

Abstract
Diadenosine 5′,5‴-P1,P2-diphosphate (Ap2A) is one of the adenylic dinucleotides stored in platelet granules. Along with proaggregant ADP, it is released upon platelet activation and is known to stimulate myocyte proliferation. We have previously demonstrated synthesis of Ap2A and of two isomers thereof, called P18 and P24, from their high pressure liquid chromatography retention time, by the ADP-ribosyl cyclase CD38 in mammalian cells. Here we show that Ap2A and its isomers are present in resting human platelets and are released during thrombin-induced platelet activation. The three adenylic dinucleotides were identified by high pressure liquid chromatography through a comparison with the retention times and the absorption spectra of purified standards. Ap2A, P18, and P24 had no direct effect on platelet aggregation, but they inhibited platelet aggregation induced by physiological agonists (thrombin, ADP, and collagen), with mean IC50 values ranging between 5 and 15 μm. Moreover, the three dinucleotides did not modify the intracellular calcium concentration in resting platelets, whereas they significantly reduced the thrombin-induced intracellular calcium increase. Through binding to the purinergic receptor P2Y11, exogenously applied Ap2A, P18, and P24 increased the intracellular cAMP concentration and stimulated platelet production of nitric oxide, the most important endogenous antiaggregant. The presence of Ap2A, P18, and P24 in resting platelets and their release during thrombin-induced platelet activation at concentrations equal to or higher than the respective IC50 value on platelet aggregation suggest a role of these dinucleotides as endogenous negative modulators of aggregation.
The dinucleoside diphosphates diadenosine 5′,5‴-P1,P2-diphosphate (Ap2A),2 adenosine guanosine diphosphate (Ap2G), and diguanosine diphosphate (Gp2G) represent a new class of growth-promoting extracellular mediators present in platelet secretory granules (1) and in cardiac myocytes (2), capable of stimulating cardiac myocyte proliferation (1) and believed to play a role in the control of cardiovascular tone (3). The intraplatelet concentration of each one of these dinucleotides has been estimated to be in the range between 30 and 100 μm (1). It has also been shown that the concentration of Ap2A, Ap2G, and Gp2G in the supernatant of platelets stimulated with 0.05 units/ml thrombin is ∼60% of the total intraplatelet amount of each dinucleoside diphosphate, suggesting that their primary function is extracellular (1). The enzyme responsible for their synthesis has not been identified, and the effect of these dinucleotides on platelet function has not yet been investigated.
We have recently demonstrated that ADP-ribosyl cyclases from Axinella polypoides, (Porifera, Demospongiae), Aplysia californica (Molluscs), and human CD38 can synthesize three adenylic dinucleotides from cyclic ADP-ribose and adenine (Ade): Ap2A and two isomers thereof, called P18 and P24, which are characterized by an unusual N-glycosidic bond between one adenine and the ribose (C1′-N1 in P18 and C1′-N3 in P24) (4). CD38 is present in human platelets (5, 6), and activation with thrombin induces an increase of CD38 activity associated with the platelet cytoskeleton (7). These results prompted us to investigate the presence of P18 and P24 in human platelets; the effect of Ap2A, P18, and P24 on platelet aggregation; and possible synergistic or antagonistic effects of these adenylic dinucleotides with thrombin, ADP, or collagen, the most potent physiological platelet agonists.
EXPERIMENTAL PROCEDURES
Materials—If not otherwise indicated, all chemicals were from Sigma and were of the highest purity grade available.
Blood Collection and Platelet Preparation—Venous blood, freshly drawn from healthy volunteers, was collected in a 130 mm aqueous trisodium citrate anticoagulant solution (9:1). The donors declared not to have taken drugs known to interfere with platelet function during 2 weeks prior to blood collection. Washed platelets (WP) were prepared as described (8). Briefly, platelet-rich plasma, obtained by centrifugation of the whole blood at 100 × g for 25 min, supplemented with 2 μg/ml apyrase and 1 μm PGE1, was centrifuged at 1000 × g for 15 min. The platelet pellet was washed once with ACD solution (75 mm trisodium citrate, 42 mm citric acid, and 136 mm glucose, pH 4.8), centrifuged at 1000 × g for 15 min, and then resuspended in Ca2+-free Hepes buffer (145 mm NaCl, 5 mm KCl, 1 mm MgSO4, 10 mm glucose, 10 mm Hepes, pH 7.4) or in Ca2+-free Tyrode's-Hepes buffer (134 mm NaCl, 12 mm NaHCO3, 2.9 mm KCl, 1 mm MgCl2, 5 mm glucose, 5 mm Hepes, pH 7.4).
Cell Culture and Transfection—HeLa cells were grown on 24-well culture dishes and maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and 4 m m l-glutamine at 37 °C in a 5% CO2 environment. Cells were transfected using FuGENE 6 (Roche Applied Science) transfection reagent 48 h prior to assay with either pcDNA 3.1 (empty vector), pcDNA3-hP2Y1, or pcDNA3-hP2Y12 together with pcDNA1-Gαq/i. pcDNA1-Gαq/i directs expression of a chimera of Gαq containing the last five amino acids of Gαi, which couples Gαi-coupled receptors to activation of phospholipase C (9).
1321N1 cells expressing the human P2Y11 receptor were generated by retroviral infection as described in Ref. 10 and cultured in Dulbecco's modified Eagle's medium with 10% fetal bovine serum and antibiotics.
Production and Purification of P18, P24, and Ap2A—P18 and P24, to be used for functional studies and as standards in the HPLC purification of the platelet dinucleotides, were produced with the purified ADP-ribosyl cyclase from A. polypoides and HPLC-purified as described by Basile et al. (4). 14C-Labeled P18, P24, and Ap2A were produced from [14C]NAD+ with the purified ADP-ribosyl cyclase from A. polypoides and HPLC-purified (4). Ap2A was synthesized and HPLC-purified as described by Rossi et al. (11).
