understand the mechanism of action of the bacterial glycosyltransferases in this family, the x-ray crystal structure of apo-GumK was solved at 1.9Å resolution. The enzyme has two well defined Rossmann domains with a catalytic cleft between them, which is a typical feature of the glycosyltransferase B superfamily. Additionally, the crystal structure of GumK complexed with UDP was solved at 2.28Å resolution. We identified a number of catalytically important residues, including Asp157, which serves as the general base in the transfer reaction. Residues Met231, Met273, Glu272, Tyr292, Met306, Lys307, and Gln310 interact with UDP, and mutation of these residues affected protein activity both in vitro and in vivo. The biological and structural data reported here shed light on the molecular basis for donor and acceptor selectivity in this glycosyltransferase family. These results also provide a rationale to obtain new polysaccharides by varying residues in the conserved α/β/α structural motif of GumK.
Glycosylation events are among the most common and important enzymatic reactions in nature. Glycosyltransferases (GTs)5 are enzymes that catalyze the transfer of a sugar moiety from a donor to an acceptor molecule. At present, there are more than 40,000 known or putative GT sequences in various data bases (1). In the CAZy (carbohydrate-active enzyme) data base (available on the World Wide Web), GTs are grouped into 90 families on the basis of sequence similarity. This impressive dispersion is due to very low sequence homology among GTs. Despite this vast sequence divergence and poor homology, the reported structures of 60 GTs in 27 families have only two folds, corresponding to superfamilies GT-A and GT-B. Other predicted folds, such as GT-C for transmembrane GTs (2, 3), remain to be described.
GT-A and -B folds are variations of Rossmann-like β/α/β domains. The GT-A members display a central sheet of 7-8 β-strands, with a DXD motif. This acidic motif coordinates the ribose and metal (divalent cation) in the catalytic center. The presence of this motif has been shown to be crucial for the catalytic activity in these GTs (4). In contrast, GT-B proteins do not bind metals and have two well defined Rossmann domains with a deep cleft between them, in which binding of substrates and catalytic activity occur (5, 6). An interesting feature of GTs is that very similar functions can be carried out by multiple sequences, which exhibit very similar folding. This situation complicates the determination of specific contacts and amino acids that affect or are directly involved in catalysis, substrate binding, and other structural functions.
GTs are involved in the biosynthesis of glycolipids, polysaccharides, glycoproteins, and a vast range of metabolites. Accordingly, GTs display a wide array of acceptor molecules, including oligosaccharides, lipids, proteins, and glycolipids (4, 7). In contrast, donor substrates are mostly activated glyconucleotides.
The numerous compounds that are synthesized by GTs have central roles in cellular biochemistry (e.g. in cell signaling, immune response, and bacterial virulence, among other processes) (8, 9). Moreover, these compounds have an enormous potential for practical applications. The chemoenzymatic synthesis of oligo/polysaccharides promises an almost infinite variety of new carbohydrate structures with as yet unknown applications (10, 11). In particular, extracellular polysaccharides have a role in bacterial virulence but also may display rheological/physical properties that would be useful for industrial applications. Such is the case for xanthan, an exopolysaccharide produced by the phytopathogen Xanthomonas campestris. Xanthan is involved in X. campestris virulence toward a substantial number of economically and agriculturally important plants (12). Also, this polysaccharide has a wide range of potential applications and functions (13).
X. campestris GumK (β-1,2 glucuronosyltransferase), a membrane-associated protein that is part of the biosynthetic machinery for xanthan, is responsible specifically for the addition of a GlcA residue from UDP-GlcA during the formation of the pentasaccharidic subunit of xanthan (Fig. 1) (14, 15).
FIGURE 1.
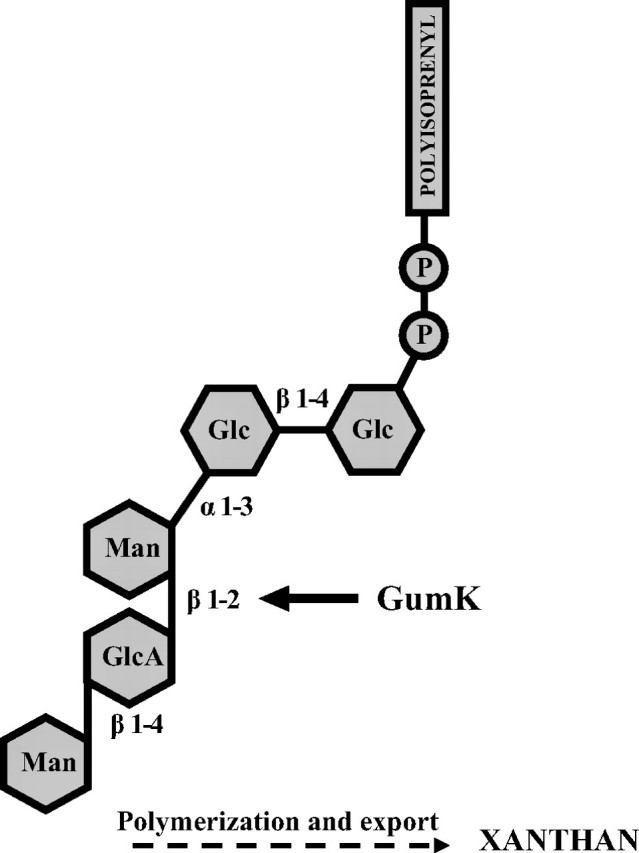
Xanthan pentasaccharidic subunit. The formation of the β-1,2-glycosidic bond catalyzed by GumK is shown.
Despite the synthetic utility and industrial/medical importance of GTs, many details of enzyme structures and mechanisms remain elusive. In particular, no structural information is available on xanthan-specific GTs, and not much is known about the GTs involved in the synthesis of other polysaccharides (16, 17). This is due to the fact that GTs are difficult to characterize because the proteins are often membrane-associated, unstable, present at very low concentrations, and difficult to express.
Herein we describe the structure of GumK in the presence and absence of UDP. We focused on the molecular contacts that anchor the donor molecule to the protein, including kinetic analyses of mutant proteins and the in vivo effects of these mutations on X. campestris polysaccharide production. Also, we mutated residues that could be directly involved in the catalytic mechanism of GumK. Determination of the catalytic mechanism and of the specific contacts with substrates could result in strategies for the exploitation of GTs as unique synthetic catalysts in the creation of unnatural polysaccharide variants.
EXPERIMENTAL PROCEDURES
Protein Purification and Crystallization—GumK protein with a C-terminal LEHHHHHH tag was expressed from plasmid pETHisKC and purified as described previously (14). Purified protein was concentrated to 20 mg/ml in storage buffer (400 mm NaCl, 0.05% Triton X-100, 50 mm Tris-HCl, pH 8.0) by ultrafiltration. The protein concentrate was stored at 4 °C until use. Crystals of the native and D157A mutated form of GumK were grown at 20 °C using the hanging drop vapor diffusion method, as described previously (18). For the UDP-GumK or UDP-GumK D157A complexes, crystals of GumK were soaked for 0.5-8 h in crystallization solution plus UDP-GlcA (10 or 100 mm) at 20 °C. Unfortunately, GlcA was readily hydrolyzed from UDP-GlcA during these soaking experiments in the native form of GumK, and we could only see the position of UDP bound to GumK. Furthermore, we were unable to co-crystallize GumK or mutant D157A in the presence of UDP-GlcA.
Data Collection and Phasing—Heavy atom soaks were carried out in crystallization buffer (35% polyethylene glycol 3350, 0.1 m Tris-HCl, 0.2 m Li2SO4, 0.1 m CsCl, pH 8.2) supplemented with 10 mm K2PtCl4 for 2 h. Single crystals of native and platinum derivative GumK were drawn out of the crystallization drop and frozen in liquid nitrogen. All data sets were collected at 110 K. Crystallization buffer was used as the cryoprotectant. A two-wavelength MAD data set (peak = 1.0718 Å and inflection = 1.0722 Å) was collected to 2.0 Å resolution from a platinum derivative in beamline X12C, National Synchrotron Light Source, Brookhaven National Laboratories (Brookhaven, NY) in an ADSC Q210 modified detector. Reflection intensities were integrated using MOSFLM, merged with SCALA, and reduced with Truncate (19). Statistics are shown in Table 1. The crystal belonged to space group P6522, in which an asymmetric unit comprised one GumK molecule. Platinum sites were found by using SHELX, and the positions, B-factors, and occupancies were refined by using Sharp (20), with the four platinum positions identified after six rounds of refinement and inspection of log likelihood gradient residual Fourier maps. Density modification was performed by using DM, and solvent flattening was performed by using Solomon (19). This processing resulted in a readily interpretable map of electronic density.
TABLE 1.
X-ray data collection and refinement statistics for wild type GumK
Values for the highest-resolution shell are shown in parentheses.
| Soaking condition | Platinum-labeled GumK 2 h, 10 mm | Native | UDP 2 h, 100 mm |
|---|---|---|---|
| Data collection | |||
| Beam line | NSLS, X12C | NSLS, X12C | LNLS, DO3B-MX1 |
| Space group | P6522 | ||
| Unit cell (Å) | a = 123.6; b = 123.6; c = 174.3 | a = 123.6; b = 123.6; c = 174.3 | a = 121.2; b = 121.2; c = 170.7 |
| Unit cell (degrees) | α = 90.0; β = 90.0; γ = 120.0 | α = 90.0; β = 90.0; γ = 120.0 | α = 90.0; β = 90.0; γ = 120.0 |
|
Platinum-labeled GumK |
Native |
UDP |
||
|---|---|---|---|---|
| Peak | Inflection | |||
| Wavelength (Å) | 1.0718 | 1.0722 | ||
| Resolution range (Å) | 33.15-2.0 (2.1-2.0) | 33.33-2.0 (2.1-2.0) | 104.8-1.9 (2.00 -1.90) | 27.0-2.28 (2.4-2.28) |
| No. of observations (F > 0) | 879,956 | 343,756 | ||
| Unique reflections | 51,441 | 51,635 | 62,435 | 33,916 |
| Completeness (%) | 95.5 (75.1) | 95.5 (74.8) | 100.0 | 98.5 |
| Anomalous completeness (%) | 91.8 (63.9) | 92.1 (74.8) | ||
| Average I/σ(I) | 17.3 (2.8) | 17.4 (2.8) | 30.0 (8.2) | 19.6 (6.4) |
|
Rmerge (%)
|
6.9 (34.4)
|
10.4 (30.3)
|
||
| Refinement | ||||
| Resolution | 104.8-1.9 | 27-2.28 | ||
| Used reflections | 59,204 | 32,197 | ||
| Rwork, Rfree (95%/5%) | 0.180/0.204 | 0.175/0.216 | ||
| No. of atoms | ||||
| Protein | 2,962 | 2,932 | ||
| Ligand/ion | 24 | |||
| Water | 474 | 420 | ||
| Average B-factors | 18.60 | 21.08 | ||
| Protein | 18.01 | 19.60 | ||
| Ligand/ion | 22.85 | |||
| Water | 30.01 | 30.87 | ||
| r.m.s. deviations | ||||
| Bond lengths (Å) | 0.013 | 0.017 | ||
| Bond angles (degrees) | 1.277 | 1.682 | ||
| Ramachandran plot (% residues) | ||||
| In most favored regions | 91.0 | 91.9 | ||
| In additional allowed regions | 8.3 | 8.1 | ||
| In generously allowed regions | 0.6 | 0.0 | ||
Single UDP-GlcA-soaked crystals (native and D157A mutated forms) were frozen in liquid nitrogen. Complete data sets of the GumK-UDP complex were collected in beamline DO3B-MX1, Laboratorio Nacional de Luz Sincrotron (Campinas, Brazil), at a wavelength of 1.427 Å. Statistics are shown in Table 1.