HPLC Determination of P18, P24, and Ap2A Content in Human Platelets—The presence of the three adenylic dinucleotides was investigated both in resting and in thrombin-stimulated human platelets. In a typical experiment, 500-μl aliquots of WP suspension (5 × 108 platelets/ml) were incubated for 20 min at 37 °C without or with 0.05 units/ml thrombin. Thereafter, aliquots were centrifuged, each supernatant was recovered, and pellets were sonicated twice for 15 s with an interval of 1 min. Lysed platelets were centrifuged (2600 × g for 5 min), and supernatants and pellets were recovered and deproteinized with trichloroacetic acid (5% final concentration). Trace amounts of 14C-labeled P18, P24, and Ap2A were added to the acid extracts to calculate the percentages of recovery of the various dinucleotides at the end of the purification procedure. Excess trichloroacetic acid was removed by diethylether extraction, and samples were analyzed by HPLC for the presence of the adenylic dinucleotides. All HPLC analyses were performed on a Hewlett-Packard 1090 instrument, equipped with a diode array spectrophotometric detector (HP 1040). Aliquots of 250 μl were subjected to the analytical phosphate HPLC (4). The column was a Delta Pak C18 column (150 × 4.6 mm, 5 μm; Waters). Solvent A was 0.1 m KH2PO4 containing 5 mm tetra-n-butylammonium, pH 5.0; solvent B was solvent A with 30% (v/v) methanol. The solvent program was a linear gradient starting at 100% A and increasing to 100% B in 30 min and then remaining at 100% B for 10 min. HPLC fractions containing P18, P24, or Ap2A were pooled, lyophilized, redissolved in deionized water, and injected into an analytical PLSAX column (50 × 4.6 mm, 8 μm; CPS). Solvent A was 20 mm ammonium bicarbonate, pH 7.6; solvent B was 500 mm ammonium bicarbonate, pH 7.6. The solvent program was the same as the one described above for reverse phase HPLC. The HPLC fractions containing the three dinucleotides were collected, dried, redissolved in deionized water, and reinjected into the analytical phosphate HPLC for final identification and quantitation (see Fig. 2 for representative chromatograms of the final purification step for each dinucleotide); P18, P24, and Ap2A were identified by comparison of their absorption spectra with computer-stored standards and were quantified by comparison of the computer-integrated peak areas with those of known amounts of the relevant dinucleotide standard. The percentage of recovery of the three adenylic dinucleotides at the end of the purification procedure was calculated from the yield of the corresponding radioactive tracer and was between 15 and 25% in the various purifications. Identification was confirmed by HPLC analysis of a nucleotide pyrophosphatase-digested parallel sample, showing conversion of the dinucleotide into the corresponding iso-AMP (or AMP for Ap2A) moiety (4). For final calculation of the intraplatelet concentration of the various dinucleotides, a platelet volume of 7 fl was considered.
FIGURE 2.

Representative chromatograms of the final purification step of P18 (A), P24 (B), and Ap2A (C) from thrombin-activated platelets.
P18, P24, and Ap2A Production and Degradation by Human Platelets—WP suspensions (1.0 × 109 platelets/ml) were sonicated and incubated at 37 °C with 0.65 mm NAD+ and 6.5 mm Ade in the presence of 2 mm MgCl2, for dinucleotide production, or with 0.02 mm Ap2A, P18, or P24, for dinucleotide degradation. At various times, aliquots of the suspension were withdrawn and deproteinized with 5% trichloroacetic acid, excess trichloroacetic acid was removed by diethylether extraction, and samples were injected into the analytical phosphate HPLC (4).
Measurement of Platelet Aggregation—Platelet aggregation, performed on a Menarini Aggrecoder PA-3210 aggregometer, was monitored according to Born's method (12) and quantified by light transmission for 3 min from the addition of the agonist. Platelet-rich plasma or WP (3.0 × 108 platelets/ml) were preincubated without (control) or with P18, P24, and Ap2A for 2 min at 37 °C before the addition of agonists (thrombin, ADP, or collagen). Measurement of Intraplatelet Calcium Levels—WP (3.0 × 108 platelets/ml), resuspended in Hepes buffer, were incubated with 1 μm Fura-2/AM for 60 min at 37 °C. Prostaglandin E1 (2 μm final concentration) and EGTA (1 mm final concentration) were added before centrifugation of the Fura-2-loaded platelets for 15 min at 1100 × g. The platelet pellet was resuspended at 2.0 × 108 platelets/ml in Hepes buffer and preincubated at 37 °C for 5 min with saline before the addition of the adenylic dinucleotides to be tested. When the effect of Ap2A, P18, and P24 was tested on platelets stimulated with thrombin, WP were preincubated with the adenylic dinucleotides for 5 min at 37 °C and then challenged with thrombin. Fluorescence of Fura-2-loaded platelets was measured at 37 °C in unstirred conditions in a PerkinElmer Life Sciences fluorescence spectrometer model LS50B with excitation at 340 and 380 nm and emission at 509 nm. The fluorescence of fully saturated Fura-2 (Fmax) was obtained by lysing platelets with 50 μm digitonin in the presence of 2 mm Ca2+, whereas Fmin was determined by the subsequent addition of 20 mm EGTA. The fluorescence was fully quenched with 5 mm MnCl2 to yield the autofluorescence value. Dedicated software converted data into cytosolic Ca2+ concentration applying a Kd value for Fura-2 and Ca2+ of 135 nm.
Inositol Phosphate Accumulation Assay in Transfected HeLa Cells—Cells were labeled overnight with 200 μl of serum-free, inositol-free Dulbecco's modified Eagle's medium containing 0.4 μCi/well 3H-labeled myo-inositol (American Radiolabeled Chemicals, St. Louis, MO). Assays were initiated with the addition of 5× concentrations of the indicated compounds in 50 μl of 50 mm LiCl, 250 mm Hepes, pH 7.25. Following incubation for 15 min at 37 °C, the medium was removed by aspiration, and the reaction was terminated by the addition of 0.75 ml of boiling 10 mm EDTA, pH 8.0. [3H]Inositol phosphates were isolated by Dowex column chromatography as described previously (13).
Measurement of Nitric Oxide (NO) Production—WP (0.5 × 108 platelets/ml), resuspended in Hepes buffer containing 1 mm CaCl2 and prewarmed at 37 °C for 10 min with saline or additions, were incubated with 40 μm l-arginine and the various dinucleoside diphosphates (10 μm), alone or with thrombin (0.05 units/ml), for 15 min at 37 °C under mild horizontal shaking. The incubation was stopped by sonicating samples on ice. To measure the NO content, 0.55-ml aliquots of supernatant were added to equal volumes of glycine buffer (15 g/liter glycine NaOH, pH 9.7) containing activated cadmium beds (Fluka AG), which catalyze the chemical reduction of nitrate to nitrite (14), and incubated overnight at room temperature under horizontal shaking. Cadmium beds were activated immediately before each experiment by three subsequent washings with 0.2 n H2SO4, bidistilled water and glycine buffer, respectively. NO formation, determined by the Griess reagent (1% sulfanylamide in 2.5% H3PO4, 0.1% naphthylenediamine dihydrochloride), was measured at 540 nm using a sodium nitrite calibration curve (15).
Determination of cAMP Levels in 1321N1 Cells and Human Platelets—Control and hP2Y11-transfected 1321N1 cells were seeded in 35 × 10-mm dishes (0.5 × 106 cells/dish). After 24 h, the medium was replaced with 0.6 ml of HBSS. P24, Ap2A, or buffer (control) were added at the indicated concentrations, and incubations were stopped after 0, 2.5, 5, or 15 min by removal of HBSS and the addition of 200 μl of ice-cold perchloric acid (PCA) (0.6 m). Cell extracts were collected and centrifuged to remove proteins; the cAMP content was measured on the neutralized cell extracts by radioimmunoassay ([3H]cAMP assay system; GE Healthcare).
Aliquots (300 μl, 1 × 109 platelets/ml) of WP from different healthy donors were preincubated or not for 30 min at 37 °C with 1 μm NF157 prior to incubation with 10 μm P18, P24, or Ap2A for 15 min. Incubations were stopped by the addition of 26 μl of 9 m PCA, and then each sample was sonicated in ice at 2.5 watts for 10 s and centrifuged at 2600 × g for 3 min at 4 °C. The supernatant from each sample was collected, and the PCA was removed as described by Graeff and Lee (16). The cAMP content was measured on neutralized platelet extracts by radioimmunoassay ([3H]cAMP assay system; GE Healthcare).