Model Building, Refinement, and Validation—Model building of the native GumK was performed with ARP/wARP. Refinement was carried out with Refmac. For nondefined regions, manual building was performed with Coot (21) alternated with Refmac. At the beginning of analysis, a fraction of the data sets (5%) was set aside for Rfree calculations. To determine the structure of the GumK-UDP complex, ARP/wARP using the native GumK as a model was used, iterated with Refmac refinement. For nondefined regions, manual building was performed with Coot. Surface electrostatic potentials were calculated using the Adaptive Poisson-Boltzmann Solver (APBS) program (22) and visualized with Pymol (DeLano Scientific LLC) (available on the World Wide Web).
Determination of Kinetic Parameters—The kinetic parameters of the enzymes were measured with a radioactive assay. The natural GumK lipid acceptor is mannose-α-1,3-glucose-β-1,4-glucose-diphosphate-polyisoprenyl (Man-Cel-P-P-lipid). This acceptor was obtained from X. campestris membranes as follows. Ten-liter cultures of X. campestris were grown in SFFM medium (4 mm K2HPO4, 1.5 mm KH2PO4, 7.5 mm (NH4)2SO4, 5 μm MnCl2, 0.5 mm MgCl2, 0.125% (w/v) tryptone, 0.125% (w/v) yeast extract, 0.125% (w/v) malt extract, 0.005% (w/v) FeSO4-7H20). These cultures were allowed to reach an A600 of 3.0, and at that point bacterial cells were collected and permeabilized as described (23). These EDTA-permeabilized cells were used in scaled up incubations with 3 mm UDP-Glc and 1.5 mm GDP-[14C]Man (specific activity 1 mCi/mol) in the presence of incubation buffer (75 mm MgCl2, 0.2 m Tris-HCl, pH 8.2) for 1 h. These incubations produced the natural acceptor (Man-Cel-P-P-lipid) as the only radioactive product species (14).
Soluble compounds were removed by three washes with deionized water, and Man-Cel-P-P-lipid was recovered by three extractions with 1:1 chloroform/methanol. The apparent Km for the donor substrate, UDP-GlcA, was determined at a fixed concentration of the acceptor Man-Cel-P-P-lipid (500 μm) because of the limited supply of this substrate. The Km for the acceptor was determined at UDP-[14C]GlcA concentrations (specific activity 30 Ci/mol) estimated to be at least 10 times greater than the Km. Incubations were performed at 20 °C for 2 min in 100-μl volume reactions with 2% Triton X-100. After the incubation, the radioactive glycolipid product was recovered by organic solvent extraction as described (14). Assays were performed in triplicate.
Site-directed Mutagenesis—Mutations were introduced into the cloned gumK gene by using the QuikChange™ site-directed mutagenesis kit (Stratagene) with the appropriate primers. Mutations were confirmed by sequence analysis. The mutated gumK genes were expressed in Escherichia coli and purified as described previously for GumK (14). Purified mutated proteins were stored at 4 °C until use. For in vivo complementation assays, the open reading frames of mutated GumK were cloned in the wide host range plasmid pBBRprom (14). Plasmids were denoted pBBRSK for the pBBRprom derivative expressing wild-type GumK and pBBRSK/mutation (e.g. pBBRSK/D157A) for pBBRprom derivatives expressing mutated GumK. Protein expression in complemented strains was verified with Western blotting using polyclonal antibodies raised against GumK.
Complementation Assays and Polysaccharide Quantification—X. campestris FC2 (wild type) and XcK (gumK- isogenic mutant) strains (15) carrying plasmid pBBRSK or pBBRSK/mutation were inoculated and incubated for 72 h in SFFM medium plus 4% glucose with the corresponding antibiotics. Xanthan was precipitated by the cetylpyridinium chloride polysaccharide precipitation method (24) with modifications. Briefly, 20 μl of culture supernatants containing xanthan or xanthan standard aqueous solutions of 0.75, 1.5, 2.25, 3, or 3.75% were mixed with 3 ml of deionized water. Three milliliters of 0.36% cetylpyridinium chloride solution was added, and the mixture was incubated at 37 °C for 1 h. This mixture was centrifuged at 5,000 × g to pellet the cetylpyridinium chloride/xanthan precipitate. The absorbance at 260 nm to measure the remaining cetylpyridinium chloride in the supernatant of the xanthan standards was used to construct a calibration curve. The amount of polysaccharide produced in the samples was estimated from the A260 by reference to the calibration curve. The amount of polysaccharide produced was measured at least three times.
RESULTS AND DISCUSSION
Structures of GumK and of the GumK-UDP Complex—X. campestris GumK is the founding member of family GT70 (see the CAZy data base on the World Wide Web), which is composed of phytopathogenic bacterial glucuronosyltransferases involved in exopolysaccharide biosynthesis. The x-ray structure of GumK was solved with a two-wavelength MAD experiment (Table 1). There was only one protein molecule in the asymmetric unit. The final 1.9-Å structure included residues 13-385 and 480 water molecules (Protein Data Bank code 2HY7). The final native GumK structure had a crystallographic R value of 0.18 and an Rfree of 0.20. The GumK-UDP complex was solved at 2.28 Å resolution by MR, using the native structure as a model. The Rfree was 0.216 for the final structure (Protein Data Bank code 2Q6V).
Fig. 2 shows that GumK is a two-domain molecule with an overall size of ∼50 × 50 × 65 Å. The N-domain is formed by residues 13-201 and the final C-terminal α-helix, Cα7 (residues 362-380), a feature observed in other GT-B enzymes (25-28). This domain is composed of 10 α-helices surrounding a core of eight mostly parallel β-sheets (Fig. 2, A and B). The C-domain is composed of residues 210-361, which consists of a core of six β-sheets shielded by six α-helices.
FIGURE 2.

Overall structure of GumK and the GumK-UDP complex. A, the apoprotein is shown as a ribbon diagram with β-strands in magenta and α-helices in cyan. B, GumK-UDP complex. UDP is drawn as a stick model.
The β-strands and α-helices of both domains are ordered as in a typical Rossmann fold (29) and exhibit high structural homology (r.m.s. deviation = 2.02 over 88 C-α), which confirms that despite the low sequence homology between these domains, the same fold is adopted. The N- and C-domains are joined by a linker (residues 202-208) between the eighth β-strand and the first α-helix of the C-domain. This interdomain linker, together with the loop connecting Cα6 and Cα7 (residues 353-361), defines the floor of the cleft between the two domains. The cleft is ∼20 Å deep and 15 Å across at its widest point. The dimensions of the cleft suggest that the enzyme crystallized in an “inactive,” open state. Recently, it was shown that a large relative rotation between the N- and C-domains is necessary for catalytic activity in GT-B MshA (30). This interdomain flexibility has also been observed or predicted for other members of the GT-B superfamily (26, 31-33). These motions of 10-25° are believed to convert the enzyme to an “active,” closed conformation, bringing critical residues from the N- and C-terminal domains together into a catalytically active conformation. We will study whether this type of movement is required for GumK activity.
Fig. 3 shows the position of UDP in its binding pocket. This pocket is located on the C-terminal face of the cleft, in a positively charged surface. The UDP-binding pocket is an α/β/α motif defined by Cα3, Cβ4, and Cα4 and the linkers between them (Figs. 2B and 3D). This structural motif is highly conserved throughout the GT-B superfamily (6, 34) as an alternative way to coordinate the negative charge of the phosphates in the nucleotide-sugar. Regardless of the low sequence homology, the degree of structural conservation with other GT-B superfamily members was evident upon calculation of the structural homology of the C-terminal globular domain. The GumK-UDP structure exhibited marked superposition with family 5 glycogen synthase from Agrobacterium tumefaciens (Protein Data Bank code 1rzu; r.m.s. deviation = 3.8 Å) and with family 4 lipopolysaccharide core biosynthesis α-1,3-glucosyltransferase (Protein Data Bank code 2iv7; r.m.s. deviation = 3.3 Å) (26, 35).
FIGURE 3.

Substrate binding in GumK. A, ribbon representation of GumK, showing UDP bound on the C-terminal face of the catalytic cleft.B, surface representation of the UDP-binding pocket. C, final (2Fo - Fc) electron density map for UDP (contoured at 1σ). Residues contacting UDP are shown as stick representations. Hydrogen bonds are depicted as dashed lines. D, GumK C-terminal α/β/α motif involved in donor substrate binding.
The most notable contacts of GumK-UDP include hydrogen bonds between the imidic NH of Met231 and α-phosphates O1 and O2 and between the imidic NH of Met306 and the β-phosphate O1. The phosphates are also coordinated by hydrogen bonding between Lys307 NH2 and β-phosphates O2 and O3 and between Tyr292 OH and O1β and O2β (Fig. 3C). Mutation of Lys307 and Tyr292 had marked effects on both the Km of UDP-GlcA and the Vmax (Table 2), indicating the importance of these contacts in the interaction with the negatively charged phosphates of the donor molecule. The ribose is bound by hydrogen bonds between its 2′- and 3′-hydroxyls and Q310 NH2. The Q310A mutation substantially increased the Kmof UDP-GlcA, despite having a minor effect on the Vmax. Finally, the uridine is bound by contacts between O4′ and the imidic NH of E272 and between the carbonilic CO of Met273 and N3′.
TABLE 2.