Determination of IP3 Levels in 1321N1 Cells and Human Platelets—Control and hP2Y11-transfected 1321N1 cells were seeded in 35 × 10-mm dishes (0.5 × 106 cells/dish). After 24 h, the medium was replaced with 0.6 ml of HBSS. 10 μm P24 or Ap2A or buffer (control) were then added, and at 0, 0.5, 1, and 2.5 min after the addition, the incubation was stopped by the removal of HBSS, the addition of 300 μl of ice-cold 0.6 m PCA, and scraping of the cells. The IP3 content was measured on the supernatants of the neutralized cell extracts by radioimmunoassay ([3H]IP3 Biotrak Assay System; Amersham Biosciences).
Aliquots (200 μl, 1 × 109 platelets/ml) of WP from different healthy donors resuspended in Hepes buffer were incubated at 37 °C for 30 s in the presence of 10 μm P24 or Ap2A or buffer (control). At the end of each incubation, the suspensions were withdrawn, and the reaction was stopped by adding 13 μl of 9 m PCA at 4 °C. PCA was removed as described (16). The intracellular IP3 concentration was determined as described above.
Statistical Analyses—All parameters were tested by paired t test. p values of <0.05 were considered significant.
RESULTS
Production of P18, P24, and Ap2A by Human Platelets—Human CD38, along with the ADP-ribosyl cyclases from the sponge A. polypoides and from the mollusc A. californica, has been shown to produce P18, P24, and Ap2A from cyclic ADP-ribose (or NAD+) and Ade (Fig. 1) (4). CD38 is expressed in human platelets (5, 6), and its activity increases during platelet aggregation (7). Thus, we preliminarily tested whether platelets also produced these adenylic dinucleotides. Indeed, P18, P24, and Ap2A production was observed upon incubation of sonicated WP with NAD+ (precursor of cyclic ADP-ribose) and Ade (Table 1). Interestingly, dinucleotide production was higher in platelets stimulated with thrombin (0.05 units/ml) as compared with resting platelets (Table 1). In the presence of 20 mm nicotinamide, an inhibitor of ADP-ribosyl cyclase activity, production of the three adenylic dinucleotides was abrogated (Table 1). No degradation of P24 and Ap2A was detectable in lysed platelets, either resting or activated; conversely, P18 was hydrolyzed to ADPR and Ade, similarly to what was observed for recombinant human CD38 (4), and the P18-hydrolyzing activity decreased in thrombin-stimulated platelets (Table 1).
FIGURE 1.

Structures of the dinucleotides Ap2A, P18, P24, Ap2G, and Gp2G. Ap2A and its isomers P18 and P24 differ in the position of the N-glycosidic bond linking one adenine to the ribose: C1′–N9 in Ap2A, C1′–N1 in P18, and C1′–N3 in P24. The structures of Ap2G and Gp2G, two dinucleotides known to be present in platelet granules, are shown for comparison.
TABLE 1.
P18, P24, and Ap2A production by human platelets
WP suspensions (1 × 109 platelets/ml), preincubated or not with 0.05 units/ml thrombin in the presence or absence of 20 mm nicotinamide, were sonicated and incubated at 37 °C with 0.65 mm NAD+ and 6.5 mm Ade for dinucleotide production or with 0.02 mm Ap2A, P18, or P24 for dinucleotide degradation. At various times, aliquots of the suspensions were withdrawn, deproteinized, and analyzed by HPLC. Production and degradation of Ap2A, P18, and P24 by human platelets are expressed as pmol/min/109 platelets. Results are the mean of three experiments, performed on platelets from different healthy donors. ND, not detectable.
|
Dinucleotide production |
Dinucleotide hydrolysis |
||||
|---|---|---|---|---|---|
| No additions | With thrombin | With thrombin and nicotinamide | No additions | With thrombin | |
| pmol/min/109 platelets | pmol/min/109 platelets | ||||
| P18 | 7.1 ± 0.3 | 12.2 ± 0.2 | ND | 6.3 ± 0.1 | 3.3 ± 0.1 |
| P24 | 18.3 ± 1.1 | 26.4 ± 1.3 | ND | ND | ND |
| Ap2A | 9.1 ± 0.5 | 75.3 ± 2.8 | ND | ND | ND |
Presence of P18, P24, and Ap2A in Resting and in Thrombin-stimulated Platelets—It has been recently demonstrated that secretory granules of human platelets contain Ap2A, which is released during platelet aggregation evoked by thrombin (1). This observation, together with the ability of platelet CD38 to synthesize the Ap2A isomers P18 and P24, in addition to Ap2A (Table 1), prompted us to investigate whether P18 and P24 were also present in human platelets and whether they were released during aggregation. HPLC analysis of acid extracts of resting platelets confirmed the presence of Ap2A at a concentration (∼20 μm) similar to that already reported (1). Both Ap2A isomers were indeed detected at a similar intraplatelet concentration (∼40 and 60 μm for P18 and P24, respectively). Neither Ap2A nor its isomers were detectable in the supernatant from resting platelets (Table 2). The intraplatelet concentration of all three adenylic dinucleotides increased in thrombin-stimulated, as compared with resting, platelets (1.5-, 1.3-, and 3-fold for P18, P24, and Ap2A, respectively; see Fig. 2 for representative chromatograms of the final purification step). All adenylic dinucleotides were released into the supernatant, where they were present at ∼40, 50, and 15 μm for P18, P24, and Ap2A, respectively (Table 2), when platelets were suspended at a concentration similar to that present in whole blood.
TABLE 2.
Presence of P18, P24, and Ap2A in platelets and supernatants from thrombin-stimulated platelets
P18, P24, and Ap2A were detected and quantified in acid extracts of platelets (Pellet) and in the supernatants (Sup) by HPLC (see details under “Experimental Procedures”). Mean results from at least three experiments are shown. ND, not detectable.
|
Resting platelets |
Thrombin-activated platelets |
|||
|---|---|---|---|---|
| Pellet | Sup | Pellet | Sup | |
| μm | μm | |||
| P18 | 41.1 ± 3.2 | ND | 60.4 ± 4.7a | 37.3 ± 2.5 |
| P24 | 63.5 ± 5.8 | ND | 82.2 ± 6.3a | 52.1 ± 4.4 |
| Ap2A | 18.3 ± 2.4 | ND | 55.1 ± 3.6a | 15.6 ± 1.1 |
p < 0.01 compared with the dinucleotide concentration in resting platelets.