Kinetic parameters for wild-type and mutant GumK
| Mutant | Km UDPGlcA | Vmax | Km aceptor | Vmax | Kcat |
|---|---|---|---|---|---|
| μM | pmol/μg min | μM | pmol/μg min | min−1 | |
| Wild type | 62 ± 5 | 116 ± 3 | 198 ± 16 | 115 ± 3 | 5.2 |
| M231A | 63 ± 9 | 116 ± 4 | 218 ± 16 | 119 ± 3 | 5.2 |
| E272A | 125 ± 10 | 50 ± 1 | 291 ± 21 | 52 ± 1.2 | 2.3 |
| E272D | 100 ± 4 | 95 ± 1 | 267 ± 13 | 99 ± 1.5 | 4.3 |
| Y292A | 210 ± 25 | 27 ± 1 | 274 ± 30 | 23.5 ± 1 | 1.2 |
| K307A | 440 ± 34 | 15.2 ± 0.2 | 390 ± 65 | 10.8 ± 0.6 | 0.68 |
| Q310A | 1,190 ± 99 | 99 ± 2 | 246 ± 20 | 98 ± 2.5 | 4.5 |
| D157A/E/N | NDa | ND | |||
| E192A | 61 ± 6 | 115 ± 3 | 199 ± 20 | 118 ± 3.5 | 5.2 |
| D207A | 60 ± 6 | 111 ± 3 | 194 ± 18 | 115 ± 3 | 5.05 |
| D234A | 63 ± 9 | 117 ± 4 | 205 ± 18 | 120 ± 3 | 5.3 |
ND, not detected, because of no or very low level activity (<0.5 pmol/μg/min).
The hydrogen bonds and atoms involved in UDP binding are detailed in Table 3. All of these interactions seem to have a cooperative effect on the binding of UDP. Mutations Y292A and K307A have marked effects on the Km of UDP-GlcA and on the catalytic efficiency (kcat), whereas other mutations, such as E272A and E272D, had smaller effects on the Km and kcat (Table 2). Despite the influences of some contacts on the efficiency of the enzyme, individual contacts proved to be relevant but not essential for binding the substrate. Interestingly, Lys307, the contact that had the most pronounced effect on the kcat of GumK, is located in the conserved Cα4 helix and is one of the residues that coordinate the phosphates (Table 3 and Fig. 4).
TABLE 3.
Hydrogen bonds between UDP and GumK residues
| GumK residues | Hydrogen bond distance of UDP |
|---|---|
| Å | |
| Met231/N | α-Phosphate O1A (2.92) |
| Met231/N | α-Phosphate O2A (2.85) |
| Tyr292/OH | β-Phosphate O1B (2.65) |
| Tyr292/OH | β-Phosphate O2B (2.42) |
| Met306/N | β-Phosphate O1B (3.06) |
| Lys307/N | β-Phosphate O2B (2.35) |
| Lys307/N | β-Phosphate O3B (2.51) |
| Gln310/NE2 | Ribose O3″ (2.50) |
| Gln310/NE2 | Ribose O2″ (2.88) |
| Met273/N | Uracil O4′ (2.66) |
| Met273/O | Uracil N3′ (2.71) |
| Glu272/OE1 | Uracil O4′ (3.36) |
FIGURE 4.

Sequence alignments of all GT70 family members. Secondary structural elements of GumK are shown above the protein sequence. Invariant residues are shaded orange. The arginine cluster proposed to interact with the membrane is shaded blue. Residues involved in UDP binding are highlighted in yellow. The invariant catalytic residue Asp157 is shaded green.
Fig. 3, C and D, shows the restrictions to which the donor substrate is exposed while entering the binding pocket. From the architecture of the α/β/α UDP-binding motif, it is clear that any purine-based nucleotide would not be able to fit in the narrow pocket created by the Cα3 and Cα4 helices, specifically because of the hydrogen bonding between Glu272 and Met273 and the uridine. All residues that contact the UDP are conserved in the GT70 family (Fig. 4), which indicates that, as expected, binding of the donor substrate is conserved. For biotechnological applications, a relaxed or even a changed specificity could be very useful for the synthesis of novel polysaccharides. We speculate that a change in the specificity of GumK is possible by mutation of residues in the conserved Cα4 helix, where some of the most important contacts for binding the donor molecule are located.
Proposed Membrane Association Site—Subcellular localization experiments showed that GumK associates with the cell membrane in X. campestris (14). The electrostatic surface potential for GumK reveals a polar protein with a positively charged N-domain (theoretical pI 9.97) and a negatively charged C-domain (theoretical pI 6.20). A cluster of basic and hydrophobic residues (in helices Nα2 and Nα4 and the linker region between Nα4 and Nβ4) lies at the tip of the N-terminal domain (Fig. 5A). The side chains of residues Arg58, Lys60, Arg86, Arg95, Arg96, Arg100, and Arg108 are solvent-exposed, forming an arginine cluster surrounded by hydrophobic residues. This arrangement suggests possible involvement of the region in membrane interactions. A model for other GTs proposes a mixed hydrophobic-electrostatic interaction between an equivalent basic region in the N-terminal domain and the membrane (36-38). In this model, there is a first contact between the negatively charged membrane and the positive charges of a cluster of basic residues. Afterward, the contact is strengthened by the interaction of the membrane lipids with the hydrophobic residues.
FIGURE 5.

Proposed membrane binding by GumK. A, the proposed binding surface between GumK and the X. campestris membrane. Helices Nα2 and Nα4 and loop 8, which contain residues Arg58, Lys60, Arg86, Arg95, Arg96, Arg100, and Arg108, are shown in blue. B, model for the proposed interaction between the inner membrane and GumK N-terminal basic patch (arginine cluster). The soluble donor substrate (UDP-GlcA) and Man-Cel-P-P protruding from the membrane are depicted as stick models. The surface representation of GumK is colored by electrostatic potential (red, -2kT; blue, +2kT; white, neutral, where k represents the Boltzmann constant and T is temperature), calculated by using the APBS program and visualized with Pymol. Membrane, GumK, and substrates are depicted in scale.