P18, P24, and Ap2A Inhibit Agonist-induced Platelet Aggregation— Production of P18, P24, and Ap2A from NAD+ and Ade by human platelet lysates, the presence of the dinucleotides in resting platelets, and their release upon platelet activation by thrombin prompted us to explore the possibility that P18, P24, and Ap2A may affect platelet aggregation. Pretreatment of platelets for 2 min at 37 °C with increasing concentrations of P18, P24, or Ap2A (1, 5, and 10 μm) had no effect on platelet aggregation, as observed during the following 3 min at 37 °C under stirring. Conversely, down-regulation of platelet aggregation was observed when platelets were pretreated with the adenylic dinucleotides and then challenged with ADP, thrombin, or collagen. As shown in Fig. 3, ADP-induced platelet aggregation was significantly reduced by all three adenylic dinucleotides, with IC50 values of 7.2, 7.9, and 6.3 μm for P18, P24, and Ap2A, respectively (Table 3). In platelets treated with suboptimal concentrations of thrombin (0.05 units/ml) or collagen (1.5 μg/ml), aggregation was also inhibited in a concentration-dependent way by each of the three dinucleotides, although IC50 values were slightly higher compared with those observed upon ADP-induced aggregation (Table 3).
FIGURE 3.

Down-regulation of ADP-induced platelet aggregation by P18, P24, and Ap2A. WP (3 × 108 platelets/ml) were preincubated for 2 min in the absence (control) or in the presence of increasing concentrations (1, 5, and 10 μm) of P18 (black squares), P24 (black rhombuses), or Ap2A (black triangles) and then incubated for 3 min with 2.5 μm ADP. Results are expressed as percentage of inhibition relative to controls and are the mean ± S.D. of at least three different experiments, performed on platelets from different donors.
TABLE 3.
IC50 values of P18, P24, and Ap2A on platelet aggregation induced by ADP, thrombin, or collagen
WP (3 × 108 platelets/ml) were preincubated for 2 min in the presence or absence (control) of increasing concentrations of P18, P24, and Ap2A and then incubated for 3 min with 2.5 μm ADP, 0.05 units/ml thrombin, or 1.5 μg/ml collagen. IC50 values were calculated from dose-response curves (see Fig. 1) of the percentage of inhibition relative to controls. Results are the mean ± S.D. of at least three different experiments, performed on platelets from different donors.
|
IC50 |
|||
|---|---|---|---|
| ADP | Thrombin | Collagen | |
| μm | |||
| P18 | 7.2 ± 0.6 | 7.3 ± 0.7 | 12.8 ± 1.2 |
| P24 | 7.9 ± 0.9 | 9.8 ± 1.1 | 16.8 ± 1.8 |
| Ap2A | 6.3 ± 0.7 | 18.9 ± 1.7 | 11.1 ± 0.8 |
P18, P24, and Ap2A Reduce the Thrombin-induced [Ca2+]i Rise in Intact Platelets—During platelet aggregation, an increase of the intracellular calcium concentration ([Ca2+]i) is known to occur downstream of the activation by ADP-, thrombin-, or collagen-specific receptors (17, 18). The inhibitory effect of P18, P24, and Ap2A on agonist-induced platelet aggregation suggested that we should investigate the effect of these dinucleotides on the [Ca2+]i. To this purpose, platelets were preincubated with Ap2A, P18, or P24 and then stimulated with thrombin. In platelets challenged with thrombin (0.05 units/ml), the [Ca2+]i increased rapidly from a resting value of 65 ± 11 nm to a maximal value of 172 ± 35 nm. At the same time point, the [Ca2+]i increase observed in platelets pretreated for 5 min with the adenylic dinucleotides (10 μm) before the addition of thrombin was significantly reduced (Table 4). P18 exerted the highest percentage of inhibition (54%), in line with the lower IC50 value observed upon thrombin-induced aggregation (Table 3). In the absence of thrombin, all three adenylic dinucleotides evoked a very limited increase of the [Ca2+]i (≤8% of the basal value), which was apparently insufficient to induce platelet aggregation, since neither dinucleotide per se had any proaggregant effect (see above).
TABLE 4.
Effect of P18, P24, and Ap2A on the thrombin-induced [Ca2+]i rise in human platelets
FURA2-loaded WP (2 × 108 platelets/ml) were preincubated in the absence (control) or in the presence of the adenylic dinucleotides for 5 min at 37 °C and then challenged with 0.05 units/ml thrombin. The percentage of inhibition (I) on thrombin-induced [Ca2+]i rise is reported. Results are the mean ± S.D. of three different experiments performed on platelets from different healthy donors.
| I | |
|---|---|
| % | |
| P18 | 54 ± 5 |
| P24 | 33 ± 2 |
| Ap2A | 36 ± 3 |
Are P18, P24, and Ap2A ADP Antagonists on P2Y1 and P2Y12 Receptors?—Platelet granules are known to contain high concentrations of adenylic nucleotides, most notably ADP and ATP, which are released during platelet aggregation and contribute to a positive feedback mechanism, by acting through specific purinergic receptors on the platelet membrane (19–21). Thus, platelet aggregation induced by collagen, thrombin, or endothelial injury induces release of ADP, which in turn binds to P2Y1 and P2Y12 receptors, further stimulating aggregation (20). The structural homology between the adenylic dinucleotides (Ap2A, P18, and P24) and ADP prompted us to investigate whether they behaved as ADP antagonists on P2Y1 or P2Y12 receptors. HeLa cells were transfected with human P2Y1 or co-transfected with human P2Y12 and Gαq/i to generate a cell system responsive to ADP with an increase of the inositol phosphate concentration. Incubation of P2Y1- or P2Y12/Gq/i-transfected cells with 30 μm P24 did not increase the inositol phosphate concentration in either cell type (Fig. 4). Conversely, 1 μm 2MeSADP, a stable analogue of ADP, strongly increased the inositol phosphate concentration both in P2Y1- and P2Y12/Gq/i-transfected cells, and this increase was not inhibited by the simultaneous addition of P24 (Fig. 4). The inability of P24 to bind to P2Y1 receptors was also confirmed by Scatchard analysis. P24 failed to displace [32P]MRS2500, a specific P2Y1 receptor radioligand (22), from the P2Y1 receptor (not shown). The fact that P24 did not interact with P2Y1 or with P2Y12 suggested that we should explore other mechanisms of action, resulting in an inhibition of platelet aggregation by Ap2A, P18, and P24.
FIGURE 4.

Inositol phosphate accumulation in HeLa cells transiently transfected with P2Y1 or with P2Y12/Gq/i. Cells transfected with pCDNA3 (empty vector) or P2Y1 or P2Y12/Gq/i were stimulated for 15 min with buffer (white bars) or 1 μm 2MeSADP (black bars) or 30 μm P24 alone (dotted bars) or with 1 μm 2MeSADP (gray bars). Inositol phosphate production was determined as described under “Experimental Procedures.” Results are the mean ± S.D. of at least three experiments.