Indirect evidence for this model was observed for GumK. When E. coli BL21(DE3)/pETHisKC cells were cultured in LB medium in the presence of added NaCl (250 mm), a substantial fraction (∼50%) of the protein became soluble. The purified soluble GumK fraction retained activity during in vitro enzymatic assays. This result might indicate that a hypothetical first electrostatic interaction was interrupted, leaving soluble, properly folded GumK. Furthermore, the location of the basic patch is consistent with the proposed acceptor binding site. Membrane association in this region would bring the middle cleft closer to the membrane surface, where the soluble UDP-GlcA donor is coupled to the membrane-anchored acceptor glycolipid, Man-Cel-P-P-lipid (Fig. 5B). The degree of this interaction and the relative importance of individual residues of the basic cluster, together with surrounding hydrophobic residues, is currently being investigated in our laboratory.
In Vivo Analysis of GumK Mutant Activities—The biosynthesis of bacterial polysaccharides is a complex process that involves several enzymes and transport proteins. In X. campestris, it is very difficult to measure intermediate glycolipids during the synthesis of xanthan, because they are present in very low amounts and do not accumulate in GT mutants.6 A simple way of assessing the effect of GumK mutations in vivo is to measure the amount of polysaccharide produced in complementation assays with a XcK (gumK-) mutant. This kind of analysis provides a powerful means of detecting minor levels of activity in GumK mutants that may have been undetected in previous in vitro assays (14, 39). Fig. 6 shows the relative percentage of xanthan production in XcK expressing mutated GumK compared with XcK mutant complemented with the wild-type gumK gene (XcK/pBBRSK). It is worth noting that mutations K307A and Y292A, involved in the coordination of the negative charge of phosphates, show a marked effect with ∼25% xanthan production compared with XcK/pBBRSK. Mutation of other residues responsible for the binding of the ribose (Q310A) or the uracil (E272A/D) show a lesser effect. Altogether, this result implies that mutations affecting the kinetic parameters of one of the enzymes in the biosynthetic machinery of xanthan have a quantifiable effect on the entire polysaccharide production system. Given that xanthan is a key virulence factor for X. campestris (15, 40), it is not surprising that the key contacts and catalytic residues are strongly conserved (Fig. 4).
FIGURE 6.

Xanthan production in XcK-complemented strains. A, xanthan production in an XcK mutant expressing mutated GumK compared with wild-type strain FC2. The polymer formed was measured by the cetylpyridinium chloride method. XcK and XcK/pBBRprom are negative controls. XcK/pBBRSK is the mutant strain complemented with wild-type GumK protein (positive control). Bars, S.D. from at least three independent experiments. B, normalization of protein quantities by Western blot.
Asp157 Is a Key Residue in the GumK Catalytic Mechanism—To identify the catalytic residue, all acidic residues lying in the catalytic cleft that could act as the general base (Asp157, Glu192, Asp207, or Asp234) were mutated. Mutations E192A, D207A, or D234A showed no effect on GumK activity both in vitro (Table 2) and in vivo (data not shown). A very interesting result of the complementation experiments described above was the lack of activity of GumK mutants D157A/Q/N, indicated by the complete absence of xanthan production in strains XcK/pBBRSKD157A, XcK/pBBRSKD157N, and XcK/pBBRSKD157E (Fig. 6). This absence of activity was also verified in the in vitro assays (Table 2). The lack of activity in Asp157 mutants after replacement of the charge (Asp to Asn mutation) or the length of the side chain (Asp to Glu) in both in vivo and in vitro assays implicates Asp157 as the catalytic residue. To check for potential folding errors, we crystallized and solved the structure of mutant D157A as apoprotein (Protein Data Bank code 3CUY; supplemental Table 1). The r.m.s. deviation for all residues between native GumK and mutant D157A is 0.28 Å, showing that the mutant structure has not suffered structural changes. Moreover, wild-type strain FC2 carrying plasmid pBBRSKD157A, pBBRSKD157N, or pBBRSKD157E showed a complete absence of xanthan production, indicating that the mutant proteins are capable of interfering with the normal xanthan biosynthetic machinery (supplemental Fig. 1). This result suggests the formation of a multienzyme complex or the modulation of GumK activity by oligomerization (41).
The position of Asp157 is structurally equivalent to the position of Asp100 in the β-glucosyltransferase BGT from T4 phage or of Glu95 in α 1-3-fucosyltransferase FucT from Helicobacter pylori, acidic residues that are responsible for the deprotonation of the acceptor substrate (28, 42). The N terminus of GumK displays a deep pocket or tunnel of 560 Å2, defined by the loops connecting Nβ1 to Nα1 (residues 22-30) and Nβ2 to Nα2 (residues 51-55). Basic and aromatic residues, such as Arg, Lys, Tyr, and Phe, are present at the boundaries of this tunnel, in line with the general features of carbohydrate-binding motifs (43, 44) (see the CAZy site on the World Wide Web).