P18, P24, and Ap2A Stimulate Platelet Nitric Oxide Production in the Presence of Thrombin—NO, a potent antagonist of platelet aggregation (23–25), is synthesized in human platelets by a constitutive NO synthase (26–28). The inhibitory effect of Ap2A, P18, and P24 on platelet aggregation induced by the physiological agonists ADP, thrombin, or collagen prompted us to investigate whether these adenylic dinucleotides could stimulate platelet NO production. WP (0.5 × 108 platelets/ml) resuspended in Hepes buffer containing 1 mm CaCl2 and prewarmed at 37 °C for 10 min were incubated with 40 μm l-arginine and the various dinucleoside diphosphates (10 μm) alone or with thrombin (0.05 units/ml) for 15 min at 37 °C under mild shaking. In platelets treated with thrombin alone, NO production was not increased over control values, measured in untreated platelets (Fig. 5, white bars), as expected due to the suboptimal thrombin concentration used for platelet activation (29). Neither adenylic dinucleotide, in the absence of thrombin, modified basal NO production (Fig. 5, striped bars). Conversely, in platelets incubated with thrombin and either of the three adenylic dinucleotides, NO production increased over control values, 3.1-, 2.1-, and 1.3-fold in P18-, P24-, and Ap2A-treated-platelets, respectively (Fig. 5, black bars).
FIGURE 5.

Effect of P18, P24, and Ap2A on NO production. WP (0.5 × 108 platelets/ml) were incubated for 15 min with 10 μm P18, P24, or Ap2A alone (striped bars) or in the presence of 0.05 units/ml thrombin without (black bars) or with 1 μm NF157 (gray bars). NO production was measured as detailed under “Experimental Procedures.” CT, control, untreated platelets (white bars); T, platelets incubated with thrombin, in the absence of dinucleotides. Results are the mean ± S.D. of five experiments, performed on platelets from different healthy donors. *, p < 0.05; **, p < 0.001 compared with control.
P18, P24, and Ap2A Increase the [cAMP]i and the [IP3]i in Human Platelets through P2Y11 Receptor Stimulation—Stimulation of NO production by P18, P24, and Ap2A suggested that we should explore the effect of the dinucleotides on the [cAMP]i, since NOS activity is known to be stimulated by PKA (30). Moreover, an increase of [cAMP]i is known to induce a complete inhibition of platelet function (31, 32). Platelet incubation for 15 min at 37 °C with 10 μm P18, P24, or Ap2A indeed increased the [cAMP]i 2.1-, 1.8-, or 1.5-fold, respectively, over values measured in untreated platelets (Fig. 6A; control, n = 5, p < 0.05). The only purinergic receptor known to stimulate adenylic cyclase is P2Y11 (10, 33), which is also expressed in human platelets (34). Preincubation of platelets for 30 min with 1 μm NF157, a specific inhibitor of P2Y11 (35), reduced cAMP overproduction induced by P18, P24, and Ap2A by ∼70, 75, and 100%, respectively (Fig. 6A).
FIGURE 6.
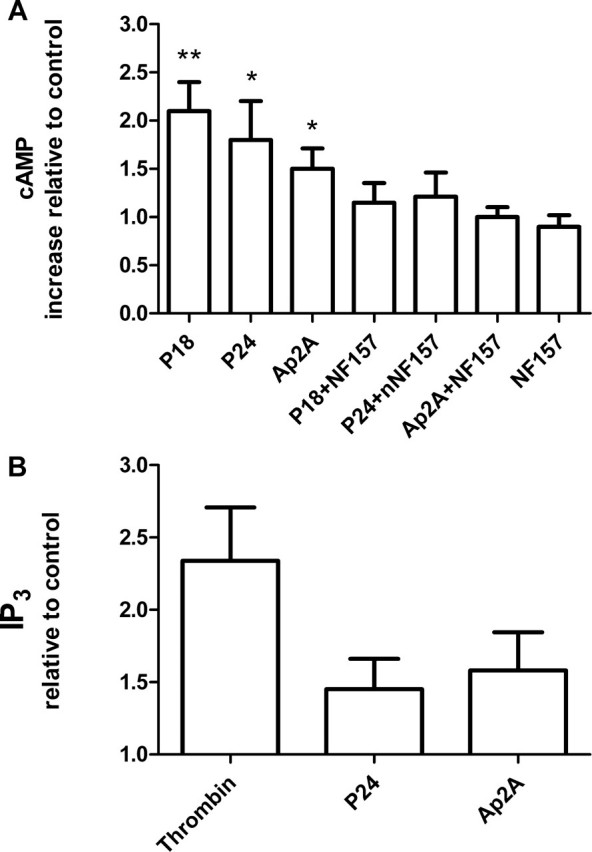
Increase of [cAMP]i and [IP3]i in platelets stimulated with P18, P24, and Ap2A. A, [cAMP]i in extracts from human platelets stimulated for 15 min at 37 °C with 10 μm P18, P24, or Ap2A or 5 μm NF157 or pretreated for 30 min with NF157 and then challenged with a 10 μm concentration of each of the three adenylic dinucleotides. cAMP production was measured as detailed under “Experimental Procedures.” The basal intraplatelet concentration of cAMP was 2.49 ± 0.77 pmol/108 platelets. B, [IP3]i in extracts from human platelets after a 30-s incubation at 37 °C with 0.1 units/ml thrombin or 10 μm P24 or Ap2A. The [IP3]i was measured as detailed under “Experimental Procedures.” The basal intraplatelet concentration of IP3 was 1.2 ± 0.2 pmol/108 platelets. Results are the mean ± S.D. of at least three experiments for each assay, performed on platelets from different healthy donors. *, p < 0.05; **, p < 0.01 compared with each dinucleotide in the presence of NF157.
Since P2Y11 is also coupled to a phospholipase C, we investigated whether the dinucleotides induced intraplatelet inositol 1,4,5-trisphospate ([IP3]i) rise. As shown in Fig. 6B, platelet stimulation with 10 μm P24 or Ap2A evoked an [IP3]i increase with the highest values (1.45 ± 0.21 and 1.58 ± 0.26 relative to control, respectively) being recorded after 30 s of incubation. The [IP3]i rise produced by the two adenylic dinucleotides was significantly less than that observed with 0.1 units/ml thrombin, in agreement with the inability of the three adenylic nucleotides to evoke aggregation. Finally, we investigated whether stimulation of NO production by Ap2A, P18, and P24 was downstream of P2Y11 activation. Pretreatment of platelets with 1μm NF157 before stimulation with 10 μm P24 or Ap2A completely abrogated the release of NO by the two adenylic dinucleotides (Fig. 5).
P24 and Ap2A Induce a [cAMP]i and [IP3]i Increase in Control and hP2Y11-transfected 1321N1—To confirm that the observed increase of the [cAMP]i and of the [IP3]i in platelets was a consequence of binding of the adenylic dinucleotides to the P2Y11 receptor, we measured the concentration of these two intracellular second messengers in control and hP2Y11-transfected 1321N1. Cells stimulated with 10 μm P24 or Ap2A showed a time-dependent increase in [cAMP]i, with the maximum value reached for both adenylic dinucleotides after 15 min of incubation (Fig. 7A). Conversely, none of the two adenylic dinucleotides generated a [cAMP]i rise in control 1321N1 (not shown).
FIGURE 7.
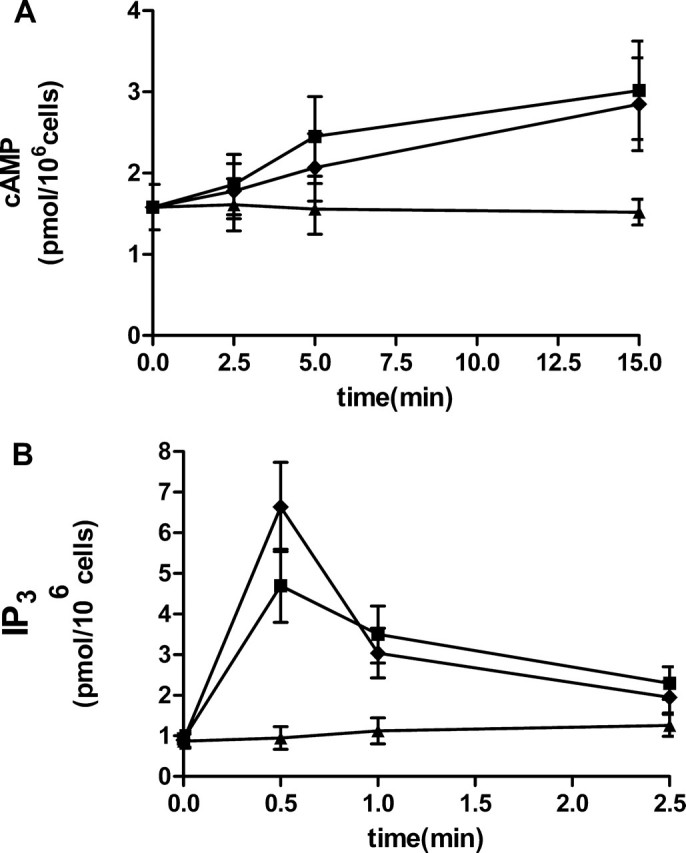
Increase of [cAMP]i and [IP3]i in hP2Y11-transfected 1321N1 cells. After the addition of 10 μm P24 (black rhombuses) or Ap2A (black squares) to control, untreated cells (black triangles), [cAMP]i (A) or [IP3]i (B) levels were determined after 15 min and 30 s, respectively, as described under “Experimental Procedures.” Results are the mean ± S.D. of at least three experiments for each assay.
Similar to what was observed for [cAMP]i, the [IP3]i was also enhanced by 10 μm P24 or Ap2A only in P2Y11-transfected cells, with the highest value reached for both adenylic dinucleotides at 30 s (Fig. 7B).
DISCUSSION
Here we show that platelets produce and release three adenylic dinucleotides, Ap2A and two isomers thereof, characterized by an unusual N-glycosidic bond between one of the adenines and the ribose: C1′-N1 and C1′-N3 in P18 and P24, respectively (Fig. 1) (4). Ap2A, P18, and P24 behave as negative modulators of platelet aggregation induced by the physiological agonists ADP, thrombin, or collagen.
The presence of Ap2A in human platelets was already reported, although the enzyme responsible for its synthesis and the effect of the dinucleotide on platelet aggregation were not investigated (1). Human CD38 has been recently demonstrated to synthesize Ap2A, P18, and P24 (Fig. 1) (4). The fact that synthesis of the adenylic dinucleotides by the platelet lysate is inhibited by nicotinamide (Table 1), a known inhibitor of ADP-ribosyl cyclase activity (36), strongly suggests that platelet CD38 is responsible for synthesis of Ap2A and of its isomers.
P18 and P24 are present in resting platelets at micromolar concentrations, similar to those already reported for Ap2A (1) (Table 2). Platelet activation by thrombin stimulates production of Ap2A, P18, and P24 (Table 2), suggesting the presence of a feedback mechanism, limiting platelet aggregation through generation of the three adenylic dinucleotides. Release of the adenylic dinucleotides from thrombin-activated platelets, at a platelet density similar to that present in human plasma, results in extracellular concentrations of each dinucleotide equal to or higher than their IC50 value upon agonist-induced platelet aggregation (Table 3). These results suggest a role for Ap2A, P18, and P24 as negative endogenous modulators of platelet aggregation. Release of antiaggregant molecules by agonist-stimulated platelets is not unprecedented; platelet activation by thrombin, ADP, or collagen also results in the production of the most potent endogenous inhibitor of platelet activation, adhesion, and aggregation (i.e. NO) (25, 23, 37). Although it was recently reported that subthreshold concentrations of thrombin (<0.02 units/ml) induce release of very low (nanomolar) concentrations of platelet-derived NO, which behaves as a proaggregant (38), there is general agreement that proaggregant agonist concentrations induce release of higher (micromolar) concentrations of NO, which then behaves as a potent endogenous antithrombotic (30).
In nucleated cells, the biological activity is markedly different among Ap2A and its isomers. On human hemopoietic progenitors (CD34+ cells), Ap2A stimulates proliferation, whereas P18 and, particularly, P24 induce apoptosis, with LD50 values on colony growth of 1.0 and 0.18 μm, respectively (4). P24 cytotoxicity, demonstrated on a wide range of different cell types (4), is due to its mitochondrial effects, which include inhibition of complex I of the respiratory chain and dissipation of the proton gradient (ΔΨm); neither P18 nor Ap2A show any effect on respiration or on ΔΨm (39). In contrast to the diverse effects observed on nucleated cells, all three adenylic dinucleotides share the same inhibitory effect on agonist-induced platelet aggregation, with P18 showing the lowest IC50 values (Table 3). Since Ap2A, P18, and P24 all share a common ADP moiety, inhibition of platelet aggregation could be due to binding of these dinucleotides to the purinergic receptors involved in endogenous ADP-induced aggregation (i.e. P2Y1 and P2Y12). However, the following experimental findings rule out the possibility that the adenylic dinucleotides behave as ADP antagonists on P2Y1 and on P2Y12. P24 neither induced an increase of the [IP3]i by itself nor antagonized the increase of the [IP3]i induced by the synthetic purinergic agonist 2MeSADP on P2Y1-transfected and on P2Y12/Gi-transfected HeLa cells (Fig. 4). Moreover, P24 failed to displace [32P]MRS2500, a specific P2Y1 receptor radioligand (22), from the P2Y1 receptor (not shown), ruling out a direct effect of this dinucleotide on P2Y1.
The mechanism through which Ap2A and its isomers inhibit agonist-induced platelet aggregation seems to depend on (i) reduction of the agonist-triggered [Ca2+]i rise (Table 4), which is causally related to platelet aggregation (17, 18); (ii) increased platelet production of NO (Fig. 5); and (iii) increased intraplatelet concentration of cAMP ([cAMP]i) (Fig. 6). The following results indicate that the receptor mediating the antiaggregant effects of Ap2A, P18, and P24 is the purinergic receptor P2Y11: (i) the P2Y11-specific inhibitor NF157 (35) prevents the increase of the [cAMP]i and of NO release induced in platelets by the adenylic dinucleotides (Figs. 5 and 6); (ii) 1321N1 cells transfected with hP2Y11 respond to the adenylic dinucleotides with an increase of the [cAMP]i and of the [IP3]i (Fig. 7), in agreement with the fact that P2Y11 activates both adenylyl cyclase and phospholipase C (10, 33). The increase of the intraplatelet [IP3]i induced by the adenylic dinucleotides is significantly less than that observed with thrombin (Fig. 6B), in agreement with the absence of a significant [Ca2+]i rise and of a proaggregant effect by the adenylic nucleotides per se.