A model of the binding of the acceptor Man-Cel-P-P-lipid can be constructed based on the location of the catalytic base and the donor substrate, as well as the shape and orientation of the active site cleft. The shape of PP-Cel-Man is complementary to the shape of the cleft (Fig. 5B). Asp157 is located immediately below this N-terminal hydrophobic pocket, where the glycolipid acceptor could be accommodated. The carboxylate of Asp157 could deprotonate the 2-OH of the mannose residue. The side chain of Asp157 is also positioned immediately adjacent to the putative location of the anomeric carbon of glucuronic acid in the GumK-UDP complex. Upon deprotonation of the C2-OH group by Asp-157, the acceptor nucleophile can attack the anomeric position of UDP-GlcA to form a new glycosidic bond with an inverted configuration (Fig. 7). The side product UDP dissociates at the same time. According to this hypothesis, the anomeric carbon is located between the acceptor nucleophile, the C2-OH of mannose, and the leaving group UDP, the geometry that is consistent with an in-line displacement mechanism (45). Indirect evidence supporting this hypothesis is that GumK showed hydrolytic activity toward UDP-GlcA after a 1-h incubation in the absence of acceptor. Under the same conditions, mutant D157A was unable to hydrolyze UDP-GlcA, even after a 24-h incubation (supplemental Fig. 2). In an attempt to find the GlcA portion of bound UDP-GlcA, we performed soaking experiments with crystals of the D157A mutant in the presence of UDP-GlcA. The position of the UDP ligand was exactly the same as in wild type GumK (Protein Data Bank code 3CV3). Unfortunately, the position of the GlcA moiety was not observed (data not shown), suggesting that the molecular motion of this portion of the molecule in the “open” conformation of GumK does not allow seeing it with crystallographic methods. RMN experiments will be carried out in the future to study this point.
FIGURE 7.

Proposed catalytic mechanism of GumK. Asp157 serves as the general base. Residues Lys307, Met306, and Tyr292 that interact with UDP phosphates are depicted in blue.
Supplementary Material
Acknowledgments
We are grateful to Susana Raffo for the preparation of NDP-[14C] sugars, Gastón Mayol for technical assistance with high pressure liquid chromatography, and Marta Bravo for technical assistance with DNA sequencing. We thank Mario Bianchet (Biophysics Department, The Johns Hopkins University, Baltimore, MD) for help with structure solving, Anand Saxena (beamline X12C, National Synchrotron Light Source) for help with data collection, and M. V. Ielmini (Fundación Instituto Leloir) for the plasmid pBBRprom.
The atomic coordinates and structure factors (codes 2HY7, 2Q6V, 3CUY, and 3CV3) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
This work was supported by Agencia Nacional de Promoción Científica y Tecnológica Grant PICT 1-11703 and Universidad de Buenos Aires (Argentina) Grant UBACyT X-193. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Table 1 and Figs. 1 and 2.
Footnotes
The abbreviations used are: GT, glycosyltransferase; Man-Cel-P-P-lipid, mannose-α-1,3-glucose-β-1,4-glucose-diphosphate-polyisoprenyl; r.m.s., root mean square.
M. Barreras, S. R. Salinas, P. L. Abdian, M. A. Kampel, and L. Ielpi, unpublished results.
References
- 1.Coutinho, P. M., Deleury, E., Davies, G. J., and Henrissat, B. (2003) J. Mol. Biol. 328 307-317 [DOI] [PubMed] [Google Scholar]
- 2.Liu, J., and Mushegian, A. (2003) Protein Sci. 12 1418-1431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosen, M. L., Edman, M., Sjostrom, M., and Wieslander, A. (2004) J. Biol. Chem. 279 38683-38692 [DOI] [PubMed] [Google Scholar]
- 4.Breton, C., Snajdrova, L., Jeanneau, C., Koca, J., and Imberty, A. (2006) Glycobiology 16 29R-37R [DOI] [PubMed] [Google Scholar]
- 5.Bourne, Y., and Henrissat, B. (2001) Curr. Opin. Struct. Biol. 11 593-600 [DOI] [PubMed] [Google Scholar]
- 6.Hu, Y., and Walker, S. (2002) Chem. Biol. 9 1287-1296 [DOI] [PubMed] [Google Scholar]
- 7.Bowles, D., Isayenkova, J., Lim, E. K., and Poppenberger, B. (2005) Curr. Opin. Plant Biol. 8 254-263 [DOI] [PubMed] [Google Scholar]
- 8.Doores, K. J., Gamblin, D. P., and Davis, B. G. (2006) Chemistry 12 656-665 [DOI] [PubMed] [Google Scholar]
- 9.Feizi, T., and Mulloy, B. (2003) Curr. Opin. Struct. Biol. 13 602-604 [DOI] [PubMed] [Google Scholar]
- 10.Faijes, M., and Planas, A. (2007) Carbohydr. Res. 342 1581-1594 [DOI] [PubMed] [Google Scholar]
- 11.Watts, A. G., and Withers, S. G. (2004) Biochem. J. 380 e9-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He, Y. Q., Zhang, L., Jiang, B. L., Zhang, Z. C., Xu, R. Q., Tang, D. J., Qin, J., Jiang, W., Zhang, X., Liao, J., Cao, J. R., Zhang, S. S., Wei, M. L., Liang, X. X., Lu, G. T., Feng, J. X., Chen, B., Cheng, J., and Tang, J. L. (2007) Genome Biol. 8 R218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Becker, A., Katzen, F., Puhler, A., and Ielpi, L. (1998) Appl. Microbiol. Biotechnol. 50 145-152 [DOI] [PubMed] [Google Scholar]