P2Y11 mRNA levels in platelets were reported to be much lower than those of P2Y1, P2Y12, and P2X1, although the level of mRNA depends on both its synthesis and degradation rates and may not linearly correlate with its protein expression (34). Also in human granulocytes, P2Y11 mRNA is present at lower levels compared with mRNA of other purinergic receptors (40). In fact, expression of the P2Y11 protein in granulocytes was found to be even lower than that observed in platelets (41). Nonetheless, ATP, NAD+, and NAADP+ have been shown to induce significant biochemical and functional effects in granulocytes through P2Y11 ligation (41– 43). At present, binding of Ap2A, P18, and P24 to other purinergic and/or adenosine receptors, in addition to P2Y11, cannot be ruled out. Adenosine is known to induce an increase of the [cAMP]i and to exert antiaggregant effects on platelets by binding to A-type receptors (44), and Ap2A has been shown to be an agonist of A1 and A2 adenosine receptors in rat kidney vasculature (45). However, the fact that the P2Y11-specific antagonist NF157 inhibits by 100, 70, and 75% the [cAMP]i increase induced by Ap2A, P18, and P24, respectively, suggests that an effect of these dinucleotides on adenosine receptors may account for ≤20–25% of the [cAMP]i increase and only for P18 and P24.
Both endogenous NO production and an intraplatelet [cAMP]i rise are known to inhibit platelet activation (25, 31, 32). NO increases intraplatelet cGMP, which inhibits cAMP phophodiesterase (46), contributing to the elevation of the [cAMP]i. Moreover, PKA is known to phosphorylate and activate platelet NOS (30). Thus, a positive feedback mechanism maintains elevated levels of [cAMP]i via NO. A high [cAMP]i inhibits IP3-induced Ca2+ release in human platelets and megakaryocytes (47, 48). The fact that Ap2A, P18, and P24 induce an increase of the [cAMP]i could result both in activation of NOS via PKA and in a reduction of the [Ca2+]i in thrombin-stimulated platelets. NO release from platelets incubated with the adenylic dinucleotides occurred only in the presence of thrombin (Fig. 5), at a concentration (0.05 units/ml) that did not induce NO release per se (Fig. 5), as already reported (29, 49). A possible synergism between the pathways triggered by thrombin and by P2Y11 and leading to NOS activation could explain this observation. A similar interpretation of their results was proposed by Radomski et al. (49), who reported that the antiaggregant effect of l-arginine on platelets stimulated with thrombin (0.03 units/ml) became evident only upon the addition of subthreshold concentrations of antiaggregant prostacyclin (a known stimulator of [cAMP]i increase).
A deficiency of bioactive NO is known to be associated with arterial thrombosis both in animal models and in humans (50, 51), and new NO donors with antithrombotic and vasodilating activities are being developed for treatment of arterial thrombosis (52). The discovery of new endogenous negative modulators of platelet aggregation that act through stimulation of platelet NO production could possibly lead to the development of a new family of antithrombotic drugs.
Acknowledgments
We are indebted to Prof. Antonio De Flora for critical review of the manuscript.
This work was supported in part by grants from the Associazione Italiana per la Ricerca sul Cancro; from the Italian Ministry of Education, University, and Scientific Research (MIUR-PRIN 2003, MIUR FIRB RBAUO19A3C, MIUR FIRB RBNE01ERXR and MIUR FIRB RBLA039LSF); and from the University of Genova and Fondazione Cassa di Risparmio di Genova e Imperia. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement”in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: Ap2A, diadenosine 5′,5‴-P1,P2-diphosphate; Ap2G, adenosine guanosine diphosphate; Gp2G, diguanosine diphosphate; NO, nitric oxide; WP, washed platelet; 2MeSADP, 2-methylthioadenosine-5′diphosphate; Ade, adenine; HPLC, high pressure liquid chromatography; PCA, perchloric acid; IP3, inositol 1,4,5-trisphosphate.
References
- 1.Jankowski, J., Hagemann, J., Tepel, M., van Der Giet, M., Stephan, N., Henning, L., Gouni-Berthold, H., Sachinidis, A., Zidek, W., and Schluter, H. (2001) J. Biol. Chem. 276 8904–8909 [DOI] [PubMed] [Google Scholar]
- 2.Schluter, H., Tepel, M., and Zidek, W. (1996) J. Auton. Pharmacol. 16 357–362 [DOI] [PubMed] [Google Scholar]
- 3.Luo, J., Jankowski, J., Knobloch, M., van der Giet, M., Gardanis, K., Russ, T., Vahlensieck, U., Neumann, J., Schmitz, W., Tepel, M., Deng, M. C., Zidek, W., and Schulter, H. (1999) FASEB J. 13 695–705 [DOI] [PubMed] [Google Scholar]
- 4.Basile, G., Taglialatela-Scafati, O., Damonte, G., Armirotti, A., Buzzone, S., Guida, L., Franco, L., Usai, C., Fattorusso, E., De Flora, A., and Zocchi, E. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 14941–14942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramaschi, G., Torti, M., Festetics, E. T., Sinigaglia, F., Malavasi, F., and Balduini, C. (1996) Blood 87 2308–2313 [PubMed] [Google Scholar]
- 6.Ohlmann, P., Leray, C., Ravanat, C., Hollia, A., Cassel, D., Cazenava, J. P., and Gachet, C. (1998) Biochem. J. 331 431– 436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Torti, M., Festetics, E. T., Bretoni, A., Sinigaglia, F., and Balduini, C. (1998) FEBS Lett. 428 200–204 [DOI] [PubMed] [Google Scholar]
- 8.Leoncini, G., Maresca, M., Buzzi, E., Piana, A., and Armani, U. (1990) Eur. J. Hematol. 44 116–120 [DOI] [PubMed] [Google Scholar]
- 9.Coward, P., Chan, S. D., Wada, H. G., Humphries, G. M., and Conklin, B. R. (1999) Anal. Biochem. 270 242–248 [DOI] [PubMed] [Google Scholar]
- 10.Qi, A. D., Kennedy, C., Harden, T., and Nicholas, R. A. (2001) Br. J. Pharmacol. 132 318–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rossi, L., Brandi, G., Schiavano, G. F., Balestra, E., Millo, E., Scarfi, S., Damonte, G., Gasparini, A., Magnani, M., Perno, C. F., Benatti, U., and De Flora, A. (1998) AIDS Res. Hum. Retroviruses 14 435– 444 [DOI] [PubMed] [Google Scholar]
- 12.Born, G. V. R. (1962) Nature 194 927–929 [DOI] [PubMed] [Google Scholar]
- 13.Nakahata, N., and Harden, T. K. (1987) Biochem. J. 241 337–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Green, L. C., Wagner, D. A., Glogowski, J., Skipper, P. L., Wishnok, J. S., and Tannembaum, S. R. (1982) Anal. Biochem. 126 131–138 [DOI] [PubMed] [Google Scholar]
- 15.Granger, D. L., Taintor, R. R., Boockvar, S. S., and Hibbs, J. B., Jr. (1996) Methods Enzymol. 268 142–151 [DOI] [PubMed] [Google Scholar]
- 16.Graeff, R., and Lee, H. C. (2002) Biochem. J. 367 163–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sage, S. O., MacKenzie, A. B., Jenner, S., and Mahaut-Smith, M. P. (1997) Prostaglandins Leukot. Essent. Fatty Acids 57 435– 438 [DOI] [PubMed] [Google Scholar]
- 18.Rao, G. H. (1993) Indian J. Physiol. Pharmacol. 37 263–275 [PubMed] [Google Scholar]
- 19.Gachet, C., and Hechler, B. (2005) Semin. Thromb. Hemost. 31 162–167 [DOI] [PubMed] [Google Scholar]
- 20.Hechler, B., Cattaneo, M., and Gachet, C. (2005) Semin. Thromb. Hemost. 31 150–161 [DOI] [PubMed] [Google Scholar]
- 21.Nylander, S., Mattsson, C., Ramstrom, S., and Lindhal, T. L. (2003) Thromb. Res. 111 65– 67 [DOI] [PubMed] [Google Scholar]
- 22.Houston, D., Ohno, M., Nicholas, R. A., Jacobson, K. A., and Harden, T. K. (2006) Br. J. Pharmacol. 147 459–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Riddell, D. R., and Owen, J. S. (1999) Vitam. Horm. 57 25– 48 [DOI] [PubMed] [Google Scholar]
- 24.Radomski, M. W., and Moncada, S. (1993) Adv. Exp. Med. Biol. 344 251–264 [DOI] [PubMed] [Google Scholar]
- 25.Radomski, M. W., Palmer, R. M., and Moncada, S. (1990) Proc. Natl. Acad. Sci. U. S. A. 87 5193–5197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bohme, E., Jung, R., and Mechler, I. (1974) Methods Enzymol. 38 199–202 [DOI] [PubMed] [Google Scholar]
- 27.Muruganandam, A., and Mutus, B. (1994) Bioch. Biophys. Acta 1200 1– 6 [DOI] [PubMed] [Google Scholar]
- 28.Leoncini, G., Pascale, R., and Signorello, M. G. (2002) Biochem. Pharmacol. 64 277–283 [DOI] [PubMed] [Google Scholar]
- 29.Malinski, T., Radomski, M. W., Taha, Z., and Moncada, S. (1993) Biochem. Biophys. Res. Commun. 194 960–965 [DOI] [PubMed] [Google Scholar]
- 30.Gkaliagkousi, E., Ritter, J., and Ferro, A. (2007) Circ. Res. 101 654–662 [DOI] [PubMed] [Google Scholar]
- 31.Salzman, E. W., and Weisenberger, H. (1972) Adv. Cyclic Nucleotide Res. 1 231–247 [PubMed] [Google Scholar]
- 32.Cole, B., Robinson, G. A., and Hartmann, R. C. (1971) Ann. N. Y. Acad. Sci. 185 477– 487 [DOI] [PubMed] [Google Scholar]
- 33.Communi, D., Govaerts, C., Parmentier, M., and Boeynaems, J. M. (1997) J. Biol. Chem. 272 31969–31973 [DOI] [PubMed] [Google Scholar]
- 34.Wang, L., Ostberg, O., Wihlborg, A. K., Brogren, H., Jern, S., and Erlinge, D. (2003) J. Thromb. Haemost. 1 330–336 [DOI] [PubMed] [Google Scholar]
- 35.Ullmann, H., Meis, S., Hongwiset, D., Marzian, C., Wiese, M., Nickel, P., Communi, D., Boeynaems, J. M., Wolf, C., Hausmann, R., Schmalzing, G., and Kassack, M. U. (2005) J. Med. Chem. 48 7040–7048 [DOI] [PubMed] [Google Scholar]
- 36.Lee, H. C. (2002) Cyclic ADP-ribose and NAADP: Structures, Metabolism, and Function, Kluwer Academic Publishers, Norwell, MA
- 37.de Graaf, J. C., Banga, J. D., Moncada, S., Palmer, R. M., de Groot, P. G., and Sixma, J. J. (1992) Circulation 85 2284–2290 [DOI] [PubMed] [Google Scholar]
- 38.Marjanovic, J. A., Li, Z., Stojanovic, A., and Du, X. (2005) J. Biol. Chem. 280 37430–37438 [DOI] [PubMed] [Google Scholar]
- 39.Bruzzone, S., Dodoni, G., Kaludercic, N., Basile, G., Millo, E., De Flora, A., Di Lisa, L., and Zocchi, E. (2007) J. Biol. Chem. 282 5045–5052 [DOI] [PubMed] [Google Scholar]
- 40.Chen, Y., Shukla, A., Mamiki, S., Insel, P. A., and Junger, W. G. (2004) J. Leukocyte Biol. 76 245–253 [DOI] [PubMed] [Google Scholar]
- 41.Vaughan, K. R., Stokes, L., Prince, L. R., Meis, S., Kassack, M. U., Bingle, C. B., Sabroe, I., Surprenant, A., and Whyte, M. K. B. (2007) J. Immunol. 179 8544–8553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moreschi, I., Bruzzone, S., Nicholas, R. A., Fruscione, F., Sturla, L., Benvenuto, F., Usai, C., Meis, S., Kassack, M. U., Zocchi, E., and De Flora, A. (2006) J. Biol. Chem. 281 31419–31429 [DOI] [PubMed] [Google Scholar]
- 43.Moreschi, I., Bruzzone, S., Bodrato, N., Usai, C., Guida, L., Nicholas, R. A., Kassack, M. U., Zocchi, E., and De Flora, A. (2007) Cell Calcium 43 344–355 [DOI] [PubMed] [Google Scholar]
- 44.Anfossi, G., Russo, I., Massucco, P., Mattiello, L., Cavalot, F., Balbo, A., and Trovati, M. (2002) Thromb. Res. 105 71–78 [DOI] [PubMed] [Google Scholar]
- 45.van der Giet, M., Khattab, M., Börgel, J., Schlüter, H., and Zidek, W. (1997) Br. J. Pharmacol. 120 1453–1460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Haslam, R. J., Dickinson, N. T., and Jang, E. K. (1999) Thromb. Haemost. 82 412– 423 [PubMed] [Google Scholar]
- 47.Tohmatsu, T., Nishida, A., Nagao, S., Nakashima, S., and Nozawa, Y. (1989) Biochim. Biophys. Acta 1013 190–193 [DOI] [PubMed] [Google Scholar]
- 48.Tertyshnikova, S., and Fein, A. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 1613–1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Radomski, M. W., Palmer, R. M. J., and Moncada, S. (1990) Br. J. Pharmacol. 101 325–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Loscalzo, J. (2001) Circ. Res. 88 756–762 [DOI] [PubMed] [Google Scholar]
- 51.Voetsch, B., Jin, R. C., and Loscalzo, J. (2004) Histochem. Cell Biol. 122 353–367 [DOI] [PubMed] [Google Scholar]
- 52.Keeble, J. E., and Moore, P. K. (2002) Br. J. Pharmacol. 137 295–310 [DOI] [PMC free article] [PubMed] [Google Scholar]