- 14.Barreras, M., Abdian, P. L., and Ielpi, L. (2004) Glycobiology 14 233-241 [DOI] [PubMed] [Google Scholar]
- 15.Katzen, F., Ferreiro, D. U., Oddo, C. G., Ielmini, M. V., Becker, A., Puhler, A., and Ielpi, L. (1998) J. Bacteriol. 180 1607-1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berg, S., Kaur, D., Jackson, M., and Brennan, P. J. (2007) Glycobiology 17 35R-56R [DOI] [PubMed] [Google Scholar]
- 17.Hancock, S. M., Vaughan, M. D., and Withers, S. G. (2006) Curr. Opin. Chem. Biol. 10 509-519 [DOI] [PubMed] [Google Scholar]
- 18.Barreras, M., Bianchet, M. A., and Ielpi, L. (2006) Acta Crystallograph. Sect. F Struct. Biol. Cryst. Commun. 62 880-883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.(1994) Acta Crystallogr. Sect. D Biol. Crystallogr. 50 760-763 [DOI] [PubMed] [Google Scholar]
- 20.Vonrhein, C., Blanc, E., Roversi, P., and Bricogne, G. (2006) Methods Mol. Biol. 364 215-230 [DOI] [PubMed] [Google Scholar]
- 21.Emsley, P., and Cowtan, K. (2004) Acta Crystallogr. Sect. D Biol. Crystallogr. 60 2126-2132 [DOI] [PubMed] [Google Scholar]
- 22.Baker, N. A., Sept, D., Joseph, S., Holst, M. J., and McCammon, J. A. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 10037-10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ielpi, L., Couso, R. O., and Dankert, M. A. (1993) J. Bacteriol. 175 2490-2500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scott, J. E. (1965) in General Polysaccharides (Whistler, R. L., ed) Vol. V, pp. 38-44, Academic Press, Inc., New York [Google Scholar]
- 25.Wrabl, J. O., and Grishin, N. V. (2001) J. Mol. Biol. 314 365-374 [DOI] [PubMed] [Google Scholar]
- 26.Buschiazzo, A., Ugalde, J. E., Guerin, M. E., Shepard, W., Ugalde, R. A., and Alzari, P. M. (2004) EMBO J. 23 3196-3205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guerin, M. E., Kordulakova, J., Schaeffer, F., Svetlikova, Z., Buschiazzo, A., Giganti, D., Gicquel, B., Mikusova, K., Jackson, M., and Alzari, P. M. (2007) J. Biol. Chem. 282 20705-20714 [DOI] [PubMed] [Google Scholar]
- 28.Sun, H. Y., Lin, S. W., Ko, T. P., Pan, J. F., Liu, C. L., Lin, C. N., Wang, A. H., and Lin, C. H. (2007) J. Biol. Chem. 282 9973-9982 [DOI] [PubMed] [Google Scholar]
- 29.Baker, P. J., Britton, K. L., Rice, D. W., Rob, A., and Stillman, T. J. (1992) J. Mol. Biol. 228 662-671 [DOI] [PubMed] [Google Scholar]
- 30.Vetting, M. W., Frantom, P. A., and Blanchard, J. S. (2008) J. Biol. Chem. 283 15834-15844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vrielink, A., Ruger, W., Driessen, H. P., and Freemont, P. S. (1994) EMBO J. 13 3413-3422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morera, S., Lariviere, L., Kurzeck, J., Aschke-Sonnenborn, U., Freemont, P. S., Janin, J., and Ruger, W. (2001) J. Mol. Biol. 311 569-577 [DOI] [PubMed] [Google Scholar]
- 33.Hu, Y., Chen, L., Ha, S., Gross, B., Falcone, B., Walker, D., Mokhtarzadeh, M., and Walker, S. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 845-849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davies, G. J., Gloster, T. M., and Henrissat, B. (2005) Curr. Opin. Struct. Biol. 15 637-645 [DOI] [PubMed] [Google Scholar]
- 35.Martinez-Fleites, C., Proctor, M., Roberts, S., Bolam, D. N., Gilbert, H. J., and Davies, G. J. (2006) Chem. Biol. 13 1143-1152 [DOI] [PubMed] [Google Scholar]
- 36.Ha, S., Walker, D., Shi, Y., and Walker, S. (2000) Protein Sci. 9 1045-1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lind, J., Ramo, T., Klement, M. L., Barany-Wallje, E., Epand, R. M., Epand, R. F., Maler, L., and Wieslander, A. (2007) Biochemistry 46 5664-5677 [DOI] [PubMed] [Google Scholar]
- 38.Edman, M., Berg, S., Storm, P., Wikstrom, M., Vikstrom, S., Ohman, A., and Wieslander, A. (2003) J. Biol. Chem. 278 8420-8428 [DOI] [PubMed] [Google Scholar]
- 39.Abdian, P. L., Lellouch, A. C., Gautier, C., Ielpi, L., and Geremia, R. A. (2000) J. Biol. Chem. 275 40568-40575 [DOI] [PubMed] [Google Scholar]
- 40.Crossman, L., and Dow, J. M. (2004) Microbes Infect. 6 623-629 [DOI] [PubMed] [Google Scholar]
- 41.Mohammadi, T., Karczmarek, A., Crouvoisier, M., Bouhss, A., Mengin-Lecreulx, D., and den Blaauwen, T. (2007) Mol. Microbiol. 65 1106-1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lariviere, L., Gueguen-Chaignon, V., and Morera, S. (2003) J. Mol. Biol. 330 1077-1086 [DOI] [PubMed] [Google Scholar]
- 43.Shoseyov, O., Shani, Z., and Levy, I. (2006) Microbiol. Mol. Biol. Rev. 70 283-295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boraston, A. B., Bolam, D. N., Gilbert, H. J., and Davies, G. J. (2004) Biochem. J. 382 769-781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pedersen, L. C., Darden, T. A., and Negishi, M. (2002) J. Biol. Chem. 277 21869-21873 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.